
Novel Chlorin e6-Curcumin Derivatives as a Potential Photosensitizer: Synthesis, Characterization, and Anticancer Activity
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthetic Chemistry
2.2. Analysis of Photophysical Properties and Singlet Oxygen Generation Capability
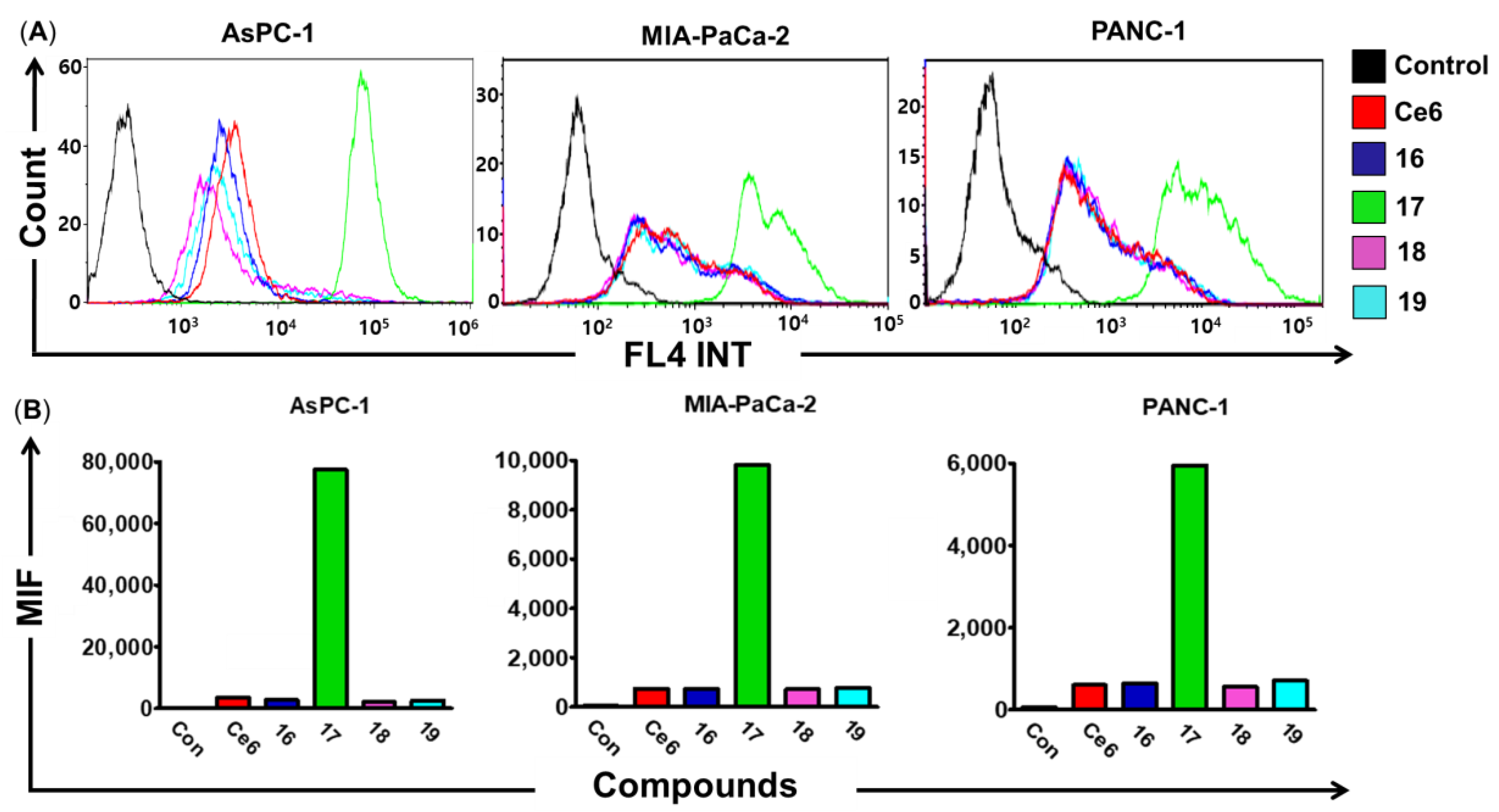
2.3. Cellular Uptake Assay
2.4. Cytotoxicity of Ce6-Curcumin Derivatives In Vitro
2.5. Compound 17 Mediates the Pancreatic Cancer Cell Death by Intrinsic Apoptosis
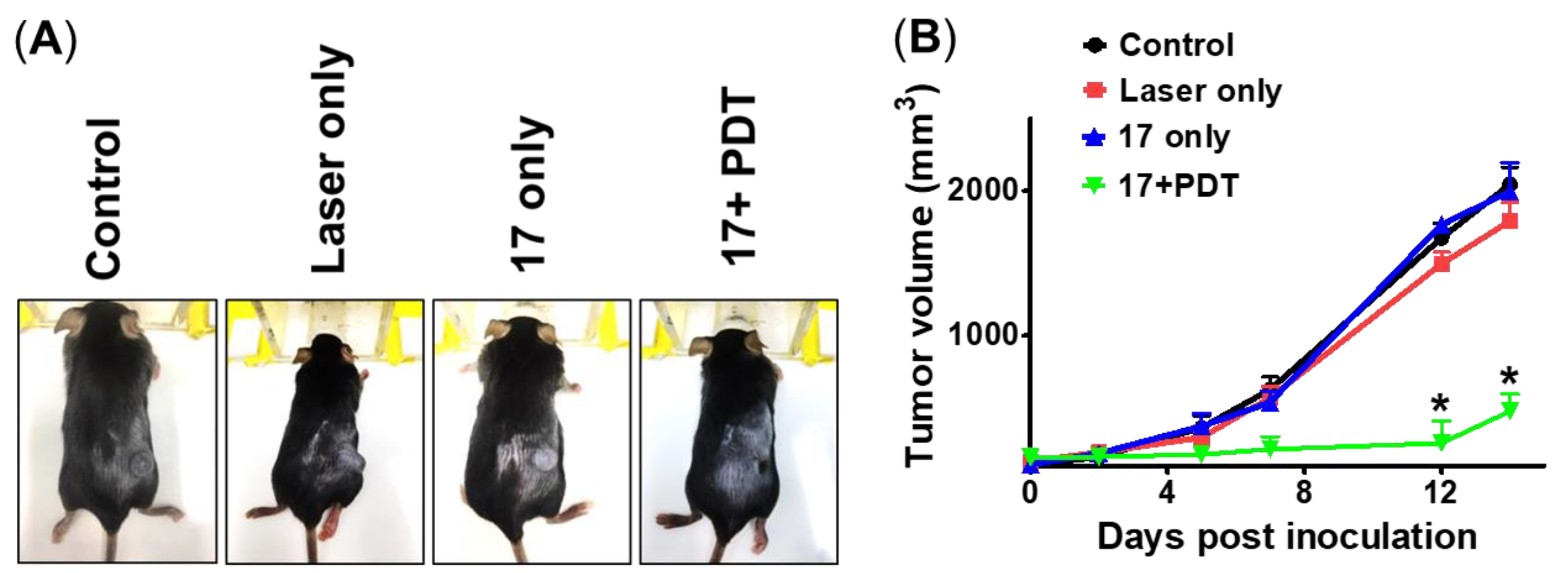
2.6. Compound 17-PDT Efficacy on Xenograft Mouse Model Using B16F10 Melanoma Cell
2.7. Effect of 17 in Mice Behavior, Growth, and Its Pharmacokinetics
2.8. Structure—Activity Relationship (SAR) Study and Molecular Docking Analysis
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. Synthesis of Ce6-DiPEG-COOtBu (5)
3.2.2. Synthesis of Ce6-DiPEG-COOH (6)
3.2.3. Synthesis of Ethyl 4-(4-((1E,3Z,6E)-3-hydroxy-7-(4-hydroxy-3-methoxyphenyl)-5-oxohepta-1,3,6-trien-1-yl)-2-methoxyphenoxy)butanoate (8a)
3.2.4. Synthesis of 4-(4-((1E,3Z,6E)-3-Hydroxy-7-(4-hydroxy-3-methoxyphenyl)-5-oxohepta-1,3,6-trien-1-yl)-2-methoxyphenoxy)butanoic Acid (8b)
3.2.5. Synthesis of Tert-butyl (3-(4-((1E,3Z,6E)-3-hydroxy-7-(4-hydroxy-3-methoxyphenyl)-5-oxohepta-1,3,6-trien-1-yl)-2-methoxyphenoxy)propyl)carbamate (9a)
3.2.6. Synthesis of (1E,4Z,6E)-7-(4-(3-Aminopropoxy)-3-methoxyphenyl)-5-hydroxy-1-(4-hydroxy-3-meth-oxyphenyl)hepta-1,4,6-trien-3-one (9b)
3.2.7. Synthesis of (1E,6E)-1,7-Bis(3,4-dimethoxyphenyl)hepta-1,6-diene-3,5-dione (11)
3.2.8. Synthesis of Ethyl 4-(((1E,3Z,6E)-1,7-bis(3,4-dimethoxyphenyl)-5-oxohepta-1,3,6-trien-3-yl)oxy) Butanoate (11a)
3.2.9. Synthesis of 4-(((1E,3Z,6E)-1,7-Bis(3,4-dimethoxyphenyl)-5-oxohepta-1,3,6-trien-3-yl)oxy)butanoic Acid (11b)
3.2.10. Synthesis of (1E,4Z)-1-(3,4-Dimethoxyphenyl)-5-hydroxyhexa-1,4-dien-3-one (12)
3.2.11. Synthesis of Tert-butyl (3-(4-formyl-2-methoxyphenoxy)propyl)carbamate (14)
3.2.12. Synthesis of Tert-butyl (3-(4-((1E,3Z,6E)-7-(3,4-dimethoxyphenyl)-3-hydroxy-5-oxohepta-1,3,6-trien-1-yl)-2-methoxyphenoxy)propyl)carbamate (15)
