

Customizable 3D Printed Implants Containing Triamcinolone Acetonide: Development, Analysis, Modification, and Modeling of Drug Release

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Hot-Melt Extrusion of Filaments
2.2.2. Triamcinolone Acetonide Assay of Filaments
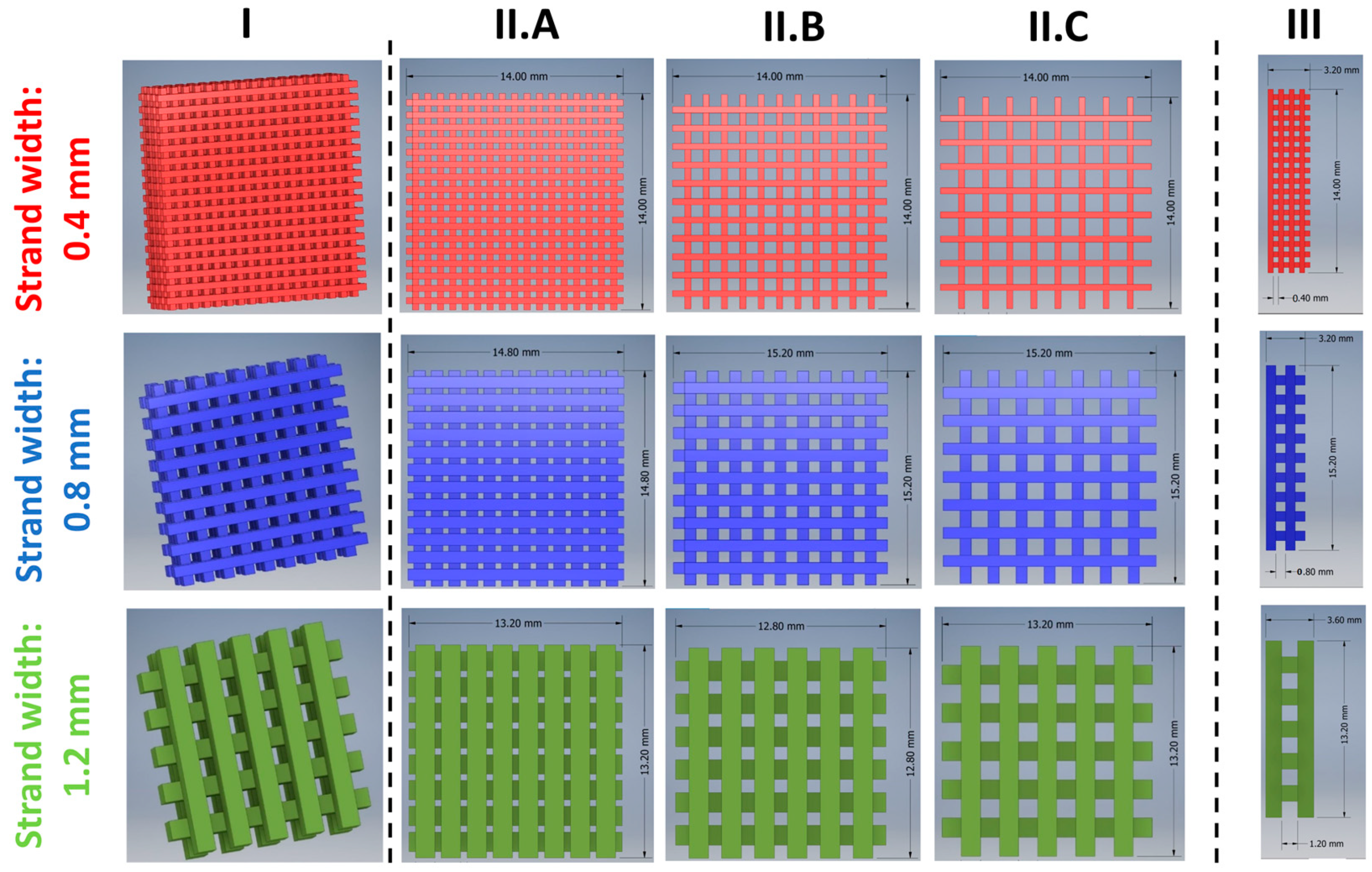
2.2.3. Design and 3D Printing of TA-loaded Implants
2.2.4. Drug Content Determination of Two-Compartment Implants
2.2.5. Determination of the Implant Surface Area Using X-ray Microcomputed Tomography
2.2.6. In Vitro Long-Term Dissolution Analysis
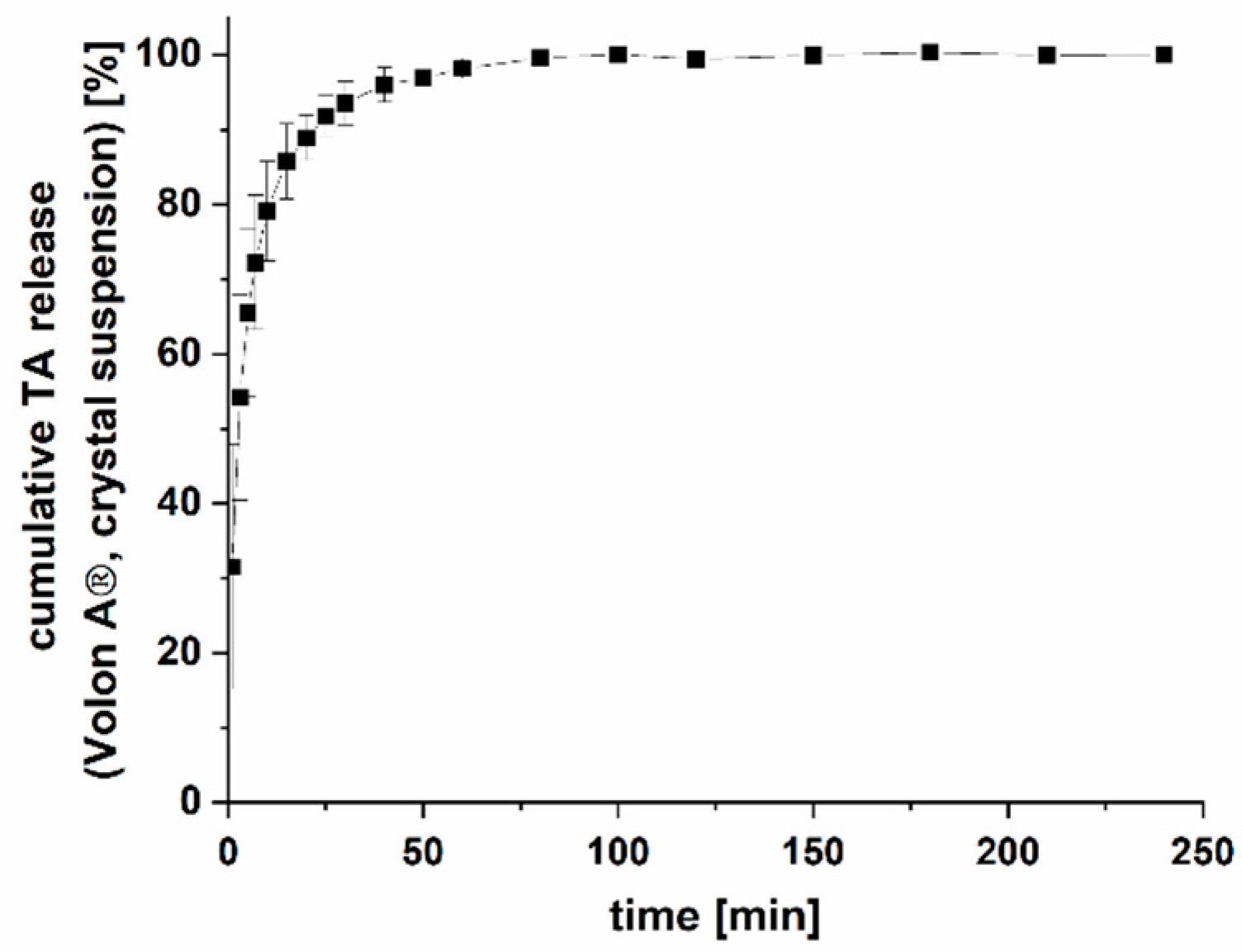
2.2.7. Drug Release of Volon® A 10-5 mL (TA Crystal Suspension)
2.2.8. Fit by the Korsmeyer–Peppas Model
2.2.9. Fitting of Dissolution Curves
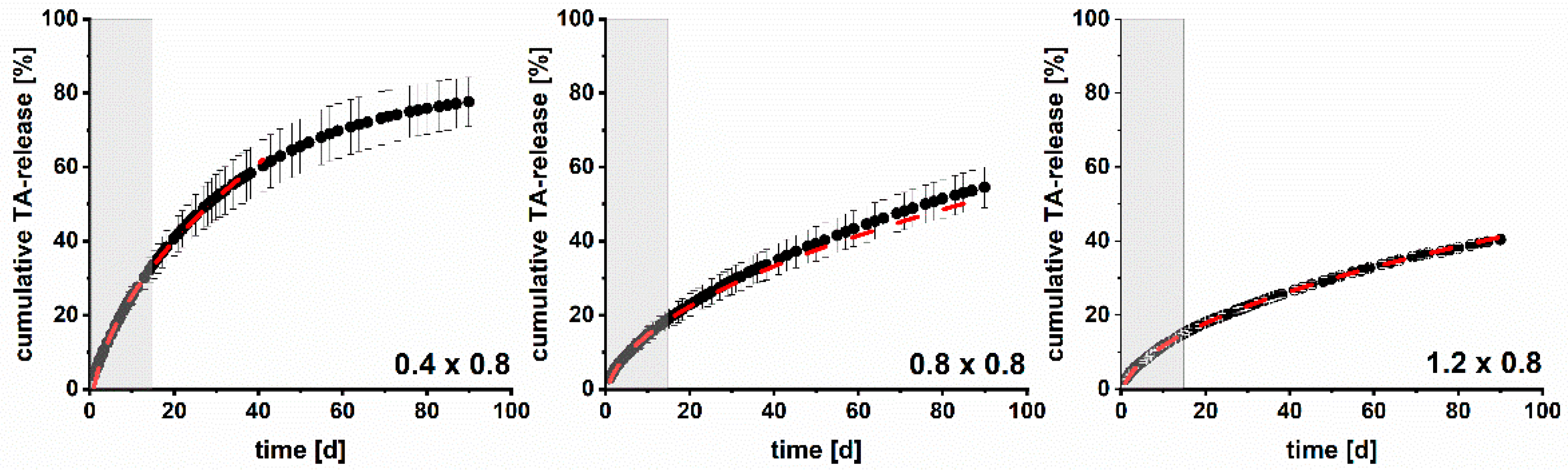
2.2.10. Prediction of Drug Release Using the Higuchi Model
2.2.11. Root Mean Square Error of Prediction (RMSEP)
3. Results and Discussion
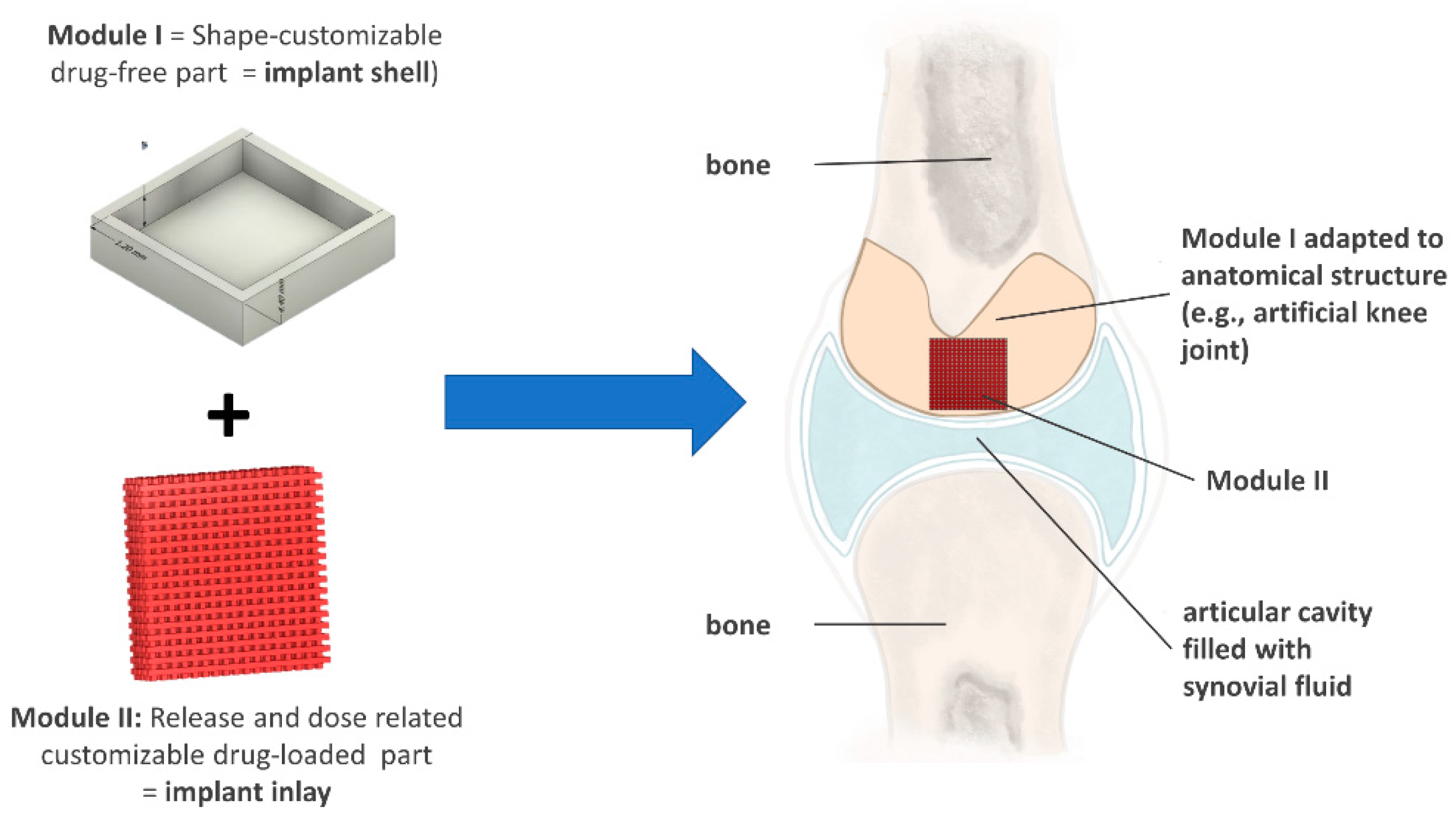
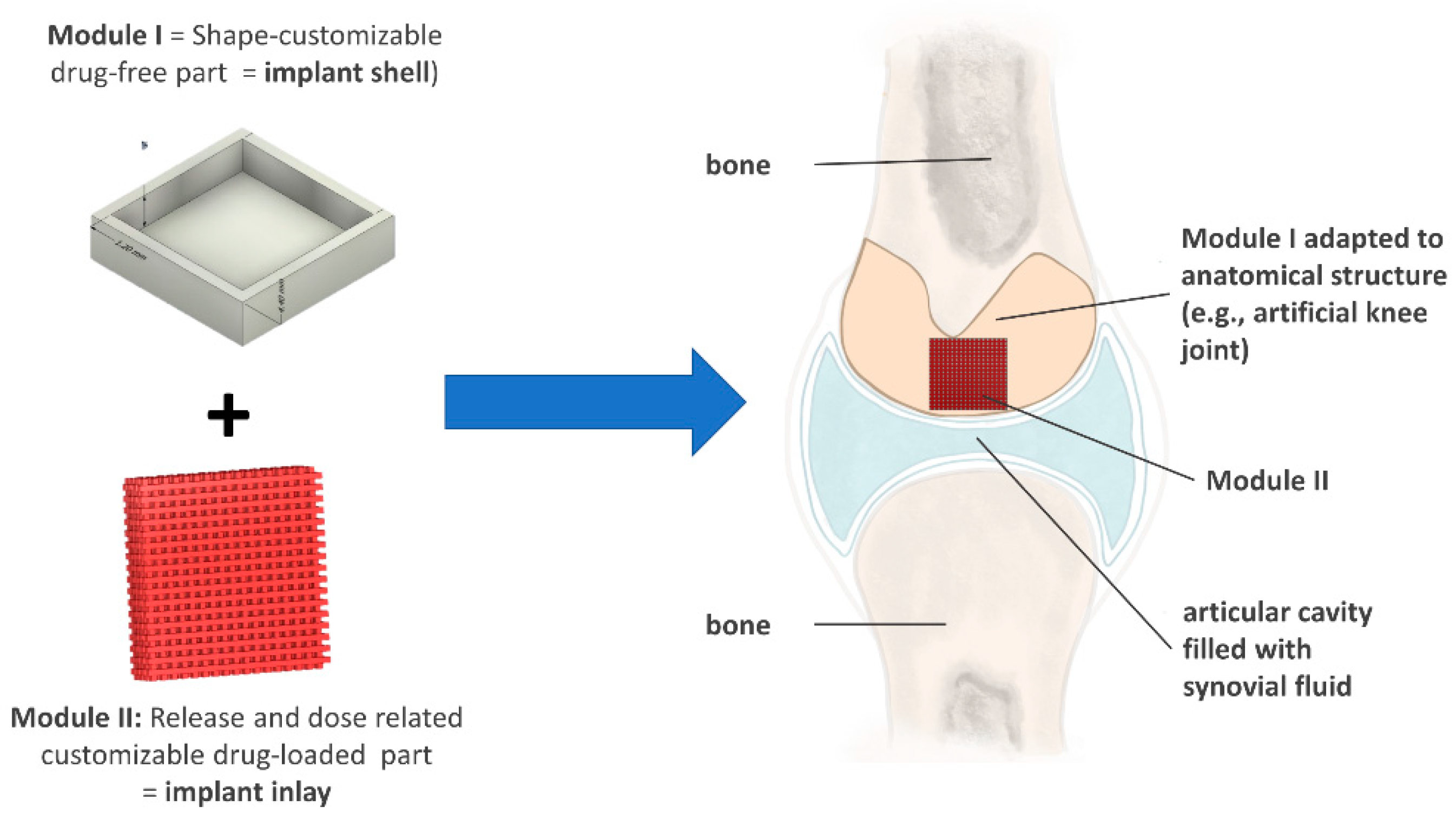
3.1. Development of a Modular Implant Concept Using Computer-Aided Design (CAD)
3.1.1. Development of a General Customizable Modular Implant Design
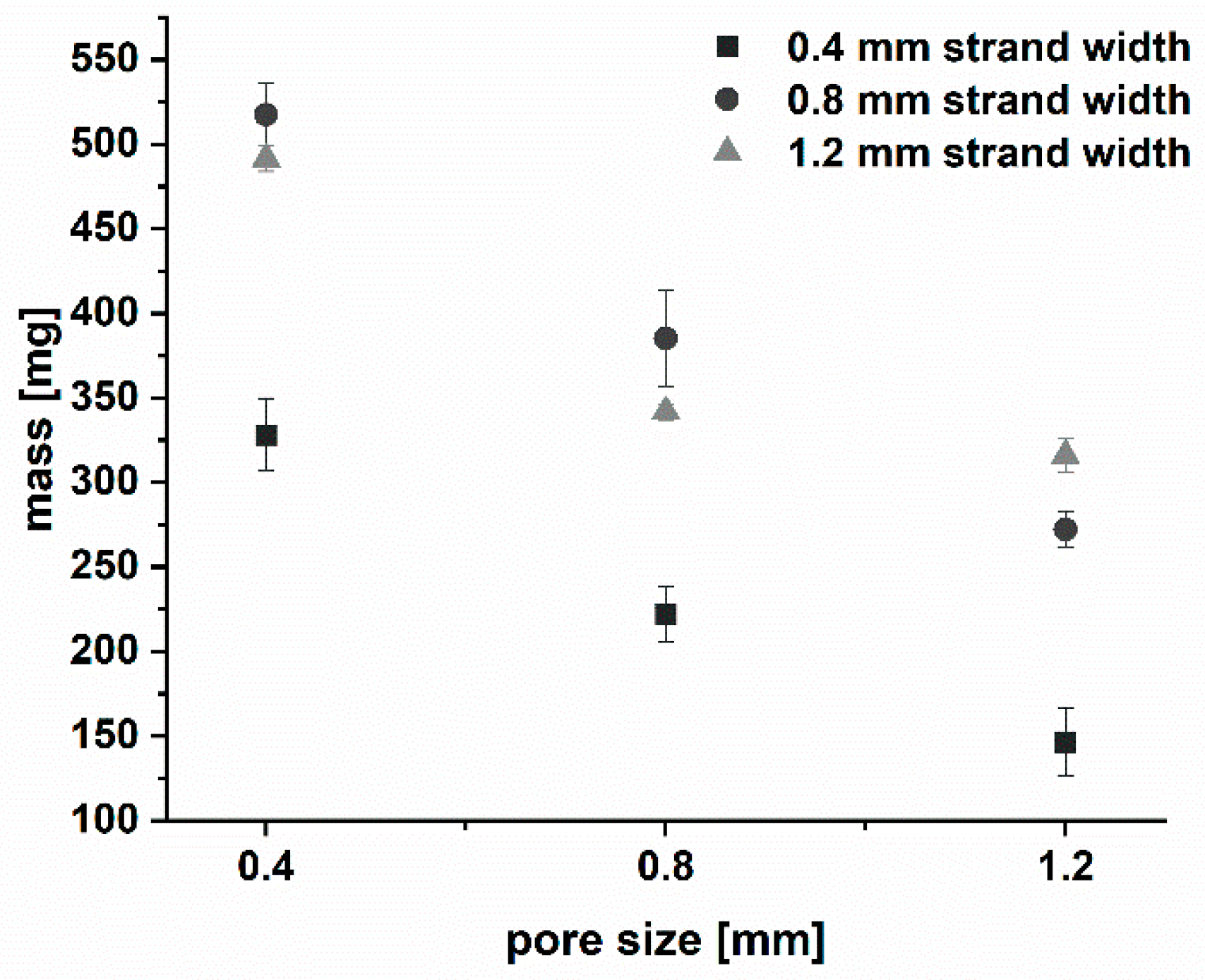
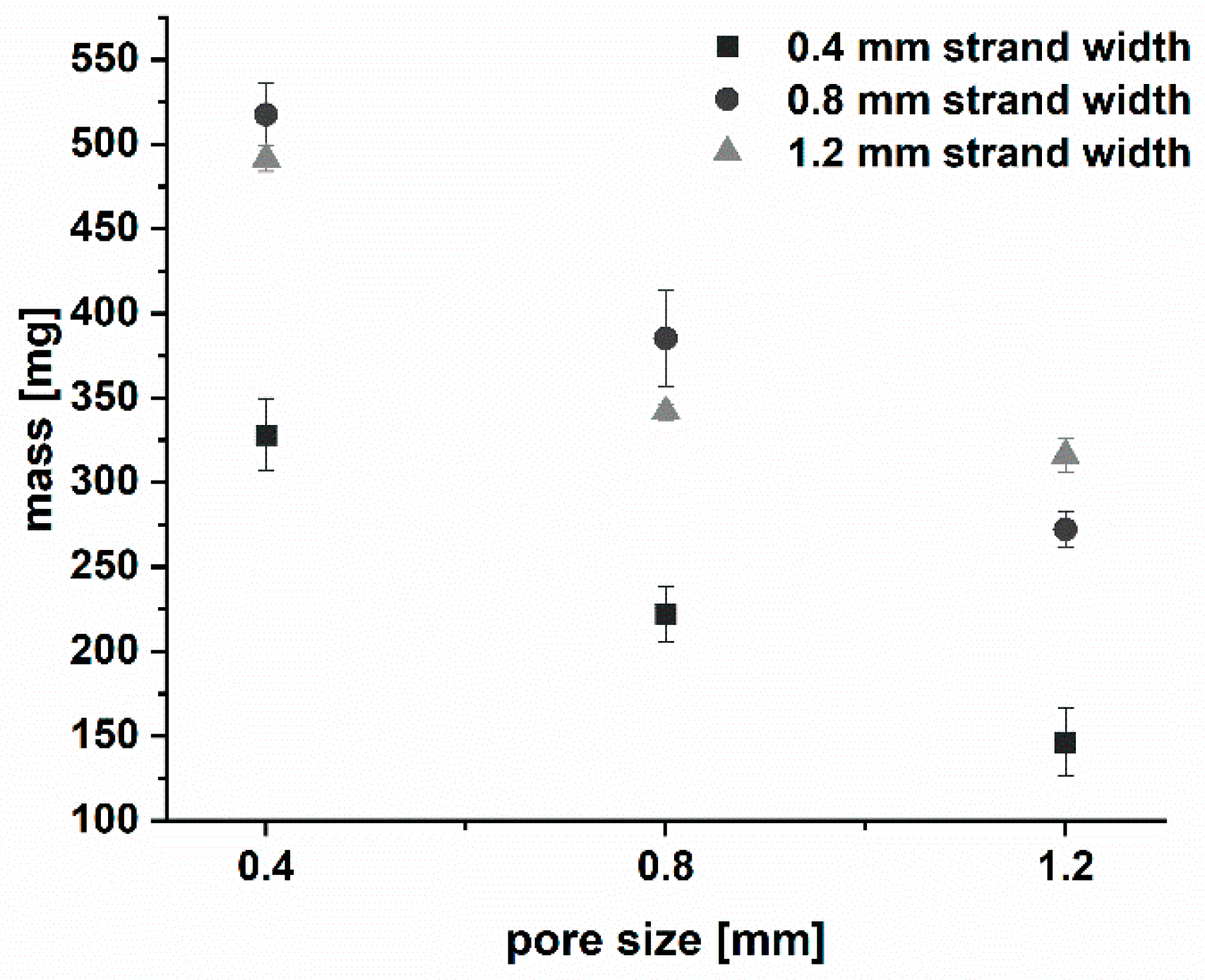
3.1.2. Production and Physical Properties of Two-Component Implants
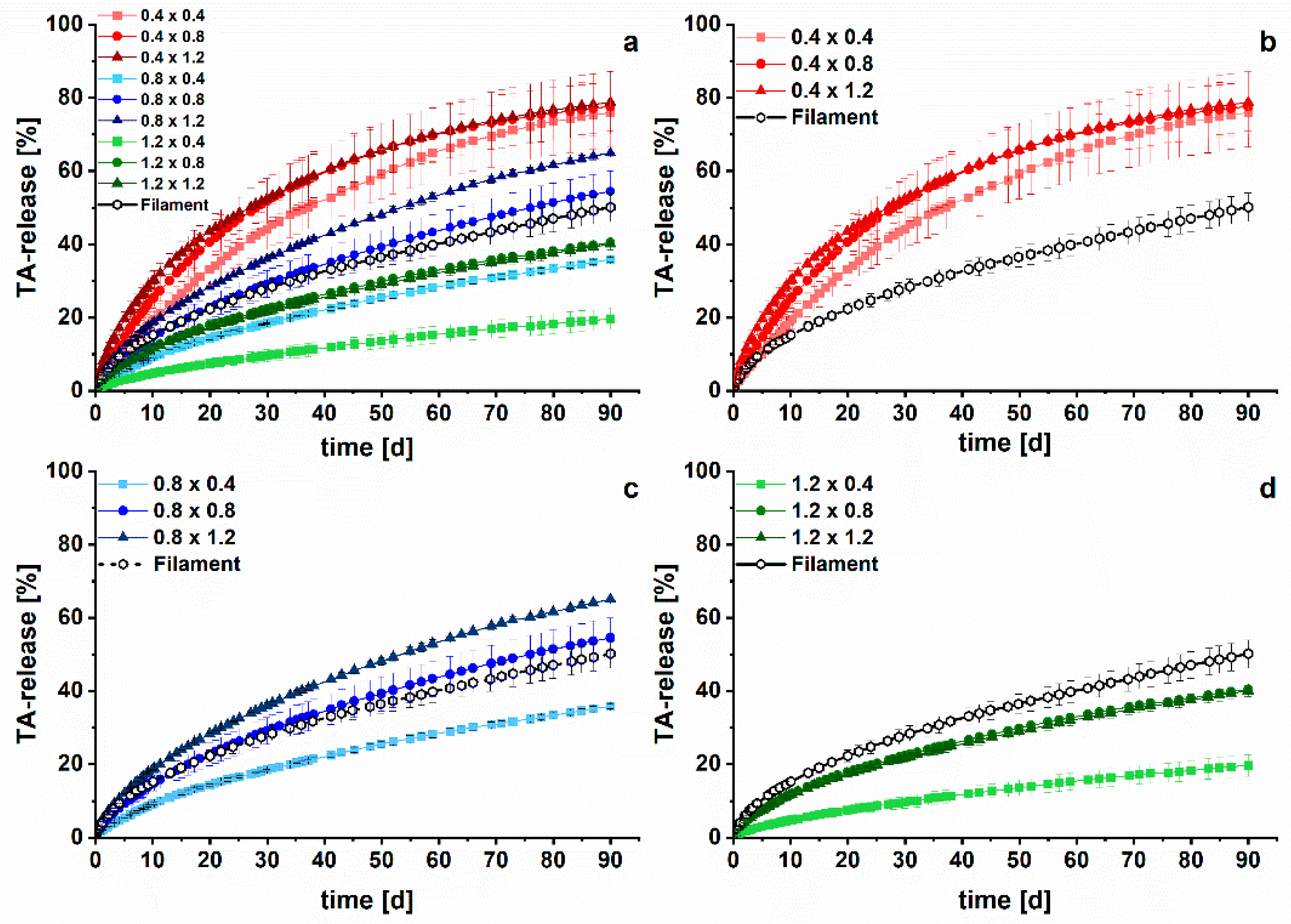
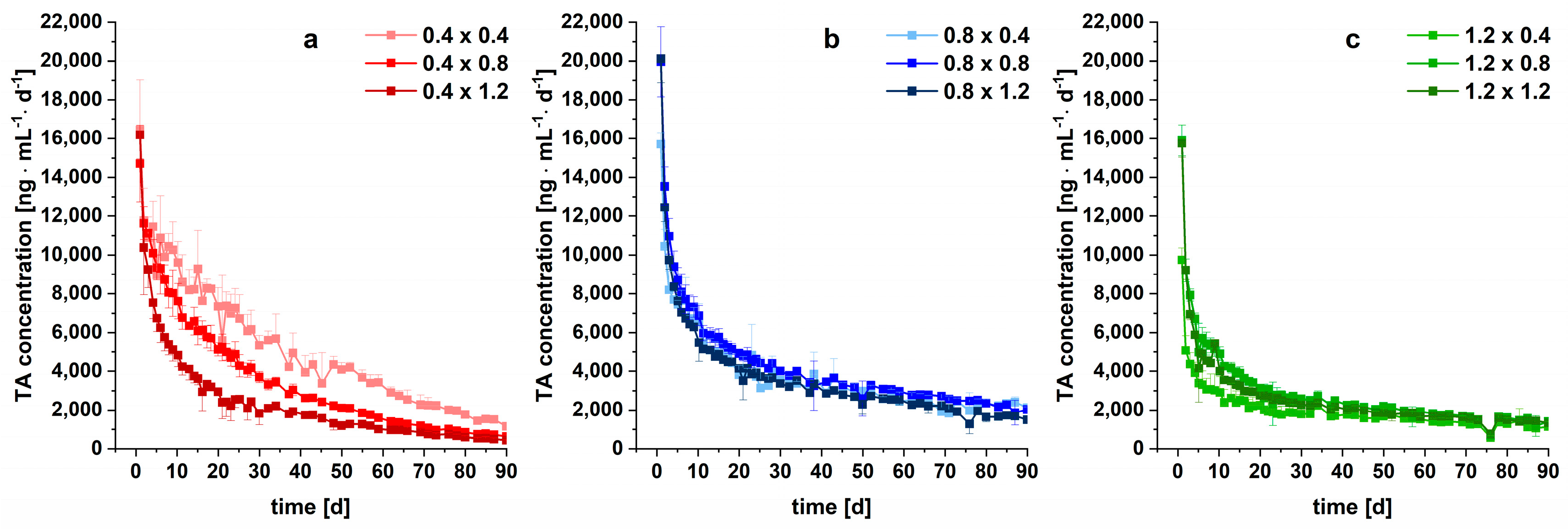
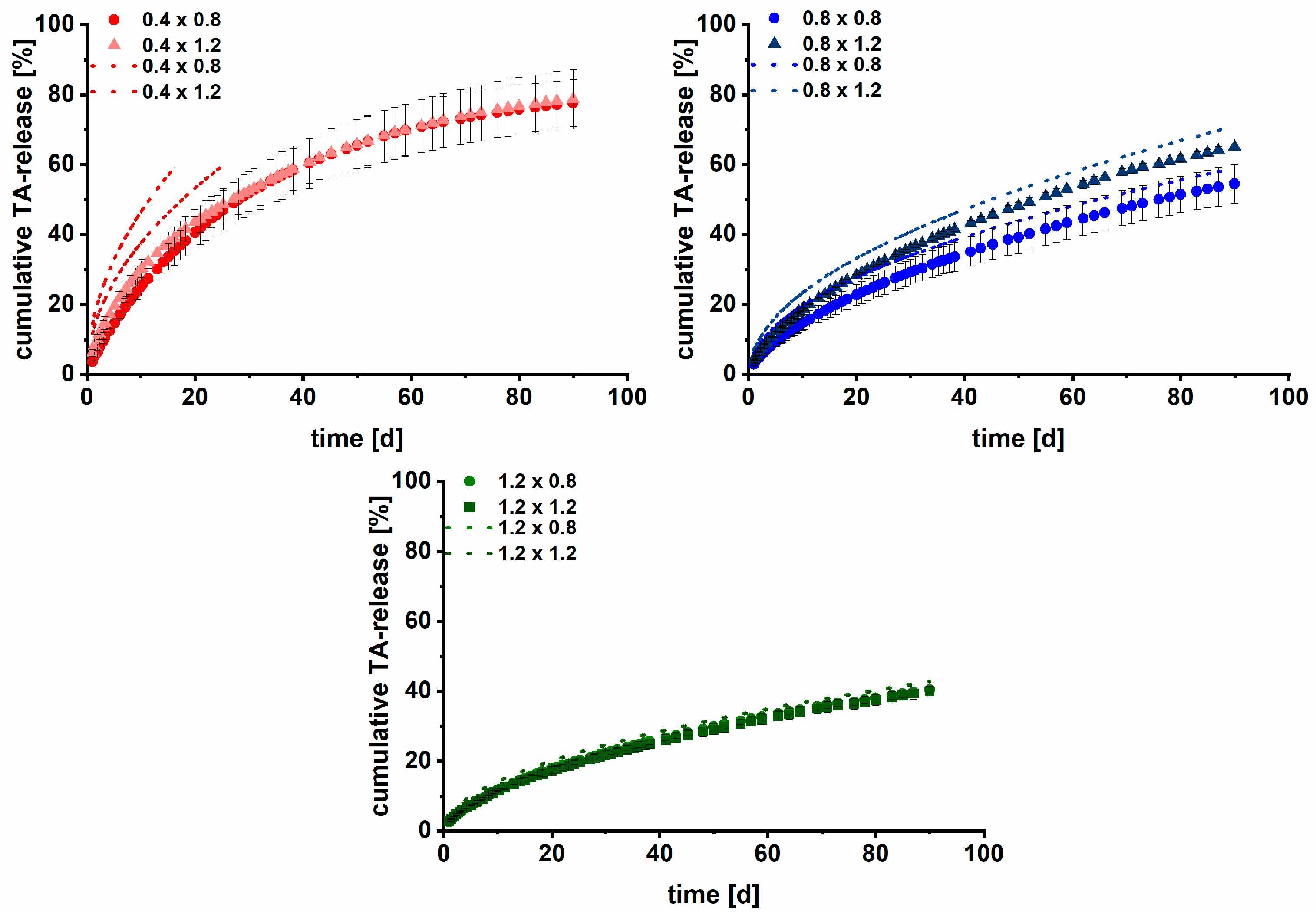
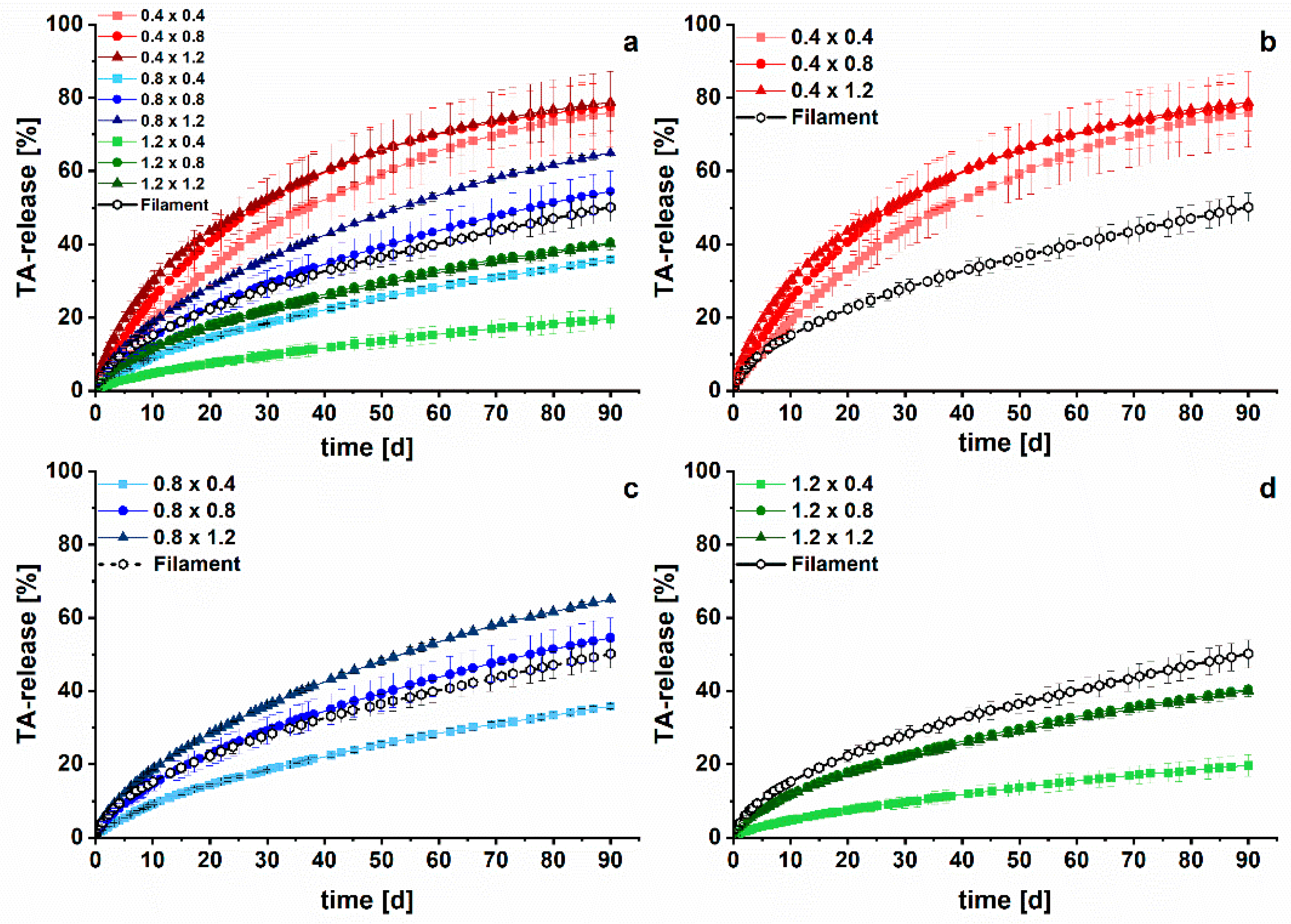
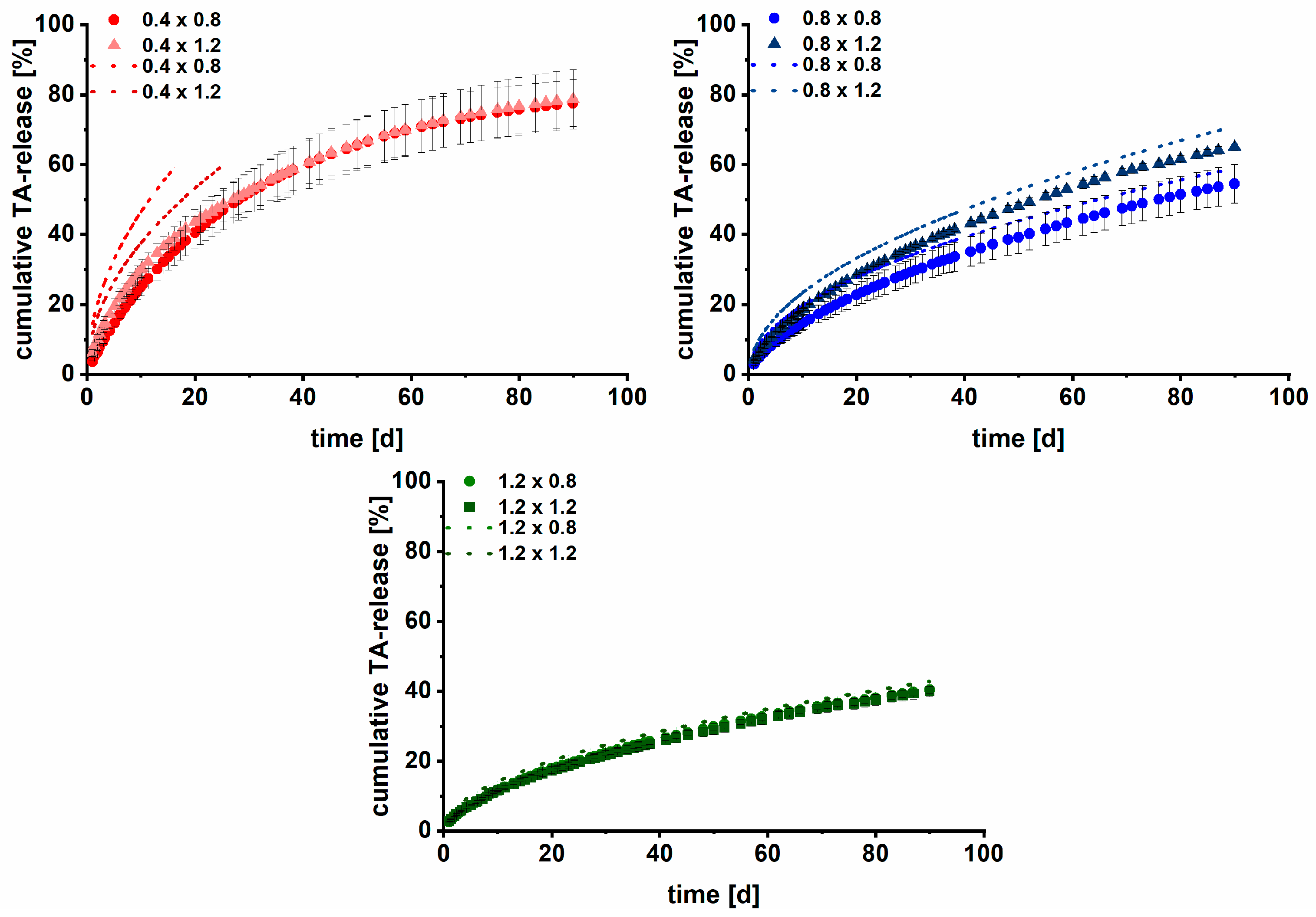
3.2. Long-Term Dissolution Studies of Implants
3.3. Kinetic Analysis
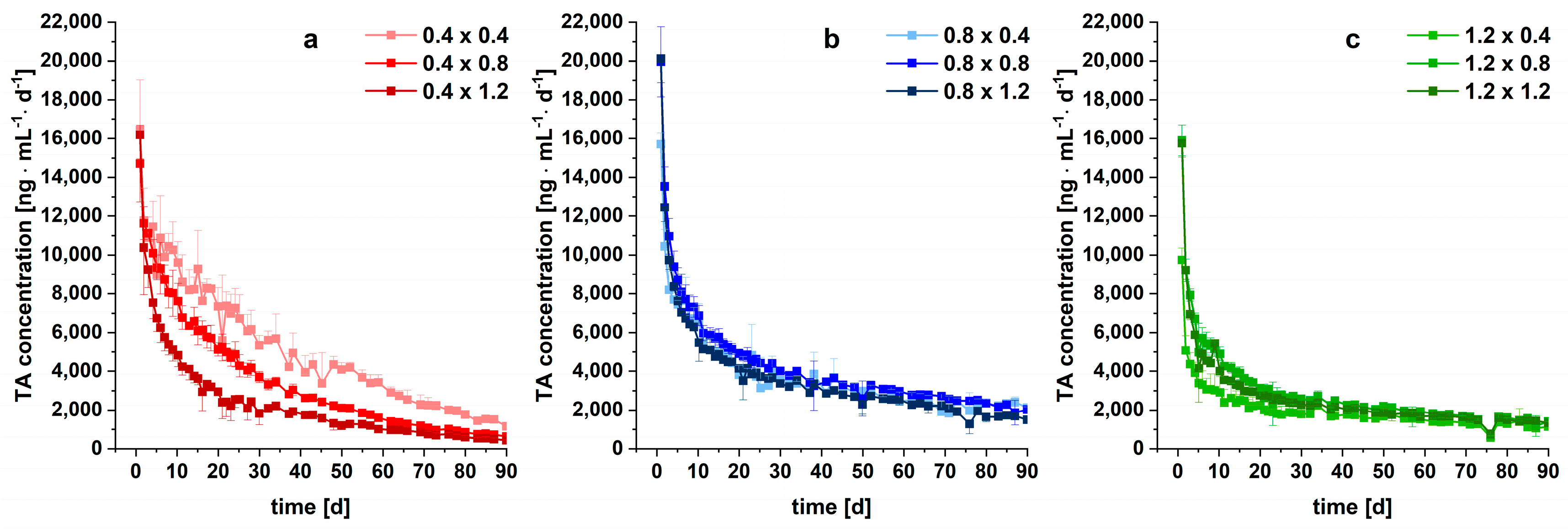
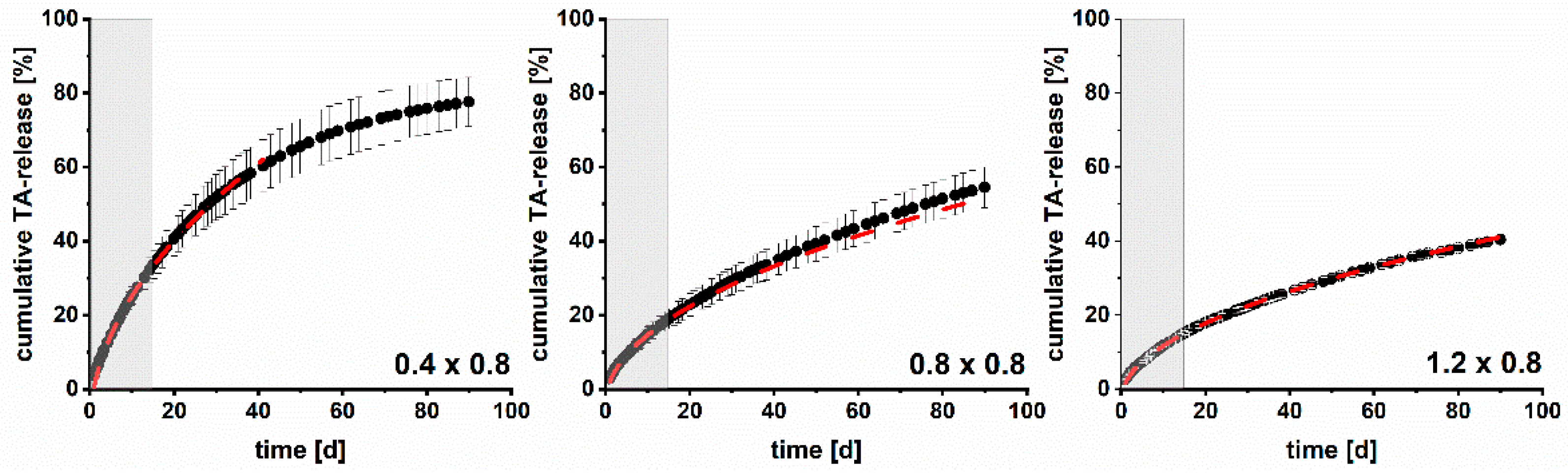
3.4. Modelling and Prediction of Drug Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.-C. A 12-Gene Pharmacogenetic Panel to Prevent Adverse Drug Reactions: An Open-Label, Multicentre, Controlled, Cluster-Randomised Crossover Implementation Study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef]
