
Glutathione-Responsive Tannic Acid-Assisted FRET Nanomedicine for Cancer Therapy


 ,
, 
 and
and 
Abstract
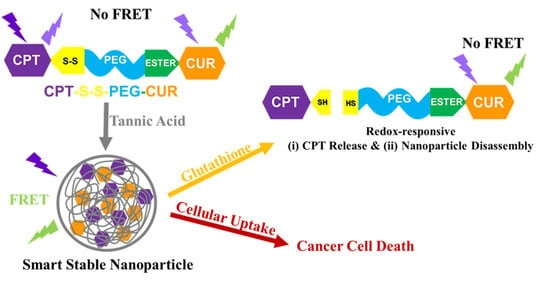
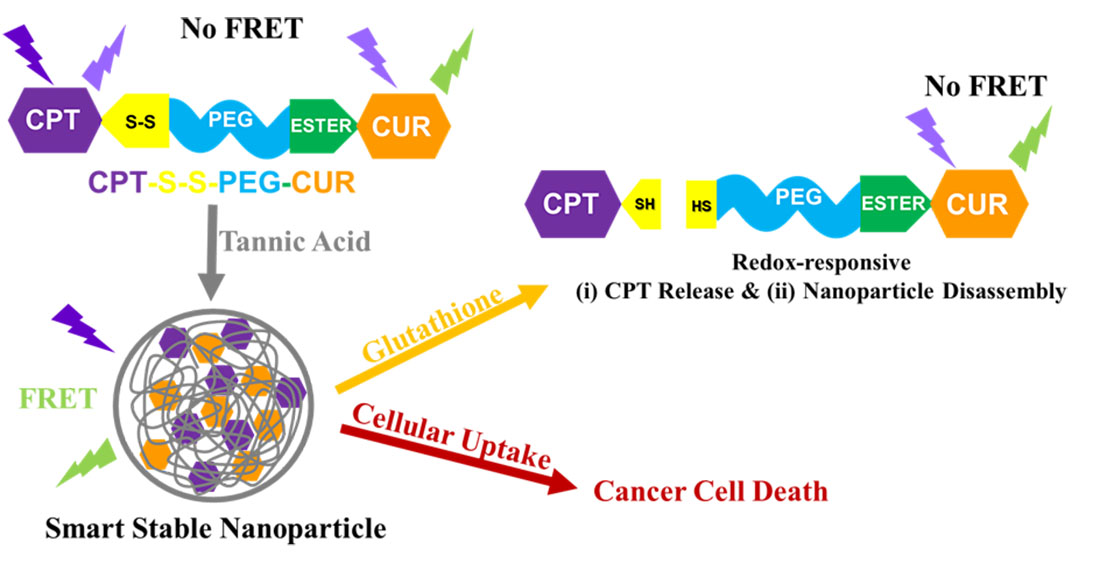
1. Introduction
2. Materials and Methods
2.1. Materials
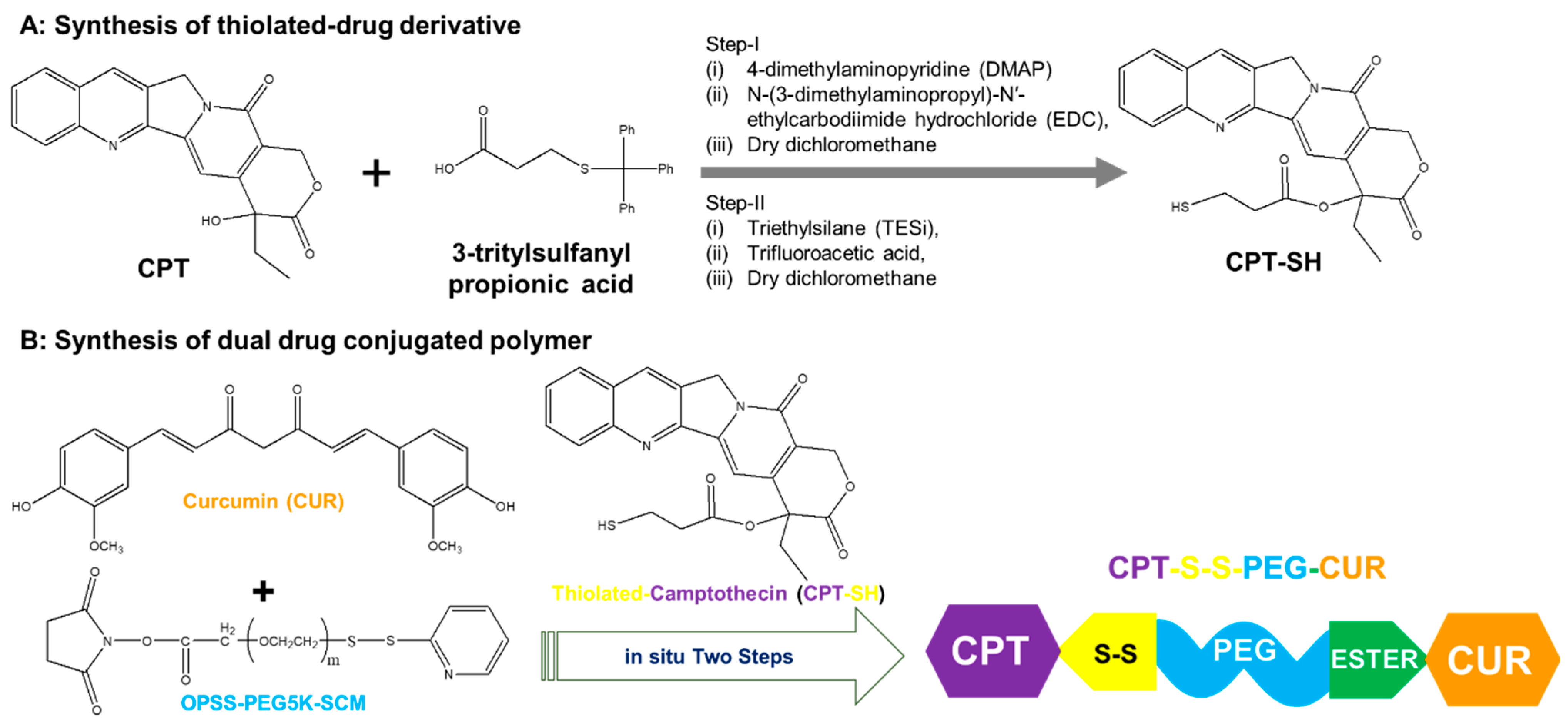
2.2. Synthesis of Thiolated Camptothecin
2.3. Synthesis of Amphiphilic Dual Drug Conjugated Polymer
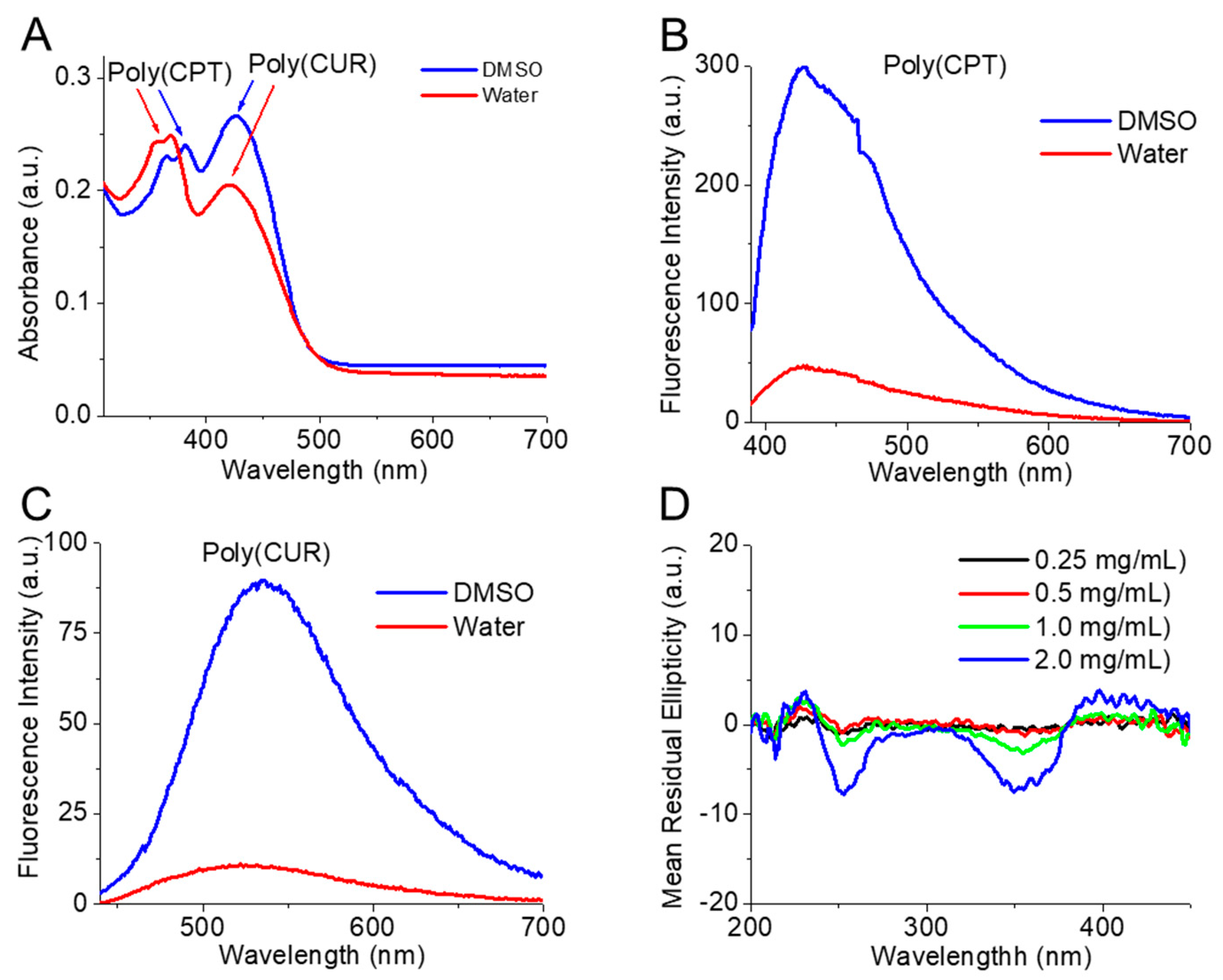
2.4. Absorbance and Fluorescence of the Conjugated and Free Drugs
2.5. Quantification of Conjugated Drugs
2.6. Hydrodynamic Size and Zeta Potential
2.7. Circular Dichroism (CD) Spectroscopy
2.8. Transmission Electron Microscopy Imaging
2.9. Redox-Sensitive CPT Release and Change in FRET Signal
- (i)
- No GSH: 75 μL of polymer (2 mg/mL) with 75 μL TA solution,
- (ii)
- 10 μM GSH: 75 μL of polymer (2 mg/mL) with 60 μL TA solution and 15 μL glutathione solution (100 μM)
- (iii)
- 10 mM GSH: 75 μL of polymer (2 mg/mL) with 60 μL TA solution and 15 μL glutathione solution (100 mM),
- (iv)
- 50 mM GSH: 75 μL of polymer (2 mg/mL) with 75 μL glutathione solution (100 mM).
