
Drug Repurposing to Circumvent Immune Checkpoint Inhibitor Resistance in Cancer Immunotherapy



Abstract
:1. Introduction
2. Application of Drug Repurposing to Overcome ICI Resistance
3. Methods for Identifying Drug Repurposing Candidates to Overcome ICI Resistance
3.1. Phenotypic Screening of Chemical Libraries for T Cell Modifying Drugs
3.2. Integrative Analysis of CRISPR/Cas9-Based Functional Screens for Repurposing
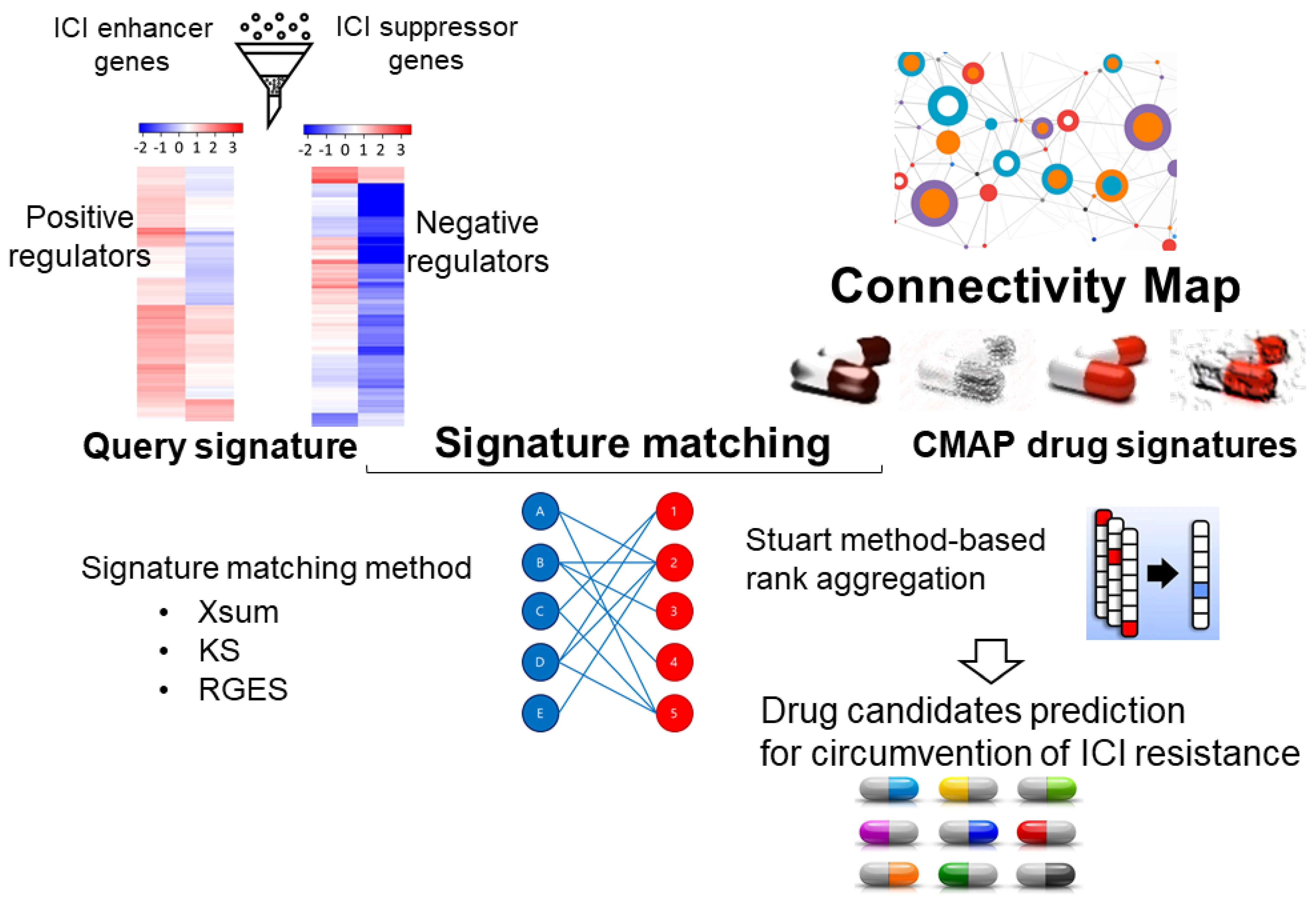
3.3. Virtual Screening and Machine Learning to Identify Novel Modulators of the Immunosuppressive TME
4. Representative Repurposed Drug Candidates to Overcome ICI Resistance
4.1. Repurposed Drug Candidates Inducing Immunostimulatory Activities
4.1.1. Metronomic Chemotherapy (Also Called Low-Dose Chemotherapy)
4.1.2. Molecular Targeted Drugs
Targeted Drugs with Anti-Angiogenic Activity
Other Small Molecule Tyrosine Kinase Inhibitors (TKIs)
Cyclin-Dependent Kinase (CDK) Inhibitors
DNA Damage Response Inhibitors (DDRIs)
4.1.3. Epigenetic Drugs
4.1.4. Drugs Promoting M1 Macrophage Polarization
4.2. Repurposed Drug Candidates Abolishing Immunosuppressive TME
4.2.1. Angiotensin II Receptor Blockers (ARBs)
4.2.2. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
4.2.3. Drugs Modulating Metabolic Pathways to Reprogram the Immunosuppressive TME
Targeting Glucose Metabolism to Enhance Anti-Tumor Efficacy of ICI Therapy
Targeting Amino Acid Catabolism to Potentiate Cancer Immunotherapy
4.3. Repurposing Traditional Chinese Medicine (TCM) to Potentiate ICI Efficacy and Overcome Drug Resistance
5. Challenges and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef]
- Bevins, N.J.; Okamura, R.; Montesion, M.; Adashek, J.J.; Goodman, A.M.; Kurzrock, R. Tumor infiltrating lymphocyte expression of PD-1 predicts response to anti-PD-1/PD-L1 immunotherapy. J. Immunother. Precis. Oncol. 2022, 5, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Vellanki, P.J.; Mulkey, F.; Jaigirdar, A.A.; Rodriguez, L.; Wang, Y.; Xu, Y.; Zhao, H.; Liu, J.; Howe, G.; Wang, J.; et al. FDA approval summary: Nivolumab with ipilimumab and chemotherapy for metastatic non-small cell lung cancer, a collaborative project Orbis review. Clin. Cancer Res. 2021, 27, 3522–3527. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Opdivo (nivolumab): Second PD-1 inhibitor receives FDA approval for unresectable or metastatic melanoma. Am. Health Drug Benefits 2015, 8, 180–183. [Google Scholar]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA approval summary: Pembrolizumab for treatment of metastatic non-small cell lung cancer: First-line therapy and beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Zhang, B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Paik, J. Nivolumab plus relatlimab: First approval. Drugs 2022, 82, 925–931. [Google Scholar] [CrossRef]
- Rousseau, A.; Parisi, C.; Barlesi, F. Anti-TIGIT therapies for solid tumors: A systematic review. ESMO Open 2023, 8, 101184. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of resistance to immune checkpoint blockade: Why does checkpoint inhibitor immunotherapy not work for all patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Scholenfeld, A.J.; Hellmann, M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adapative, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, A.B.; Wang, Q.Q.; Joseph, P.; Zheng, C.; Chen, Y.; Kovalenko, O.; Singh, S.; Armstrong, A.; Resnick, K.; Zanotti, K.; et al. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene 2018, 37, 403–414. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Konc, J.; Janezic, D. ProBiS tools (algorithm, database, and web servers) for predicting and modeling of biologically interesting proteins. Prog. Biophys. Mol. Biol. 2017, 128, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, X.; Chen, J.; Qi, X. Decoding connectivity map-based drug repurposing for oncotherapy. Brief. Bioinform. 2023, 24, bbad142. [Google Scholar] [CrossRef]
- Koleti, A.; Terryn, R.; Stathias, V.; Chung, C.; Cooper, D.J.; Turner, J.P.; Vidovic, D.; Forlin, M.; Kelley, T.T.; D’Urso, A.; et al. Data portal for the Library of Integrated Network-based Cellular Signatures (LINCS) program: Integrated access to diverse large-scale cellular perturbations response data. Nucleic Acids Res. 2018, 46, D558–D566. [Google Scholar] [CrossRef]
- Gao, S.; Han, L.; Luo, D.; Xiao, Z.; Liu, G.; Zhang, Y.; Zhou, W. Deep learning applications for the accurate identification of low-transcriptional activity drug and their mechanism of actions. Pharmacol. Res. 2022, 180, 106225. [Google Scholar] [CrossRef]
- Corsello, S.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef]
- Schneider, L.; Kehl, T.; Thedinga, K.; Grammes, N.L.; Backes, C.; Mohr, C.; Schubert, B.; Lenhof, K.; Gerstner, N.; Hartkopf, A.D.; et al. ClinOmicsTrailbc: A visual analytics tool for breast cancer treatment stratification. Bioinformatics 2019, 35, 5171–5181. [Google Scholar] [CrossRef]
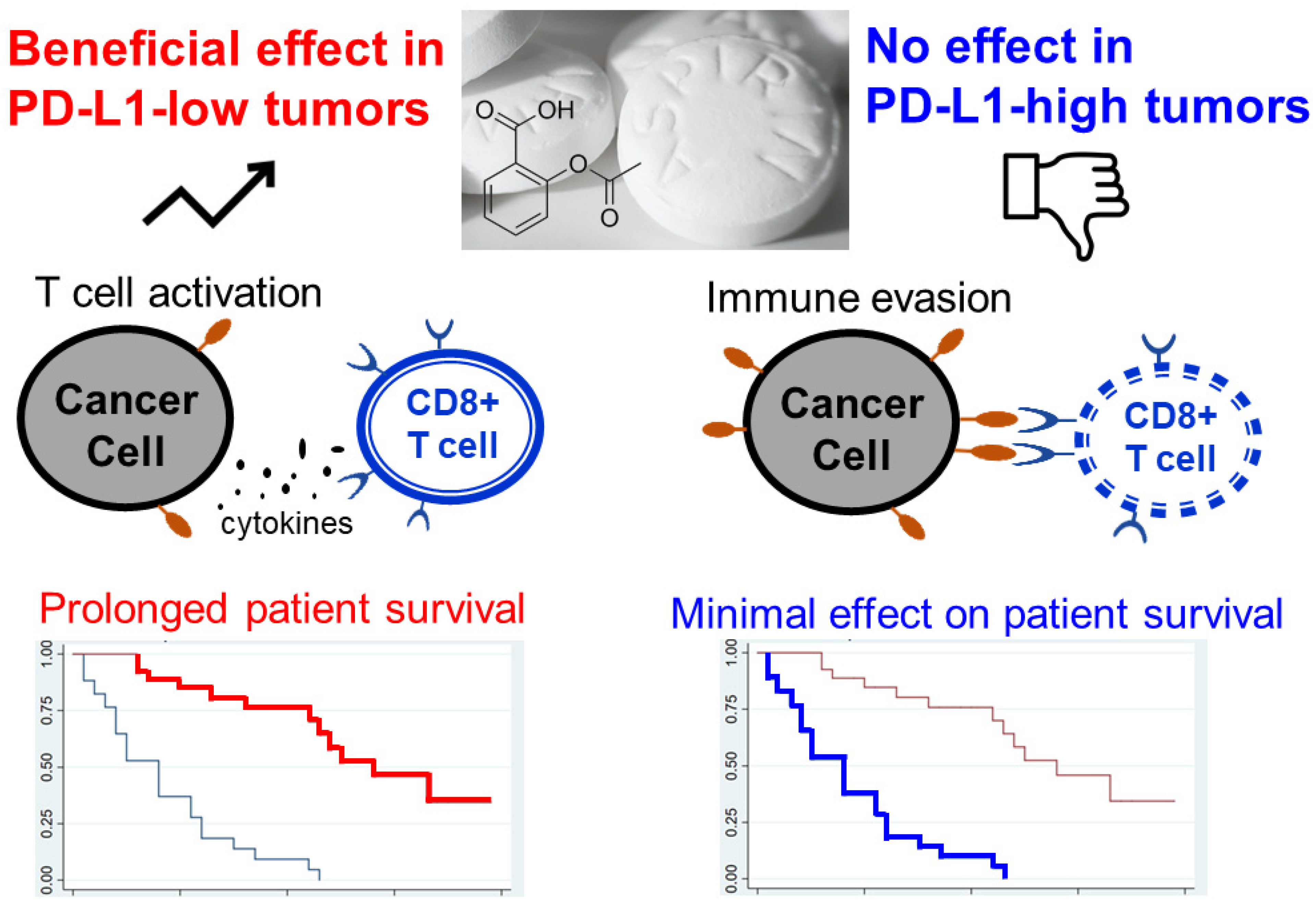
- Hamada, T.; Cao, Y.; Qian, Z.R.; Masugi, Y.; Nowak, J.A.; Yang, J.; Song, M.; Mima, K.; Kosumi, K.; Liu, L.; et al. Aspirin use and colorectal cancer survival according to tumor CD274 (Programmed cell death 1 ligand 1) expression status. J. Clin. Oncol. 2017, 35, 1836–1844. [Google Scholar] [CrossRef]
- Jiang, H.; Suo, H.; Gao, L.; Liu, Y.; Chen, B.; Lu, S.; Jin, F.; Cao, Y. Metformin plays an antitumor role by downregulating inhibitory cells and immune checkpoint molecules while activating protective immune responses in breast cancer. Int. Immunopharmacol. 2023, 118, 110038. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Andre, N. “Hard” drug repurposing for precision oncology: The missing link? Front. Pharmacol. 2018, 9, 637. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Qiu, X.; Timmerman, C.; Fu, Y.X. Targeting tertiary lymphoid structures for tumor immunotherapy. In Tertiary Lymphoid Structures. Methods in Molecular Biology; Dieu-Nosjean, M.C., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1845. [Google Scholar] [CrossRef]
