
Neurodegenerative Proteinopathies Induced by Environmental Pollutants: Heat Shock Proteins and Proteasome as Promising Therapeutic Tools
 , ,
, ,
Abstract
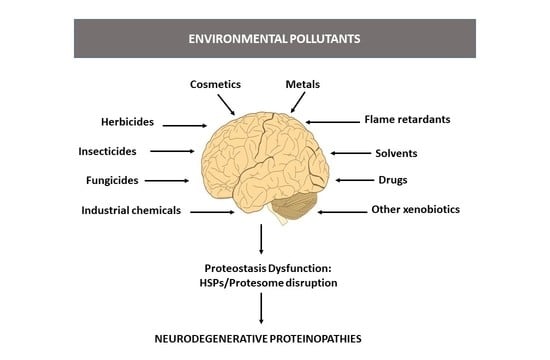
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
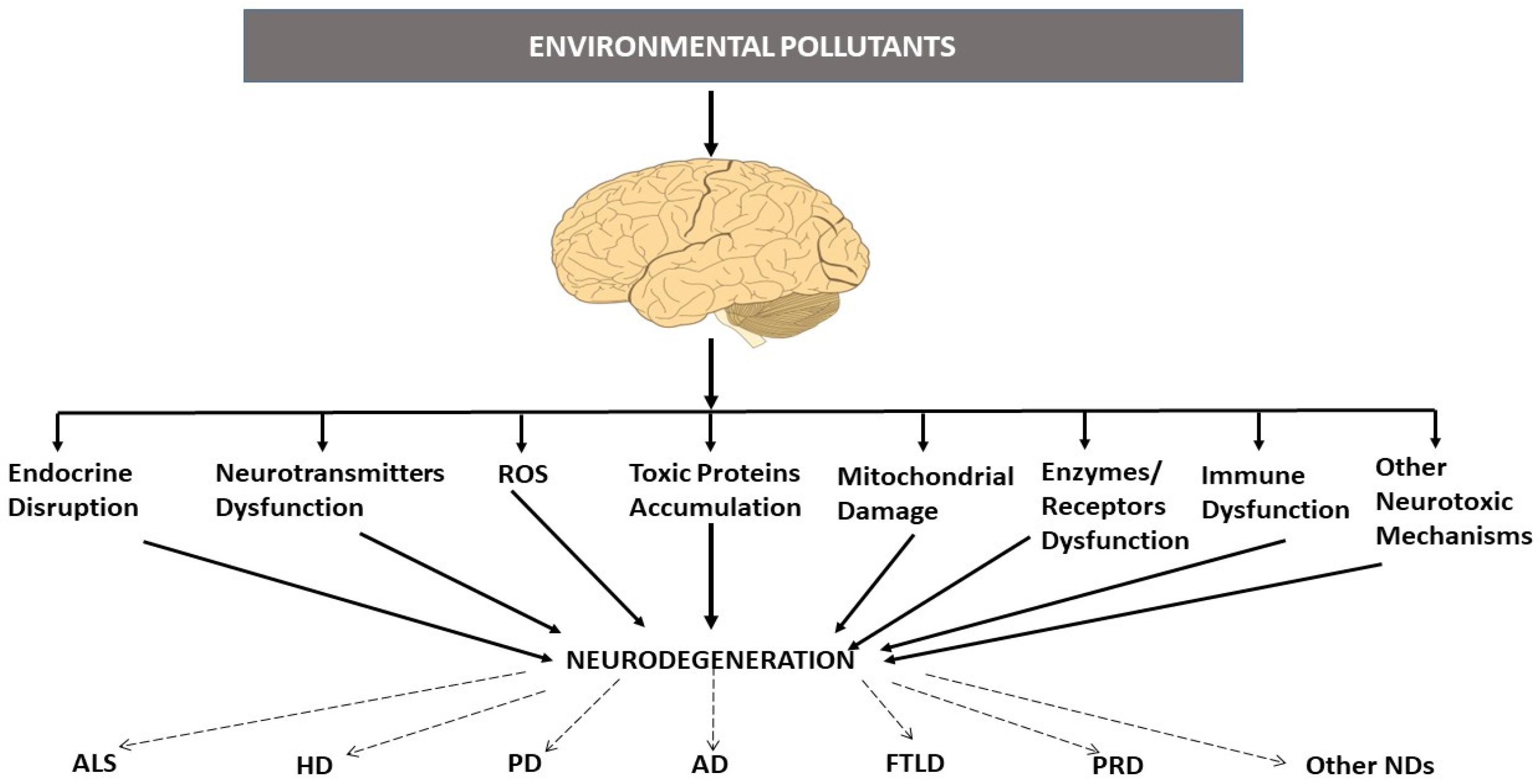
1. Environmental Pollutants and Neurodegenerative Diseases
2. Neurodegenerative Proteinopathies and Environmental Pollutants
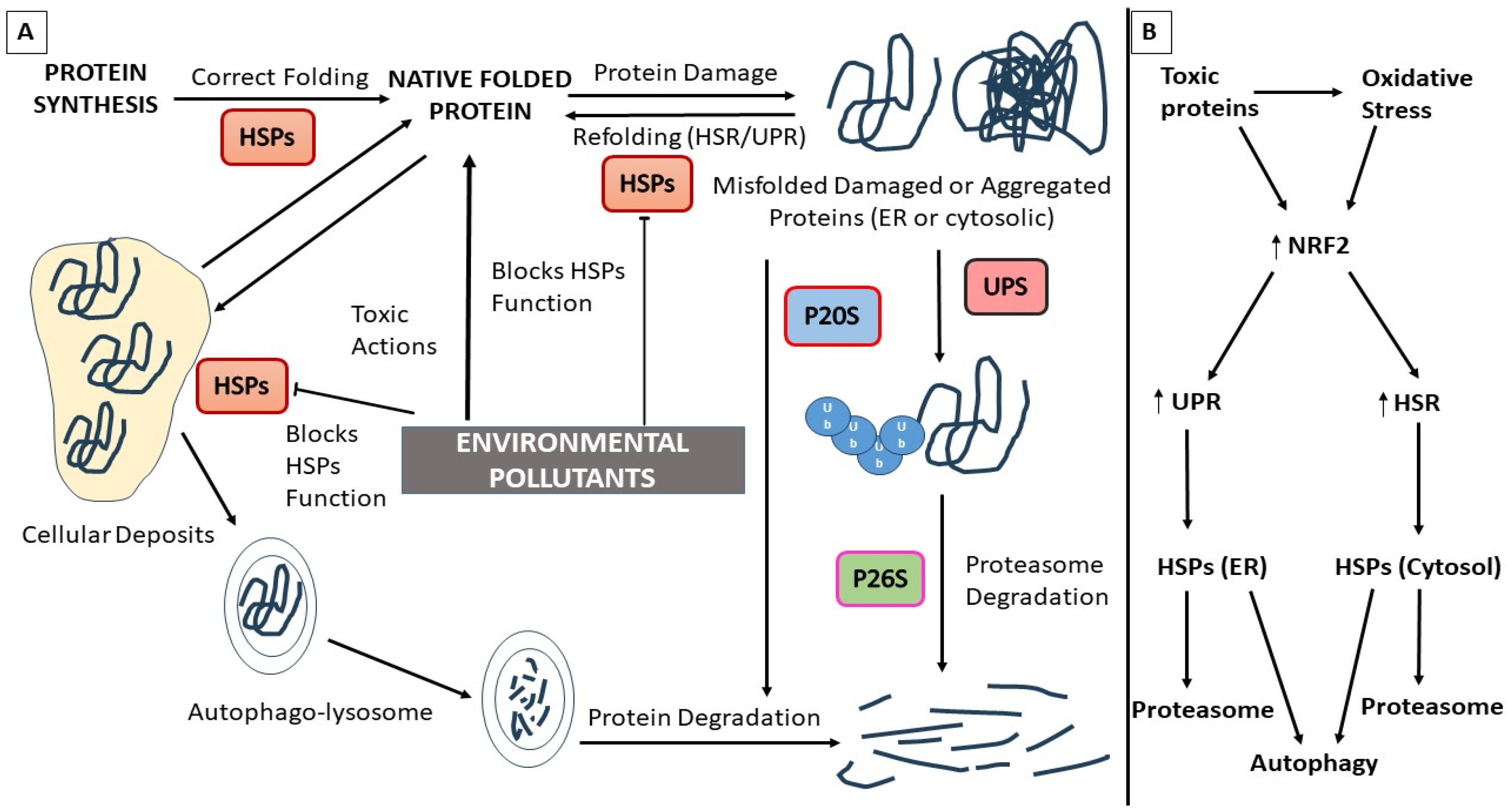
3. Heat Shock Proteins (HSPs) and EPs
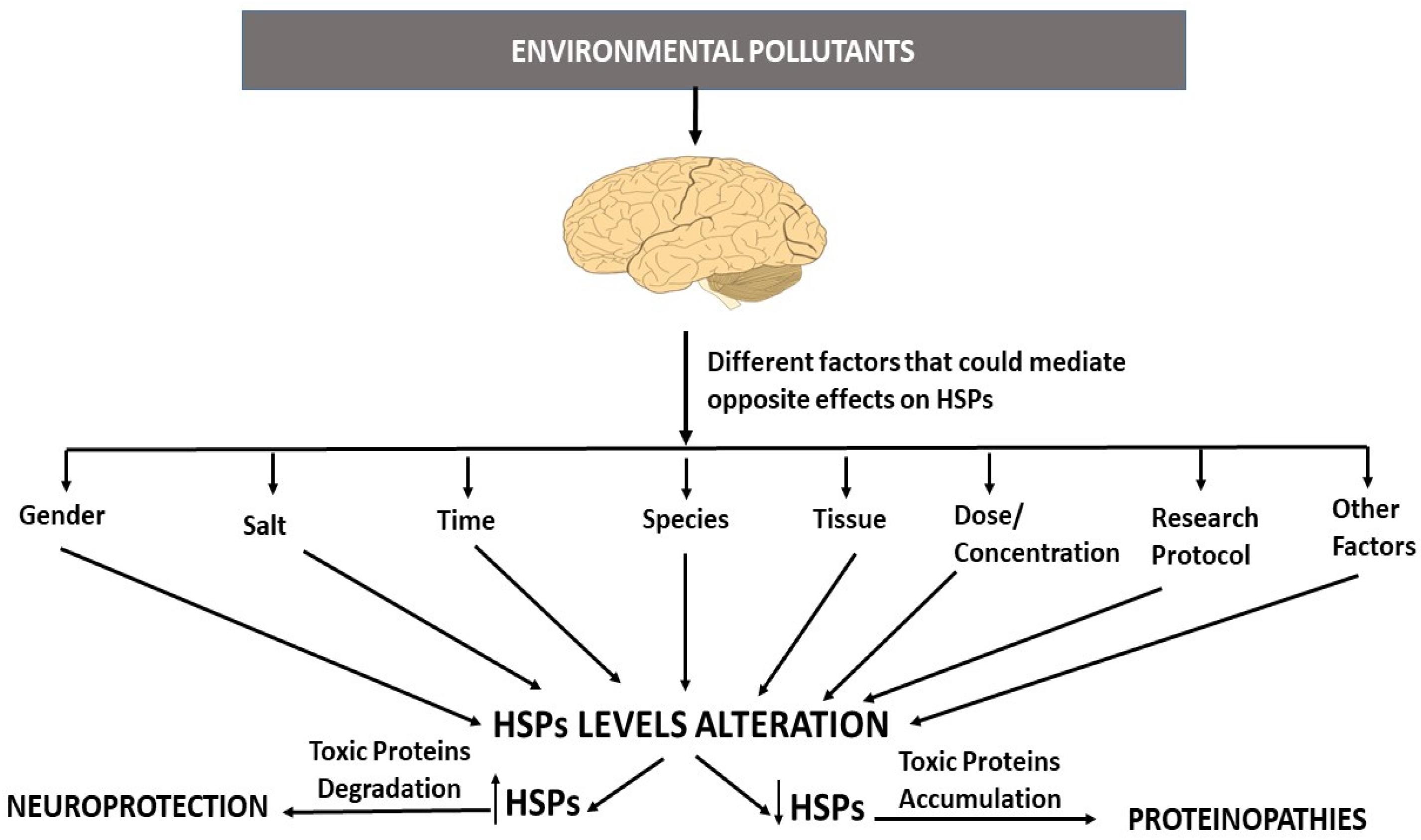
3.1. Environmental Pollutants and HSP Dysfunction
3.2. Heat Shock Protein Dysfunction, Neurodegenerative Proteinopathies, and EPs
4. Proteasome and EPs
4.1. Environmental Pollutants and Proteasome Dysfunction
4.2. Proteasome Dysfunction, Neurodegenerative Proteinopathies, and EPs
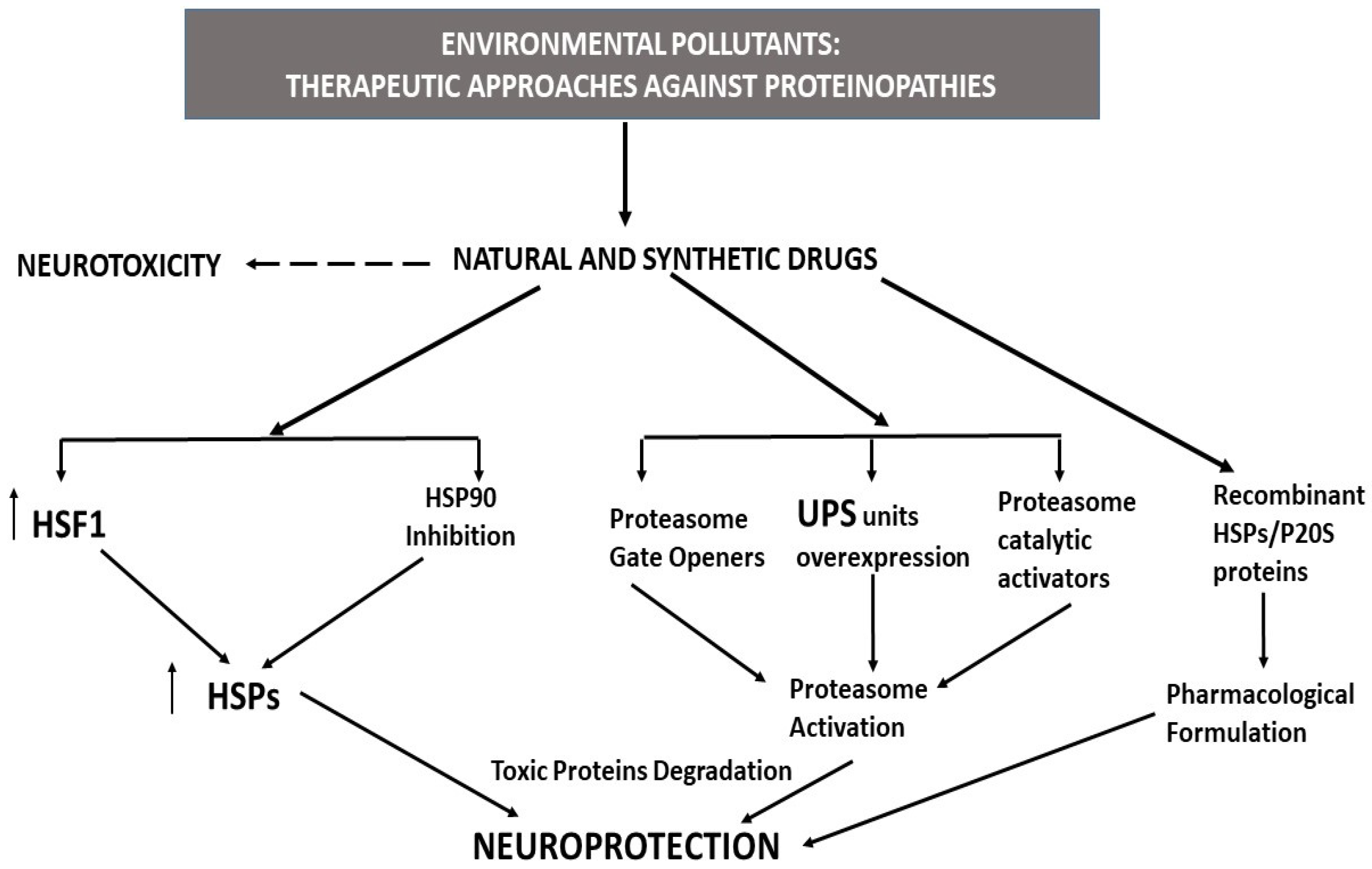
5. Therapeutic Strategies against Proteinopathies
5.1. HSP Activators/Inductors as Therapeutic Tools for the Neurodegenerative Proteinopathies Induced by Environmental Pollutants
5.2. Proteasome Activators as Therapeutic Tools for Neurodegenerative Proteinopathies Induced by Environmental Pollutants
5.3. Recombinant HSPs and P20 Proteins as Therapeutic Tools against Neurodegenerative Effects Induced by Environmental Pollutants
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acevedo, K.; Masaldan, S.; Opazo, C.M.; Bush, A.I. Redox active metals in neurodegenerative diseases. JBIC J. Biol. Inorg. Chem. 2019, 24, 1141–1157. [Google Scholar] [CrossRef]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Research 2016, 5, F1000 Faculty Rev-366. [Google Scholar] [CrossRef] [Green Version]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disor-ders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.; Goyal, K.; Pandey, A.K.; Benjamin, M.; Khan, F.; Pandey, P.; Mittan, S.; Iqbal, D.; Alsaweed, M.; Alturaiki, W.; et al. Elucidations of Molecular Mechanism and Mechanistic Effects of Environmental Toxicants in Neurological Disorders. CNS Neurol. Disord. Drug. Targets 2023, 22, 84–97. [Google Scholar]
- Nabi, M.; Tabassum, N. Role of Environmental Toxicants on Neurodegenerative Disorders. Front. Toxicol. 2022, 4, 837579. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Reed, W.; Maronpot, R.R.; Henríquez-Roldán, C.; Delgado-Chavez, R.; Calderón-Garcidueñas, A.; Dragustinovis, I.; Franco-Lira, M.; Aragón-Flores, M.; Solt, A.C.; et al. Brain inflammation and Alzheimer’s-like pa-thology in individuals exposed to severe air pollution. Toxicol. Pathol. 2004, 32, 650–658. [Google Scholar] [CrossRef]
- del Pino, J.; Zeballos, G.; Anadón, M.J.; Moyano, P.; Díaz, M.J.; García, J.M.; Frejo, M.T. Cadmium-induced cell death of basal forebrain cholinergic neurons mediated by muscarinic M1 receptor blockade, increase in GSK-3β enzyme, β-amyloid and tau protein levels. Arch. Toxicol. 2016, 90, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, J.; Zeballos, G.; Anadon, M.J.; Capo, M.A.; Díaz, M.J.; García, J.; Frejo, M.T. Higher sensitivity to cadmium induced cell death of basal forebrain cholinergic neurons: A cholinesterase dependent mechanism. Toxicology 2014, 325, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Moyano, P.; García, J.M.; Anadon, M.J.; Lobo, M.; García, J.; Frejo, M.T.; Sola, E.; Pelayo, A.; del Pino, J. Manganese induced ROS and AChE variants alteration leads to SN56 basal forebrain cholinergic neuronal loss after acute and long-term treatment. Food Chem. Toxicol. 2019, 125, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A. The potential role of aluminum in Alzheimer’s disease. Nephrol. Dial. Transpl. 2002, 17, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, R.A.; Hu, H.; Weisskopf, M.G.; Schwartz, B.S. Cumulative Lead Dose and Cognitive Function in Adults: A Review of Studies That Measured Both Blood Lead and Bone Lead. Environ. Health Perspect. 2007, 115, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, F.; Hoppin, J.A. Association of Pesticide Exposure with Neurologic Dysfunction and Disease. Environ. Health Perspect. 2004, 112, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Del Pino, J.; Moyano, P.; Díaz, G.G.; Anadon, M.J.; Diaz, M.J.; García, J.M.; Lobo, M.; Pelayo, A.; Sola, E.; Frejo, M.T. Primary hippocampal neuronal cell death induction after acute and repeated paraquat exposures mediated by AChE variants alteration and cholinergic and glutamatergic transmission disruption. Toxicology 2017, 390, 88–99. [Google Scholar] [CrossRef]
- Moyano, P.; Frejo, M.T.; Anadon, M.J.; García, J.M.; Díaz, M.J.; Lobo, M.; Sola, E.; García, J.; Del Pino, J. SN56 neuronal cell death after 24 h and 14 days chlorpyrifos exposure through glutamate transmission dysfunction, increase of GSK-3β enzyme, β-amyloid and tau protein levels. Toxicology 2018, 402–403, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Iteire, K.A.; Sowole, A.T.; Ogunlade, B. Exposure to pyrethroids induces behavioral impairments, neurofibrillary tangles and tau pathology in Alzheimer’s type neurodegeneration in adult Wistar rats. Drug Chem. Toxicol. 2020, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bali, Y.A.; Kaikai, N.E.; Ba-M’hamed, S.; Bennis, M. Learning and memory impairments associated to acetylcholinesterase inhibition and oxidative stress following glyphosate based-herbicide exposure in mice. Toxicology 2019, 415, 18–25. [Google Scholar] [CrossRef]
- Yegambaram, M.; Manivannan, B.; Beach, T.; Halden, R. Role of Environmental Contaminants in the Etiology of Alzheimer’s Disease: A Review. Curr. Alzheimer Res. 2015, 12, 116–146. [Google Scholar] [CrossRef] [Green Version]
- Viberg, H.; Fredriksson, A.; Eriksson, P. Neonatal exposure to polybrominated diphenyl ether (PBDE 153) disrupts spontaneous behavior, impairs learning and memory, and decreases hippocampal cholinergic receptors in adult mice. Toxicol. Appl. Phar-macol. 2003, 192, 95–106. [Google Scholar] [CrossRef]
- Zaman, T. The Prevalence and Environmental Impact of Single Use Plastic Products. In Public Health Management & Policy: An Online Textbook, Retrieved November, 11th ed.; Health Administration Press: Chicago, IL, USA, 2010; Volume 23, p. 2011. [Google Scholar]
- Barse, A.V.; Chakrabarti, T.; Ghosh, T.K.; Pal, A.K.; Kumar, N.; Raman, R.P.; Jadhao, S.B. Vitellogenin Induction and Histo-metabolic Changes Following Exposure of Cyprinuscarpio to Methyl Paraben. Asian-Australas. J. Anim. Sci. 2010, 23, 1557–1565. [Google Scholar] [CrossRef]
- Tran, D.N.; Jung, E.-M.; Yoo, Y.-M.; Lee, J.-H.; Jeung, E.-B. Perinatal Exposure to Triclosan Results in Abnormal Brain Development and Behavior in Mice. Int. J. Mol. Sci. 2020, 21, 4009. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, R.; Jahan, I.; Ali, C.; Mitra, S.; Munni, Y.A.; Timalsina, B.; Hannan, A.; Moon, I.S. Potential roles of natural products in the targeting of proteinopathic neurodegenerative diseases. Neurochem. Int. 2021, 145, 105011. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Shabir, S.; Yousuf, S.; Kauts, S.; Bhat, S.A.; Mir, A.H.; Singh, M.P. Proteinopathies: Deciphering Physiology and Mechanisms to Develop Effective Therapies for Neurodegenerative Diseases. Mol. Neurobiol. 2022, 59, 7513–7540. [Google Scholar] [CrossRef]
- Devi, S.; Chaturvedi, M.; Fatima, S.; Priya, S. Environmental factors modulating protein conformations and their role in protein aggregation diseases. Toxicology 2021, 465, 153049. [Google Scholar] [CrossRef]