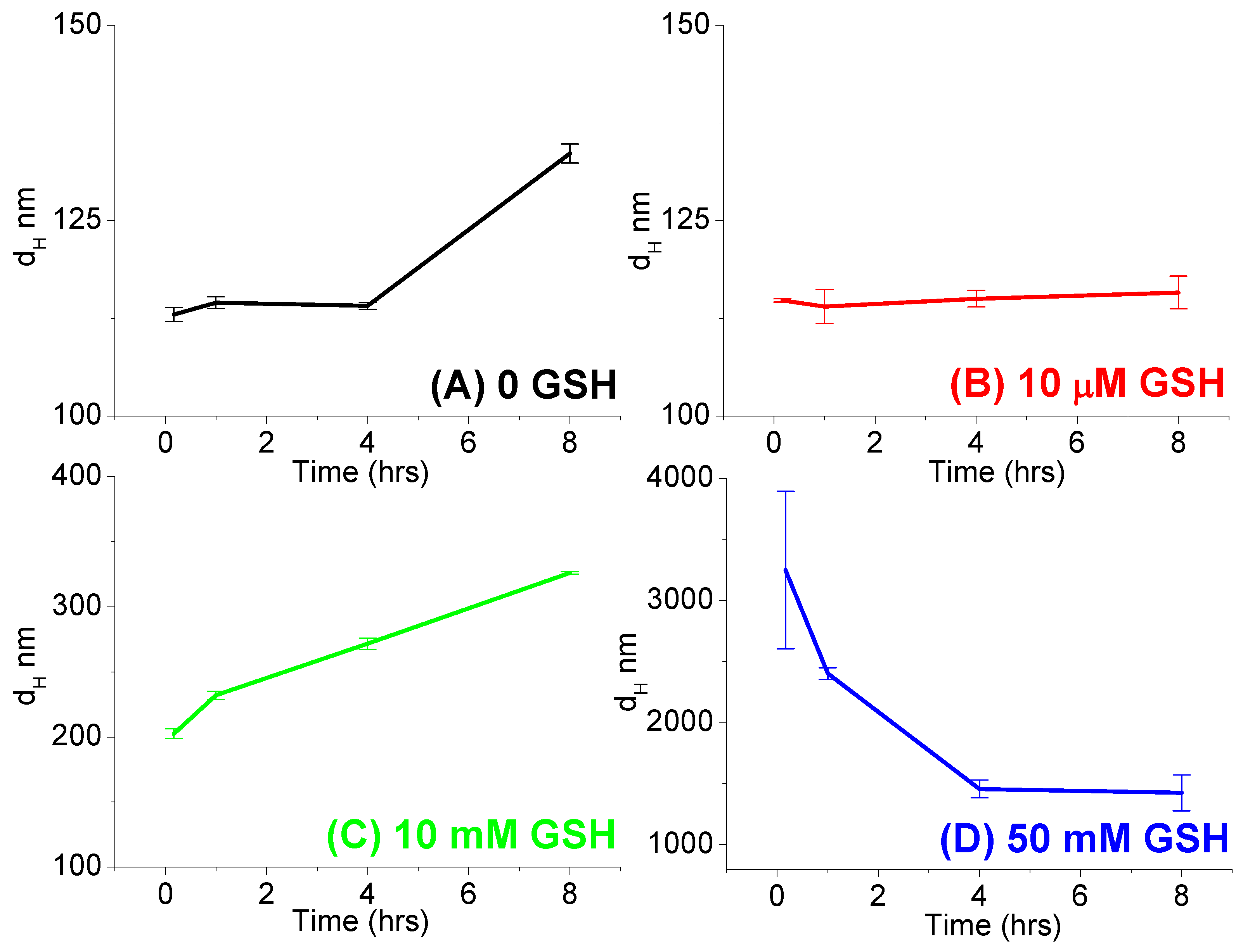
2.10. Redox-Responsive Change in the Hydrodynamic Size of Self-Assemblies
- (i)
- No GSH: 600 μL of polymer (2 mg/mL) with 600 μL TA solution,
- (ii)
- 10 μM GSH: 600 μL of polymer (2 mg/mL) with 480 μL TA solution and 120 μL glutathione solution (100 μM),
- (iii)
- 10 mM GSH: 600 μL of polymer (2 mg/mL) with 480 μL TA solution and 120 μL glutathione solution (100 mM),
- (iv)
- 50 mM GSH: 600 μL of polymer (2 mg/mL) with 600 μL glutathione solution (100 mM).