- Marro, B.S.; Zak, J.; Zavareh, R.B.; Teijaro, J.R.; Lairson, L.L.; Oldstone, M.B.A. Discovery of small molecules for the reversal of T cell exhaustion. Cell Rep. 2019, 29, 3293–3302. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020, 586, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Dubrot, J.; Lane-Reticker, S.K.; Kessler, E.A.; Ayer, A.; Mishra, G.; Wolfe, C.H.; Zimmer, M.D.; Du, P.P.; Mahapatra, A.; Ockerman, K.M.; et al. In vivo screens using a selective CRISPR antigen removal lentiviral vector system reveal immune dependencies in renal cell carcinoma. Immunity 2021, 54, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Garippa, R.J.; Poirier, J.T. Design, execution, and analysis of pooled in vitro CRISPR/Cas9 screens. FEBS J. 2016, 283, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhao, X.; Tang, A.; Xu, X.; Liu, S.; Zha, L.; Ma, W.; Zheng, J.; Shi, M. CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188378. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, C.; Liu, Z.; Du, S.; Can, S.; Zhang, H.; Zhang, L.; Huang, X.; Xiao, Z.; Li, X.; et al. Integrative analysis of CRISPR screening data uncovers new opportunities for optimizing cancer immunotherapy. Mol. Cancer 2022, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Sugimoto, M.; Wakita, D.; Yorozu, K.; Kurasawa, M.; Yamamoto, K. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and upregulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti-PD-L1 antibodies. Oncotarget 2018, 9, 31411–31421. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, L.; Li, X.; Song, W.; Liu, Y.; Huang, L. Nanoformulated codelivery of quercetin and alantolactone promotes an antitumor response through synergistic immunogenic cell death for microsatellite-stable colorectal cancer. ACS Nano 2019, 13, 12511–12524. [Google Scholar] [CrossRef]
- Suzuki, N.; Tsukihara, H.; Nakagawa, F.; Kobunai, T.; Takechi, T. Synergistic anticancer activity of a novel oral chemotherapeutic agent containing trifluridine and tipiracil in combination with anti-PD-1 blockade in microsatellite stable-type murine colorectal cancer cells. Am. J. Cancer Res. 2017, 7, 2032–2040. [Google Scholar]
- Chen, L.; Yang, S.; Liao, W.; Xiong, Y. Modification of antitumor immunity and tumor microenvironment by resveratrol in mouse renal tumor model. Cell Biochem. Biophys. 2015, 72, 617–625. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Xia, C.; Yin, S.; To, K.K.; Fu, L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol. Cancer 2023, 22, 44. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, W.; Liu, Z.; Ju, Y.; Xu, M.; Zhang, Y.; Wu, X.; Gu, Q.; Wang, Z.; Xu, J. Discovery of indoleamine 2,3-dioxygenase inhibitors using machine learning based virtual screening. MedChemComm 2018, 9, 837–945. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Fujieda, K.; Senju, S.; Ikeda, T.; Oshiumi, H.; Nishimura, Y. Immune-suppressive effects of interleukin-6 on T-cell-mediated anti-tumor immunity. Cancer Sci. 2018, 109, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumor progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef]
- Chen, X.; Fu, S.; Tian, J.; Cao, Y.; Li, C.; Lin, J. Abstract 864: Repurposing FDA-approved drug bazedoxifene as a novel inhibitor of IL-6 signaling for triple-negative breast cancer. Cancer Res. 2018, 78, 864. [Google Scholar] [CrossRef]
- Tomar, S.; Zumbrun, E.E.; Nagarkatti, M.; Nagarkatti, P.S. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J. Pharm. Exp. Ther. 2015, 353, 369–379. [Google Scholar] [CrossRef]
- Pairet, N.; Mang, S.; Fois, G.; Keck, M.; Kühnbach, M.; Gindele, J.; Frick, M.; Dietl, P.; Lamb, D.J. TRPV4 inhibition attenuates stretch-induced inflammatory cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS ONE 2018, 13, e0196055. [Google Scholar] [CrossRef]
- Kong, D.; Zhou, C.; Guo, H.; Wang, W.; Qiu, J.; Liu, X.; Liu, J.; Wang, L.; Wang, Y. Praziquantel targets M1 macrophages and ameliorates splenomegaly in chronic schistosomiasis. Antimicrob. Agents Chemother. 2018, 62, e00005-17. [Google Scholar] [CrossRef]
- Bok, E.; Chung, Y.C.; Kim, K.S.; Baik, H.H.; Shin, W.H.; Jin, B.K. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the immune response by pro-angiogenic factors. Front. Oncol. 2014, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Pang, X.; Lian, W.; Xu, L.; Wang, J.; Jia, H.; Zhang, B.; Liu, A.L.; Du, G.H. Discovery of VEGFR2 inhibitors by integrating naïve Bayesian classification, molecular docking and drug screening approaches. RSC Adv. 2018, 8, 5286–5297. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Chew, H.Y.; Cruz, J.G.; Leggatt, G.R.; Wells, J.W. Investigating T cells immunity in cancer: Achievements and prospects. Int. J. Mol. Sci. 2021, 22, 2907. [Google Scholar] [CrossRef]
- Chi, X.; Luo, S.; Ye, P.; Hwang, W.L.; Cha, J.H.; Yan, X.; Yang, W.H. T-cell exhaustion and stemness in antitumor immunity: Characteristics, mechanism, and implications. Front. Immunol. 2023, 14, 1104771. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Kareva, I.; Waxman, D.J.; Klement, G.L. Metronomic chemotherapy: An attractive alternative to maximum tolerated dose therapy that can activate anti-tumor immunity and minimize therapeutic resistance. Cancer Lett. 2015, 358, 100–106. [Google Scholar] [CrossRef]
- Bocci, G.; Kerbel, R.S. Pharmacokinetics of metronomic chemotherapy: A neglected but crucial aspect. Nat. Rev. Clin. Oncol. 2016, 13, 659–673. [Google Scholar] [CrossRef]
- Banissi, C.; Ghiringhelli, F.; Chen, L.; Carpentier, A.F. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother. 2009, 58, 1627–1634. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Tanaka, H.; Matsushima, H.; Mizumoto, N.; Takashima, A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009, 69, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Nars, M.S.; Kaneno, R. Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy. Int. J. Cancer 2013, 132, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Dominant effect of antiangiogenesis in combination therapy involving cyclophosphamide and axitinib. Clin. Cancer Res. 2009, 15, 578–588. [Google Scholar] [CrossRef]
- Tu, N.; Trinh, T.L.; Zhou, J.M.; Gilvary, D.L.; Coppola, D.; Wei, S.; Djeu, J.Y. Abstract 3666: Chemotherapeutic sensitivity of myeloid-derived suppressor cells during cancer therapy is dictated by selective expression of clusterin. Am. Assoc. Cancer Res. (AACR) 2017, 77, 3666. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 Trap. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef] [PubMed]