- Devi, S.; Kim, J.J.; Singh, A.P.; Kumar, S.; Dubey, A.K.; Singh, S.K.; Singh, R.S.; Kumar, V. Proteotoxicity: A Fatal Consequence of Environmental Pollutants-Induced Impairments in Protein Clearance Machinery. J. Pers. Med. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Leak, R.K. Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 2014, 8, 293–310. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of cur-cumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [Green Version]
- Esser, C.; Alberti, S.; Höhfeld, J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1695, 171–188. [Google Scholar] [CrossRef] [Green Version]
- Türker, F.; Cook, E.K.; Margolis, S.S. The proteasome and its role in the nervous system. Cell Chem. Biol. 2021, 28, 903–917. [Google Scholar] [CrossRef]
- Deshmukh, F.K.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The Contribution of the 20S Proteasome to Proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.J.; Fort, P.E. Heat Shock Proteins Regulatory Role in Neurodevelopment. Front. Neurosci. 2018, 12, 821. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. Medcomm 2020, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Prajapati, K.S.; Swamy, M.; Pachauri, V. Heat shock proteins: A therapeutic target worth to consider. Veter-World 2015, 8, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alz-heimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I.; Santoro, G.M. Stress-inducible responses and heat shock proteins: New pharmacologic targets for cytopro-tection. Nat. Biotechnol. 2018, 16, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D. Regulation of Hsf1 and the Heat Shock Response. Adv. Exp. Med. Biol. 2020, 1243, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Steurer, C.; Eder, N.; Kerschbaum, S.; Wegrostek, C.; Gabriel, S.; Pardo, N.; Ortner, V.; Czerny, T.; Riegel, E. HSF1 mediated stress response of heavy metals. PLoS ONE 2018, 13, e0209077. [Google Scholar] [CrossRef] [Green Version]
- Carver, J.A.; Ecroyd, H.; Truscott, R.J.W.; Thorn, D.C.; Holt, C. Proteostasis and the Regulation of Intra- and Extracellular Protein Aggregation by ATP-Independent Molecular Chaperones: Lens α-Crystallins and Milk Caseins. Acc. Chem. Res. 2018, 51, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Mambula, S.S.; Gray, P.J.; Theriault, J.R. Extracellular heat shock proteins in cell signaling. FEBS Lett. 2007, 581, 3689–3694. [Google Scholar] [CrossRef] [Green Version]
- Marsh, A.P. Molecular mechanisms of proteinopathies across neurodegenerative disease: A review. Neurol. Res. Pract. 2019, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.A.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Chang, S.C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the proteasomal ATPases’ carboxyl ter-mini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.M.; Fraga, H.; Reis, C.; Kafri, G.; Goldberg, A.L. ATP Binds to Proteasomal ATPases in Pairs with Distinct Functional Effects, Implying an Ordered Reaction Cycle. Cell 2011, 144, 526–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.-C.; Goldberg, A.L.; Cheng, Y. Mechanism of Gate Opening in the 20S Proteasome by the Proteasomal ATPases. Mol. Cell 2008, 30, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Del Razo, L.M.; Quintanilla-Vega, B.; Brambila-Colombres, E.; Calderón-Aranda, E.S.; Manno, M.; Albores, A. Stress proteins in-duced by arsenic. Toxicol. Appl. Pharmacol. 2001, 177, 132–148. [Google Scholar] [CrossRef]
- Giusi, G.; Alò, R.; Crudo, M.; Facciolo, R.M.; Canonaco, M. Specific cerebral heat shock proteins and histamine receptor cross-talking mechanisms promote distinct lead-dependent neurotoxic responses in teleosts. Toxicol. Appl. Pharmacol. 2008, 227, 248–256. [Google Scholar] [CrossRef]
- Moyano, P.; García, J.M.; García, J.; Anadon, M.J.; Naval, M.V.; Frejo, M.T.; Sola, E.; Pelayo, A.; Del Pino, J. Manganese increases Aβ and Tau protein levels through proteasome 20S and heat shock proteins 90 and 70 alteration, leading to SN56 cholinergic cell death following single and repeated treatment. Ecotoxicol. Environ. Saf. 2020, 203, 110975. [Google Scholar] [CrossRef]
- Verma, S.; Sharma, S.; Ranawat, P.; Nehru, B. Modulatory Effects of Ginkgo biloba Against Amyloid Aggregation Through In-duction of Heat Shock Proteins in Aluminium Induced Neurotoxicity. Neurochem. Res. 2020, 45, 465–490. [Google Scholar] [CrossRef]
- Moyano, P.; Garcia, J.M.; Frejo, M.T.; Lobo, M.; Garcia, J.; del Pino, J. Proteasome 20S and Rab5 Alteration after 24 h and 14 Days Chlorpyrifos Exposure Lead to β-Amyloid and Tau Protein Level Increases and SN56 Neuronal Cell Death. Chem. Res. Toxicol. 2019, 32, 1920–1924. [Google Scholar] [CrossRef]
- Moyano, P.; Sanjuan, J.; Garcia, J.M.; Garcia, J.; Frejo, M.T.; Naval, M.V.; Del Pino, J. Paraquat Treatment Compromises the Clearance of β-Amyloid and Tau Proteins and Induces Primary Hippocampal Neuronal Cell Death through HSP70, P20S, and TFEB Disruption. Chem. Res. Toxicol. 2021, 34, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Moyano, P.; García, J.M.; Lobo, M.; Anadón, M.J.; Sola, E.; Pelayo, A.; García, J.; Frejo, M.T.; del Pino, J. Cadmium alters heat shock protein pathways in SN56 cholinergic neurons, leading to Aβ and phosphorylated Tau protein generation and cell death. Food Chem. Toxicol. 2018, 121, 297–308. [Google Scholar] [CrossRef]
- Wills, J.; Credle, J.; Oaks, A.W.; Duka, V.; Lee, J.H.; Jones, J.; Sidhu, A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS ONE 2012, 7, e30745. [Google Scholar] [CrossRef]
- Betarbet, R.; Canet-Aviles, R.M.; Sherer, T.B.; Mastroberardino, P.G.; McLendon, C.; Kim, J.H.; Lund, S.; Na, H.M.; Taylor, G.; Bence, N.F.; et al. Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol. Dis. 2006, 22, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Cholanians, A.B.; Phan, A.V.; Ditzel, E.J.; Camenisch, T.D.; Lau, S.S.; Monks, T.J. From the Cover: Arsenic 197. Induces Accumulation of α-Synuclein: Implications for Synucleinopathies and Neurodegeneration. Toxicol. Sci. 2016, 153, 271–281. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Cordaro, M.; Genovese, T.; Peritore, A.F.; Crupi, R.; Interdonato, L.; Di Paola, D.; Cuzzocrea, S.; et al. Toxic Exposure to Endocrine Disruptors Worsens Parkinson’s Disease Progression through NRF2/HO-1 Alteration. Biomedicines 2022, 10, 1073. [Google Scholar] [CrossRef]
- Izco, M.; Vettorazzi, A.; Forcen, R.; Blesa, J.; de Toro, M.; Alvarez-Herrera, N.; Cooper, J.M.; Gonzalez-Peñas, E.; Lopez de Cerain, A.; Alvarez-Erviti, L. Oral subchronic exposure to the mycotoxin ochratoxin A induces key pathological features of Parkinson’s disease in mice six months after the end of the treatment. Food Chem. Toxicol. 2021, 152, 112164. [Google Scholar] [CrossRef]
- Cheng, S.; Trombetta, L. The induction of amyloid precursor protein and α-synuclein in rat hippocampal astrocytes by diethyldithiocarbamate and copper with or without glutathione. Toxicol. Lett. 2004, 146, 139–149. [Google Scholar] [CrossRef]