- Schlender, J.-F.; Meyer, M.; Thelen, K.; Krauss, M.; Willmann, S.; Eissing, T.; Jaehde, U. Development of a Whole-Body Physiologically Based Pharmacokinetic Approach to Assess the Pharmacokinetics of Drugs in Elderly Individuals. Clin. Pharmacokinet. 2016, 55, 1573–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamburg, M.A.; Collins, F.S. The Path to Personalized Medicine. N. Engl. J. Med. 2010, 363, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J.; Crommelin, D.J.; Hussain, A.S.; Chen, M.-L. Assuring Quality and Performance of Sustained and Controlled Release Parenterals: Eufeps Workshop Report. AAPS PharmSci 2004, 6, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Fialho, S.L.; Silva Cunha, A.D. Manufacturing Techniques of Biodegradable Implants Intended for Intraocular Application. Drug Deliv. 2005, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Aw, M.S.; Bariana, M.; Kumeria, T.; Wang, Y.; Losic, D. Drug-Releasing Implants: Current Progress, Challenges and Perspectives. J. Mater. Chem. B 2014, 2, 6157–6182. [Google Scholar] [CrossRef]
- Stewart, S.A.; Domínguez-Robles, J.; Donnelly, R.F.; Larrañeta, E. Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials; Clinical Applications. Polymers 2018, 10, 1379. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.; Madurawe, R.D.; Moore, C.M.V.; Khan, M.A.; Khairuzzaman, A. A New Chapter in Pharmaceutical Manufacturing: 3D printed Drug Products. Adv. Drug Deliv. Rev. 2017, 108, 39–50. [Google Scholar] [CrossRef]
- Goyanes, A. Effect of Geometry on Drug Release from 3d Printed Tablets. Int. J. Pharm. 2015, 494, 657. [Google Scholar] [CrossRef]
- Alhnan, M.A.; Okwuosa, T.C.; Sadia, M.; Wan, K.W.; Ahmed, W.; Arafat, B. Emergence of 3d Printed Dosage Forms: Opportunities and Challenges. Pharm. Res. 2016, 33, 1817–1832. [Google Scholar] [CrossRef]
- Sutradhar, A.; Park, J.; Carrau, D.; Miller, M.J. Experimental Validation of 3d Printed Patient-Specific Implants Using Digital Image Correlation and Finite Element Analysis. Comput. Biol. Med. 2014, 52, 8–17. [Google Scholar] [CrossRef]
- Gronet, P.M.; Waskewicz, G.A.; Richardson, C. Preformed Acrylic Cranial Implants Using Fused Deposition Modeling: A Clinical Report. J. Prosthet. Dent. 2003, 90, 429–433. [Google Scholar] [CrossRef]
- Nyberg, E.L.; Farris, A.L.; Hung, B.P.; Dias, M.; Garcia, J.R.; Dorafshar, A.H.; Grayson, W.L. 3D printing Technologies for Craniofacial Rehabilitation, Reconstruction; Regeneration. Ann. Biomed. Eng. 2017, 45, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrzak, K.; Isreb, A.; Alhnan, M.A. A Flexible-Dose Dispenser for Immediate and Extended Release 3d Printed Tablets. Eur. J. Pharm. Biopharm. 2015, 96, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Korte, C.; Quodbach, J. 3D printed Network Structures as Controlled-Release Drug Delivery Systems: Dose Adjustment, Api Release Analysis and Prediction. AAPS PharmSciTech 2018, 19, 3333–3342. [Google Scholar] [CrossRef] [PubMed]