- Merlano, M.C.; Merlotti, A.M.; Licitra, L.; Denaro, N.; Fea, E.; Galizia, D.; Di Maio, M.; Fruttero, C.; Curcio, P.; Vecchio, S.; et al. Activation of immune responses in patients with relapsed-metastatic head and neck cancer (CONFRONT phase I-II trial): Multimodality immunotherapy with avelumab, short-course radiotherapy, and cyclophosphamide. Clin. Transl. Radiat. Oncol. 2018, 12, 47–52. [Google Scholar] [CrossRef]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicenter, open-label, phase 3, randomized, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulieres, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomized, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Kim, D.W.; Tan, E.; Zhou, J.M.; Schell, M.J.; Martinez, M.; Yu, J.; Carballido, E.; Mehta, R.; Strosberg, J.; Imanirad, I.; et al. A phase 1/2 trial of ibrutinib in combination with pembrolizumab in patients with mismatch repair proficient metastatic colorectal cancer. Br. J. Cancer 2021, 124, 1803–1808. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase 1 results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Zhou, J.; Li, W.; Cheng, P.; Fu, P. Co-targeting tumor angiogenesis and immunosuppressive tumor microenvironment: A perspective in ethnopharmacology. Front. Pharmacol. 2022, 13, 886198. [Google Scholar] [CrossRef]
- Chu, T.; Zhong, R.; Zhong, H.; Zhang, B.; Zhang, W.; Shi, C.; Qian, J.; Zhang, Y.; Chang, Q.; Zhang, X.; et al. Phase 1b study of sintilimab plus anlotinib as first-line therapy in patients with advanced NSCLC. J. Thorac. Oncol. 2021, 16, 643–652. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Eto, M.; Motzer, R.; De Giorgi, U.; Buchler, T.; Basappa, N.S.; Méndez-Vidal, M.J.; Tjulandin, S.; Park, S.H.; Melichar, B.; et al. Lenvatinib plus pembrolizumab versus sunitinib as first-line treatment of patients with advanced renal cell carcinoma (CLEAR): Extended follow-up from the phase 3, randomized, open-label study. Lancet Oncol. 2023, 24, 228–238. [Google Scholar] [CrossRef]
- Huang, J.; Guo, Y.; Huang, W.; Hong, X.; Quan, Y.; Lin, L.; Zhou, J.; Liang, L.; Zhang, Y.; Zhou, J.; et al. Regorafenib combined with PD-1 blockade immunotherapy versus regorafenib as second-line treatment for advanced hepatocellular carcinoma: A multicenter retrospective study. J. Hepatocell. Carcinoma 2022, 9, 157–170. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF inhibitors: Molecular targeting and immunomodulatory actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination dabrafenib and trametinib versus combination nivolumab and ipilimumab for patients with advanced BRAF-mutant melanoma: The DREAMseq Trial-ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Keam, B.; Ahn, Y.-O.; Park, H.-R.; Kim, M.; Kim, T.M.; Kim, D.-W.; Heo, D.S. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology 2018, 8, e1515057. [Google Scholar] [CrossRef]
- Prasad, M.; Zorea, J.; Jagadeeshan, S.; Shnerb, A.B.; Mathukkada, S.; Bouaoud, J.; Michon, L.; Novoplansky, O.; Badarni, M.; Cohen, L.; et al. MEK1/2 inhibition transiently alters the tumor immune microenvironment to enhance immunotherapy efficacy against head and neck cancer. J. Immunother. Cancer 2022, 10, e003917. [Google Scholar] [CrossRef]
- Chi, K.H.; Wang, Y.S.; Huang, Y.C.; Chiang, H.C.; Chi, M.S.; Chi, C.H.; Wang, H.E.; Kao, S.J. Simultaneous activation and inhibition of autophagy sensitizes cancer cells to chemotherapy. Oncotarget 2016, 7, 58075–58088. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.U.; Sangai, T.; Akcakanat, A.; Chen, H.; Wei, C.; Meric-Bernstam, F. Vertical inhibition of the PI3K-Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis 2017, 6, e385. [Google Scholar] [CrossRef]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.; Liu, Y.; Pandeswara, S.; Svatek, R.S.; Gelfond, J.A.; Liu, A.; Hurez, V.; Curiel, T.J. Immune-stimulatory effects of rapamycin are mediated by stimulation of antitumor gammadelta T cells. Cancer Res. 2016, 76, 5970–5982. [Google Scholar] [CrossRef]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, K.; Haratani, K.; Hayashi, H.; Shimizu, S.; Tomida, S.; Niwa, T.; Yokoyama, T.; Fukuda, Y.; Chiba, Y.; Kato, R.; et al. Impact of EGFR-TKI treatment on the tumor immune microenvironment in mutation-positive non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 2037–2046. [Google Scholar] [CrossRef]
- Creelan, B.C.; Yeh, T.C.; Kim, S.W.; Nogami, N.; Kim, D.W.; Chow LQ, M.; Kanda, S.; Taylor, R.; Tang, W.; Tang, M.; et al. A Phase I study of gefitinib combined with durvalumab in EGFR TKI-naïve patients with EGFR mutation-positive locally advanced/metastatic non-small-cell lung cancer. Br. J. Cancer 2021, 124, 383–390. [Google Scholar] [CrossRef]
- Riudavets, M.; Naigeon, M.; Texier, M.; Dorta, M.; Barlesi, F.; Mazieres, J.; Varga, A.; Cassard, L.; Boselli, L.; Grivel, J.; et al. Gefitinib plus tremelimumab combination in refractory non-small cell lung cancer patients harboring EGFR mutations: The GEFTREM phase I trial. Lung Cancer 2022, 166, 255–264. [Google Scholar] [CrossRef]
- YYang, J.C.-H.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.-L.; Papadimitrakopoulou, V.A.; Su, W.-C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in combination with erlotinib or gefitinib as first-line therapy for advanced NSCLC with sensitizing EGFR mutation. J. Thorac. Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef]
- Yamaguchi, O.; Kaira, K.; Kawasaki, T.; Mouri, A.; Hashimoto, K.; Shiono, A.; Shinomiya, S.; Miura, Y.; Nishihara, F.; Murayama, Y.; et al. Severe hepatotoxicity due to osimertinib after nivolumab therapy in patients with non-small cell lung cancer harboring EGFR mutation. Thorac. Cancer 2020, 11, 1045–1051. [Google Scholar] [CrossRef]
- Sacco, A.G.; Chen, R.; Worden, F.P.; Wong, D.J.L.; Adkins, D.; Swiecicki, P.; Chai-Ho, W.; Oppelt, P.; Ghosh, D.; Bykowski, J.; et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: An open-label, multi-arm, non-randomized, multicenter, phase 2 trial. Lancet Oncol. 2021, 22, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Boland, P.M.; Hutson, A.; Maguire, O.; Minderman, H.; Fountzilas, C.; Iyer, R.V. A phase Ib/II study of cetuximab and pembrolizumab in RAS-wt mCRC. J. Clin. Oncol. 2018, 36, 834. [Google Scholar] [CrossRef]
- Lee, M.S.; Loehrer, P.J.; Imanirad, I.; Cohen, S.; Ciombor, K.K.; Moore, D.T.; Carlson, C.A.; Sanoff, H.K.; McRee, A.J. Phase II study of ipilimumab, nivolumab, and panitumumab in patients with KRAS/NRAS/BRAF wide-type (WT) microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39, 7. [Google Scholar] [CrossRef]
- Besse, B.; Garrido, P.; Cortot, A.B.; Johnson, M.; Murakami, H.; Gazzah, A.; Gil, M.; Bennouna, J. Efficacy and safety of necitumumab and pembrolizumab combination therapy in patients with Stage IV non-small cell lung cancer. Lung Cancer 2020, 142, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.M.; Lee, S.C.; Filho, P.A.A.; Lord, C.A.; Jie, H.-B.; Davidson, H.C.; López-Albaitero, A.; Gibson, S.P.; Gooding, W.E.; Ferrone, S.; et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin. Cancer Res. 2013, 19, 1858–1872. [Google Scholar] [CrossRef]