- Li, Y.; Sun, L.; Cai, T.; Zhang, Y.; Lv, S.; Wang, Y.; Ye, L. α-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain Res. Bull. 2010, 81, 428–433. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Stanford, E.A.; AIAbdulatif, A.; Ramirez-Cardenas, A.; Ballance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Meyerowitz, J.; Parker, S.J.; Vella, L.J.; Ng, D.C.; Price, K.A.; Liddell, J.R.; Caragounis, A.; Li, Q.X.; Masters, C.L.; Nonaka, T.; et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol. Neurodegener. 2011, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, P.E.A.; Dhawan, U.; Boudeau, S.; Lei, S.; Carlomagno, Y.; Knobel, M.; Al Mohanna, L.F.A.; Boomhower, S.R.; Newland, M.C.; Sherr, D.H.; et al. Heavy Metal Neurotoxicants Induce ALS-Linked TDP-43 Pathology. Toxicol. Sci. 2019, 167, 105–115. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett. 2001, 500, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Kanthasamy, A.; Choi, C.; Jin, H.; Harischandra, D.; Anantharam, V. Effect of divalent metals on the neuronal proteasomal system, prion protein ubiquitination and aggregation. Toxicol. Lett. 2012, 214, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafon, P.-A.; Imberdis, T.; Wang, Y.; Torrent, J.; Robitzer, M.; Huetter, E.; Alvarez-Martinez, M.-T.; Chevallier, N.; Givalois, L.; Desrumaux, C.; et al. Low doses of bioherbicide favour prion aggregation and propagation in vivo. Sci. Rep. 2018, 8, 8023. [Google Scholar] [CrossRef] [Green Version]
- Heldens, L.; Hensen, S.M.M.; Onnekink, C.; van Genesen, S.T.; Dirks, R.P.; Lubsen, N.H. An Atypical Unfolded Protein Response in Heat Shocked Cells. PLoS ONE 2011, 6, e23512. [Google Scholar] [CrossRef] [Green Version]
- Weindling, E.; Bar-Nun, S. Sir2 links the unfolded protein response and the heat shock response in a stress response network. Biochem. Biophys. Res. Commun. 2015, 457, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Radanović, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. [Google Scholar] [CrossRef] [PubMed]
- Terrab, L.; Wipf, P. Hsp70 and the Unfolded Protein Response as a Challenging Drug Target and an Inspiration for Probe Mol-ecule Development. ACS Med. Chem. Lett. 2020, 11, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70-a master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef] [Green Version]
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.E.; Vígh, L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. Int. J. Mol. Sci. 2018, 19, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, H.; Handa, K.; Murakami, T.; Aizawa, T.; Ozawa, H. Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System. Cells 2022, 11, 1205. [Google Scholar] [CrossRef]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neuro-degeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med. 2003, 4, 21–36. [Google Scholar] [CrossRef]
- Sola, E.; Moyano, P.; Flores, A.; García, J.M.; García, J.; Anadon, M.J.; Frejo, M.T.; Pelayo, A.; Fernandez, M.d.l.C.; del Pino, J. Cadmium-promoted thyroid hormones disruption mediates ROS, inflammation, Aβ and Tau proteins production, gliosis, spongiosis and neurodegeneration in rat basal forebrain. Chem. Biol. Interact. 2023, 375, 110428. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zeng, Q.; Sun, B.; Wei, S.; Wang, Q.; Zhang, A. Assessing the Role of Nrf2/GPX4-Mediated Oxidative Stress in Arsenic-Induced Liver Damage and the Potential Application Value of Rosa roxburghii Tratt [Rosaceae]. Oxid. Med. Cell. Longev. 2022, 2022, 9865606. [Google Scholar] [CrossRef]
- Baiyun, R.; Li, S.; Liu, B.; Lu, J.; Lv, Y.; Xu, J.; Wu, J.; Li, J.; Lv, Z.; Zhang, Z. Luteolin-mediated PI3K/AKT/Nrf2 signaling pathway ameliorates inorganic mercury-induced cardiac injury. Ecotoxicol. Environ. Saf. 2018, 161, 655–661. [Google Scholar] [CrossRef]
- Liu, W.; Xu, Z.; Li, H.; Guo, M.; Yang, T.; Feng, S.; Xu, B.; Deng, Y. Protective effects of curcumin against mercury-induced hepatic injuries in rats, involvement of oxidative stress antagonism, and Nrf2-ARE pathway activation. Hum. Exp. Toxicol. 2017, 36, 949–966. [Google Scholar] [CrossRef]
- He, C.; Zhao, X.; Lei, Y.; Du, J.; Niu, Q. The role of Nrf2/HO-1 signal pathway in regulating aluminum-induced apoptosis of PC12 cells. J. Trace Elem. Med. Biol. 2023, 79, 127232. [Google Scholar] [CrossRef]
- Sedik, A.A.; Hassan, S.A.; Shafey, H.I.; Khalil, W.K.B.; Mowaad, N.A. Febuxostat attenuates aluminum chloride-induced hepatorenal injury in rats with the impact of Nrf2, Crat, Car3, and MNK-mediated apoptosis. Environ. Sci. Pollut. Res. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Roede, J.R.; Hansen, J.M.; Go, Y.M.; Jones, D.P. Maneb and paraquat-mediated neurotoxicity: Involvement of peroxiredox-in/thioredoxin system. Toxicol. Sci. 2011, 121, 368–375. [Google Scholar] [CrossRef]
- Moyano, P.; Sanjuan, J.; García, J.M.; Anadon, M.J.; Lobo, M.; Pelayo, A.; García, J.; Frejo, M.T.; Del Pino, J. Primary hippocampal estrogenic dysfunction induces synaptic proteins alteration and neuronal cell death after single and repeated paraquat exposure. Food Chem. Toxicol. 2020, 136, 110961. [Google Scholar] [CrossRef]
- Zhang, C.; Qin, L.; Dou, D.-C.; Li, X.-N.; Ge, J.; Li, J.-L. Atrazine induced oxidative stress and mitochondrial dysfunction in quail (Coturnix C. coturnix) kidney via modulating Nrf2 signaling pathway. Chemosphere 2018, 212, 974–982. [Google Scholar] [CrossRef]
- Müller, S.G.; Jardim, N.S.; Quines, C.B.; Nogueira, C.W. Diphenyl diselenide regulates Nrf2/Keap-1 signaling pathway and counteracts hepatic oxidative stress induced by bisphenol A in male mice. Environ. Res. 2018, 164, 280–287. [Google Scholar] [CrossRef]
- Chepelev, N.L.; Enikanolaiye, M.I.; Chepelev, L.L.; Almohaisen, A.; Chen, Q.; Scoggan, K.A.; Coughlan, M.C.; Cao, X.L.; Jin, X.; Willmore, W.G. Bisphenol A activates the Nrf1/2-antioxidant response element pathway in HEK 293 cells. Chem. Res. Toxicol. 2013, 26, 498–506. [Google Scholar] [CrossRef]
- Liu, W.; Yang, B.; Wu, L.; Zou, W.; Pan, X.; Zou, T.; Liu, F.; Xia, L.; Wang, X.; Zhang, D. Involvement of NRF2 in Perfluorooctanoic Acid-Induced Testicular Damage in Male Mice. Biol. Reprod. 2015, 93, 41. [Google Scholar] [CrossRef] [PubMed]
- Boesch-Saadatmandi, C.; Wagner, A.E.; Graeser, A.C.; Hundhausen, C.; Wolffram, S.; Rimbach, G. Ochratoxin A impairs Nrf2-dependent gene expression in porcine kidney tubulus cells. J. Anim. Physiol. Anim. Nutr. 2009, 93, 547–554. [Google Scholar] [CrossRef]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef] [PubMed]
- El-Ghaiesh, S.H.; Bahr, H.I.; Ibrahiem, A.T.; Ghorab, D.; Alomar, S.Y.; Farag, N.E.; Zaitone, S.A. Metformin Protects From Rotenone–Induced Nigrostriatal Neuronal Death in Adult Mice by Activating AMPK-FOXO3 Signaling and Mitigation of Angiogenesis. Front. Mol. Neurosci. 2020, 13, 84. [Google Scholar] [CrossRef]
- Pan, E.; Chen, H.; Wu, X.; He, N.; Gan, J.; Feng, H.; Sun, Y.; Dong, J. Protective effect of quercetin on avermectin induced splenic toxicity in carp: Resistance to inflammatory response and oxidative damage. Pestic. Biochem. Physiol. 2023, 193, 105445. [Google Scholar] [CrossRef] [PubMed]
- El-Saied, Y.E.; Mostafa, M.E.; Refaat, M.; El-Senduny, F.F.; Alsharif, F.M.; El-Khawaga, O.Y. The Hepatoprotective Role of Balanites Aegyptiaca Extract and its Nano-Formulation against Methomyl-Induced Toxicity and Oxidative Stress in Mice via Overexpression of Nrf2. J. Appl. Biotechnol. Rep. 2021, 8, 263–274. [Google Scholar]