3.2.13. Synthesis of Synthesis of (1E,4Z,6E)-7-(4-(3-Aminopropoxy)-3-methoxyphenyl)-1-(3,4-dimethoxyphenyl)-5-hydroxyhepta-1,4,6-trien-3-one (15a)
3.2.14. Synthesis of Methyl 3-((7S,8S)-18-ethyl-3-((2-(2-(2-(4-(4-((1E,4Z,6E)-5-hydroxy-7-(4-hydroxy-3-methoxyphenyl)-3-oxohepta-1,4,6-trien-1-yl)-2-methoxyphenoxy)butanamido)et-hoxy)ethoxy)ethyl)carbamoyl)-5-(2-methoxy-2-oxoethyl)-2,8,12,17-tetramethyl-13-vinyl-7H,8H-porphyrin-7-yl)propanoate (16)
3.2.15. Synthesis of Methyl 3-(3-(((15Z,18E)-19-(3,4-dimethoxyphenyl)-15-((E)-3,4-dimethoxystyryl)-10,17-dioxo-3,6,14-trioxa-9-azanonadeca-15,18-dien-1-yl)carbamoyl)-18-ethyl-5-(2-methoxy-2-oxoethyl)-2,8,12,17-tetramethyl-13-vinyl-7H,8H-porphyrin-7-yl)propanoate (17)
3.2.16. Synthesis of Methyl 3-((7S,8S)-18-ethyl-3-((16-(4-((1E,4Z,6E)-5-hydroxy-7-(4-hydroxy-3-methoxy-phenyl)-3-oxohepta-1,4,6-trien-1-yl)-2-methoxyphenoxy)-12-oxo-3,6,9-trioxa-13-azahexadecyl)carbamoyl)-5-(2-methoxy-2-oxoethyl)-2,8,12,17-tetramethyl-13-vinyl-7H,8H-porphyrin-7-yl)propanoate (18)
3.2.17. Synthesis of Methyl 3-((7S,8S)-3-((16-(4-((1E,3Z,6E)-7-(3,4-dimethoxyphenyl)-3-hydroxy-5-oxohepta-1,3,6-trien-1-yl)-2-methoxyphenoxy)-12-oxo-3,6,9-trioxa-13-azahexadecyl)carbamoyl)-18-ethyl-5-(2-methoxy-2-oxoethyl)-2,8,12,17-tetramethyl-13-vinyl-7H,8H-porphyrin-7-yl)propanoate (19)
3.3. Photophysical Properties and Singlet Oxygen Photogeneration Analysis
3.4. Biological Analysis
3.4.1. Measurement of Cellular Uptake
3.4.2. Cell Viability Assay
3.4.3. Annexin V and Propidium Iodide Staining
3.4.4. Western Blot
3.4.5. Animal Model
3.4.6. Xenograft Mouse Model Using B16F10 Melanoma Cell
3.4.7. PDT in Animal Model
3.4.8. Pharmacokinetics
3.4.9. Animals and Treatment
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Guan, W.; Cao, Z.; Guo, W.; Xiong, G.; Zhao, F.; Feng, M.; Qiu, J.; Liu, Y.; Zhang, M.Q.; et al. Integrative Genomic Analysis of Gemcitabine Resistance in Pancreatic Cancer by Patient-Derived Xenograft Models. Clin. Cancer Res. 2021, 27, 3383–3396. [Google Scholar] [CrossRef]
- Jia, Y.; Xie, J. Promising Molecular Mechanisms Responsible for Gemcitabine Resistance in Cancer. Genes Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef]