2.11. In Vitro Cell Culture
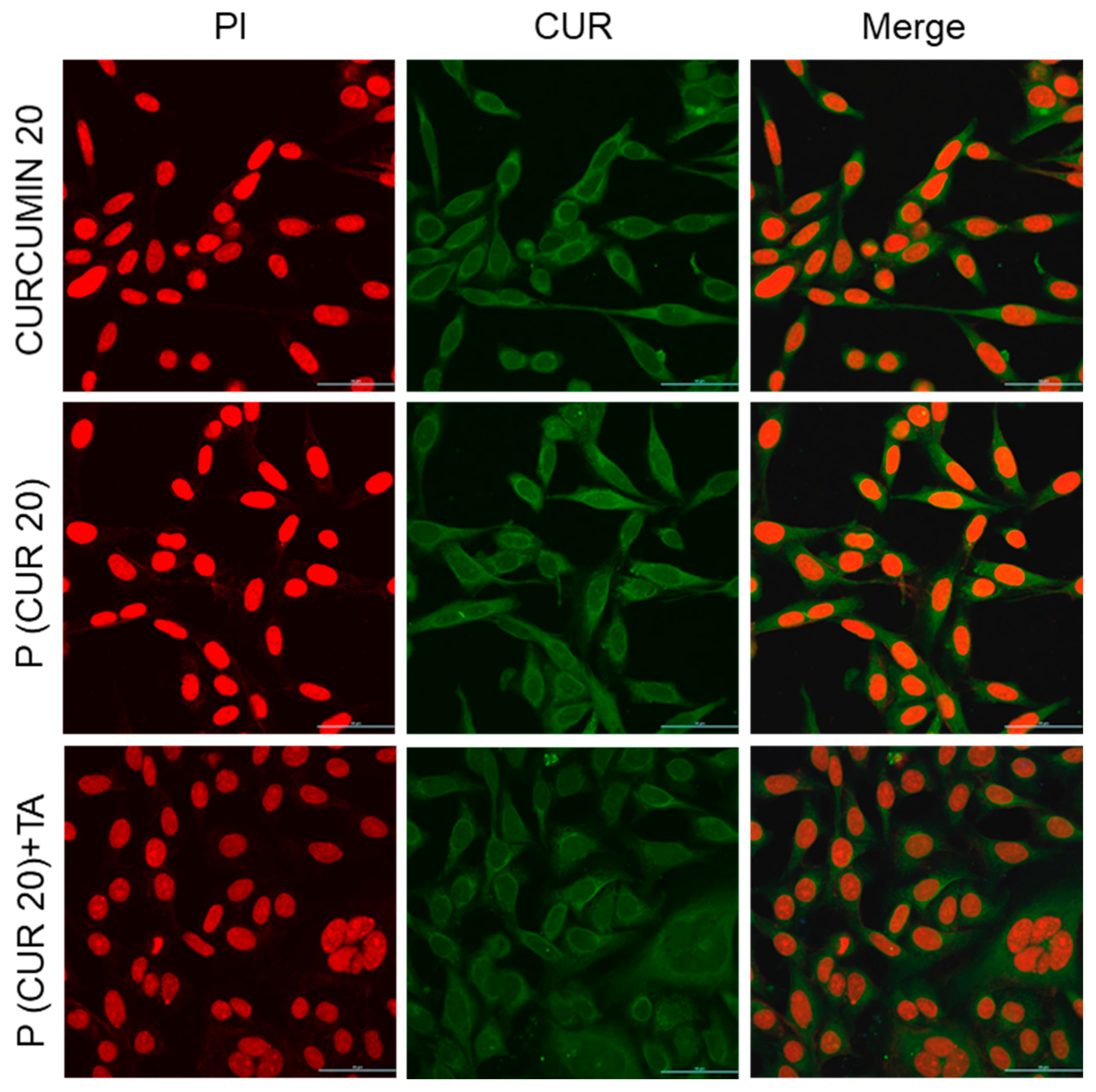
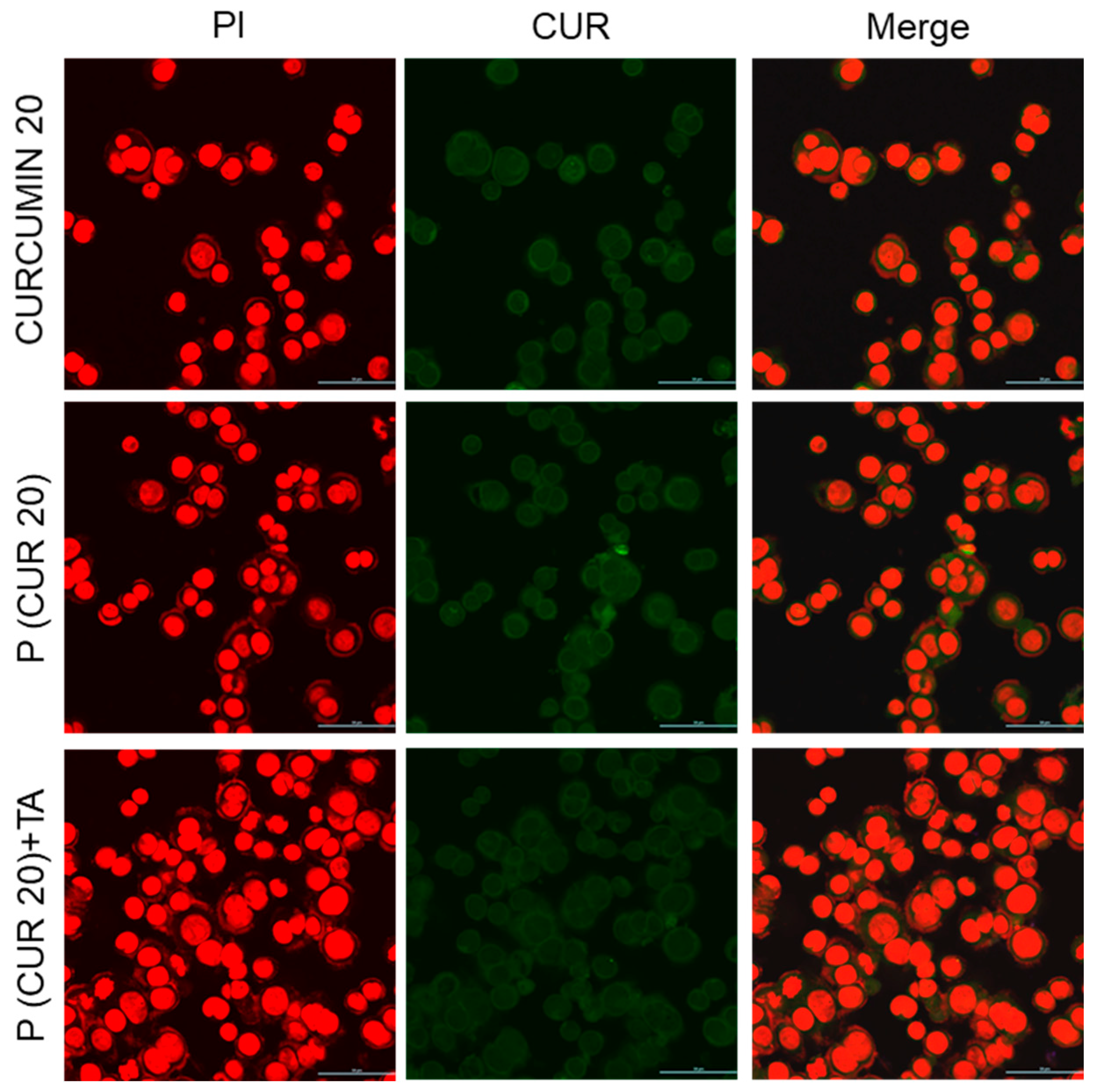
2.12. Confocal Microscopy
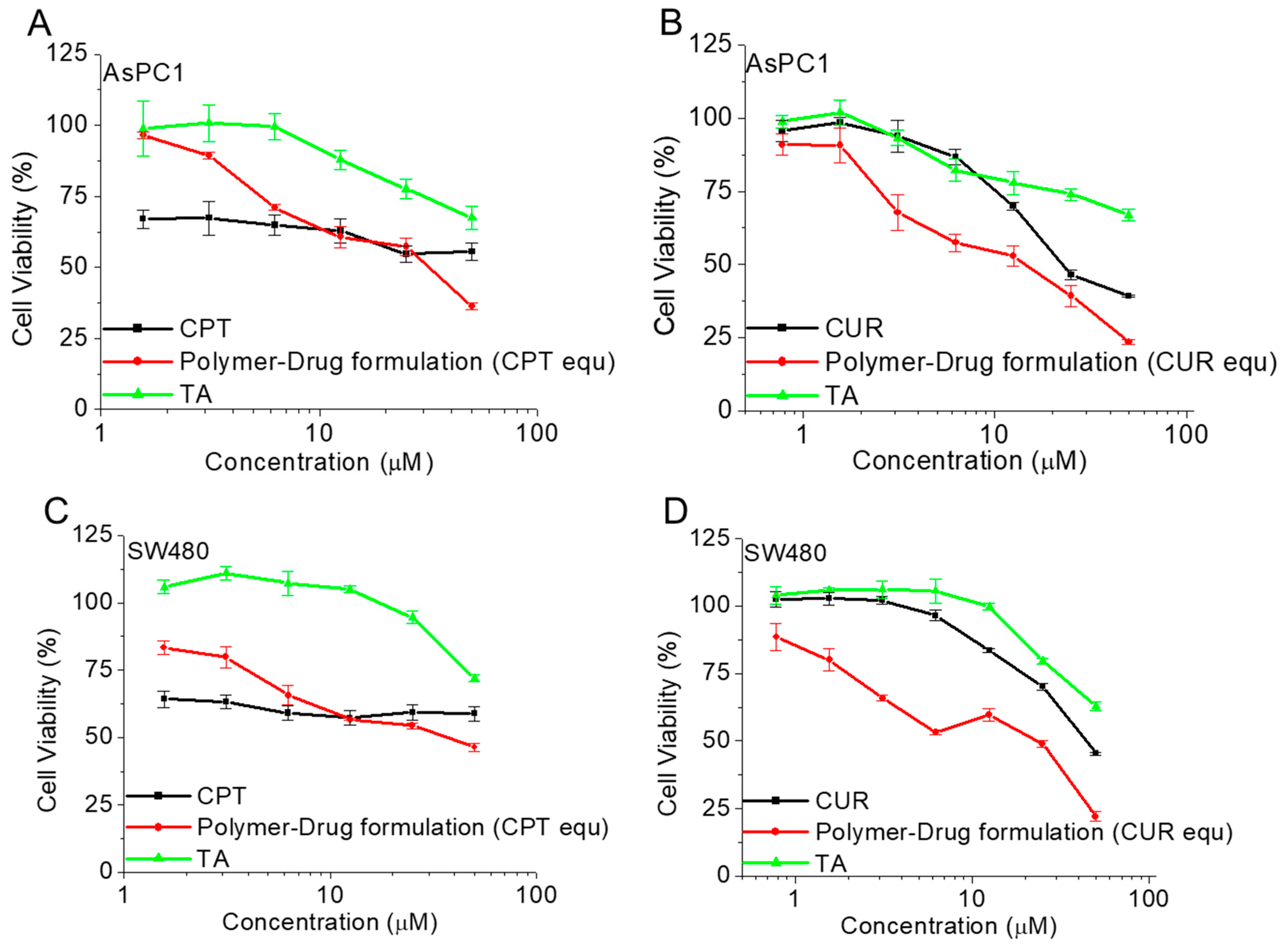
2.13. Cell Proliferation Assay
2.14. Bioinformatics Analyses
3. Results
3.1. Molecular Characterization
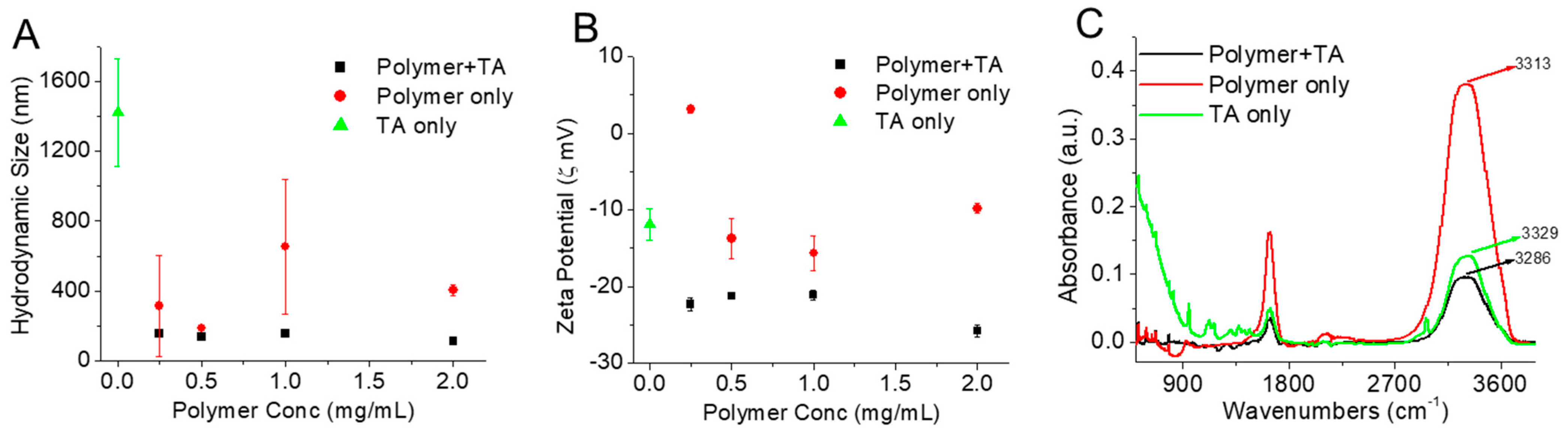
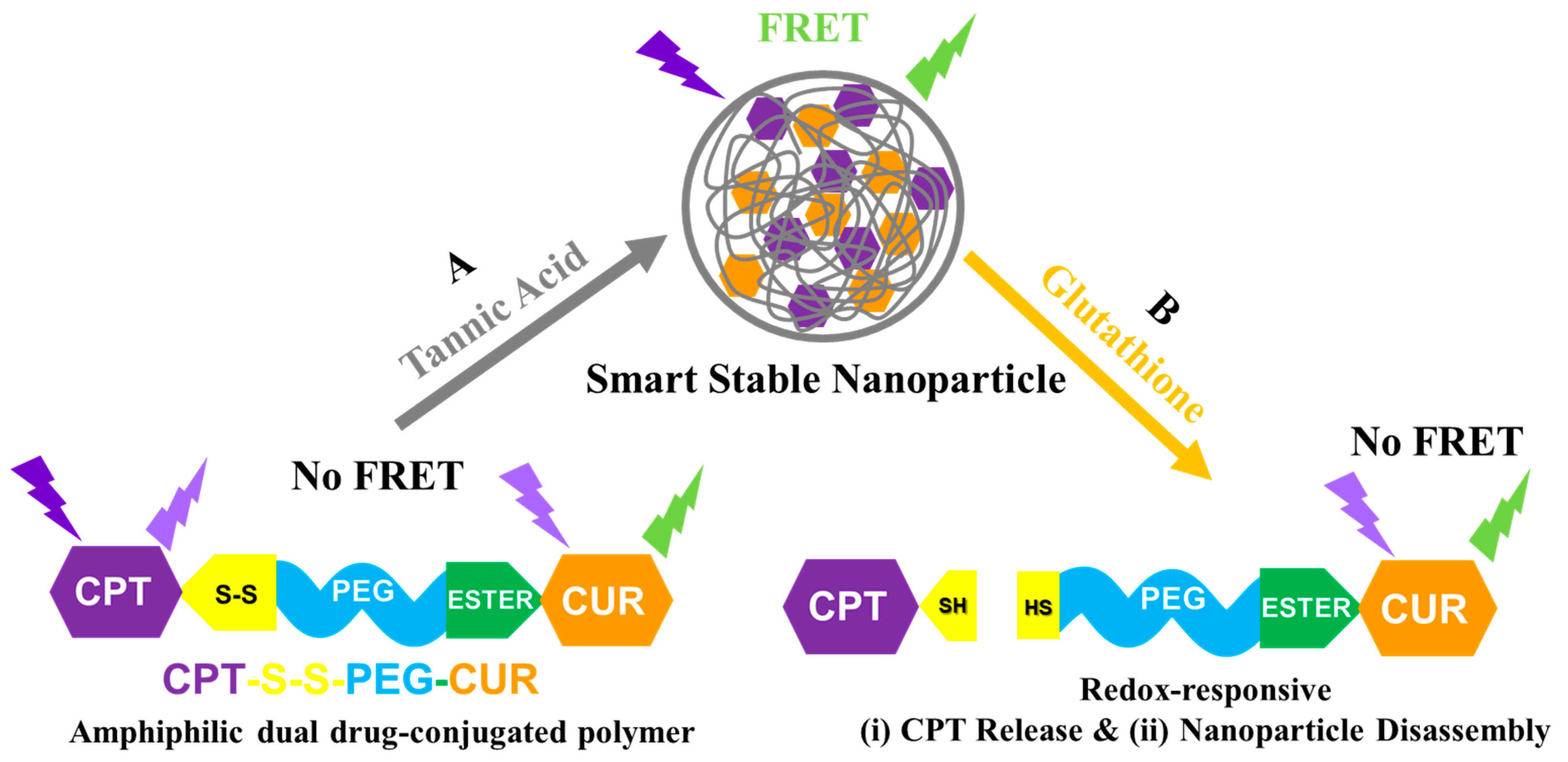
3.2. Self-Assembly Behavior of CPT-S-S-PEG-CUR in Presence and Absence of TA
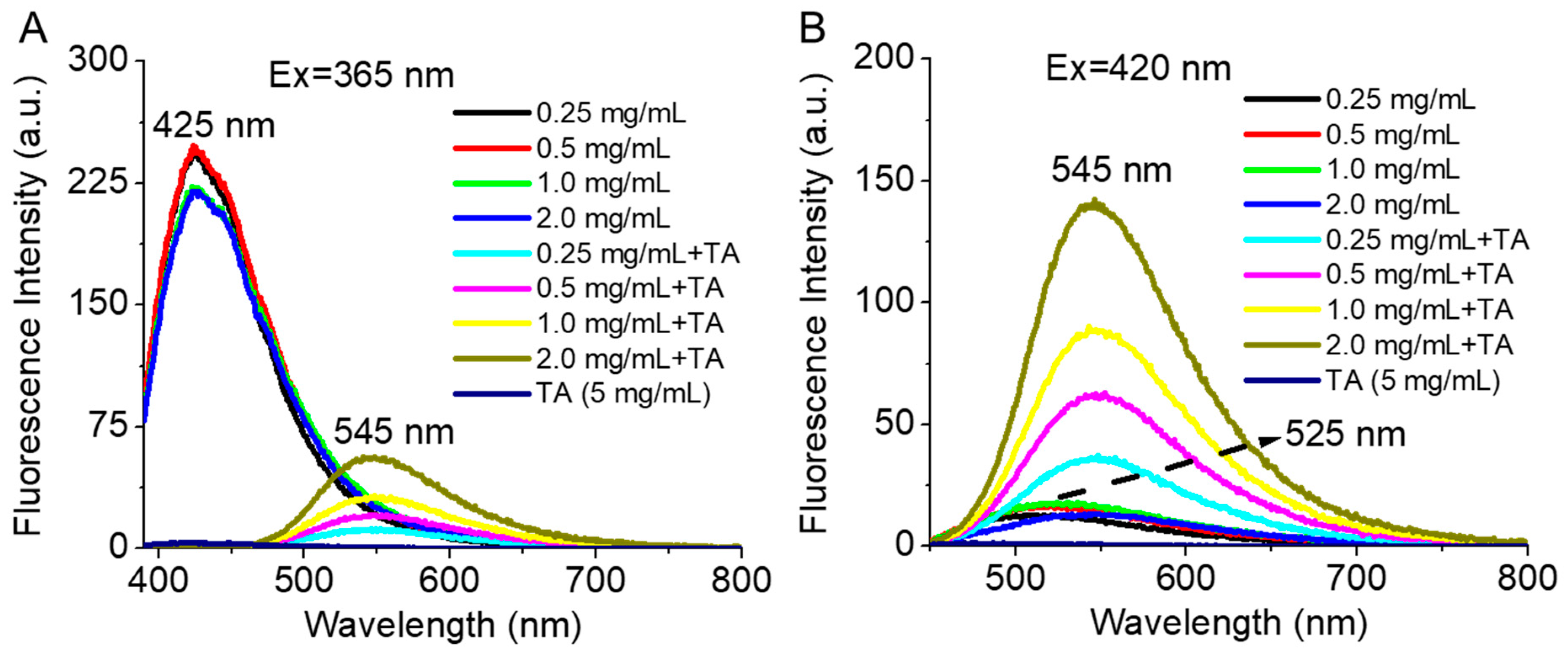
3.3. Occurrence of FRET in Presence of TA
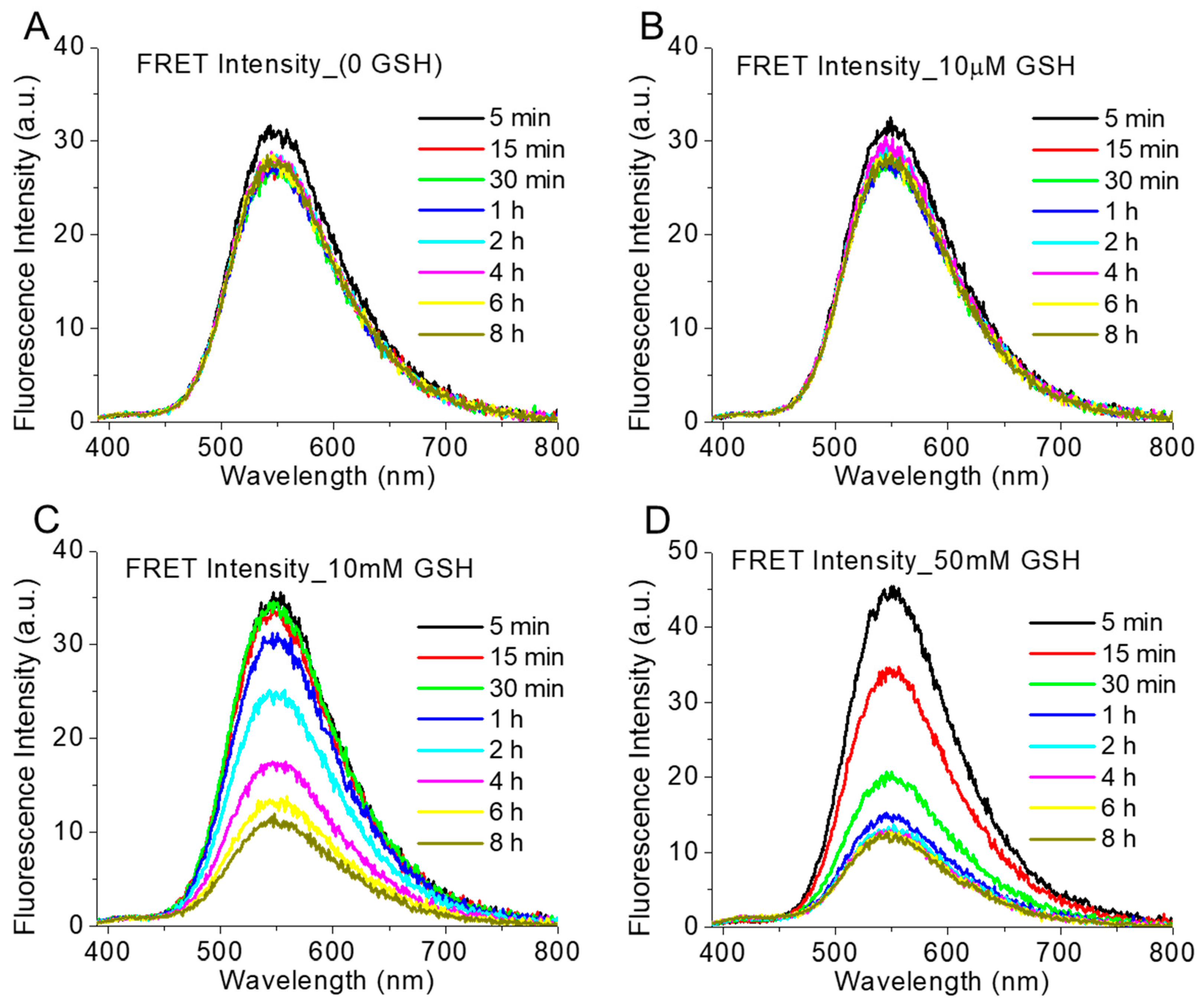
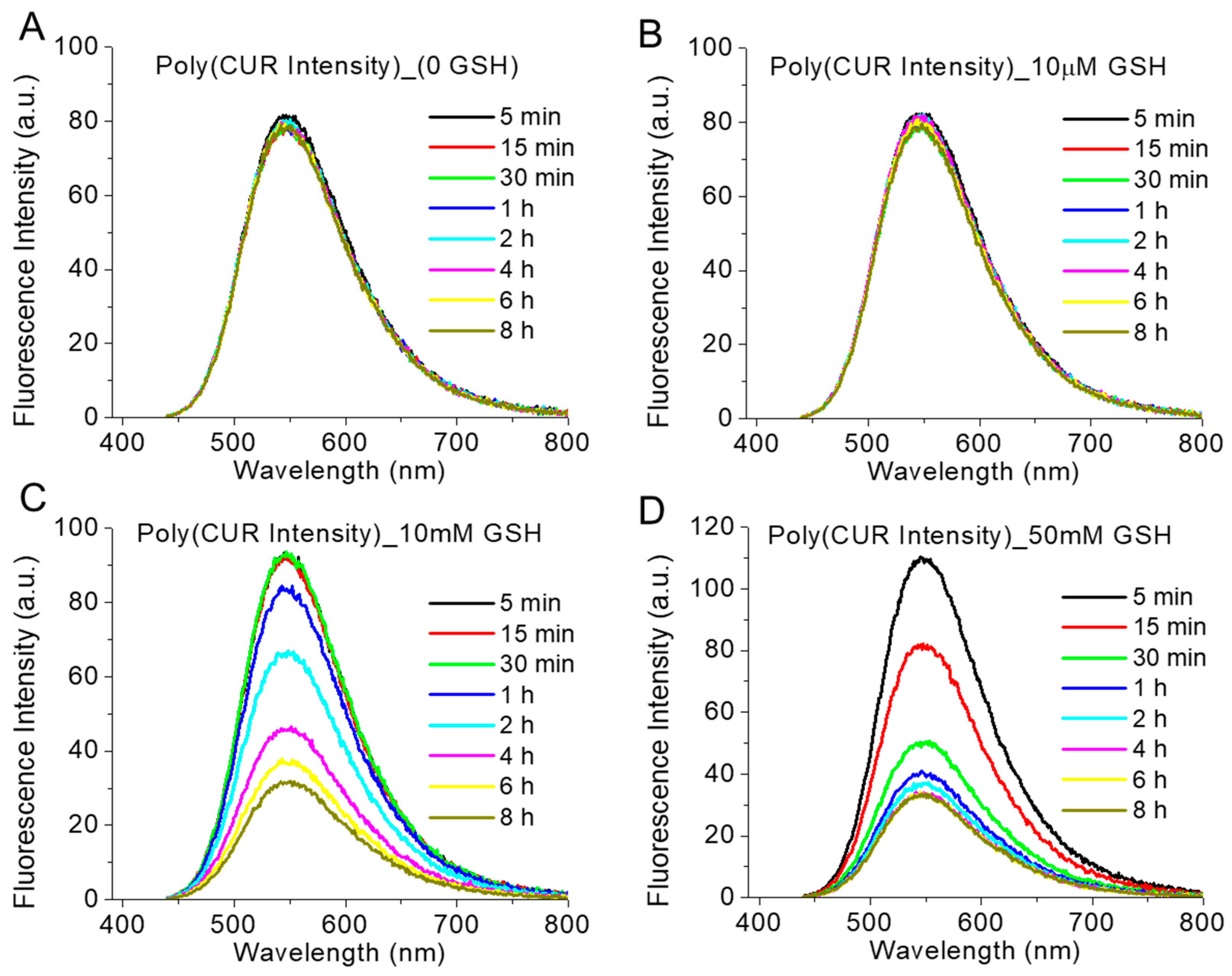
3.4. Reduction-Responsive Drug Release and Disintegration of Nanoparticles
3.5. Cellular Uptake of the Polymer CPT-S-S-PEG-CUR
3.6. Antiproliferative Activity of Polymer CPT-S-S-PEG-CUR
3.7. Bioinformatics Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, W.; Yung, B.; Huang, P.; Chen, X. Nanotechnology for Multimodal Synergistic Cancer Therapy. Chem. Rev. 2017, 117, 13566–13638. [Google Scholar] [CrossRef]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef]
- Zhao, G.; Sun, Y.; Dong, X. Zwitterionic Polymer Micelles with Dual Conjugation of Doxorubicin and Curcumin: Synergistically Enhanced Efficacy against Multidrug-Resistant Tumor Cells. Langmuir 2020, 36, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Huang, P.; Wang, Y.; Su, Y.; Zhou, L.; Zhu, X.; Yan, D. Synergistic Combination Chemotherapy of Camptothecin and Floxuridine through Self-Assembly of Amphiphilic Drug-Drug Conjugate. Bioconjug. Chem. 2015, 26, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Wang, D.; Su, Y.; Huang, W.; Zhou, Y.; Cui, D.; Zhu, X.; Yan, D. Combination of small molecule prodrug and nanodrug delivery: Amphiphilic drug-drug conjugate for cancer therapy. J. Am. Chem. Soc. 2014, 136, 11748–11756. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, J.; Ma, J.; Song, L.; Lin, H.; Tang, B.; Chen, D.; Su, G.; Ye, S.; Zhu, X.; et al. Methotrexate-Camptothecin Prodrug Nanoassemblies as a Versatile Nanoplatform for Biomodal Imaging-Guided Self-Active Targeted and Synergistic Chemotherapy. ACS Appl. Mater. Interfaces 2017, 9, 34650–34665. [Google Scholar] [CrossRef]
- Xiao, B.; Si, X.; Han, M.K.; Viennois, E.; Zhang, M.; Merlin, D. Co-delivery of camptothecin and curcumin by cationic polymeric nanoparticles for synergistic colon cancer combination chemotherapy. J. Mater. Chem. B 2015, 3, 7724–7733. [Google Scholar] [CrossRef]
- Li, Y.; Thambi, T.; Lee, D.S. Co-Delivery of Drugs and Genes Using Polymeric Nanoparticles for Synergistic Cancer Therapeutic Effects. Adv. Healthc. Mater 2018, 7. [Google Scholar] [CrossRef]
- Su, Z.; Dong, S.; Zhao, S.C.; Liu, K.; Tan, Y.; Jiang, X.; Assaraf, Y.G.; Qin, B.; Chen, Z.S.; Zou, C. Novel nanomedicines to overcome cancer multidrug resistance. Drug Resist. Updat. 2021, 58, 100777. [Google Scholar] [CrossRef]
- Laskar, P.; Dey, J.; Banik, P.; Mandal, M.; Ghosh, S.K. In Vitro Drug and Gene Delivery Using Random Cationic Copolymers Forming Stable and pH-Sensitive Polymersomes. Macromol. Biosci. 2017, 17, 1600324. [Google Scholar] [CrossRef]