- Gargouri, B.; Yousif, N.M.; Attaai, A.; Bouchard, M.; Chtourou, Y.; Fiebich, B.L.; Fetoui, H. Pyrethroid bifenthrin induces oxidative stress, neuroinflammation, and neuronal damage, associated with cognitive and memory impairment in murine hippocampus. Neurochem. Int. 2018, 120, 121–133. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, H.-T.; Cao, Y.-M.; Jiang, Z.-G.; Liu, J.; Fan, Q.-Y. Biphasic Dose-Response of Mn-Induced Mitochondrial Damage, PINK1/Parkin Expression, and Mitophagy in SK-N-SH Cells. Dose-Response 2023, 21, 15593258231169392. [Google Scholar] [CrossRef]
- Cai, T.; Che, H.; Yao, T.; Chen, Y.; Huang, C.; Zhang, W.; Du, K.; Zhang, J.; Cao, Y.; Chen, J.; et al. Manganese induces tau hyperphosphorylation through the activation of ERK MAPK pathway in PC12 cells. Toxicol. Sci. 2011, 119, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Yang, H.; Tian, X.; Wang, H.; Zhou, T.; Zhang, S.; Yu, J.; Zhang, T.; Fan, D.; Guo, X.; et al. High manganese, a risk for Alzheimer’s disease: High manganese induces amyloid-β related cognitive impairment. J. Alzheimers Dis. 2014, 42, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.J.; Anantharam, V.; Martin, D.P.; Nicholson, E.M.; Richt, J.A.; Kanthasamy, A.; Kanthasamy, A.G. Manganese Upregulates Cellular Prion Protein and Contributes to Altered Stabilization and Proteolysis: Relevance to Role of Metals in Pathogenesis of Prion Disease. Toxicol. Sci. 2010, 115, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Lu, X.; Wu, D.; Cai, S.; Li, S.; Teng, X. The Effect of Manganese-induced Cytotoxicity on mRNA Expressions of HSP27, HSP40, HSP60, HSP70 and HSP90 in Chicken Spleen Lymphocytes in Vitro. Biol. Trace Element Res. 2013, 156, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-W.; Lu, L.; Li, W.-X.; Zhang, L.-Y.; Ji, C.; Lin, X.; Liu, H.-C.; Odle, J.; Luo, X.-G. Effect of dietary manganese on antioxidant status and expression levels of heat-shock proteins and factors in tissues of laying broiler breeders under normal and high environmental temperatures. Br. J. Nutr. 2015, 114, 1965–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Im, A.R.; Lee, S.; Chae, S. Dual Protective Effects of Flavonoids from Petasites japonicus against UVB-Induced Apoptosis Mediated via HSF-1 Activated Heat Shock Proteins and Nrf2-Activated Heme Oxygenase-1 Pathways. Biol. Pharm. Bull. 2017, 40, 765–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, E.; Romero, A.; Marco-Contelles, J.; López-Muñoz, F.; Del Pino, J. Modulation of Heat Shock Response Proteins by ASS234, Targeted for Neurodegenerative Diseases Therapy. Chem. Res. Toxicol. 2018, 31, 839–842. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Bower, K.; Fink, A.L. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: Implications for Parkinson’s disease. Neurotoxicology 2002, 23, 527–536. [Google Scholar] [CrossRef]
- Liegro, D.; Gerspacher, C.; Scheuber, U.; Schiera, G.; Proia, P.; Gygax, D.; Di Liegro, I. The effect of cadmium on brain cells in culture. Int. J. Mol. Med. 2009, 24, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Olmedo, D.G.; Biaggio, V.S.; Koumbadinga, G.A.; Gómez, N.N.; Shi, C.; Ciocca, D.R.; Batulan, Z.; Fanelli, M.A.; O’Brien, E.R. Recombinant heat shock protein 27 (HSP27/HSPB1) protects against cadmium-induced oxidative stress and toxicity in human cervical cancer cells. Cell Stress Chaperones 2017, 22, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhu, Y.-H.; Cheng, X.-Y.; Zhang, Z.-W.; Xu, S.-W. The Protection of Selenium against Cadmium-Induced Cytotoxicity via the Heat Shock Protein Pathway in Chicken Splenic Lymphocytes. Molecules 2012, 17, 14565–14572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Folgar, R.; Martínez-Guitarte, J.-L. Cadmium alters the expression of small heat shock protein genes in the aquatic midge Chironomus riparius. Chemosphere 2017, 169, 485–492. [Google Scholar] [CrossRef]
- E Selim, M.; A Rashed, E.H.; A Aleisa, N.; Daghestani, M.H. The protection role of heat shock protein 70 (HSP-70) in the testes of cadmium-exposed rats. Bioinformation 2012, 8, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkai, Y.; Masuda, A.; Akiyama, M.; Xian, M.; Kumagai, Y. Cadmium-mediated activation of the HSP90/HSF1 pathway regu-lated by reactive persulfides/polysulfides. Toxicol. Sci. 2017, 156, 412–421. [Google Scholar] [PubMed] [Green Version]
- Boujelben, M.; Ghorbel, F.; Vincent, C.; Makni-Ayadi, F.; Guermazi, F.; Croute, F.; El-Feki, A. Lipid peroxidation and HSP72/73 expression in rat following cadmium chloride administration: Interactions of magnesium supplementation. Exp. Toxicol. Pathol. 2006, 57, 437. [Google Scholar] [CrossRef]
- De Furia, J.; Shea, T.B. Arsenic inhibits neurofilament transport and induces perikaryal accumulation of phosphorylated neuro-filaments: Roles of JNK and GSK-3beta. Brain Res. 2007, 1181, 74–82. [Google Scholar] [CrossRef]
- Jacobson, T.; Navarrete, C.; Sharma, S.K.; Sideri, T.C.; Ibstedt, S.; Priya, S.; Grant, C.M.; Christen, P.; Goloubinoff, P.; Tamás, M.J. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci. 2012, 125, 5073–5083. [Google Scholar] [CrossRef] [Green Version]
- Papaconstantinou, A.D.; Brown, K.M.; Noren, B.T.; McAlister, T.; Fisher, B.R.; Goering, P.L. Mercury, cadmium, and arsenite enhance heat shock protein synthesis in chick embryos prior to embryotoxicity. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2003, 68, 456–464. [Google Scholar] [CrossRef]
- Oshima, H.; Hatayama, T.; Nakamura, M. A possibility for new evaluating method of cytotoxicity by using heat shock protein assay. J. Mater. Sci. Mater. Med. 1997, 8, 143–147. [Google Scholar] [CrossRef]
- Goering, P.L.; Fisher, B.R.; Noren, B.T.; Papaconstantinou, A.; Rojko, J.L.; Marler, R.J. Mercury induces regional and cell-specific stress protein expression in rat kidney. Toxicol. Sci. 2000, 53, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernyak, Y.; Merinova, A. HSP70 (HSPA1) polymorphisms for former workers with chronic mercury vapor exposure. Int. J. Occup. Med. Environ. Health 2017, 30, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Hwang, G.W.; Ryoke, K.; Lee, J.Y.; Takahashi, T.; Naganuma, A. siRNA-mediated silencing of the gene for heat shock transcription factor 1 causes hypersensitivity to methylmercury in HEK293 cells. J. Toxicol. Sci. 2011, 36, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Caito, S.; Zeng, H.; Aschner, J.L.; Aschner, M. Methylmercury Alters the Activities of Hsp90 Client Proteins, Prostaglandin E Synthase/p23 (PGES/23) and nNOS. PLoS ONE 2014, 9, e98161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, M.; Kato-Negishi, M. Link between Aluminum and the Pathogenesis of Alzheimer’s Disease: The Integration of the Aluminum and Amyloid Cascade Hypotheses. Int. J. Alzheimers Dis. 2011, 2011, 276393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exley, C. Human exposure to aluminium. Environ. Sci. Process. Impacts 2013, 15, 1807–1816. [Google Scholar] [CrossRef] [Green Version]
- Sood, P.K.; Nahar, U.; Nehru, B. Stress Proteins and Glial Cell Functions During Chronic Aluminium Exposures: Protective Role of Curcumin. Neurochem. Res. 2012, 37, 639–646. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Zhao, Y.; Pogue, A.I.; Tarr, M.A.; Kruck, T.P.; Percy, M.E.; Cui, J.-G.; Lukiw, W.J. Synergistic effects of iron and aluminum on stress-related gene expression in primary human neural cells. J. Alzheimer’s Dis. 2005, 8, 117–127. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Rodella, L.F.; Ricci, F.; Rezzani, R.; Lavazza, A.; Bianchi, R. Stress proteins expression in rat kidney and liver chronically exposed to aluminium sulphate. Histol. Histopathol. 2006, 21, 131–140. [Google Scholar]