- Juarranz, Á.; Gilaberte, Y.; González, S. Photodynamic Therapy (PDT) in Oncology. Cancers 2020, 12, 3341. [Google Scholar] [CrossRef]
- Allison, R.R.; Moghissi, K. Photodynamic Therapy (PDT): PDT Mechanisms. Clin. Endosc. 2013, 46, 24–29. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Aniogo, E.C.; George, B.P.A.; Abrahamse, H. Role of Bcl-2 Family Proteins in Photodynamic Therapy Mediated Cell Survival and Regulation. Molecules 2020, 25, 5308. [Google Scholar] [CrossRef]
- Mishchenko, T.; Balalaeva, I.; Gorokhova, A.; Vedunova, M.; Krysko, D.V. Which Cell Death Modality Wins the Contest for Photodynamic Therapy of Cancer? Cell Death Dis. 2022, 13, 455. [Google Scholar] [CrossRef]
- Yaqoob, M.D.; Xu, L.; Li, C.; Leong, M.M.L.; Xu, D.D. Targeting Mitochondria for Cancer Photodynamic Therapy. Photodiagn. Photodyn. Ther. 2022, 38, 102830. [Google Scholar] [CrossRef]
- Mokoena, D.R.; George, B.P.; Abrahamse, H. Photodynamic Therapy Induced Cell Death Mechanisms in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 10506. [Google Scholar] [CrossRef]
- Santos, M.L.; D’Ambrosio, M.; Rodrigo, A.P.; Parola, A.J.; Costa, P.M. A Transcriptomic Approach to the Metabolism of Tetrapyrrolic Photosensitizers in a Marine Annelid. Molecules 2021, 26, 3924. [Google Scholar] [CrossRef]
- Pucci, C.; Martinelli, C.; Degl’Innocenti, A.; Desii, A.; De Pasquale, D.; Ciofani, G. Light-Activated Biomedical Applications of Chlorophyll Derivatives. Macromol. Biosci. 2021, 21, 2100181. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, I.-W. P62 Manipulation Affects Chlorin E6-mediated Photodynamic Therapy Efficacy in Colorectal Cancer Cell Lines. Oncol. Lett. 2020, 19, 3907–3916. [Google Scholar] [CrossRef]
- Sheleg, S.V.; Zhavrid, E.A.; Khodina, T.V.; Kochubeev, G.A.; Istomin, Y.P.; Chalov, V.N.; Zhuravkin, I.N. Photodynamic Therapy with Chlorin E6 for Skin Metastases of Melanoma. Photodermatol. Photoimmunol. Photomed. 2004, 20, 21–26. [Google Scholar] [CrossRef]