- Chauhan, D.S.; Dhasmana, A.; Laskar, P.; Prasad, R.; Jain, N.K.; Srivastava, R.; Jaggi, M.; Chauhan, S.C.; Yallapu, M.M. Nanotechnology synergized immunoengineering for cancer. Eur. J. Pharm. Biopharm. 2021, 163, 72–101. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Somani, S.; Campbell, S.J.; Mullin, M.; Keating, P.; Tate, R.J.; Irving, C.; Leung, H.Y.; Dufes, C. Camptothecin-based dendrimersomes for gene delivery and redox-responsive drug delivery to cancer cells. Nanoscale 2019, 11, 20058–20071. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, A.G.; Zhang, P.; Lin, Y.A.; Lock, L.L.; Cui, H. Supramolecular nanostructures formed by anticancer drug assembly. J. Am. Chem. Soc. 2013, 135, 2907–2910. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; He, B.; Tao, J.; He, Y.; Deng, H.; Wang, X.; Zheng, Y. Application of Forster Resonance Energy Transfer (FRET) technique to elucidate intracellular and In Vivo biofate of nanomedicines. Adv. Drug. Deliv. Rev. 2019, 143, 177–205. [Google Scholar] [CrossRef]
- Nhien, P.Q.; Chou, W.L.; Cuc, T.T.K.; Khang, T.M.; Wu, C.H.; Thirumalaivasan, N.; Hue, B.T.B.; Wu, J.I.; Wu, S.P.; Lin, H.C. Multi-Stimuli Responsive FRET Processes of Bifluorophoric AIEgens in an Amphiphilic Copolymer and Its Application to Cyanide Detection in Aqueous Media. ACS Appl. Mater. Interfaces 2020, 12, 10959–10972. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; De Paoli, V.; Rosenzweig, N.; Rosenzweig, Z. Synthesis and application of quantum dots FRET-based protease sensors. J. Am. Chem. Soc. 2006, 128, 10378–10379. [Google Scholar] [CrossRef]
- Dong, Z.; Bi, Y.; Cui, H.; Wang, Y.; Wang, C.; Li, Y.; Jin, H.; Wang, C. AIE Supramolecular Assembly with FRET Effect for Visualizing Drug Delivery. ACS Appl. Mater. Interfaces 2019, 11, 23840–23847. [Google Scholar] [CrossRef]
- Taemaitree, F.; Fortuni, B.; Koseki, Y.; Fron, E.; Rocha, S.; Hofkens, J.; Uji, I.H.; Inose, T.; Kasai, H. FRET-based intracellular investigation of nanoprodrugs toward highly efficient anticancer drug delivery. Nanoscale 2020, 12, 16710–16715. [Google Scholar] [CrossRef]
- Zhang, X.; Xiong, J.; Wang, K.; Yu, H.; Sun, B.; Ye, H.; Zhao, Z.; Wang, N.; Wang, Y.; Zhang, S.; et al. Erythrocyte membrane-camouflaged carrier-free nanoassembly of FRET photosensitizer pairs with high therapeutic efficiency and high security for programmed cancer synergistic phototherapy. Bioact. Mater. 2021, 6, 2291–2302. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, J.; Kang, T.; Chen, Y.; Liu, Y.; Huang, Y.; Luo, Y.; Huang, M.; Gou, M. Co-assembling FRET nanomedicine with self-indicating drug release. Chem. Commun. 2018, 54, 11618–11621. [Google Scholar] [CrossRef]
- Mi, P. Stimuli-responsive nanocarriers for drug delivery, tumor imaging, therapy and theranostics. Theranostics 2020, 10, 4557–4588. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Dey, J.; Ghosh, S.K. Evaluation of zwitterionic polymersomes spontaneously formed by pH-sensitive and biocompatible PEG based random copolymers as drug delivery systems. Colloids Surf. B Biointerfaces 2016, 139, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Dey, J.; Ghosh, S.K. Spontaneously formed redox- and pH-sensitive polymersomes by mPEG based cytocompatible random copolymers. J. Colloid Interface Sci. 2017, 501, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Dufes, C. Emergence of cationic polyamine dendrimersomes: Design, stimuli sensitivity and potential biomedical applications. Nanoscale Adv. 2021, 3, 6007–6026. [Google Scholar] [CrossRef]