- Skowyra, J.; Pietrzak, K.; Alhnan, M.A. Fabrication of Extended-Release Patient-Tailored Prednisolone Tablets Via Fused Deposition Modelling (Fdm) 3d Printing. Eur. J. Pharm. Sci. 2015, 68, 11–17. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3d Printing of Tablets Containing Multiple Drugs with Defined Release Profiles. Int. J. Pharm. 2015, 494, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Maroni, A.; Melocchi, A.; Parietti, F.; Foppoli, A.; Zema, L.; Gazzaniga, A. 3d Printed Multi-Compartment Capsular Devices for Two-Pulse Oral Drug Delivery. J. Control. Release 2017, 268, 10–18. [Google Scholar] [CrossRef]
- Domsta, V.; Seidlitz, A. 3D printing of Drug-Eluting Implants: An Overview of the Current Developments Described in the Literature. Molecules 2021, 26, 4066. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Domínguez-Robles, J.; McIlorum, V.J.; Mancuso, E.; Lamprou, D.A.; Donnelly, R.F.; Larrañeta, E. Development of a Biodegradable Subcutaneous Implant for Prolonged Drug Delivery Using 3d Printing. Pharmaceutics 2020, 12, 105. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.; Moore, C.; Seyler, T.; Gall, K. Modulating Antibiotic Release from Reservoirs in 3D printed Orthopedic Devices to Treat Periprosthetic Joint Infection. J. Orthop. Res. 2020, 38, 2239–2249. [Google Scholar] [CrossRef] [PubMed]
- Weisman, J.A.; Ballard, D.H.; Jammalamadaka, U.; Tappa, K.; Sumerel, J.; D’Agostino, H.B.; Mills, D.K.; Woodard, P.K. 3d Printed Antibiotic and Chemotherapeutic Eluting Catheters for Potential Use in Interventional Radiology: In Vitro Proof of Concept Study. Acad. Radiol. 2019, 26, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Water, J.J.; Bohr, A.; Boetker, J.; Aho, J.; Sandler, N.; Nielsen, H.M.; Rantanen, J. Three-Dimensional Printing of Drug-Eluting Implants: Preparation of an Antimicrobial Polylactide Feedstock Material. J. Pharm. Sci. 2015, 104, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Derendorf, H.; Hochhaus, G.; Rohatagi, S.; Möllmann, H.; Barth, J.; Sourgens, H.; Erdmann, M. Pharmacokinetics of Triamcinolone Acetonide after Intravenous, Oral; Inhaled Administration. J. Clin. Pharmacol. 1995, 35, 302–305. [Google Scholar] [CrossRef]