- Lee, L.S.; Thong, P.S.P.; Olivo, M.; Chin, W.W.L.; Ramaswamy, B.; Kho, K.W.; Lim, P.L.; Lau, W.K.O. Chlorin E6-Polyvinylpyrrolidone Mediated Photodynamic Therapy—A Potential Bladder Sparing Option for High Risk Non-Muscle Invasive Bladder Cancer. Photodiagn. Photodyn. Ther. 2010, 7, 213–220. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, T.; Shen, J.; Karges, J.; Jin, C.; Zhao, Y.; Ji, L.; Chao, H. A Mitochondria-Localized Iridium(III)–Chlorin E6 Conjugate for Synergistic Sonodynamic and Two-Photon Photodynamic Therapy against Melanoma. Inorg. Chem. Front. 2022, 9, 3034–3046. [Google Scholar] [CrossRef]
- Kataoka, H.; Nishie, H.; Tanaka, M.; Sasaki, M.; Nomoto, A.; Osaki, T.; Okamoto, Y.; Yano, S. Potential of Photodynamic Therapy Based on Sugar-Conjugated Photosensitizers. J. Clin. Med. 2021, 10, 841. [Google Scholar] [CrossRef]
- Guo, X.; Wang, S.; Zhang, F.; Li, G.; Li, Y.; Zhao, W. Derivatization of Chlorin E6 with Maleimide Enhances Its Photodynamic Efficacy in HepG2 Cells. J. Porphyr. Phthalocyanines 2020, 24, 1093–1098. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Han, G.-Y.; Guo, C.-Y.; Ma, Z.-Q.; Lin, M.-Y.; Wang, Y.; Miao, Z.-Y.; Zhang, W.-N.; Sheng, C.-Q.; Yao, J.-Z. Design, Synthesis and Biological Evaluation of Novel 31-Hexyloxy Chlorin E6-Based 152- or 131-Amino Acid Derivatives as Potent Photosensitizers for Photodynamic Therapy. Eur. J. Med. Chem. 2020, 207, 112715. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, T.; Farooqui, A.A. Chapter 2—Curcumin: Historical Background, Chemistry, Pharmacological Action, and Potential Therapeutic Value. In Curcumin for Neurological and Psychiatric Disorders; Farooqui, T., Farooqui, A.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 23–44. ISBN 978-0-12-815461-8. [Google Scholar]
- Jyotirmayee, B.; Mahalik, G. A Review on Selected Pharmacological Activities of Curcuma Longa L. Int. J. Food Prop. 2022, 25, 1377–1398. [Google Scholar] [CrossRef]
- Urošević, M.; Nikolić, L.; Gajić, I.; Nikolić, V.; Dinić, A.; Miljković, V. Curcumin: Biological Activities and Modern Pharmaceutical Forms. Antibiotics 2022, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [PubMed]