- Laskar, P.; Somani, S.; Altwaijry, N.; Mullin, M.; Bowering, D.; Warzecha, M.; Keating, P.; Tate, R.J.; Leung, H.Y.; Dufes, C. Redox-sensitive, cholesterol-bearing PEGylated poly(propylene imine)-based dendrimersomes for drug and gene delivery to cancer cells. Nanoscale 2018, 10, 22830–22847. [Google Scholar] [CrossRef]
- Laskar, P.; Somani, S.; Mullin, M.; Tate, R.J.; Warzecha, M.; Bowering, D.; Keating, P.; Irving, C.; Leung, H.Y.; Dufes, C. Octadecyl chain-bearing PEGylated poly(propyleneimine)-based dendrimersomes: Physicochemical studies, redox-responsiveness, DNA condensation, cytotoxicity and gene delivery to cancer cells. Biomater. Sci. 2021, 9, 1431–1448. [Google Scholar] [CrossRef]
- Laskar, P.; Samanta, S.; Ghosh, S.K.; Dey, J. In vitro evaluation of pH-sensitive cholesterol-containing stable polymeric micelles for delivery of camptothecin. J. Colloid Interface Sci. 2014, 430, 305–314. [Google Scholar] [CrossRef]
- Batra, H.; Pawar, S.; Bahl, D. Curcumin in combination with anti-cancer drugs: A nanomedicine review. Pharm. Res. 2019, 139, 91–105. [Google Scholar] [CrossRef]
- Khan, S.; Setua, S.; Kumari, S.; Dan, N.; Massey, A.; Hafeez, B.B.; Yallapu, M.M.; Stiles, Z.E.; Alabkaa, A.; Yue, J.; et al. Superparamagnetic iron oxide nanoparticles of curcumin enhance gemcitabine therapeutic response in pancreatic cancer. Biomaterials 2019, 208, 83–97. [Google Scholar] [CrossRef]
- Su, P.; Yang, Y.; Wang, G.; Chen, X.; Ju, Y. Curcumin attenuates resistance to irinotecan via induction of apoptosis of cancer stem cells in chemoresistant colon cancer cells. Int. J. Oncol. 2018, 53, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E. Curcumin Combination Chemotherapy: The Implication and Efficacy in Cancer. Molecules 2019, 24, 2527. [Google Scholar] [CrossRef]
- Fleming, A.B.; Haverstick, K.; Saltzman, W.M. In vitro cytotoxicity and in vivo distribution after direct delivery of PEG-camptothecin conjugates to the rat brain. Bioconjug. Chem. 2004, 15, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Omar, R.; Bardoogo, Y.L.; Corem-Salkmon, E.; Mizrahi, B. Amphiphilic star PEG-Camptothecin conjugates for intracellular targeting. J. Control. Release 2017, 257, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Lin, L.; Hao, K.; Wang, D.; Liu, F.; Sun, P.; Yu, H.; Tang, Z.; Chen, M.; Tian, H.; et al. Helix Self-Assembly Behavior of Amino Acid-Modified Camptothecin Prodrugs and Its Antitumor Effect. Acs Appl. Mater. Interfaces 2020, 12, 7466–7476. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Jaggi, M.; Chauhan, S.C. beta-Cyclodextrin-curcumin self-assembly enhances curcumin delivery in prostate cancer cells. Colloids Surf. B Biointerfaces 2010, 79, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Othman, S.F.; Curtis, E.T.; Bauer, N.A.; Chauhan, N.; Kumar, D.; Jaggi, M.; Chauhan, S.C. Curcumin-loaded magnetic nanoparticles for breast cancer therapeutics and imaging applications. Int. J. Nanomed. 2012, 7, 1761–1779. [Google Scholar] [CrossRef]