- Nixon, M.; Andrew, R.; Chapman, K.E. It Takes Two to Tango: Dimerisation of Glucocorticoid Receptor and Its Anti-Inflammatory Functions. Steroids 2013, 78, 59–68. [Google Scholar] [CrossRef]
- Fernández, L.S.; Castro, M.F.; Sánchez, J.L.A. Use of Glucocorticosteroids in Rheumatoid Arthritis. How and When Should Steroids Be Used in Rheumatoid Arthritis? Reum. Clin. 2007, 3, 262–269. [Google Scholar] [CrossRef]
- Østergaard, M.; Halberg, P. Intra-Articular Corticosteroids in Arthritic Disease. Biodrugs 1998, 9, 95–103. [Google Scholar] [CrossRef]
- Kraus, V.B.; Conaghan, P.G.; Aazami, H.A.; Mehra, P.; Kivitz, A.J.; Lufkin, J.; Hauben, J.; Johnson, J.R.; Bodick, N. Synovial and Systemic Pharmacokinetics (Pk) of Triamcinolone Acetonide (Ta) Following Intra-Articular (Ia) Injection of an Extended-Release Microsphere-Based Formulation (Fx006) or Standard Crystalline Suspension in Patients with Knee Osteoarthritis (Oa). Osteoarthr. Cart. 2018, 26, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Derendorf, H.; Möllmann, H.; Grüner, A.; Haack, D.; Gyselby, G. Pharmacokinetics and Pharmacodynamics of Glucocorticoid Suspensions after Intra-Articular Administration. Clin. Pharmacol. Ther. 1986, 39, 313–317. [Google Scholar] [CrossRef]
- Gerwin, N.; Hops, C.; Lucke, A. Intraarticular Drug Delivery in Osteoarthritis. Adv. Drug Deliv. Rev. 2006, 58, 226–242. [Google Scholar] [CrossRef]
- Jüni, P.; Hari, R.; Rutjes, A.W.S.; Fischer, R.; Silletta, M.G.; Reichenbach, S.; da Costa, B.R. Intra-Articular Corticosteroid for Knee Osteoarthritis. Cochrane Database Syst. Rev. 2015, 11, CD005328. [Google Scholar] [CrossRef] [Green Version]
- Godwin, M.; Dawes, M. Intra-Articular Steroid Injections for Painful Knees. Systematic Review with Meta-Analysis. Can. Fam. Physician 2004, 50, 241–248. [Google Scholar]