- Kasi, P.D.; Tamilselvam, R.; Skalicka-Woźniak, K.; Nabavi, S.F.; Daglia, M.; Bishayee, A.; Pazoki-toroudi, H.; Nabavi, S.M. Molecular Targets of Curcumin for Cancer Therapy: An Updated Review. Tumor Biol. 2016, 37, 13017–13028. [Google Scholar] [CrossRef]
- Xue, X.; Yu, J.-L.; Sun, D.-Q.; Kong, F.; Qu, X.-J.; Zou, W.; Wu, J.; Wang, R.-M. Curcumin Induces Apoptosis in SGC-7901 Gastric Adenocarcinoma Cells via Regulation of Mitochondrial Signaling Pathways. Asian Pac. J. Cancer Prev. 2014, 15, 3987–3992. [Google Scholar] [CrossRef]
- Wu, L.; Guo, L.; Liang, Y.; Liu, X.; Jiang, L.; Wang, L. Curcumin Suppresses Stem-like Traits of Lung Cancer Cells via Inhibiting the JAK2/STAT3 Signaling Pathway. Oncol. Rep. 2015, 34, 3311–3317. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, C.; Xi, H.; Gao, Y.; Xu, D. Curcumin Induces Apoptosis in Pancreatic Cancer Cells through the Induction of Forkhead Box O1 and Inhibition of the PI3K/Akt Pathway. Mol. Med. Rep. 2015, 12, 5415–5422. [Google Scholar] [CrossRef]
- Kwon, Y. Curcumin as a Cancer Chemotherapy Sensitizing Agent. Appl. Biol. Chem. 2014, 57, 273–280. [Google Scholar] [CrossRef]
- Chen, S.; Gao, W.; Zhang, M.-J.; Chan, J.Y.-W.; Wong, T.-S. Curcumin Enhances Cisplatin Sensitivity by Suppressing NADPH Oxidase 5 Expression in Human Epithelial Cancer. Oncol. Lett. 2019, 18, 2132–2139. [Google Scholar] [CrossRef]
- Jalde, S.S.; Chauhan, A.K.; Lee, J.H.; Chaturvedi, P.K.; Park, J.-S.; Kim, Y.-W. Synthesis of Novel Chlorin E6-Curcumin Conjugates as Photosensitizers for Photodynamic Therapy against Pancreatic Carcinoma. Eur. J. Med. Chem. 2018, 147, 66–76. [Google Scholar] [CrossRef]
- Liu, W.; Ma, X.; Jin, Y.; Zhang, J.; Li, Y.; Tang, Y.; Song, Y.; Wang, S. Chlorin E6-Biotin Conjugates for Tumor-Targeting Photodynamic Therapy. Molecules 2021, 26, 7342. [Google Scholar] [CrossRef]
- Kimani, S.; Ghosh, G.; Ghogare, A.; Rudshteyn, B.; Bartusik, D.; Hasan, T.; Greer, A. Synthesis and Characterization of Mono-, Di-, and Tri-Poly(Ethylene Glycol) Chlorin E6 Conjugates for the Photokilling of Human Ovarian Cancer Cells. J. Org. Chem. 2012, 77, 10638–10647. [Google Scholar] [CrossRef]