- Zaman, M.S.; Chauhan, N.; Yallapu, M.M.; Gara, R.K.; Maher, D.M.; Kumari, S.; Sikander, M.; Khan, S.; Zafar, N.; Jaggi, M.; et al. Curcumin Nanoformulation for Cervical Cancer Treatment. Sci. Rep. 2016, 6, 20051. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, M.H.; Cheng, A.L.; Lin, L.I.; Ho, Y.S.; Hsieh, C.Y.; Lin, J.K. Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Pendri, A.; Conover, C.D.; Lee, C.; Choe, Y.H.; Gilbert, C.; Martinez, A.; Xia, J.; Wu, D.; Hsue, M. Camptothecin-20-PEG ester transport forms: The effect of spacer groups on antitumor activity. Bioorg. Med. Chem. 1998, 6, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, A.V.; Hiller, A.; Syed, S.; Yin, M.; Lu, X.M.; Fischman, A.J.; Papisov, M.I. Synthesis of a macromolecular camptothecin conjugate with dual phase drug release. Mol. Pharm. 2004, 1, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sreenivasan, K. Conjugating curcumin to water soluble polymer stabilized gold nanoparticles via pH responsive succinate linker. J. Mater. Chem. B 2015, 3, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Bordenave, N.; Ferruzzi, M.G.; Safavy, A.; Kim, K.H. Modification of curcumin with polyethylene glycol enhances the delivery of curcumin in preadipocytes and its antiadipogenic property. J. Agric. Food Chem. 2011, 59, 1012–1019. [Google Scholar] [CrossRef]
- Zhang, G.; Li, X.; Liao, Q.; Liu, Y.; Xi, K.; Huang, W.; Jia, X. Water-dispersible PEG-curcumin/amine-functionalized covalent organic framework nanocomposites as smart carriers for in vivo drug delivery. Nat. Commun. 2018, 9, 2785. [Google Scholar] [CrossRef]
- Somani, S.; Laskar, P.; Altwaijry, N.; Kewcharoenvong, P.; Irving, C.; Robb, G.; Pickard, B.S.; Dufes, C. PEGylation of polypropylenimine dendrimers: Effects on cytotoxicity, DNA condensation, gene delivery and expression in cancer cells. Sci. Rep. 2018, 8, 9410. [Google Scholar] [CrossRef]
- Laskar, P.; Saha, B.; Ghosh, S.K.; Dey, J. PEG based random copolymer micelles as drug carriers: The effect of hydrophobe content on drug solubilization and cytotoxicity. Rsc Adv. 2015, 5, 16265–16276. [Google Scholar] [CrossRef]
- Meewan, J.; Somani, S.; Laskar, P.; Irving, C.; Mullin, M.; Woods, S.; Roberts, C.W.; Alzahrani, A.R.; Ferro, V.A.; McGill, S.; et al. Limited impact of the protein Corona on the cellular uptake of PEGylated zein micelles by melanoma cancer cells. Pharmaceutics 2022, 14, 439. [Google Scholar] [CrossRef]
- Chowdhury, P.; Nagesh, P.K.B.; Hatami, E.; Wagh, S.; Dan, N.; Tripathi, M.K.; Khan, S.; Hafeez, B.B.; Meibohm, B.; Chauhan, S.C.; et al. Tannic acid-inspired paclitaxel nanoparticles for enhanced anticancer effects in breast cancer cells. J. Colloid Interface Sci. 2019, 535, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.K.B.; Hatami, E.; Chowdhury, P.; Kashyap, V.K.; Khan, S.; Hafeez, B.B.; Chauhan, S.C.; Jaggi, M.; Yallapu, M.M. Tannic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Prostate Cancer. Cancers 2018, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Bang, J.B.; Na, Y.G.; Lee, J.Y.; Cho, C.W.; Baek, J.S.; Lee, H.K. Development and Evaluation of Tannic Acid-Coated Nanosuspension for Enhancing Oral Bioavailability of Curcumin. Pharmaceutics 2021, 13, 1460. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.K.B.; Chowdhury, P.; Hatami, E.; Kumari, S.; Kashyap, V.K.; Tripathi, M.K.; Wagh, S.; Meibohm, B.; Chauhan, S.C.; Jaggi, M.; et al. Cross-Linked Polyphenol-Based Drug Nano-Self-Assemblies Engineered to Blockade Prostate Cancer Senescence. ACS Appl. Mater. Interfaces 2019, 11, 38537–38554. [Google Scholar] [CrossRef] [PubMed]