- Nkanga, C.I.; Fisch, A.; Rad-Malekshahi, M.; Romic, M.D.; Kittel, B.; Ullrich, T.; Wang, J.; Krause, R.W.M.; Adler, S.; Lammers, T. Clinically Established Biodegradable Long Acting Injectables: An Industry Perspective. Adv. Drug Deliv. Rev. 2020, 167, 19–46. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S.T.; Keam, S.J. Triamcinolone Acetonide Extended-Release: A Review in Osteoarthritis Pain of the Knee. Drugs 2019, 79, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Conaghan, P.G.; Hunter, D.J.; Cohen, S.B.; Kraus, V.B.; Berenbaum, F.; Lieberman, J.R.; Jones, D.G.; Spitzer, A.I.; Jevsevar, D.S.; Katz, N.P.; et al. Effects of a Single Intra-Articular Injection of a Microsphere Formulation of Triamcinolone Acetonide on Knee Osteoarthritis Pain: A Double-Blinded, Randomized, Placebo-Controlled, Multinational Study. J. Bone Jt. Surg. 2018, 100, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Bodick, N.; Lufkin, J.; Willwerth, C.; Kumar, A.; Bolognese, J.; Schoonmaker, C.; Ballal, R.; Hunter, D.; Clayman, M. An Intra-Articular, Extended-Release Formulation of Triamcinolone Acetonide Prolongs and Amplifies Analgesic Effect in Patients with Osteoarthritis of the Knee: A Randomized Clinical Trial. J. Bone Jt. Surg. 2015, 97, 877–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.F.; Puga, A.M.; Diaz-Gomez, L.; Concheiro, A.; Busch, D.H.; Alvarez-Lorenzo, C. Additive Manufacturing of Scaffolds with Dexamethasone Controlled Release for Enhanced Bone Regeneration. Int. J. Pharm. 2015, 496, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Farmer, Z.-L.; Utomo, E.; Domínguez-Robles, J.; Mancinelli, C.; Mathew, E.; Larrañeta, E.; Lamprou, D.A. 3d Printed Estradiol-Eluting Urogynecological Mesh Implants: Influence of Material and Mesh Geometry on Their Mechanical Properties. Int. J. Pharm. 2021, 593, 120145. [Google Scholar] [CrossRef]
- Holländer, J.; Hakala, R.; Suominen, J.; Moritz, N.; Yliruusi, J.; Sandler, N. 3d Printed Uv Light Cured Polydimethylsiloxane Devices for Drug Delivery. Int. J. Pharm. 2018, 544, 433–442. [Google Scholar] [CrossRef]
- Sadia, M.; Arafat, B.; Ahmed, W.; Forbes, R.T.; Alhnan, M.A. Channelled Tablets: An Innovative Approach to Accelerating Drug Release from 3d Printed Tablets. J. Control. Release 2018, 269, 355–363. [Google Scholar] [CrossRef]