- Ryu, J.H.; Jeong, Y.-I.; Kim, H.Y.; Son, G.M.; Lee, H.L.; Chung, C.-W.; Chu, C.W.; Kang, D.H. Enhanced Photosensing and Photodynamic Treatment of Colon Cancer Cells Using Methoxy Poly(Ethylene Glycol)-Conjugated Chlorin E6. J. Nanosci. Nanotechnol. 2018, 18, 1131–1136. [Google Scholar] [CrossRef]
- Ailioaie, L.M.; Ailioaie, C.; Litscher, G. Latest Innovations and Nanotechnologies with Curcumin as a Nature-Inspired Photosensitizer Applied in the Photodynamic Therapy of Cancer. Pharmaceutics 2021, 13, 1562. [Google Scholar] [CrossRef]
- Luo, H.; Lu, L.; Liu, N.; Li, Q.; Yang, X.; Zhang, Z. Curcumin Loaded Sub-30 Nm Targeting Therapeutic Lipid Nanoparticles for Synergistically Blocking Nasopharyngeal Cancer Growth and Metastasis. J. Nanobiotechnol. 2021, 19, 224. [Google Scholar] [CrossRef]
- He, G.; Mu, T.; Yuan, Y.; Yang, W.; Zhang, Y.; Chen, Q.; Bian, M.; Pan, Y.; Xiang, Q.; Chen, Z.; et al. Effects of Notch Signaling Pathway in Cervical Cancer by Curcumin Mediated Photodynamic Therapy and Its Possible Mechanisms in Vitro and in Vivo. J. Cancer 2019, 10, 4114–4122. [Google Scholar] [CrossRef]
- Şueki, F.; Ruhi, M.K.; Gülsoy, M. The Effect of Curcumin in Antitumor Photodynamic Therapy: In Vitro Experiments with Caco-2 and PC-3 Cancer Lines. Photodiagn. Photodyn. Ther. 2019, 27, 95–99. [Google Scholar] [CrossRef]
- Gao, Y.-H.; Lovreković, V.; Kussayeva, A.; Chen, D.-Y.; Margetić, D.; Chen, Z.-L. The Photodynamic Activities of Dimethyl 131-[2-(Guanidinyl)Ethylamino] Chlorin E6 Photosensitizers in A549 Tumor. Eur. J. Med. Chem. 2019, 177, 144–152. [Google Scholar] [CrossRef]
- Narumi, A.; Rachi, R.; Yamazaki, H.; Kawaguchi, S.; Kikuchi, M.; Konno, H.; Osaki, T.; Okamoto, Y.; Shen, X.; Kakuchi, T. Maltotriose–Chlorin E6 Conjugate Linked via Tetraethyleneglycol as an Advanced Photosensitizer for Photodynamic Therapy. Synthesis and Antitumor Activities against Canine and Mouse Mammary Carcinoma Cells. ACS Omega 2021, 6, 7023–7033. [Google Scholar] [CrossRef]
- Llambi, F.; Green, D.R. Apoptosis and Oncogenesis: Give and Take in the BCL-2 Family. Curr. Opin. Genet. Dev. 2011, 21, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Gavathiotis, E. BAX Unleashed: The Biochemical Transformation of an Inactive Cytosolic Monomer into a Toxic Mitochondrial Pore. Trends Biochem. Sci. 2011, 36, 642–652. [Google Scholar] [CrossRef]