- Al Nakeeb, N.; Nischang, I.; Schmidt, B. Tannic Acid-Mediated Aggregate Stabilization of Poly(N-vinylpyrrolidone)-b-poly(oligo (ethylene glycol) methyl ether methacrylate) Double Hydrophilic Block Copolymers. Nanomaterials 2019, 9, 662. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fan, H.; Li, W.; Bai, G.; Li, X.; Zhao, N.; Xu, J.; Zhou, F.; Guo, X.; Dai, B.; et al. Competitive self-assembly driven as a route to control the morphology of poly(tannic acid) assemblies. Nanoscale 2019, 11, 4751–4758. [Google Scholar] [CrossRef]
- Smith, R.A.; Walker, R.C.; Levit, S.L.; Tang, C. Single-Step Self-Assembly and Physical Crosslinking of PEGylated Chitosan Nanoparticles by Tannic Acid. Polymers 2019, 11, 749. [Google Scholar] [CrossRef]
- Liu, X.; O, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef]
- Dhasmana, A.; Uniyal, S.; Anukriti; Kashyap, V.K.; Somvanshi, P.; Gupta, M.; Bhardwaj, U.; Jaggi, M.; Yallapu, M.M.; Haque, S.; et al. Topological and system-level protein interaction network (PIN) analyses to deduce molecular mechanism of curcumin. Sci. Rep. 2020, 10, 12045. [Google Scholar] [CrossRef]
- Dhasmana, A.; Uniyal, S.; Somvanshi, P.; Bhardwaj, U.; Gupta, M.; Haque, S.; Lohani, M.; Kumar, D.; Ruokolainen, J.; Kesari, K.K. Investigation of Precise Molecular Mechanistic Action of Tobacco-Associated Carcinogen ‘NNK’ Induced Carcinogenesis: A System Biology Approach. Genes 2019, 10, 564. [Google Scholar]
- Chen, Y.N.; Peng, L.; Liu, T.; Wang, Y.; Shi, S.; Wang, H. Poly(vinyl alcohol)-Tannic Acid Hydrogels with Excellent Mechanical Properties and Shape Memory Behaviors. Acs Appl. Mater. Interfaces 2016, 8, 27199–27206. [Google Scholar] [CrossRef]
- Souza, T.G.; Ciminelli, V.S.; Mohallem, N.D.S. A comparison of TEM and DLS methods to characterize size distribution of ceramic nanoparticles. In Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2016; Volume 733, p. 012039. [Google Scholar]
- Sikander, M.; Malik, S.; Khan, S.; Kumari, S.; Chauhan, N.; Khan, P.; Halaweish, F.T.; Chauhan, B.; Yallapu, M.M.; Jaggi, M.; et al. Novel mechanistic insight into the anticancer activity of cucurbitacin D against pancreatic cancer (Cuc D attenuates pancreatic cancer). Cells 2019, 9, 103. [Google Scholar] [CrossRef]
- Gupta, B.K.; Maher, D.M.; Ebeling, M.C.; Stephenson, P.D.; Puumala, S.E.; Koch, M.R.; Aburatani, H.; Jaggi, M.; Chauhan, S.C. Functions and regulation of MUC13 mucin in colon cancer cells. J. Gastroenterol. 2014, 49, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Zhao, H.; Mehlig, M.; Malaby, J.; Kraft, P.; Longley, C.; Greenberger, L.M.; Horak, I.D. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin. Cancer Res. 2008, 14, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.; Lin, L.; Ball, S.; Bekaii-Saab, T.; Fuchs, J.; Li, P.K.; Li, C.; Lin, J. Curcumin analogues exhibit enhanced growth suppressive activity in human pancreatic cancer cells. Anti-Cancer Drugs 2009, 20, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Wen, H.Y.; Dong, H.Q.; Xue, W.M.; Pauletti, G.M.; Cai, X.J.; Xia, W.J.; Shi, D.; Li, Y.Y. Self-assembling nanomicelles of a novel camptothecin prodrug engineered with a redox-responsive release mechanism. Chem. Commun. 2011, 47, 8647–8649. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Cheetham, A.G.; Cui, H. Building Nanostructures with Drugs. Nano Today 2016, 11, 13–30. [Google Scholar] [CrossRef]
- Zhou, Y.; Fan, S.; Feng, L.; Huang, X.; Chen, X. Manipulating intratumoral fenton chemistry for enhanced chemodynamic and chemodynamic-synergized multimodal therapy. Adv. Mater. 2021, 33, 2104223. [Google Scholar] [CrossRef]