- Wu, C.; Luo, Y.; Cuniberti, G.; Xiao, Y.; Gelinsky, M. Three-Dimensional Printing of Hierarchical and Tough Mesoporous Bioactive Glass Scaffolds with a Controllable Pore Architecture, Excellent Mechanical Strength and Mineralization Ability. Acta Biomater. 2011, 7, 2644–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Qiao, X.; Huang, R.; Chen, H.; Shi, X.; Wang, J.; Tan, W.; Tan, Z. E-Jet 3d Printed Drug Delivery Implants to Inhibit Growth and Metastasis of Orthotopic Breast Cancer. Biomaterials 2020, 230, 119618. [Google Scholar] [CrossRef] [PubMed]
- Hilgeroth, P.S.; Thümmler, J.F.; Binder, W.H. 3d Printing of Triamcinolone Acetonide in Triblock Copolymers of Styrene-Isobutylene-Styrene as a Slow-Release System. Polymers 2022, 14, 3742. [Google Scholar] [CrossRef] [PubMed]
- European Directorate for the Quality of Medicines & Healthcare. Triamcinolone Acetonide. In European Pharmacopoeia 9.0; Deutscher Apotheker Verlag: Stuttgart, Germany, 2017. [Google Scholar]
- Matysova, L.; Hajkova, R.; Šícha, J.; Solich, P. Determination of Methylparaben, Propylparaben, Triamcinolone Acetonide and Its Degradation Product in a Topical Cream by Rp-Hplc. Anal. Bioanal. Chem. 2003, 376, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research (CDER). Validation of Chromatographic Methods. In Reviewer Guidance; US Food and Drug Adminstration: Rockville, MD, USA, 1994. [Google Scholar]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Validation of Analytical Procedures: Text and Methodology Q2 (R1); ICH: Geneva, Switzerland, 2005. [Google Scholar]
- European Directorate for the Quality of Medicines & Healthcare. 5.17.1 Recommendations on Dissolution Testing. In European Pharmacopoeia 10.0; Deutscher Apotheker Verlag: Stuttgart, Germany, 2020. [Google Scholar]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of Solute Release from Porous Hydrophilic Polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Windolf, H.; Chamberlain, R.; Quodbach, J. Predicting Drug Release from 3d Printed Oral Medicines Based on the Surface Area to Volume Ratio of Tablet Geometry. Pharmaceutics 2021, 13, 1453. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of Sustained-Action Medication. Theoretical Analysis of Rate of Release of Solid Drugs Dispersed in Solid Matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A Simple Equation for Description of Solute Release Ii. Fickian and Anomalous Release from Swellable Devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Tack, P.; Victor, J.; Gemmel, P.; Annemans, L. 3D printing Techniques in a Medical Setting: A Systematic Literature Review. Biomed. Eng. Online 2016, 15, 115. [Google Scholar] [CrossRef] [Green Version]