- Baysal, H.; De Pauw, I.; Zaryouh, H.; De Waele, J.; Peeters, M.; Pauwels, P.; Vermorken, J.B.; Smits, E.; Lardon, F.; Jacobs, J.; et al. Cetuximab-induced natural killer cell cytotoxicity in head and neck squamous cell carcinoma cell lines: Investigation of the role of cetuximab sensitivity and HPV status. Br. J. Cancer 2020, 123, 752–761. [Google Scholar] [CrossRef]
- Baysal, H.; De Pauw, I.; Zaryouh, H.; Peeters, M.; Vermorken, J.B.; Lardon, F.; De Waele, J.; Wouters, A. The right partner in crime: Unlocking the potential of the anti-EGFR antibody cetuximab via combination with natural killer cell chartering immunotherapeutic strategies. Front. Immunol. 2021, 12, 2021. [Google Scholar] [CrossRef]
- Mughal, M.J.; Bhadresha, K.; Kwok, H.F. CDK inhibitors from past to present: A new wave of cancer therapy. Semin. Cancer Biol. 2023, 88, 106–122. [Google Scholar] [CrossRef]
- Lukasik, P.; Zaluski, M.; Gutowska, I. Cyclin-dependent kinases (CDK) and their role in diseases development—Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.; Koolen, S.L.; Jager, A. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: Similarities and differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Minton, K. Cell cycle inhibitors boost tumor immunogenicity. Nat. Rev. Immunol. 2017, 17, 529. [Google Scholar] [CrossRef] [PubMed]
- Uzhachenko, R.V.; Bharti, V.; Ouyang, Z.; Blevins, A.; Mont, S.; Saleh, N.; Lawrence, H.A.; Shen, C.; Chen, S.C.; Ayers, G.D.; et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 2021, 35, 108944. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; Sheppard, K.E.; McArthur, G.A. Harnessing the immunotherapeutic potential of CDK4/6 inhibitors in melanoma: Is timing everything? NPJ Precis. Oncol. 2022, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Heckler, M.; Ali, L.R.; Clancy-Thompson, E.; Qiang, L.; Ventre, K.S.; Lenehan, P.; Roehle, K.; Luoma, A.; Boelaars, K.; Peters, V.; et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer Discov. 2021, 11, 2564–2581. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; Kong, I.Y.; Zethoven, M.; Ramsbottom, K.M.; Martelotto, L.G.; Meyran, D.; Zhu, J.J.; Costacurta, M.; Kirby, L.; Sandow, J.J.; et al. CDK4/6 inhibition promotes anti-tumor immunity through the induction of T cell memory. Cancer Discov. 2021, 11, 2582–2601. [Google Scholar] [CrossRef]
- Yuan, Y.; Lee, J.S.; Yost, S.E.; Frankel, P.H.; Ruel, C.; Egelston, C.A.; Guo, W.; Padam, S.; Tang, A.; Martinez, N.; et al. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur. J. Cancer 2021, 154, 11–20. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Kabos, P.; Dickler, M.N.; Gianni, L.; Jansen, V.; Lu, Y.; Young, S.; Rugo, H.S. Updated efficacy, safety, & PD-L1 status of patients with HR+, HER2- metastatic breast cancer administered abemaciclib plus pembrolizumab. J. Clin. Oncol. 2018, 36, 1059. [Google Scholar]
- Pujol, J.-L.; Vansteenkiste, J.; Rodríguez, L.P.-A.; Gregorc, V.; Mazieres, J.; Awad, M.; Jänne, P.A.; Chisamore, M.; Hossain, A.M.; Chen, Y.; et al. Abemaciclib in combination with pembrolizumab for stage IV KRAS-mutant or squamous NSCLC: A phase 1b study. JTO Clin. Res. Rep. 2021, 2, 100234. [Google Scholar] [CrossRef]
- Yu, X.; Zhu, L.; Wang, T.; Li, L.; Liu, J.; Che, G.; Zhou, Q. Enhancing the anti-tumor response by combining DNA damage repair inhibitors in the treatment of solid tumors. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188910. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Qin, K.; Lin, A.; Jiang, A.; Cheng, Q.; Liu, Z.; Zhang, J.; Luo, P. The role of DNA damage repair (DDR) system in response to immune checkpoint inhibitor (ICI) therapy. J. Exp. Clin. Cancer Res. 2022, 41, 268. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018, 25, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Philie, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP inhibitors: Extending benefit beyond BRCA-mutant cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Revythis, A.; Limbu, A.; Mikropoulos, C.; Ghose, A.; Sanchez, E.; Sheriff, M.; Boussios, S. Recent insights into PARP and immuno-checkpoint inhibitors in epithelial ovarian cancer. Int. J. Environ. Res. Public Health 2022, 19, 8577. [Google Scholar] [CrossRef]
- Krebs, M.G.; Delord, J.-P.; Evans, T.R.J.; De Jonge, M.; Kim, S.-W.; Meurer, M.; Postel-Vinay, S.; Lee, J.-S.; Angell, H.K.; Rocher-Ros, V.; et al. Olaparib and durvalumab in patients with relapsed small cell lung cancer (MEDIOLA): An open-label, multicenter, phase ½, basket study. Lung Cancer 2023, 180, 107216. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.S.; Padget, M.R.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP inhibitors olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: A proof-of-concept phase II study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunothe. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Wong, W.K.; Yin, B.; Lam, C.Y.K.; Huang, Y.; Yan, J.; Tan, Z.; Wong, S.H.D. The interplay between epigenetic regulation and CD8+ T cell differentiation/exhaustion for T cell immunotherapy. Front. Cell Dev. Biol. 2022, 9, 2021. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T-cell rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, C.; Li, X.; Dong, L.; Yang, Q.; Chen, M.; Shi, F.; Brock, M.; Liu, M.; Mei, Q.; et al. Improved clinical outcome in a randomized phase II study of anti-PD-1 camrelizumab plus decitabine in relapsed/refractory Hodgkin lymphoma. J. Immunother. Cancer 2021, 9, e002347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumor potential via epigenetic reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef]
- Covre, A.; Coral, S.; Nicolay, H.; Parisi, G.; Fazio, C.; Colizzi, F.; Fratta, E.; Di Giacomo, A.M.; Sigalotti, L.; Natali, P.G.; et al. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. Oncoimmunology 2015, 4, 1019978. [Google Scholar] [CrossRef]
- Pieniawska, M.; Izykowska, K. Role of histone deacetylases in T-cell development and function. Int. J. Mol. Sci. 2022, 23, 7828. [Google Scholar] [CrossRef]
- McCaw, T.R.; Randall, T.D.; Forero, A.; Buchsbaum, D.J. Modulation of antitumor immunity with histone deacetylase inhibitors. Immunotherapy 2017, 9, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Saltos, A.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.; Antonia, S.J.; Shafique, M.; Zheng, H.; Dai, W.; Saller, J.J.; et al. Phase I/Ib study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin. Cancer Res. 2019, 25, 6623–6632. [Google Scholar] [CrossRef]