- Feng, L.; Zhao, R.; Yang, L.; Liu, B.; Dong, S.; Qian, C.; Liu, J.; Zhao, Y. Tumor-Specific NIR-Activatable Nanoreactor for Self-Enhanced Multimodal Imaging and Cancer Phototherapy. ACS Nano 2023, 17, 1622–1637. [Google Scholar] [CrossRef]
- Yang, L.; Shi, R.; Zhao, R.; Zhu, Y.; Liu, B.; Gai, S.; Feng, L. Near-infrared upconversion mesoporous tin dioxide theranostic nanocapsules for synergetic cancer chemophototherapy. ACS Appl. Mater. Interfaces 2022, 14, 2650–2662. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
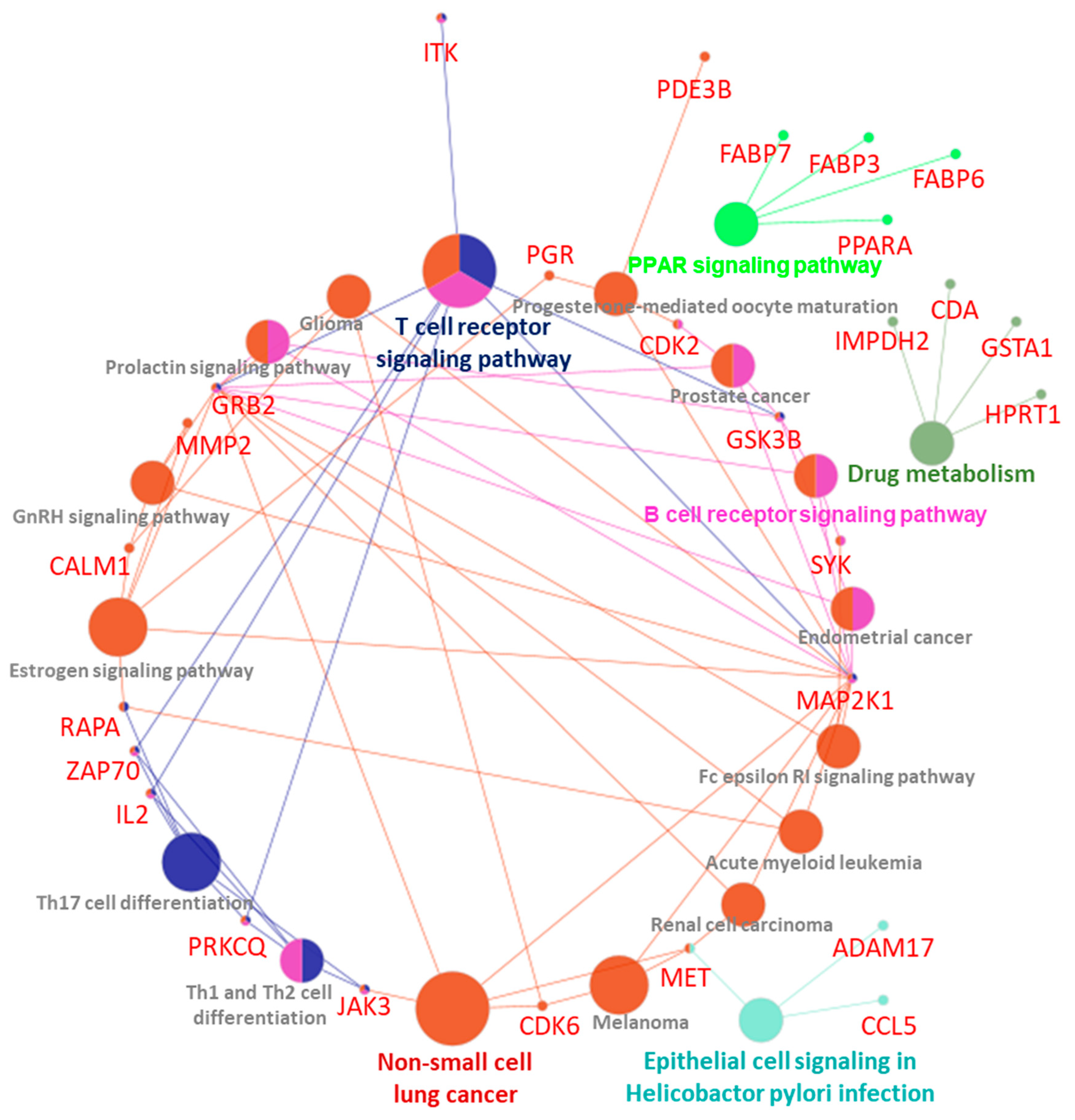
| GOTerm | Ontology Source | Nr. Genes | Associated Genes Found |
|---|---|---|---|
| Epithelial cell signaling in Helicobacter pylori infection | KEGG_10.03.2021 | 3 | [ADAM17, CCL5, MET] |
| Endometrial cancer | KEGG_10.03.2021 | 3 | [GRB2, GSK3B, MAP2K1] |
| Fc epsilon RI signaling pathway | KEGG_10.03.2021 | 3 | [GRB2, MAP2K1, SYK] |
| Prolactin signaling pathway | KEGG_10.03.2021 | 3 | [GRB2, GSK3B, MAP2K1] |
| Renal cell carcinoma | KEGG_10.03.2021 | 3 | [GRB2, MAP2K1, MET] |
| Melanoma | KEGG_10.03.2021 | 3 | [CDK6, MAP2K1, MET] |
| Acute myeloid leukemia | KEGG_10.03.2021 | 3 | [GRB2, MAP2K1, RARA] |
| Drug metabolism | KEGG_10.03.2021 | 4 | [CDA, GSTA1, HPRT1, IMPDH2] |
| PPAR signaling pathway | KEGG_10.03.2021 | 4 | [FABP3, FABP6, FABP7, PPARA] |
| Th1 and Th2 cell differentiation | KEGG_10.03.2021 | 4 | [IL2, JAK3, PRKCQ, ZAP70] |
| B cell receptor signaling pathway | KEGG_10.03.2021 | 4 | [GRB2, GSK3B, MAP2K1, SYK] |
| Prostate cancer | KEGG_10.03.2021 | 4 | [CDK2, GRB2, GSK3B, MAP2K1] |
| GnRH signaling pathway | KEGG_10.03.2021 | 4 | [CALM1, GRB2, MAP2K1, MMP2] |
| Progesterone-mediated oocyte maturation | KEGG_10.03.2021 | 4 | [CDK2, MAP2K1, PDE3B, PGR] |
| Glioma | KEGG_10.03.2021 | 4 | [CALM1, CDK6, GRB2, MAP2K1] |
| Th17 cell differentiation | KEGG_10.03.2021 | 5 | [IL2, JAK3, PRKCQ, RARA, ZAP70] |
| Non-small cell lung cancer | KEGG_10.03.2021 | 5 | [CDK6, GRB2, JAK3, MAP2K1, MET] |
| Estrogen signaling pathway | KEGG_10.03.2021 | 6 | [CALM1, GRB2, MAP2K1, MMP2, PGR, RARA] |
| T cell receptor signaling pathway | KEGG_10.03.2021 | 7 | [GRB2, GSK3B, IL2, ITK, MAP2K1, PRKCQ, ZAP70] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laskar, P.; Dhasmana, A.; Kotnala, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Glutathione-Responsive Tannic Acid-Assisted FRET Nanomedicine for Cancer Therapy. Pharmaceutics 2023, 15, 1326. https://doi.org/10.3390/pharmaceutics15051326
Laskar P, Dhasmana A, Kotnala S, Jaggi M, Yallapu MM, Chauhan SC. Glutathione-Responsive Tannic Acid-Assisted FRET Nanomedicine for Cancer Therapy. Pharmaceutics. 2023; 15(5):1326. https://doi.org/10.3390/pharmaceutics15051326
Chicago/Turabian StyleLaskar, Partha, Anupam Dhasmana, Sudhir Kotnala, Meena Jaggi, Murali M. Yallapu, and Subhash C. Chauhan. 2023. "Glutathione-Responsive Tannic Acid-Assisted FRET Nanomedicine for Cancer Therapy" Pharmaceutics 15, no. 5: 1326. https://doi.org/10.3390/pharmaceutics15051326
APA StyleLaskar, P., Dhasmana, A., Kotnala, S., Jaggi, M., Yallapu, M. M., & Chauhan, S. C. (2023). Glutathione-Responsive Tannic Acid-Assisted FRET Nanomedicine for Cancer Therapy. Pharmaceutics, 15(5), 1326. https://doi.org/10.3390/pharmaceutics15051326