- Kempin, W.; Franz, C.; Koster, L.C.; Schneider, F.; Bogdahn, M.; Weitschies, W.; Seidlitz, A. Assessment of Different Polymers and Drug Loads for Fused Deposition Modeling of Drug Loaded Implants. Eur. J. Pharm. Biopharm. 2017, 115, 84–93. [Google Scholar] [CrossRef]
- Pillai, O.; Panchagnula, R. Polymers in Drug Delivery. Curr. Opin. Chem. Biol. 2001, 5, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, E. Influence of Formulation and Process Parameters on the Release Characteristics of Ethylcellulose Sustained-Release Mini-Matrices Produced by Hot-Melt Extrusion. Eur. J. Pharm. Biopharm. 2008, 69, 312–319. [Google Scholar] [CrossRef]
- Meena, A.; Parikh, T.; Gupta, S.S.; Serajuddin, A.T.M. Investigation of Thermal and Viscoelastic Properties of Polymers Relevant to Hot Melt Extrusion-Ii: Cellulosic Polymers. J. Excip. Food Chem. 2016, 5, 1002. [Google Scholar]
- Pinto, E.; Dürig, T. Cellulose Ethers for Extrusion Applications. In Melt Extrusion: Materials, Technology and Drug Product Design; Springer: New York, NY, USA, 2013; pp. 123–144. [Google Scholar]
- Goyanes, A.; Fernandez-Ferreiro, A.; Majeed, A.; Gomez-Lado, N.; Awad, A.; Luaces-Rodriguez, A.; Gaisford, S.; Aguiar, P.; Basit, A.W. Pet/Ct Imaging of 3d Printed Devices in the Gastrointestinal Tract of Rodents. Int. J. Pharm. 2018, 536, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, X.; Patil, H.; Tiwari, R.; Repka, M. Coupling 3d Printing with Hot-Melt Extrusion to Produce Controlled-Release Tablets. Int. J. Pharm. 2017, 519, 186–197. [Google Scholar] [CrossRef]
- Gunder, W.; Lippold, B.; Lippold, B. Release of Drugs from Ethyl Cellulose Microcapsules (Diffusion Pellets) with Pore Formers and Pore Fusion. Eur. J. Pharm. Sci. 1995, 3, 203–214. [Google Scholar] [CrossRef]
- Ho, M.J.; Kim, S.R.; Choi, Y.W.; Kang, M.J. A Novel Stable Crystalline Triamcinolone Acetonide-Loaded Plga Microsphere for Prolonged Release after Intra-Articular Injection. Bull. Korean Chem. Soc. 2016, 37, 1496–1500. [Google Scholar] [CrossRef]
- Volon A 40/A 40-5 ml Summary of Product Characteristics (Fachinformation); Dermapharm: Grünwald, Germany, 2021.