- Hourani, T.; Holden, J.A.; Li, W.; Lenzo, J.C.; Hadjigol, S.; O’Brien-Simpson, N.M. Tumor associated macrophages: Origin, recruitment, phenotypic diversity, and targeting. Front. Oncol. 2021, 11, 788365. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Gao, J.; Liang, Y.; Wang, L. Shaping polarization of tumor-associated macrophages in cancer immunotherapy. Front. Immunol. 2022, 13, 888713. [Google Scholar] [CrossRef]
- Zou, Z.; Lin, H.; Li, M.; Lin, B. Tumor-associated macrophage polarization in the inflammatory tumor microenvironment. Front. Oncol. 2023, 13, 1103149. [Google Scholar] [CrossRef]
- Mallardo, D.; Simeone, E.; Vanella, V.; Vitale, M.G.; Palla, M.; Scarpato, L.; Paone, M.; De Cristofaro, T.; Borzillo, V.; Cortellini, A.; et al. Concomitant medication of cetirizine in advanced melanoma could enhance anti-PD-1 efficacy by promoting M1 macrophages polarization. J. Trans. Med. 2022, 20, 436. [Google Scholar] [CrossRef]
- Uguz, A.; Sanlioglu, S.; Yuzbey, S.; Coskun, M.; Yegin, O. The effect of cetirizine on IFN-gamma and IL-10 production in children with allergic rhinitis. Turk. J. Pediatr. 2005, 47, 111–115. [Google Scholar]
- Lin, X.; Zhang, J.; Wang, X.; Lin, G.; Chen, T. Pre-activation with TLR7 in combination with thioridazine and loratadine promotes tumoricidal T-cell activity in colorectal cancer. Anticancer Drugs 2020, 31, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, T.; Lin, J.; Zhang, L.; Zheng, J.; Liu, Y.; Xie, G.; Wang, B.; Yuan, Y. Local angiotensin II contributes to tumor resistance to checkpoint immunotherapy. J. Immunother. Cancer 2018, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hou, H.; Liang, B.; Guo, X.; Chen, L.; Yang, Y.; Wang, Y. Effect of renin-angiotensin-aldosterone system inhibitors on survival outcomes in cancer patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1155104. [Google Scholar] [CrossRef]
- Xie, G.; Liu, Y.; Yao, Q.; Zheng, R.; Zhang, L.; Lin, J.; Guo, Z.; Du, S.; Ren, C.; Yuan, Q.; et al. Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells. Sci. Rep. 2017, 7, 42396. [Google Scholar] [CrossRef]
- Nakamura, K.; Yaguchi, T.; Ohmura, G.; Kobayashi, A.; Kawamura, N.; Iwata, T.; Kiniwa, Y.; Okuyama, R.; Kawakami, Y. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. 2018, 109, 54–64. [Google Scholar] [CrossRef]
- Nakamura, K.; Kiniwa, Y.; Okuyama, R. CCL5 production by fibroblasts through a local renin-angiotensin system in malignant melanoma affects tumor immune responses. J. Cancer Res. Clin. Oncol. 2021, 147, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Takahara, N.; Ito, Y.; Matsubara, S.; Uchino, R.; Yagioka, H.; Arizumi, T.; et al. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Investig. New Drugs 2013, 31, 1294–1299. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Chen, I.X.; Tong, R.; Ng, M.R.; Martin, J.D.; Naxerova, K.; Wu, M.W.; Huang, P.; Boucher, Y.; Kohane, D.S.; et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 10674–10680. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and cancer: Insight into tumor progression and immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- An, Y.; Yao, J.; Niu, X. The signaling pathway of PGE2 and its regulatory role in T cell differentiation. Mediat. Inflamm. 2021, 2021, 9087816. [Google Scholar] [CrossRef]
- Marzbani, E.; Instsuka, C.; Lu, H.; Disis, M.L. The invisible arm of immunity in common cancer chemoprevention agents. Cancer Prev. Res. 2013, 6, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Elwood, P.; Protty, M.; Morgan, G.; Pickering, J.; Delon, C.; Watkins, J. Aspirin and cancer: Biological mechanisms and clinical outcomes. Open Biol. 2022, 12, 220124. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Zhou, Z.; Fang, J.; Sun, Z.; He, M.; He, B.; Chen, Q.; Paek, C.; Chen, P.; Zhou, J.; et al. Aspiring induces immunogenic cell death and enhances cancer immunotherapy in colorectal cancer. Int. Immunopharmacol. 2023, 121, 110350. [Google Scholar] [CrossRef] [PubMed]
- Aiad, M.; Tahir, A.; Fresco, K.; Prenatt, Z.; Ramos-Feliciano, K.; Walia, J.; Stoltzfus, J.; Albandar, H.J.; Ramos-Feliciano, K.M.; Albandar, H. Does the combined use of aspirin and immunotherapy result in better outcomes in non-small cell lung cancer than immunotherapy alone? Cureus 2022, 14, e25891. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Nakatsuji, M.; Seno, H.; Ishizu, S.; Akitake-Kawano, R.; Kanda, K.; Ueo, T.; Komekado, H.; Kawada, M.; Minami, M.; et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis 2011, 32, 1333–1339. [Google Scholar] [CrossRef]
- Pi, C.; Jing, P.; Li, B.; Feng, Y.; Xu, L.; Xie, K.; Huang, T.; Xu, X.; Gu, H.; Fang, J. Reversing PD-1 resistance in B16F10 cells and recovering tumor immunity using a COX2 inhibitor. Cancers 2022, 14, 4134. [Google Scholar] [CrossRef]
- Cecil, D.; Gad, E.A.; Corulli, L.R.; Drovetto, N.; Lubet, R.A.; Disis, M.L. COX-2 inhibitors decrease expression of PD-L1 in colon tumors and increase the influx of type I tumor-infiltrating lymphocytes. Cancer Prev. Res. 2022, 15, 225–231. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumor immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Shevhenko, I.; Bazhin, A.V. Metabolic checkpoints: Novel avenues for immunotherapy of cancer. Front. Immunol. 2018, 9, 1816. [Google Scholar] [CrossRef]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting tumor metabolism: A new chellenge to improve immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef]
- Urbano, A.M. Otto Warburg: The journey towards the seminal discovery of tumor cell bioenergetic reprogramming. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165965. [Google Scholar] [CrossRef]
- McCarty, M.F.; Whitaker, J. Manipulating tumor acidification as a cancer treatment strategy. Altern. Med. Rev. 2010, 15, 264–272. [Google Scholar]
- Nakagawa, Y.; Negishi, Y.; Shimizu, M.; Takahashi, M.; Ichikawa, M.; Takahashi, H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol Lett. 2015, 167, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.; Prado-Garcia, H.; Sanchez-Garcia, F.J. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumor microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Bianchini, F.; Calorini, L. Extracellular acidity, a “reappreciated” trait of tumor environment driving malignancy: Perspectives in diagnosis and therapy. Cancer Metastasis Rev. 2014, 33, 823–832. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43 (Suppl. C), 74–89. [Google Scholar] [CrossRef]