- Shen, Y.-J.; Cao, J.; Sun, F.; Cai, X.-L.; Li, M.-M.; Zheng, N.-N.; Qu, C.-Y.; Zhang, Y.; Shen, F.; Zhou, M.; et al. Effect of Photodynamic Therapy with (17R,18R)-2-(1-Hexyloxyethyl)-2-Devinyl Chlorine E6 Trisodium Salt on Pancreatic Cancer Cells in Vitro and in Vivo. World J. Gastroenterol. 2018, 24, 5246–5258. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-M.; Shim, Y.H.; Kwon, H.; Kim, J.-P.; Park, J.-I.; Kim, D.H.; Kim, D.-H.; Kim, J.H.; Jeong, Y.-I. CD44 Receptor–Specific and Redox-Sensitive Nanophotosensitizers of Hyaluronic Acid–Chlorin E6 Tetramer Having Diselenide Linkages for Photodynamic Treatment of Cancer Cells. J. Pharm. Sci. 2019, 108, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-I.; Yoo, S.Y.; Heo, J.; Kang, D.H. Chlorin E6-Conjugated and PEGylated Immune Checkpoint Inhibitor Nanocomposites for Pulmonary Metastatic Colorectal Cancer. ACS Omega 2019, 4, 18593–18599. [Google Scholar] [CrossRef]
- Sakagami, K.; Masuda, T.; Kawano, K.; Futaki, S. Importance of Net Hydrophobicity in the Cellular Uptake of All-Hydrocarbon Stapled Peptides. Mol. Pharm. 2018, 15, 1332–1340. [Google Scholar] [CrossRef]
- Jori, G. Low-Density Lipoproteins-Liposome Delivery Systems for Tumor Photosensitizers in Vivo. In Photodynamic Therapy; Henderson, B.W., Dougherty, T.J., Eds.; CRC Press: Boca Raton, FL, USA, 2020; pp. 173–186. ISBN 1-00-306689-5. [Google Scholar]
- Liu, Y.; Grimm, M.; Dai, W.; Hou, M.; Xiao, Z.-X.; Cao, Y. CB-Dock: A Web Server for Cavity Detection-Guided Protein–Ligand Blind Docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Gurung, P.; Lim, J.; Shrestha, R.; Kim, Y.-W. Chlorin E6-Associated Photodynamic Therapy Enhances Abscopal Antitumor Effects via Inhibition of PD-1/PD-L1 Immune Checkpoint. Sci. Rep. 2023, 13, 4647. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Dark Toxicity (IC50, µM) | Phototoxicity (IC50, µM) | Ratio * (AsPC-1, MIA-PaCa-2, PANC-1) | ||||
|---|---|---|---|---|---|---|---|
| AsPC-1 | MIA-PaCa-2 | PANC-1 | AsPC-1 | MIA-PaCa-2 | PANC-1 | ||