- Kraus, V.B.; Stabler, T.V.; Kong, S.Y.; Varju, G.; McDaniel, G. Measurement of Synovial Fluid Volume Using Urea. Osteoarthr. Cart. 2007, 15, 1217–1220. [Google Scholar] [CrossRef] [Green Version]
- Pinto, F.C.H.; Junior, A.D.S.-C.; Oréfice, R.L.; Ayres, E.; Andrade, S.P.; Lima, L.D.C.; Moura, S.A.L.; Da Silva, G.R. Controlled Release of Triamcinolone Acetonide from Polyurethane Implantable Devices: Application for Inhibition of Inflammatory-Angiogenesis. J. Mater. Sci. Mater. Med. 2012, 23, 1431–1445. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A Simple Equation for Description of Solute Release I. Fickian and Non-Fickian Release from Non-Swellable Devices in the Form of Slabs, Spheres, Cylinders or Discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of Drug Release from Delivery Systems Based on Hydroxypropyl Methylcellulose (Hpmc). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Modeling of Diffusion Controlled Drug Delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | TA | EC | TEC | HPMC | Fumed Silica | |
|---|---|---|---|---|---|---|
| Function | API | Thermoplastic Polymer | Plasticizer | Pore Former | Glidant | |
| Formulation | ||||||
| F1 (drug-loaded implant inlay) | 10 | 54.64 | 10 | 25 | 0.36 | |
| F2 (drug-free shell) | - | 89.64 | 10 | - | 0.36 | |
| Implant | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strand width [mm] | 0.4 | 0.4 | 0.4 | 0.8 | 0.8 | 0.8 | 1.2 | 1.2 | 1.2 |
| Pore size [mm] | 0.4 | 0.8 | 1.2 | 0.4 | 0.8 | 1.2 | 0.4 | 0.8 | 1.2 |
| Dimensions (x, y) [mm] | 14 | 14 | 14 | 14.8 | 15.2 | 15.2 | 13.2 | 12.8 | 13.2 |
| Height [mm] | 3.2 | 3.2 | 3.2 | 3.2 | 3.2 | 3.2 | 3.6 | 3.6 | 3.6 |
| Mass [mg] (mean ± s) | 327.0 ± 21.5 | 222.1 ± 16.0 | 146.4 ± 19.9 | 517.3 ± 18.3 | 385.12 ± 28.8 | 272.1 ± 10.3 | 491.4 ± 7.7 | 241.7 ± 4.7 | 316.0 ± 9.7 |
| TA [mg] (mean ± s) | 29.1 ± 1.9 | 19.7 ± 1.4 | 13.0 ± 1.8 | 45.8 ± 1.6 | 34.1 ± 2.6 | 24.1 ± 0.9 | 43.5 ± 0.7 | 30.5 ± 0.4 | 28.0 ± 0.9 |
| Acal [cm2] | 23.02 | 16.93 | 12.45 | 15.78 | 13.31 | 10.89 | 10.25 | 8.06 | 7.27 |
| Atrue [cm2] | 20.54 | 18.54 | 13.08 | 12.95 | 15.30 | 13.08 | 9.80 | 8.69 | 7.91 |
| Atrue/Acal [-] | 0.89 | 1.10 | 1.05 | 0.82 | 1.15 | 1.20 | 0.96 | 1.08 | 1.09 |
| SSA [cm2/g] | 62.6 | 83.5 | 89.3 | 25.0 | 39.7 | 48.1 | 19.9 | 25.4 | 25.0 |
| Implant | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strand width [mm] | 0.4 | 0.4 | 0.4 | 0.8 | 0.8 | 0.8 | 1.2 | 1.2 | 1.2 |
| Pore size [mm] | 0.4 | 0.8 | 1.2 | 0.4 | 0.8 | 1.2 | 0.4 | 0.8 | 1.2 |
| n [mean ± s] | 0.76 ± 0.02 | 0.69 ± 0.05 | 0.54 ± 0.01 | 0.66 ± 0.02 | 0.68 ± 0.03 | 0.60 ± 0.01 | 0.65 ± 0.01 | 0.59 ± 0.02 | 0.58 ± 0.00 |
| R2 | >0.9933 | >0.9924 | >0.9887 | >0.9952 | >0.9991 | >0.9992 | >0.9986 | >0.9964 | >0.9988 |
| Implant (Strand Width × Pore Size) | [mg·cm−2·d−0.5] Based on Measured A | R2 |
|---|---|---|
| 0.4 × 0.4 | 0.1401 | 0.9935 |
| 0.4 × 0.8 | 0.1182 | 0.9977 |
| 0.4 × 1.2 | 0.1031 | 0.9981 |
| 0.8 × 0.4 | 0.1460 | 0.9988 |
| 0.8 × 0.8 | 0.1386 | 0.9993 |
| 0.8 × 1.2 | 0.1371 | 0.9994 |
| 1.2 × 0.4 | 0.1005 | 0.9952 |
| 1.2 × 0.8 | 0.1592 | 0.9997 |
| 1.2 × 1.2 | 0.1575 | 0.9994 |
| Implant (Strand Width × Pore Size) | Prediction Based on Dk and the Determined Surface Area According to [15] | Prediction Based on Initial Drug Release Data until Day 15 | ||
|---|---|---|---|---|
| Data Set Used for Prediction | RMSEP [%] | R2 | RMSEP [%] | |
| 0.4 × 0.4 | - | - | 0.9974 | 2.19 |
| 0.4 × 0.8 | 0.4 × 1.2 | 19.44 | 0.9989 | 0.62 |
| 0.4 × 1.2 | 0.4 × 0.8 | 8.24 | 0.9999 | 3.15 |
| 0.8 × 0.4 | - | - | 0.9982 | 0.86 |
| 0.8 × 0.8 | 0.8 × 1.2 | 4.29 | 0.9989 | 1.85 |
| 0.8 × 1.2 | 0.8 × 0.8 | 4.67 | 0.9998 | 1.21 |
| 1.2 × 0.4 | - | - | 0.9988 | 1.41 |
| 1.2 × 0.8 | 1.2 × 1.2 | 2.32 | 0.9979 | 0.21 |
| 1.2 × 1.2 | 1.2 × 0.8 | 2.65 | 0.9962 | 0.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponsar, H.; Quodbach, J. Customizable 3D Printed Implants Containing Triamcinolone Acetonide: Development, Analysis, Modification, and Modeling of Drug Release. Pharmaceutics 2023, 15, 2097. https://doi.org/10.3390/pharmaceutics15082097
Ponsar H, Quodbach J. Customizable 3D Printed Implants Containing Triamcinolone Acetonide: Development, Analysis, Modification, and Modeling of Drug Release. Pharmaceutics. 2023; 15(8):2097. https://doi.org/10.3390/pharmaceutics15082097
Chicago/Turabian StylePonsar, Hanna, and Julian Quodbach. 2023. "Customizable 3D Printed Implants Containing Triamcinolone Acetonide: Development, Analysis, Modification, and Modeling of Drug Release" Pharmaceutics 15, no. 8: 2097. https://doi.org/10.3390/pharmaceutics15082097
APA StylePonsar, H., & Quodbach, J. (2023). Customizable 3D Printed Implants Containing Triamcinolone Acetonide: Development, Analysis, Modification, and Modeling of Drug Release. Pharmaceutics, 15(8), 2097. https://doi.org/10.3390/pharmaceutics15082097