- Yang, L.; Zha, J.; Li, W.; Li, Z.; Wang, Z. Atrazine affects kidney and adrenal hormones (AHs) related genes expressions of rare minnow (Gobiocypris rarus). Aquat. Toxicol. 2010, 97, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Li, S.; Wang, X.; Gao, X.; Xu, S.; Wang, X. Effects of atrazine and chlorpyrifos on the mRNA levels of HSP70 and HSC70 in the liver, brain, kidney and gill of common carp (Cyprinus carpio L.). Chemosphere 2012, 90, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.; Moyano, P.; Sola, E.; García, J.M.; García, J.; Anadon, M.J.; Frejo, M.T.; Naval, M.V.; Fernadez, M.C.; Pino, J.D. Single and repeated bisphenol A treatment induces ROS, Aβ and hyperphosphorylated-tau accumulation, and insulin pathways disruption, through HDAC2 and PTP1B overexpression, leading to SN56 cholinergic apoptotic cell death. Food Chem. Toxicol. 2022, 170, 113500. [Google Scholar] [CrossRef]
- Tan, L.; Wang, S.; Wang, Y.; He, M.; Liu, D. Bisphenol A exposure accelerated the aging process in the nematode Caenorhabditis elegans. Toxicol. Lett. 2015, 235, 75–83. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, H.; Liu, W.; Zhang, Z.; Qin, H.; Luo, F.; Leng, S. Developmental perfluorooctane sulfonate exposure results in tau hyperphosphorylation and β-amyloid aggregation in adults rats: Incidence for link to Alzheimer’s disease. Toxicology 2016, 347–349, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Johansson, N.; Eriksson, P.; Viberg, H. Neonatal Exposure to PFOS and PFOA in Mice Results in Changes in Proteins which are Important for Neuronal Growth and Synaptogenesis in the Developing Brain. Toxicol. Sci. 2009, 108, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Zhang, J.; Martin, F.L.; Peng, S.; Tian, M.; Mu, X.; Shen, H. Perfluorooctanoic acid induces apoptosis through the p53-dependent mitochondrial pathway in human hepatic cells: A proteomic study. Toxicol. Lett. 2013, 223, 211–220. [Google Scholar] [CrossRef] [PubMed]
- El Golli Bennour, E.; Bouaziz, C.; Ladjimi, M.; Renaud, F.; Bacha, H. Comparative mechanisms of zearalenone and ochratoxin A toxicities on cultured HepG2 cells: Is oxidative stress a common process? Environ. Toxicol. 2009, 24, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Raghubeer, S.; Nagiah, S.; Chuturgoon, A. Ochratoxin A upregulates biomarkers associated with hypoxia and transfor-mation in human kidney cells. Toxicol. Vitr. 2019, 57, 211–216. [Google Scholar] [CrossRef]
- Brownjohn, P.W.; Smith, J.; Portelius, E.; Serneels, L.; Kvartsberg, H.; De Strooper, B.; Blennow, K.; Zetterberg, H.; Livesey, F.J. Phenotypic Screening Identifies Modulators of Amyloid Precursor Protein Processing in Human Stem Cell Models of Alzheimer’s Disease. Stem Cell Rep. 2017, 8, 870–882. [Google Scholar] [CrossRef] [Green Version]
- Sala, G.; Marinig, D.; Riva, C.; Arosio, A.; Stefanoni, G.; Brighina, L.; Formenti, M.; Alberghina, L.; Colangelo, A.M.; Ferrarese, C. Rote-none down-regulates HSPA8/hsc70 chaperone protein in vitro: A new possible toxic mechanism contributing to Parkinson’s disease. Neurotoxicology 2016, 54, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Misra, H.P. Developmental exposure to pesticides zineb and/or endosulfan renders the nigrostriatal dopamine system more susceptible to these environmental chemicals later in life. Neurotoxicology 2007, 28, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Gezer, A.O.; Kochmanski, J.; VanOeveren, S.E.; Cole-Strauss, A.; Kemp, C.J.; Patterson, J.R.; Miller, K.M.; Kuhn, N.C.; Herman, D.E.; McIn-tire, A.; et al. Developmental exposure to the organochlorine pesticide dieldrin causes male-specific exacerbation of α-synuclein-preformed fibril-induced toxicity and motor deficits. Neurobiol. Dis. 2020, 141, 104947. [Google Scholar] [CrossRef] [PubMed]
- Heys, K.A.; Shore, R.F.; Pereira, M.G.; Martin, F.L. Levels of Organochlorine Pesticides Are Associated with Amyloid Aggregation in Apex Avian Brains. Environ. Sci. Technol. 2017, 51, 8672–8681. [Google Scholar] [CrossRef] [Green Version]
- Sonia Angeline, M.; Chaterjee, P.; Anand, K.; Ambasta, R.K.; Kumar, P. Rotenone-induced parkinsonism elicits behavioral impairments and differential expression of parkin, heat shock proteins and caspases in the rat. Neuroscience 2012, 220, 291–301. [Google Scholar] [CrossRef]
- Jang, J.; Oh, H.; Nam, D.; Seol, W.; Seo, M.K.; Park, S.W.; Kim, H.G.; Seo, H.; Son, I.; Ho, D.H. Increase in anti-apoptotic molecules, nucleolin, and heat shock protein 70, against upregulated LRRK2 kinase activity. Anim. Cells Syst. 2018, 22, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Skandrani, D.; Gaubin, Y.; Vincent, C.; Beau, B.; Claude Murat, J.; Soleilhavoup, J.P.; Croute, F. Relationship between toxicity of selected insecticides and expression of stress proteins (HSP, GRP) in cultured human cells: Effects of commercial formulations versus pure active molecules. Biochim. Biophys. Acta 2006, 1760, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Skandrani, D.; Gaubin, Y.; Beau, B.; Murat, J.C.; Vincent, C.; Croute, F. Effect of selected insecticides on growth rate and stress protein expression in cultured human A549 and SH-SY5Y cells. Toxicol. Vitro 2006, 20, 1378–1386. [Google Scholar] [CrossRef]
- da Rosa, J.G.S.; Barcellos, H.H.D.A.; Fagundes, M.; Variani, C.; Rossini, M.; Kalichak, F.; Koakoski, G.; Oliveira, T.A.; Idalencio, R.; Frandoloso, R.; et al. Muscarinic receptors mediate the endocrine-disrupting effects of an organophosphorus insecticide in zebrafish. Environ. Toxicol. 2017, 32, 1964–1972. [Google Scholar] [CrossRef]
- Alak, G.; Yeltekin, A.Ç.; Tas, I.H.; Ucar, A.; Parlak, V.; Topal, A.; Kocaman, E.M.; Atamanalp, M. Investigation of 8-OHdG, CYP1A, HSP70 and transcriptional analyses of antioxidant defense system in liver tissues of rainbow trout exposed to eprinomectin. Fish Shellfish Immunol. 2017, 65, 136–144. [Google Scholar] [CrossRef]
- Nappi, L.; Aguda, A.H.; Al Nakouzi, N.; Lelj-Garolla, B.; Beraldi, E.; Lallous, N.; Thi, M.; Moore, S.; Fazli, L.; Battsogt, D.; et al. Ivermectin inhibits HSP27 and potentiates efficacy of oncogene targeting in tumor models. J. Clin. Investig. 2020, 130, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Lee, M.A.; Ki, J.S. Different transcriptional responses of heat shock protein 70/90 in the marine diatom Ditylum brightwellii exposed to metal compounds and endocrine-disrupting chemicals. Chemosphere 2013, 92, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, I.; Saxena, D.K.; Chowdhuri, D.K. Hazardous effects of effluent from the chrome plating industry: 70 kDa heat shock protein expression as a marker of cellular damage in transgenic Drosophila melanogaster (hsp70-lacZ). Environ. Health Perspect. 2003, 111, 1926–1932. [Google Scholar] [CrossRef]
- ATSDR. Toxicological Profile for Manganese [Internet]; U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2012. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp151.pdf (accessed on 15 April 2023).