| I | ≥50 | ≥50 | ≥50 | 0.04 | 0.035 | 0.04 | ND, ND, ND |
| II | ≥50 | ≥50 | ≥50 | ≥3.1 | ≥3.1 | ≥3.1 | ND, ND, ND |
| 16 | ≥50 | ≥50 | ≥50 | ≥3.1 | 2.05 | 2.15 | ND, ND, ND |
| 17 | 27.32 | 11.03 | 18.73 | 0.27 | 0.42 | 0.21 | 101.18, 26.26, 89.19 |
| 18 | ≥50 | ≥50 | ≥50 | ≥3.1 | 2.33 | 2.41 | ND, ND, ND |
| 19 | ≥50 | ≥50 | 24.4 | ≥3.1 | 2.19 | 2.03 | ND, ND, 12.01 |
| Ce6 | ≥50 | ≥50 | ≥50 | ≥3.1 | ≥3.1 | ≥3.1 | ND, ND, ND |
| Cell Line | Concentration (µM) | Q1 (NE) | Q2 (LA) | Q3 (Live) | Q4 (EA) | Q2 + Q4 |
|---|---|---|---|---|---|---|
| AsPC-1 | Control | 3.51% | 2.83% | 91.51% | 2.15% | 4.98% |
| 0.2 | 2.67% | 50.65% | 41.34% | 5.34% | 55.99% | |
| 0.4 | 4.39% | 58.59% | 27.30% | 9.72% | 68.31% | |
| 0.8 | 0.10% | 84.35% | 7.92% | 7.63% | 91.98% | |
| MIA-PaCa-2 | Control | 0.15% | 0.12% | 99.11% | 0.62% | 0.74% |
| 0.1 | 7.94% | 52.17% | 29.91% | 9.98% | 62.15% | |
| 0.2 | 7.70% | 46.77% | 34.70% | 10.83% | 57.60% | |
| 0.4 | 0.24% | 57.85% | 7.81% | 34.10% | 91.95% | |
| PANC-1 | Control | 26.09% | 3.00% | 69.94% | 0.97% | 3.97% |
| 0.1 | 10.09% | 21.21% | 42.76% | 25.94% | 47.15% | |
| 0.2 | 5.37% | 26.83% | 38.49% | 29.31% | 56.14% | |
| 0.4 | 1.25% | 31.96% | 13.41% | 53.38% | 85.34% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa Magar, T.B.; Lee, J.; Lee, J.H.; Jeon, J.; Gurung, P.; Lim, J.; Kim, Y.-W. Novel Chlorin e6-Curcumin Derivatives as a Potential Photosensitizer: Synthesis, Characterization, and Anticancer Activity. Pharmaceutics 2023, 15, 1577. https://doi.org/10.3390/pharmaceutics15061577
Thapa Magar TB, Lee J, Lee JH, Jeon J, Gurung P, Lim J, Kim Y-W. Novel Chlorin e6-Curcumin Derivatives as a Potential Photosensitizer: Synthesis, Characterization, and Anticancer Activity. Pharmaceutics. 2023; 15(6):1577. https://doi.org/10.3390/pharmaceutics15061577
Chicago/Turabian StyleThapa Magar, Til Bahadur, Jusuk Lee, Ji Hoon Lee, Juhee Jeon, Pallavi Gurung, Junmo Lim, and Yong-Wan Kim. 2023. "Novel Chlorin e6-Curcumin Derivatives as a Potential Photosensitizer: Synthesis, Characterization, and Anticancer Activity" Pharmaceutics 15, no. 6: 1577. https://doi.org/10.3390/pharmaceutics15061577