- Hong, C.S.; Graham, N.A.; Gu, W.; Camacho, C.E.; Mah, V.; Maresh, E.L.; Alavi, M.; Bagryanova, L.; Krotee, P.A.; Gardner, B.K.; et al. MCT1 modulates cancer cell pyruvate export and growth of tumors that co-express MCT1 and MCT4. Cell Rep. 2016, 14, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Eichner, R.; Heider, M.; Fernández-Sáiz, V.; van Bebber, F.; Garz, A.-K.; Lemeer, S.; Rudelius, M.; Targosz, B.-S.; Jacobs, L.; Knorn, A.-M.; et al. Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Görgün, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.-T.; et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010, 116, 3227–3237. [Google Scholar] [CrossRef]
- Chirasani, S.R.; Leukel, P.; Gottfried, E.; Hochrein, J.; Stadler, K.; Neumann, B.; Oefner, P.J.; Gronwald, W.; Bogdahn, U.; Hau, P.; et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int. J. Cancer 2013, 132, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)—Diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef]
- Lacroix, R.; Rozeman, E.A.; Kreutz, M.; Renner, K.; Blank, C.U. Targeting tumor-associated acidity in cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1331–1348. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T. Cancer: Fundamentals behind pH targeting and the double-edged approach. OncoTargets Ther. 2016, 9, 6343–6360. [Google Scholar] [CrossRef]
- Lopes, S.; Pabst, L.; Dory, A.; Klotz, M.; Gourieux, B.; Michel, B.; Mascaux, C. Do proton pump inhibitors alter the response to immune checkpoint inhibitors in cancer patients? A meta-analysis. Front. Immunol. 2023, 14, 1070076. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, S.; Chun, K.H.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kim, Y.S.; Woo, J.-T.; Nam, M.-S.; Baik, S.H.; et al. Metformin reduces the risk of cancer in patients with type 2 diabetes. Medicine 2018, 97, e0036. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin promotes antitumor immunity via endoplasmic reticulum associated degradation of PD-L1. Mol. Cell 2018, 71, 606–620. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Lequeux, A.; Xiao, M.; Duhem, C.; Chouaib, S.; Berchem, G.; Janji, B. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: New opportunities and challenges. Cells 2019, 8, 1083. [Google Scholar] [CrossRef]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, C.; Najafi, M. Targeting of the tumor immune microenvironment by metformin. J. Cell Commun. Signal. 2022, 16, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, H.-J.; Kim, C.W.; Kim, H.C.; Jung, Y.; Lee, H.-S.; Lee, Y.; Ju, Y.S.; Oh, J.E.; Park, S.-H.; et al. Tumor hypoxia represses T cell-mediated antitumor immunity against brain tumors. Nat. Immunol. 2021, 22, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef]
- Finisguerra, V.; Dvorakova, T.; Formenti, M.; Van Meerbeeck, P.; Mignion, L.; Gallez, B.; Eynde, B.J.V.D. Metformin improves cancer immunotherapy by directly rescuing tumor-infiltrating CD8 T lymphocytes from hypoxia-induced immunosuppression. J. Immunother. Cancer 2023, 11, e005719. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Chen, S.; Li, Z.; Chen, J.; Li, W. The effect of concomitant use of statins, NSAIDs, low-dose aspirin, metformin and beta-blockers on outcomes in patients receiving immune checkpoint inhibitors: A systematic review and meta-analysis. Oncoimmunology 2021, 10, 1957605. [Google Scholar] [CrossRef]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino acid depletion therapies: Starving cancer cells to death. Trends Endorinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef] [PubMed]
- van Baren, N.; van den Eynde, B.J. Tumoral immune resistance mediated by enzymes that degrade tryptophan. Cancer Immunol. Res. 2015, 3, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DcHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Yen, M.C.; Lin, C.C.; Chen, Y.L.; Huang, S.S.; Yang, H.J.; Chang, C.P.; Lei, H.Y.; Lai, M.D. A novel cancer therapy by skin delivery of indoleamine 2,3-dioxygenase siRNA. Clin. Cancer Res. 2009, 15, 641–649. [Google Scholar] [CrossRef]
- Brown, Z.J.; Yu, S.J.; Heinrich, B.; Ma, C.; Fu, Q.; Sandhu, M.; Agdashian, D.; Zhang, Q.; Korangy, F.; Greten, T.F. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 2018, 67, 1305–1315. [Google Scholar] [CrossRef]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by targeting IDO1/TDO and their downstream effectors. Front. Immunol. 2015, 5, 673. [Google Scholar] [CrossRef]
- Zhai, L.; Spranger, S.; Binder, D.C.; Gritsina, G.; Lauing, K.L.; Giles, F.J.; Wainwright, D.A. Molecular pathways: Targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 5427–5433. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates anti-tumor T cell responses in gastrointestinal stromal tumor through the inhibition of IDO. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef]
- Reilley, M.J.; Bailey, A.; Subbiah, V.; Janku, F.; Naing, A.; Falchook, G.; Karp, D.; Piha-Paul, S.; Tsimberidou, A.; Fu, S.; et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J. Immunother. Cancer 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, S.; Li, Y.; Yuan, C.; Zhang, J.; Xu, Z.; Hu, Y.; Shi, H.; Wang, S. Simultaneous glutamine metabolism and PD-L1 inhibition to enhance suppression of triple-negative breast cancer. J. Nanobiotechnol. 2022, 20, 216. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Byun, J.K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.J.; Lee, I.K.; Choi, Y.K.; et al. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol. Cell 2020, 80, 592–606.e8. [Google Scholar] [CrossRef]
- Xiang, Y.; Guo, Z.; Zhu, P.; Chen, J.; Huang, Y. Traditional Chinse medicine as a cancer treatment: Modern perspectives of ancient but advanced science. Cancer Med. 2019, 8, 1958–1975. [Google Scholar] [CrossRef]
- Yu, Y.-X.; Wang, S.; Liu, Z.-N.; Zhang, X.; Hu, Z.-X.; Dong, H.-J.; Lu, X.-Y.; Zheng, J.-B.; Cui, H.-J. Traditional Chinese medicine in the era of immune checkpoint inhibitor: Theory, development, and future directions. Chin. Med. 2023, 18, 59. [Google Scholar] [CrossRef]
- Liu, K.; Sun, Q.; Liu, Q.; Li, H.; Zhang, W.; Sun, C. Focus on immune checkpoint PD-1/PD-L1 pathway: New advances of polyphenol phytochemicals in tumor immunotherapy. Biomed. Pharmacother. 2022, 154, 113618. [Google Scholar] [CrossRef]