- Bonham, R.T.; Fine, M.R.; Pollock, F.M.; A Shelden, E. Hsp27, Hsp70, and metallothionein in MDCK and LLC-PK1 renal epithelial cells: Effects of prolonged exposure to cadmium. Toxicol. Appl. Pharmacol. 2003, 191, 63–73. [Google Scholar] [CrossRef]
- Croute, F.; Beau, B.; Murat, J.-C.; Vincent, C.; Komatsu, H.; Obata, F.; Soleilhavoup, J.-P. Expression of Stress-related Genes in a Cadmium-resistant A549 Human Cell Line. J. Toxicol. Environ. Health Part A 2005, 68, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Roccheri, M.C.; Agnello, M.; Bonaventura, R.; Matranga, V. Cadmium induces the expression of specific stress proteins in sea urchin embryos. Biochem. Biophys. Res. Commun. 2004, 321, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Aït-Aïssa, S.; Ausseil, O.; Palluel, O.; Vindimian, E.; Garnier-Laplace, J.; Porcher, J.M. Biomarker responses in juvenile rainbow trout (Oncorhynchus mykiss) after single and combined exposure to low doses of cadmium, zinc, PCB77 and 17beta-oestradiol. Biomarkers 2003, 8, 491–508. [Google Scholar] [CrossRef] [Green Version]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; E Boczek, E.; Maharana, S.; O Lee, H.; Carra, S.; A Hyman, A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Madnani, R.S. Alzheimer’s disease: A mini-review for the clinician. Front. Neurol. 2023, 14, 1178588. [Google Scholar] [CrossRef]
- Prodromou, C.; Aran-Guiu, X.; Oberoi, J.; Perna, L.; Chapple, J.P.; van der Spuy, J. HSP70-HSP90 Chaperone Networking in Protein-Misfolding Disease. Subcell Biochem. 2023, 101, 389–425. [Google Scholar]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A. The Hsp70/Hsp90 chaper-one machinery in neurodegenerative diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutledge, B.S.; Choy, W.Y.; Duennwald, M.L. Folding or holding?-Hsp70 and Hsp90 chaperoning of misfolded proteins in neuro-degenerative disease. J. Biol. Chem. 2022, 298, 101905. [Google Scholar] [CrossRef] [PubMed]
- Baughman, H.E.R.; Clouser, A.F.; Klevit, R.E.; Nath, A. HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 2018, 293, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Ann. Rev. Pharmacol. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, M.S.; Milligan, C. Extracellular heat shock proteins in neurodegenerative diseases: New perspectives. Neurosci. Lett. 2019, 711, 134462. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar] [CrossRef]
- Chen, S.; Brown, I.R. Neuronal expression of constitutive heat shock proteins: Implications for neurodegenerative diseases. Cell Stress Chaperon 2007, 12, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged Expression of Molecular Chaperones in Brains of Patients with Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258. [Google Scholar] [CrossRef]
- Noguchi-Shinohara, M.; Ono, K. The Mechanisms of the Roles of α-Synuclein, Amyloid-β, and Tau Protein in the Lewy Body Diseases: Pathogenesis, Early Detection, and Therapeutics. Int. J. Mol. Sci. 2023, 24, 10215. [Google Scholar] [CrossRef]
- Tao, J.; Berthet, A.; Citron, Y.R.; Tsiolaki, P.L.; Stanley, R.; Gestwicki, J.E.; Agard, D.A.; McConlogue, L. Hsp70 chaperone blocks α-synuclein oligomer formation via a novel engagement mechanism. J. Biol. Chem. 2021, 296, 100613. [Google Scholar] [CrossRef]
- Huang, C.; Cheng, H.; Hao, S.; Zhou, H.; Zhang, X.; Gao, J.; Sun, Q.-H.; Hu, H.; Wang, C.-C. Heat Shock Protein 70 Inhibits α-Synuclein Fibril Formation via Interactions with Diverse Intermediates. J. Mol. Biol. 2006, 364, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hulette, C.; Wang, Y.; Zhang, T.; Pan, C.; Wadhwa, R.; Zhang, J. Proteomic identification of a stress protein, mor-talin/mthsp70/GRP75, relevance to Parkinson disease. Mol. Cell. Proteom. 2006, 5, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, V.; Meister-Broekema, M.; Minoia, M.; Carra, S.; Kampinga, H.H. Barcoding heat shock proteins to human diseases: Look-ing beyond the heat shock response. Dis. Model Mech. 2014, 7, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, P.; Nehru, B. Long-term heat shock proteins (HSPs) induction by carbenoxolone improves hallmark features of Parkin-son’s disease in a rotenone-based model. Neuropharmacology 2014, 79, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Shannon, K.M.; Kordower, J.H. Huntington’s disease: Pathological mechanisms and therapeutic strategies. Cell Transplant. 2011, 16, 301–312. [Google Scholar] [CrossRef]
- Söti, C.; Csermely, P. Chaperones and aging: Role in neurodegeneration and in other civilizational diseases. Neurochem. Int. 2002, 41, 383–389. [Google Scholar] [CrossRef]
- Hughes, R.E.; Olson, J.M. Therapeutic opportunities in polyglutamine disease. Nat. Med. 2001, 7, 419–423. [Google Scholar] [CrossRef]
- Jana, N.R.; Tanaka, M.; Wang, G.; Nukina, N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaper-ones with truncated N-terminal huntingtin: Their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 2000, 9, 2009–2018. [Google Scholar] [CrossRef] [Green Version]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Nollen, E.A.; Garcia, S.M.; van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; MacRae, T.H. The small heat shock proteins and their role in human disease. FEBS J. 2005, 272, 2613–2627. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small Heat Shock Proteins Potentiate Amyloid Dissolution by Protein Disaggregases from Yeast and Humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorter, J. Hsp104, A Weapon to Combat Diverse Neurodegenerative Disorders. Neurosignals 2008, 16, 63–74. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- He, Y.; Ruganzu, J.B.; Lin, C.; Ding, B.; Zheng, Q.; Wu, X.; Ma, R.; Liu, Q.; Wang, Y.; Jin, H.; et al. Tanshinone IIA ameliorates cognitive deficits by inhibiting endoplasmic reticulum stress-induced apoptosis in APP/PS1 transgenic mice. Neurochem. Int. 2020, 133, 104610. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alz-heimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar] [CrossRef]
- Schipper, H. Heme oxygenase-1, role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1 in Alzheimer disease: A tribute to Moussa Youdim. J. Neural Transm. 2011, 118, 381–387. [Google Scholar] [CrossRef]
- Yokota, T.; Mishra, M.; Akatsu, H.; Tani, Y.; Miyauchi, T.; Yamamoto, T.; Kosaka, K.; Nagai, Y.; Sawada, T.; Heese, K. Brain site-specific gene expression analysis in Al-zheimer’s disease patients. Eur. J. Clin. Investig. 2006, 36, 820–830. [Google Scholar] [CrossRef]
- Putcha, P.; Danzer, K.M.; Kranich, L.R.; Scott, A.; Silinski, M.; Mabbett, S.; Hicks, C.D.; Veal, J.M.; Steed, P.M.; Hyman, B.T.; et al. Brain-Permeable Small-Molecule Inhibitors of Hsp90 Prevent α-Synuclein Oligomer Formation and Rescue α-Synuclein-Induced Toxicity. J. Pharmacol. Exp. Ther. 2010, 332, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.J.; Tandon, A.; Phoolmala Srivastava, T.; Singh, N.; Goyal, S.; Priya, S.; Chaturvedi, R.K. Bisphenol-A (BPA) Impairs Hippo-campal Neurogenesis via Inhibiting Regulation of the Ubiquitin Proteasomal System. Mol. Neurobiol. 2023, 60, 3277–3298. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Suzuki, N.; Ikenaka, Y.; Hoshi, N.; Tabuchi, Y. Neurotoxicity of a pyrethroid pesticide deltamethrin is associated with the imbalance in proteolytic systems caused by mitophagy activation and proteasome inhibition. Toxicol. Appl. Pharmacol. 2021, 430, 115723. [Google Scholar] [CrossRef]
- Moyano, P.; Ruiz, M.; García, J.M.; Frejo, M.T.; Baselga, M.J.A.; Lobo, M.; García, J.; Del Pino, J. Oxidative stress and cell death induction by amitraz and its metabolite BTS-27271 mediated through cytochrome P450 and NRF2 pathway alteration in primary hippocampal cell. Food Chem. Toxicol. 2019, 129, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Chen, D.; Zhai, G.; Chen, M.S.; Cui, Q.C.; Zhou, Q.; He, B.; Dou, Q.P.; Jiang, G. The proteasome is a molecular target of environmental toxic organotins. Environ. Health Perspect. 2009, 117, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipen, H.M.; Gandhi, S.; Rich, D.Q.; Ohman-Strickland, P.; Laumbach, R.; Fan, Z.-H.; Chen, L.; Laskin, D.L.; Zhang, J.; Madura, K. Acute Decreases in Proteasome Pathway Activity after Inhalation of Fresh Diesel Exhaust or Secondary Organic Aerosol. Environ. Health Perspect. 2011, 119, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Dianat, M.; Radan, M.; Badavi, M.; Mard, S.A.; Bayati, V.; Ahmadizadeh, M. Crocin attenuates cigarette smoke-induced lung injury and cardiac dysfunction by anti-oxidative effects: The role of Nrf2 antioxidant system in preventing oxidative stress. Respir. Res. 2018, 19, 58. [Google Scholar] [CrossRef] [Green Version]
- Frias, D.; Nunes, R.; Matsuda, M.; Yoshizaki, K.; Carvalho-Oliveira, R.; Pereira, D.; Vasconcellos, P.; Mauad, T.; Macchione, M. Relationship between Nrf2-Keap1 system and cell death in BEAS-2B exposed to Diesel Exhaust Particles. Eur. Respir. J. 2017, 50 (Suppl. 61), PA4449. [Google Scholar]