- Kumari, A.; Karnatak, M.; Singh, D.; Shankar, R.; Jat, J.L.; Sharma, S.; Yadav, D.; Shrivastava, R.; Verma, V.P. Current scenario of artemisinin and its analogues for antimalarial activity. Eur. J. Med. Chem. 2019, 163, 804–829. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, N.; Yang, T.; Wang, W.; Li, X. The potential mechanisms by which Artemisin and its derivatives induce ferroptosis in the treatment of cancer. Oxidative Med. Cell Longev. 2022, 2022, 1458143. [Google Scholar] [CrossRef]
- Huang, Z.; Gan, S.; Zhuang, X.; Chen, Y.; Lu, L.; Wang, Y.; Qi, X.; Feng, Q.; Huang, Q.; Du, B.; et al. Artesunate inhibits the cell growth in colorectal cancer by promoting ROS dependent cell senescence and autophagy. Cells 2022, 11, 2472. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Liu, W.; Wang, T.; De Sanctis, F.; Zhu, L.; Zhang, G.; Cheng, J.; Cao, Q.; Zhou, J.; et al. Targeting inhibition of accumulation and function of myeloid-derived suppressor cells by artemisinin via PI3K/AKT, mTOR, and MAPK pathways enhances anti-PD-L1 immunotherapy in melanoma and liver tumors. J. Immunol. Res. 2022, 2022, 2253436. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Morris-Natschke, S.L.; Lee, K.H. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med. Res. Rev. 2007, 27, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Han, Z.; Trivett, A.L.; Lin, H.; Hannifin, S.; Yang, D.; Oppenheim, J.J. Crytotanshinone has curative dual anti-proliferative and immunotherapeutic effects on mouse Lewis lung carcinoma. Cancer Immunol. Immunother. 2019, 68, 1059–1071. [Google Scholar] [CrossRef]
- Su, C.Y.; Ming, Q.L.; Rahman, K.; Han, T.; Qin, L.P. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 2015, 13, 163–182. [Google Scholar] [CrossRef]
- Chen, Q.; Hong, Y.; Weng, S.; Guo, P.; Li, B.; Zhang, Y.; Yu, C.; Wang, S.; Mo, P. Traditional Chinese medicine Pien-Tze-Huang inhibits colorectal cancer growth and immune evasion by reducing-catenin transcriptional activity and PD-L1 expression. Front. Pharmacol. 2022, 13, 828440. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ren, W.; Zhang, L.; Zhang, Y.; Liu, D.; Liu, Y. A review of the pharmacological action of Astragalus polysaccharide. Front. Pharmacol. 2020, 11, 2020. [Google Scholar] [CrossRef]
- Hwang, J.; Zhang, W.; Dhananjay, Y.; An, E.-K.; Kwak, M.; You, S.; Lee, P.C.-W.; Jin, J.-O. Astragalus membranaceus polysaccharides potentiate the growth-inhibitory activity of immune checkpoint inhibitors against pulmonary metastatic melanoma in mice. Int. J. Biol. Macromol. 2021, 182, 1292–1300. [Google Scholar] [CrossRef]
- Han, X.; Wei, Q.; Lv, Y.; Weng, L.; Huang, H.; Wei, Q.; Li, M.; Mao, Y.; Hua, D.; Cai, X.; et al. Ginseng-derived nanoparticles potentiate immune checkpoint antibody efficacy by reprogramming the cold tumor microenvironment. Mol. Ther. 2022, 30, 327–340. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Ma, S.; Zhao, Q.; Wu, J.; Duan, L.; Xie, Y.; Wang, S. Traditional uses, phytochemistry, and pharmacology of Ailanthus altissima (Mill.) Swingle bark: A comprehensive review. J. Ethnopharmacol. 2021, 275, 114121. [Google Scholar] [CrossRef]
- Yu, P.; Wei, H.; Li, K.; Zhu, S.; Li, J.; Chen, C.; Zhang, D.; Li, Y.; Zhu, L.; Yi, X.; et al. The traditional Chinese medicine monomer Ailanthone improves the therapeutic efficacy of anti-PD-L1 in melanoma cells by targeting c-Jun. J. Exp. Clin. Cancer Res. 2022, 41, 346. [Google Scholar] [CrossRef] [PubMed]
- Vernocchi, P.; Gili, T.; Conte, F.; Del Chierico, F.; Conta, G.; Miccheli, A.; Botticelli, A.; Paci, P.; Caldarelli, G.; Nuti, M.; et al. Network analysis of gut microbiome and metabolome to discover microbiota-linked biomarkers in patients affected by non-small cell lung cancer. Int. J. Mol. Sci. 2020, 21, 8730. [Google Scholar] [CrossRef]
- Kovtonyuk, L.V.; McCoy, K.D. Microbial metabolites and immunotherapy: Basic rationale and clinical indications. Semin. Immunol. 2023, 67, 101755. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, D.; Wang, Y.; Liu, L.; Li, J.; Yuan, J.; Jiang, Z.; Jiang, Z.; Hsiao, W.W.; Liu, H.; et al. Ginseng polysaccharides alter the gut microbiota and kynurenine/tryptophan ratio, potentiating the antitumor effect of antiprogrammed cell death 1/programmed cell death ligand 1 (anti-PD-1/PD-L1) immunotherapy. Gut 2022, 71, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Jia, Y.; Li, J.; Kuai, W.; Li, Y.; Guo, F.; Xu, X.; Zhao, Z.; Lv, J.; Li, Z. Gegen Qinlian decoction enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by remodeling the gut microbiota and the tumor microenvironment. Cell Death Dis. 2019, 10, 415. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Yuan, M.-Y.; Chen, Y.L.; Zhang, X.; Xu, X.-T.; Liu, S.-L.; Zou, X.; Tao, J.-L.; Qiang, Y.-H.; Wu, J.; et al. The gut microbiota and traditional Chinese medicine: A new clinical frontier on cancer. Curr. Drug Targets 2021, 22, 1222–1231. [Google Scholar] [CrossRef]
- Li, X.; Wu, D.; Niu, J.; Sun, Y.; Wang, Q.; Yang, B.; Kuang, H. Intestinal flora: A pivotal role in investigation of traditional Chinese medicine. Am. J. Chin. Med. 2021, 49, 237–268. [Google Scholar] [CrossRef]
- Wirsdorfer, F.; de Leve, S.; Jenfrossek, V. Combining radiotherapy and immunotherapy in lung cancer: Can we expect limitations due to altered normal tissue toxicity? Int. J. Mol. Sci. 2018, 20, 24. [Google Scholar] [CrossRef]
- Cremolini, C.; Vitale, E.; Rastaldo, R.; Giachino, C. Advanced nanotechnology for enhancing immune checkpoint blockade therapy. Nanomaterials 2021, 11, 661. [Google Scholar] [CrossRef]
- Gawali, P.; Saraswat, A.; Bhide, S.; Gupta, S.; Patel, K. Human solid tumors and clinical relevance of the enhanced permeation and retention effect: A ‘golden gate’ for nanomedicine in preclinical studies? Nanomedicine 2023, 18, 169–190. [Google Scholar] [CrossRef]
- Batool, S.; Sohail, S.; Din, F.U.; Alamri, A.H.; Alqahtani, A.S.; Alshahrani, M.A.; Alshehri, M.A.; Choi, H.G. A detailed insight of the tumor targeting using nanocarrier drug delivery system. Drug Deliv. 2023, 30, 2183815. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F. Gut microbiome in cancer immunotherapy: Current trends, translational challenges and future possibilities. Biochim. Biophys. Acta Gen. Subj. 2022, 82, 104163. [Google Scholar] [CrossRef] [PubMed]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef] [PubMed]



 = increased level;
= increased level;  = decreased level.
= increased level; = decreased level.
= decreased level.