- Pardo, M.; Qiu, X.; Zimmermann, R.; Rudich, Y. Particulate Matter Toxicity Is Nrf2 and Mitochondria Dependent: The Roles of Metals and Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2020, 33, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shie, F.-S.; Piccardo, P.; Montine, T.; Zhang, J. Proteasomal inhibition induced by manganese ethylene-bis-dithiocarbamate: Relevance to parkinson′s disease. Neuroscience 2004, 128, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Yao, T.; Li, Y.; Chen, Y.; Du, K.; Chen, J.; Luo, W. Proteasome inhibition is associated with manganese-induced oxidative injury in PC12 cells. Brain Res. 2007, 1185, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Asanuma, M.; Miyazaki, I.; Hattori, N.; Mizuno, Y.; Ogawa, N. Parkin attenuates manganese-induced dopaminergic cell death. J. Neurochem. 2004, 89, 1490–1497. [Google Scholar] [CrossRef]
- Golovine, K.; Makhov, P.; Uzzo, R.G.; Kutikov, A.; Kaplan, D.J.; Fox, E.; Kolenko, V.M. Cadmium down-regulates expression of XIAP at the post-transcriptional level in prostate cancer cells through an NF-kappaB-independent, proteasome-mediated mechanism. Mol. Cancer 2010, 9, 183. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Frezza, M.; Shakya, R.; Cui, Q.C.; Milacic, V.; Verani, C.N.; Dou, Q.P. Inhibition of the Proteasome Activity by Gallium(III) Complexes Contributes to Their Anti–Prostate Tumor Effects. Cancer Res 2007, 67, 9258–9265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Structural characterization of copper (II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef]
- Arrigoni, F.; Prosdocimi, T.; Mollica, L.; De Gioia, L.; Zampella, G.; Bertini, L. Copper reduction and dioxygen activation in Cu–amyloid beta peptide complexes: Insight from molecular modelling. Metallomics 2018, 10, 1618–1630. [Google Scholar] [CrossRef]
- Santoro, A.M.; Monaco, I.; Attanasio, F.; Lanza, V.; Pappalardo, G.; Tomasello, M.F.; Cunsolo, A.; Rizzarelli, E.; De Luigi, A.; Salmona, M.; et al. Copper(II) ions affect the gating dynamics of the 20S proteasome: A molecular and in cell study. Sci. Rep. 2016, 6, 33444. [Google Scholar] [CrossRef] [Green Version]
- Tam, L.M.; Wang, Y. Arsenic Exposure and Compromised Protein Quality Control. Chem. Res. Toxicol. 2020, 33, 1594–1604. [Google Scholar] [CrossRef]
- Wang, X.F.; Li, S.; Chou, A.P.; Bronstein, J.M. Inhibitory effects of pesticides on proteasome activity: Implication in Parkinson’s disease. Neurobiol. Dis. 2006, 23, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Niu, P. Low doses of single or combined agrichemicals induces α-synuclein aggregation in nigrostriatal system of mice through inhibition of proteasomal and autophagic pathways. Int. J. Clin. Exp. Med. 2015, 8, 20508–20515. [Google Scholar] [PubMed]
- Moyano, P.; García, J.M.; García, J.; Pelayo, A.; Muñoz-Calero, P.; Frejo, M.T.; Anadon, M.J.; Naval, M.V.; Flores, A.; Mirat, V.A.; et al. Chlorpyrifos induces cell proliferation in MCF-7 and MDA-MB-231 cells, through cholinergic and Wnt/β-catenin signaling disruption, AChE-R upregulation and oxidative stress generation after single and repeated treatment. Food Chem. Toxicol. 2021, 152, 112241. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Le Guerroué, F.; Youle, R.J. Ubiquitin signaling in neurodegenerative diseases: An autophagy and proteasome perspective. Cell Death Differ. 2021, 28, 439–454. [Google Scholar] [CrossRef]
- Keller, J.; Hanni, K.B.; Markesbery, W.R. Impaired Proteasome Function in Alzheimer’s Disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- Almeida, C.G.; Takahashi, R.H.; Gouras, G.K. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 2006, 26, 4277–4288. [Google Scholar] [CrossRef] [Green Version]
- Davidson, K.; Pickering, A.M. The proteasome: A key modulator of nervous system function, brain aging, and neurodegenerative disease. Front. Cell Dev. Biol. 2023, 11, 1124907. [Google Scholar] [CrossRef]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [Green Version]
- Chocron, E.S.; Munkácsy, E.; Kim, H.S.; Karpowicz, P.; Jiang, N.; Van Skike, C.E.; DeRosa, N.; Banh, A.Q.; Palavicini, J.P.; Wityk, P.; et al. Genetic and pharmacologic proteasome augmentation ameliorates Alzheimer’s-like pathology in mouse and fly APP overexpression models. Sci. Adv. 2022, 8, eabk2252. [Google Scholar] [PubMed]
- Fuentes, J.J.; Genescà, L.; Kingsbury, T.J.; Cunningham, K.W.; Pérez-Riba, M.; Estivill, X.; de la Luna, S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 2000, 9, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Liu, J.; Zhu, G.; Zhuang, Y.; Suo, H.; Wang, P.; Huang, D.; Xu, J.; Huang, Y.; Yu, M.; et al. Parkin overexpression ameliorates hippocampal long-term potentiation and β-amyloid load in an Alzheimer’s disease mouse model. Hum. Mol. Genet. 2014, 23, 1056–1072. [Google Scholar]
- Rosen, K.M.; Moussa, C.E.H.; Lee, H.K.; Kumar, P.; Kitada, T.; Qin, G.; Fu, Q.; Querfurth, H.W. Parkin reverses intracellular beta-amyloid accumula-tion and its negative effects on proteasome function. J. Neurosci. Res. 2010, 88, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar]
- Gong, B.; Cao, Z.; Zheng, P.; Vitolo, O.V.; Liu, S.; Staniszewski, A.; Moolman, D.; Zhang, H.; Shelanski, M.; Arancio, O. Ubiquitin hydro-lase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 2006, 126, 775–788. [Google Scholar]
- Zhang, M.; Deng, Y.; Luo, Y.; Zhang, S.; Zou, H.; Cai, F.; Wada, K.; Song, W. Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J. Neurochem. 2012, 120, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphos-phorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Sharoar, M.G.; Shi, Q.; Ge, Y.; He, W.; Hu, X.; Perry, G.; Zhu, X.; Yan, R. Dysfunctional tubular endoplasmic reticulum constitutes a pathological feature of Alzheimer’s disease. Mol. Psychiatry 2016, 21, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Shi, Q.; Zhou, X.; He, W.; Yi, H.; Yin, X.; Gearing, M.; Levey, A.; Yan, R. Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J. 2007, 26, 2755–2767. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.H.P.; Reits, E.A.J.; Hol, E.M. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front. Mol. Neurosci. 2014, 7, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhatwa, S.; Zeng, B.Y.; Rose, S.; Jenner, P. A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. Brain Res. 2010, 1326, 174–183. [Google Scholar] [CrossRef]
- Arduino, D.M.; Esteves, A.R.; Silva, D.F.; Martins-Branco, D.; Santos, D.; Pimentel, D.F.; Cardoso, S.M. Therapeutic intervention at cellular quality control systems in Alzheimer’s and Parkinson’s diseases. Curr. Pharm. Des. 2011, 17, 3446–3459. [Google Scholar] [CrossRef]
- Chang, K.-H.; Lee-Chen, G.-J.; Huang, C.-C.; Lin, J.-L.; Chen, Y.-J.; Wei, P.-C.; Lo, Y.-S.; Yao, C.-F.; Kuo, M.-W.; Chen, C.-M. Modeling Alzheimer’s Disease by Induced Pluripotent Stem Cells Carrying APP D678H Mutation. Mol. Neurobiol. 2019, 56, 3972–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by α-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegen-erative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [Green Version]
- Rao, G.; Croft, B.; Teng, C.; Awasthi, V. Ubiquitin-Proteasome System in Neurodegenerative Disorders. J. Drug Metab. Toxicol. 2015, 6, 187. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Jin, Z.; Tan, H.; Xu, Q.; Peng, T.; Li, H. Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated degra-dation of mutant huntingtin. Brain Res. 2016, 1643, 103–112. [Google Scholar] [CrossRef]
- Bhat, K.P.; Yan, S.; Wang, C.-E.; Li, S.; Li, X.-J. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc. Natl. Acad. Sci. USA 2014, 111, 5706–5711. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Southwell, A.L.; Ehrnhoefer, D.E.; Skotte, N.H.; Metzler, M.; Franciosi, S.; Leprivier, G.; Somasekharan, S.P.; Barokas, A.; Deng, Y.; et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA 2014, 111, 3032–3037. [Google Scholar] [CrossRef] [PubMed]
- Al-Ramahi, I.; Lam, Y.C.; Chen, H.K.; de Gouyon, B.; Zhang, M.; Pérez, A.M.; Branco, J.; de Haro, M.; Patterson, C.; Zoghbi, H.Y.; et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degrada-tion. J. Biol. Chem. 2006, 281, 26714–26724. [Google Scholar] [CrossRef] [Green Version]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP associates with expand-ed polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhong, X.; Ballar, P.; Luo, S.; Shen, Y.; Rubinsztein, D.C.; Monteiro, M.J.; Fang, S. Ubiquitin ligase Hrd1 enhances the degradation and sup-presses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007, 313, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Rubio, I.; Rodríguez-Navarro, J.A.; Tomás-Zapico, C.; Ruíz, C.; Casarejos, M.J.; Perucho, J.; Gómez, A.; Rodal, I.; Lucas, J.J.; Mena, M.A.; et al. Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res. 2009, 1281, 91–100. [Google Scholar] [CrossRef]
- Yang, J.; Hao, X.; Cao, X.; Liu, B.; Nyström, T. Spatial sequestration and detoxification of Huntingtin by the ribosome quality con-trol complex. eLife 2016, 5, e11792. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Anantharam, V.; Latchoumycandane, C.; Kanthasamy, A.; Kanthasamy, A.G. Dieldrin induces ubiquitin-proteasome dys-function in alpha-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. J. Pharmacol. Exp. Ther. 2005, 315, 69–79. [Google Scholar] [CrossRef]
- Moyano, P.; Vicente-Zurdo, D.; Blázquez-Barbadillo, C.; Menéndez, J.C.; González, J.F.; Rosales-Conrado, N.; Pino, J.D. Neuroprotec-tive mechanisms of multitarget 7-aminophenanthridin-6(5H)-one derivatives against metal-induced amyloid proteins genera-tion and aggregation. Food Chem. Toxicol. 2022, 167, 113264. [Google Scholar] [CrossRef]
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 2015, 25, 713–724. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug and gene delivery to the brain: The vascular route. Neuron 2002, 36, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Friesen, E.L.; De Snoo, M.L.; Rajendran, L.; Kalia, L.V.; Kalia, S.K. Chaperone-Based Therapies for Disease Modification in Parkin-son’s Disease. Park. Dis. 2017, 2017, 5015307. [Google Scholar]
- Myeku, N.; Duff, K.E. Targeting the 26S Proteasome To Protect Against Proteotoxic Diseases. Trends Mol. Med. 2018, 24, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sõti, C.; Nagy, E.; Giricz, Z.; Vígh, L.; Csermely, P.; Ferdinandy, P. Heat shock proteins as emerging therapeutic targets. Br. J. Pharmacol. 2005, 146, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, B.N.; Kawahara, T.L.; Matsumoto, G.; Kim, S.; Gu, W.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as Inducers of the Heat Shock Response and Cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar] [CrossRef] [Green Version]
- Pesonen, L.; Svartsjö, S.; Bäck, V.; de Thonel, A.; Mezger, V.; Sabéran-Djoneidi, D.; Roos-Mattjus, P. Gambogic acid and gambogenic acid induce a thiol-dependent heat shock response and disrupt the interaction between HSP90 and HSF1 or HSF2. Cell Stress Chaperones 2021, 26, 819–833. [Google Scholar] [PubMed]
- Chirumamilla, C.S.; Pérez-Novo, C.; Van Ostade, X.; Berghe, W.V. Molecular insights into cancer therapeutic effects of the dietary medicinal phytochemical withaferin A. Proc. Nutr. Soc. 2017, 76, 96–105. [Google Scholar] [CrossRef]
- Yan, D.; Saito, K.; Ohmi, Y.; Fujie, N.; Ohtsuka, K. Paeoniflorin, a novel heat shock protein-inducing compound. Cell Stress Chaperones 2004, 9, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Qian, C.; Zheng, Z.G.; Qian, F.; Wang, Y.; Thu, P.M.; Zhang, X.; Zhou, Y.; Tu, L.; Liu, Q.; et al. Jujuboside A promotes Aβ clearance and ameliorates cognitive deficiency in Alzheimer’s disease through activating Axl/HSP90/PPARγ pathway. Theranostics 2018, 8, 4262–4278. [Google Scholar]
- Hargitai, J.; Lewis, H.; Boros, I.; Rácz, T.; Fiser, A.; Kurucz, I.; Benjamin, I.; Vígh, L.; Pénzes, Z.; Csermely, P.; et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 2003, 307, 689–695. [Google Scholar] [CrossRef]
- Ren, Z.; Dong, Z.; Xie, P.; Lv, J.; Hu, Y.; Guan, Z.; Zhang, C.; Yu, W. PNU282987 inhibits amyloid-β aggregation by upregulating astrocytic endogenous αB-crystallin and HSP-70 via regulation of the α7AChR, PI3K/Akt/HSF-1 signaling axis. Mol. Med. Rep. 2020, 22, 201–208. [Google Scholar]
- Hahm, K.B.; Park, I.S.; Kim, Y.S.; Kim, J.H.; Cho, S.W.; Lee, S.I.; Youn, J.K. Role of Rebamipide on Induction of Heat-Shock Proteins and Protection Against Reactive Oxygen Metabolite-Mediated Cell Damage in Cultured Gastric Mucosal Cells. Free. Radic. Biol. Med. 1997, 22, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Kasza, Á.; Hunya, Á.; Frank, Z.; Fülöp, F.; Török, Z.; Balogh, G.; Sántha, M.; Bálind, Á.; Bernáth, S.; Blundell, K.L.; et al. Dihydropyridine Derivatives Modulate Heat Shock Responses and have a Neuroprotective Effect in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 557–571. [Google Scholar] [PubMed]
- Turturici, G.; Sconzo, G.; Geraci, F. Hsp70 and Its Molecular Role in Nervous System Diseases. Biochem. Res. Int. 2011, 2011, 618127. [Google Scholar] [CrossRef] [Green Version]
- Glaze, E.R.; Lambert, A.L.; Smith, A.C.; Page, J.G.; Johnson, W.D.; McCormick, D.L.; Brown, A.P.; Levine, B.S.; Covey, J.M.; Egorin, M.J.; et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: Potential clinical relevance. Cancer Chemother. Pharmacol. 2005, 56, 637–647. [Google Scholar] [CrossRef]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [PubMed]
- Kalmar, B.; Greensmith, L. Activation of the heat shock response in a primary cellular model of motoneuron neurodegenera-tion-evidence for neuroprotective and neurotoxic effects. Cell. Mol. Biol. Lett. 2009, 14, 319–335. [Google Scholar]
- Gass, P.; Prior, P.; Kiessling, M. Correlation between seizure intensity and stress protein expression after limbic epilepsy in the rat brain. Neuroscience 1995, 65, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Hsu, C.; Liao, W.; Chuang, J.S. Heat shock protein 70 expression in epilepsy suggests stress rather than protection. Acta Neuropathol. 2008, 115, 219–230. [Google Scholar] [CrossRef]
- Guzhova, I.; Kislyakova, K.; Moskaliova, O.; Fridlanskaya, I.; Tytell, M.; Cheetham, M.; Margulis, B. In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res. 2001, 914, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hightower, L.E.; Guidon, P.T. Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J. Cell. Physiol. 1989, 138, 257–266. [Google Scholar] [CrossRef]
- Tytell, M.; Lasek, R.J.; Gainer, H. Axonal maintenance, glia, exosomes, and heat shock proteins. F1000Research 2016, 5, F1000 Faculty Rev-205. [Google Scholar]
- Chen, T.; Tan, J.; Wan, Z.; Zou, Y.; Afewerky, H.K.; Zhang, Z.; Zhang, T. Effects of Commonly Used Pesticides in China on the Mito-chondria and Ubiquitin-Proteasome System in Parkinson’s Disease. Int. J. Mol. Sci. 2017, 18, 2507. [Google Scholar] [PubMed] [Green Version]
- Njomen, E.; Tepe, J.J. Proteasome Activation as a New Therapeutic Approach To Target Proteotoxic Disorders. J. Med. Chem. 2019, 62, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Tepe, J.J. Proteasome Activation to Combat Proteotoxicity. Molecules 2019, 24, 2841. [Google Scholar] [CrossRef] [Green Version]
- Vechio, F.H.D.; Cerqueira, F.; Augusto, O.; Lopes, R.; Demasi, M. Peptides that activate the 20S proteasome by gate opening increased oxidized protein removal and reduced protein aggregation. Free. Radic. Biol. Med. 2014, 67, 304–313. [Google Scholar] [CrossRef]
- Guo, X.; Huang, X.; Chen, M.J. Reversible phosphorylation of the 26S proteasome. Protein Cell 2017, 8, 255–272. [Google Scholar] [CrossRef] [Green Version]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar]
- George, D.E.; Tepe, J.J. Advances in Proteasome Enhancement by Small Molecules. Biomolecules 2021, 11, 1789. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Tsai, C.-W. Carnosic acid protects SH-SY5Y cells against 6-hydroxydopamine-induced cell death through upregulation of parkin pathway. Neuropharmacology 2016, 110 (Pt A), 109–117. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, S.; Li, Q.; Lang, W.; Li, W.; Jiang, X.; Wan, Z.; Chen, J.; Wang, H. Epigallocatechin-3-gallate: A phytochemical as a promising drug candidate for the treatment of Parkinson’s disease. Front. Pharmacol. 2022, 13, 977521. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Li, L.; Dong, L.; Wang, S.; Jiang, X.; Zhang, T.; Jin, P.; Fan, J.; Mao, S.; Fan, X.; et al. Epigallocatechin-3-gallate (EGCG) inhibits aggregation of pulmonary fibrosis associated mutant surfactant protein A2 via a proteasomal degradation pathway. Int. J. Biochem. Cell Biol. 2019, 116, 105612. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.B.; Sodero, A.C.R.; Cordeiro, Y. Green Tea Epigallocatechin-3-gallate (EGCG) Targeting Protein Misfolding in Drug Discovery for Neurodegenerative Diseases. Biomolecules 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- Maher, P. The flavonoid fisetin promotes nerve cell survival from trophic factor withdrawal by enhancement of proteasome activity. Arch. Biochem. Biophys. 2008, 476, 139–144. [Google Scholar] [CrossRef]
- Hassan, S.S.U.; Samanta, S.; Dash, R.; Karpiński, T.M.; Habibi, E.; Sadiq, A.; Ahmadi, A.; Bunagu, S. The neuroprotective effects of fisetin, a natural flavonoid in neurodegenerative diseases: Focus on the role of oxidative stress. Front. Pharmacol. 2022, 13, 1015835. [Google Scholar]
- Boado, R.J. IgG Fusion Proteins for Brain Delivery of Biologics via Blood–Brain Barrier Receptor-Mediated Transport. Pharmaceutics 2022, 14, 1476. [Google Scholar] [CrossRef]
- Alotaibi, B.S.; Buabeid, M.; Ibrahim, N.A.; Kharaba, Z.J.; Ijaz, M.; Noreen, S.; Murtaza, G. Potential of Nanocarrier-Based Drug Delivery Systems for Brain Targeting: A Current Review of Literature. Int. J. Nanomed. 2021, 16, 7517–7533. [Google Scholar]
- Ahlawat, J.; Barroso, G.G.; Asil, S.M.; Alvarado, M.; Armendariz, I.; Bernal, J.; Carabaza, X.; Chavez, S.; Cruz, P.; Escalante, V.; et al. Nanocarriers as Potential Drug Delivery Candidates for Overcoming the Blood–Brain Barrier: Challenges and Possibilities. ACS Omega 2020, 5, 12583–12595. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moyano, P.; Sola, E.; Naval, M.V.; Guerra-Menéndez, L.; Fernández, M.D.l.C.; del Pino, J. Neurodegenerative Proteinopathies Induced by Environmental Pollutants: Heat Shock Proteins and Proteasome as Promising Therapeutic Tools. Pharmaceutics 2023, 15, 2048. https://doi.org/10.3390/pharmaceutics15082048
Moyano P, Sola E, Naval MV, Guerra-Menéndez L, Fernández MDlC, del Pino J. Neurodegenerative Proteinopathies Induced by Environmental Pollutants: Heat Shock Proteins and Proteasome as Promising Therapeutic Tools. Pharmaceutics. 2023; 15(8):2048. https://doi.org/10.3390/pharmaceutics15082048
Chicago/Turabian StyleMoyano, Paula, Emma Sola, María Victoria Naval, Lucia Guerra-Menéndez, Maria De la Cabeza Fernández, and Javier del Pino. 2023. "Neurodegenerative Proteinopathies Induced by Environmental Pollutants: Heat Shock Proteins and Proteasome as Promising Therapeutic Tools" Pharmaceutics 15, no. 8: 2048. https://doi.org/10.3390/pharmaceutics15082048