= increased level; = decreased level.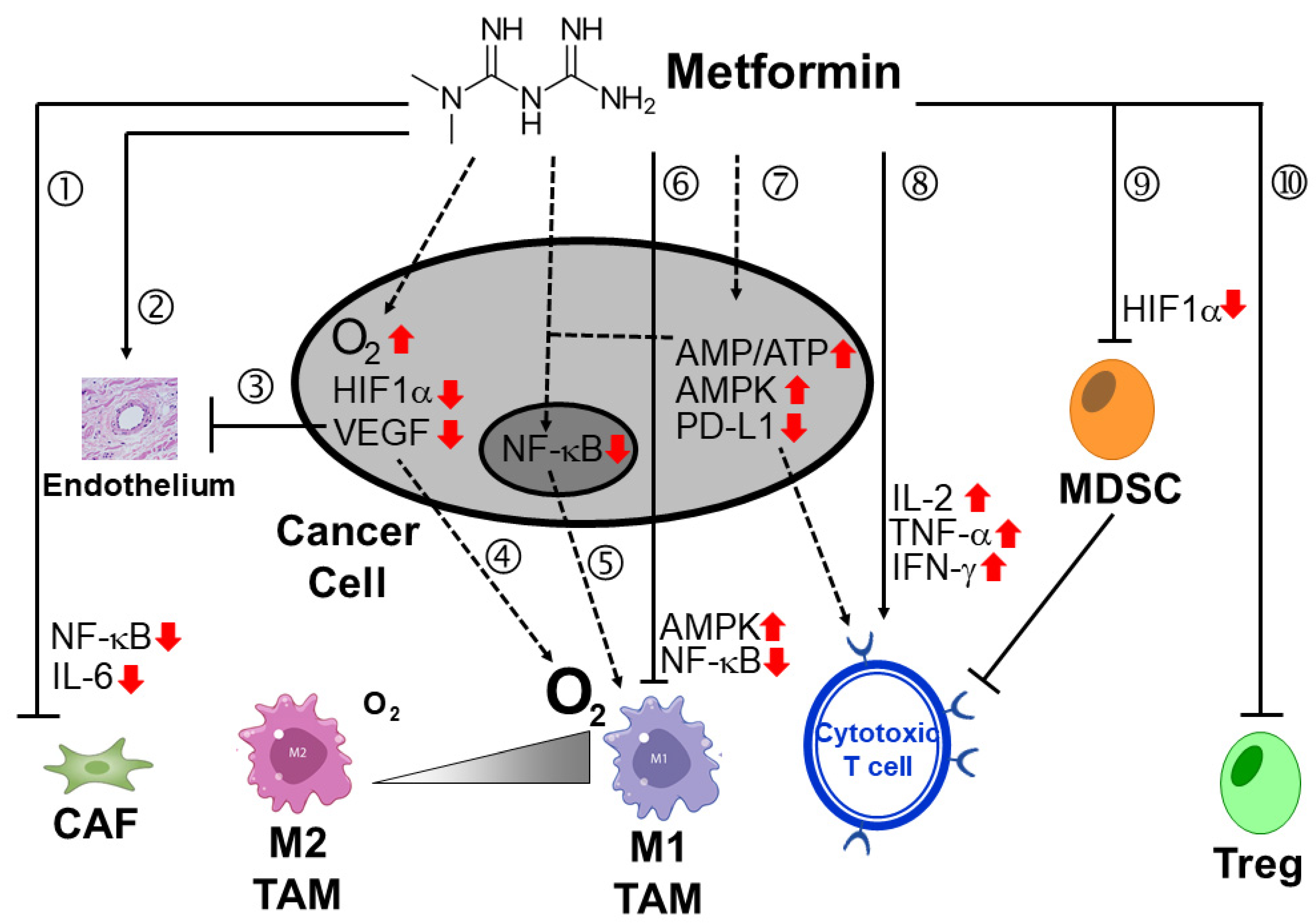

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Combination | Cancer Type | ClinicaTrials.gov Identifier (Phase) | Status | |
|---|---|---|---|---|
| ICI | Other Treatment Modalities | |||
| Combination with chemotherapy | ||||
| Alezolizumab (anti-PD-L1 mAb) | Carboplatin, Etoposide | Untreated extensive-stage SCLC | NCT04028050 (Phase 3) | Active; not recruiting |
| Atezolizumab (anti-PD-L1 mAb) | Pegylated liposomal doxorubicin, Cyclophosphamide | Metastatic triple-negative breast cancer | NCT03164993 (Phase 2) | Active; not recruiting |
| Nivolumab (anti-PD-1 mAb) | Lenalidomide (thalidomide analogue) | Relapsed or refractory non-Hodgkin or Hodgkin lymphoma | NCT03015896 (Phase 2) | Active; not recruiting |
| Anti-PD-1 mAb | Lenalidomide (thalidomide analogue) and azacytidine (epigenetic drug) | Relapsed/refractory peripheral T cell lymphoma | NCT05182957 (Phase 2) | Recruiting |
| Nivolumab (anti-PD-1 mAb), Ipilimumab (anti CTLA-4 mAb) | Trabectedin (marine derived and DNA-binding chemotherapeutic drug) | Advanced soft tissue sarcoma | NCT03138161 (Phase 2) | Recruiting |
| Pembrolizumab (anti-PD-1 mAb) | Metronomic cyclophosphamide | Metastatic breast cancer | NCT03139851 (Phase 2) | Completed; results not yet published |
| Combination with targeted therapy | ||||
| Atezolizumab (anti-PD-L1 mAb) | Cobimetinib (MEK inhibitor) | Previously treated unresectable locally advanced or metastatic CRC | NCT02788279 (Phase 3) | Completed; Median OS (8.87 months combination versus 7.10 months regorafenib); HR 1.00 (combination versus regorafenib) [69] |
| Atezolizumab (anti-PD-L1 mAb) | Cobimetinib (MEK inhibitor), Alectinib (ALK inhibitor), Entrectinib (ROS1 inhibitor), Vemurafenib (BRAF inhibitor), GDC-6036 (KRAS inhibitor) | Advanced or metastatic NSCLC (multiple trial arms including different combinations; estimated to recruit 1000 participants) | NCT03178552 (Phase 2/3) | Recruiting |
| Atezolizumab (anti-PD-L1 mAb) | Entinostat (Class I HDACI), Fulvestrant (Anti-estrogen), Ipatasertib (Akt inhibitor), Exemestane (steroidal aromatase inhibitor), Tamoxifen (SERM), Abemaciclib (CDK4/6 inhibitor) | HR-positive HER2-negative breast cancer | NCT03280563 (Phase 2) | Active; not recruiting |
| Avelumab (anti-PD-L1 mAb) | Axitinib (VEGFR, PDGFR, c-Kit inhibitor) | Advanced RCC | NCT02684006 (Phase 3) | Active; not recruiting |
| Carelizumab (anti-PD-1 mAb) | Apatinib (VEGFR, RET, c-Kit inhibitor) | Breast cancer | NCT04335006 (Phase 3) | Terminated (sponsor R&D strategy adjustment) |
| Ipilimumab (anti-CTLA-4 mAb); Nivolumab (anti-PD-1 mAb) | Cabozantinib (VEGFR2, Met inhibitor) | HCC | NCT01658878 (Phase 2) | Active; not recruiting |
| Nivolumab (anti-PD-1 mAb) | Ibrutinib (BTK inhibitor), Cetuximab (anti-EGFR mAb) | Metastatic HNSCC | NCT03646461 (Phase 2) | Active; not recruiting |
| Nivolumab (anti-PD-1 mAb) | Regorafenib (dual targeted VEGFR2-TIE2 TKI) | Gastro-oesophageal cancer | NCT04879368 (Phase 3) | Recruiting |
| Nivolumab (anti-PD-1 mAb) | Tivozanib (VEGFR, PDGFR, c-Kit inhibitor) | Renal cell carcinoma | NCT04987203 (Phase 3) | Recruiting |
| Pembrolizumab (anti-PD-L1 mAb) | Axitinib (VEGFR, c-Kit, PDGFR inhibitor) | Renal cell carcinoma | NCT02853331 (Phase 3) | Completed; median PFS (15.1 months combination versus 11.1 months sunitinib monotherapy) [70] |
| Pembrolizumab (anti-PD-L1 mAb) | Dasatinib (Abl, Src, c-Kit inhibitor), Imatinib mesylate (Abl, c-Kit, PDGFR inhibitor), Nilotinib (Bc-Abl inhibitor) | CML; patients with detectable minimal residual disease | NCT03516279 (Phase 2) | Recruiting |
| Pembrolizumab (anti-PD-L1 mAb) | Ibrutinib (BTK inhibitor) | Advanced colorectal cancer | NCT03332498 (Phase 1/2) | Completed; among 31 evaluable patients, 8 (26%) achieved stable disease; no objective response was observed [71] |
| Pembrolizumab (anti-PD-L1 mAb) | Letrozole (aromatase inhibitor), Palbociclib (CDK4/6 inhibitor) | Newly diagnosed metastatic stage IV ER-positive breast cancer | NCT02778685 (Phase 2) | Suspended (accrual on hold)—last update posted on 20 May 2023 |
| Pembrolizumab (anti-PD-1 mAb) | Lenvatinib (VEGFR, FGFR, PDGFR, c-Kit, RET inhibitor) | Treatment naïve, metastatic NSCLC | NCT03829332 (Phase 3) | Active; not recruiting |
| Tislelizumab (anti-PD-1 mAb) | Sitravatinib (TAM family of receptors and VEGFR2 inhibitor) | Metastatic NSCLC | NCT04921358 (Phase 3) | Active; not recruiting |
| Combination with epigeneticmodifying drugs | ||||
| Pembrolizumab (anti-PD-1 mAb) | Azacitidine (DNA demethylating agent) | Pancreatic cancer | NCT03264404 (Phase 2) | Active; not recruiting |
| Pembrolizumab (anti-PD-L1 mAb) | Vorinostat (HDACI) | Stage IV NSCLC | NCT02638090 (Phase 2) | Active; not recruiting |
| Pembrolizumab (anti-PD-L1 mAb) | Vorinostat (HDACI), Tamoxifen (SERM) | Breast neoplasms | NCT02395627 (Phase 2) | Terminated; insufficient efficacy in an unselected patient population |
| Pembrolizumab (anti-PD-L1 mAb) | Decitabine (DNA demethylating agent), Radiation therapy | Pediatric and young adult cancer patients with solid tumor or lymphoma | NCT03445858 (Phase 2) | Active; not recruiting |
| Combination with DNA damage response inhibitors | ||||
| Atezolizumab (anti-PD-L1 mAb) | Niraparib (PARP inhibitor) | Recurrent ovarian cancer | NCT03598270 (Phase 3) | Active; not recruiting |
| Dostarlimab (anti-PD-1 mAb) | Niraparib (PARP inhibitor) | Metastatic endometrial or ovarian carcinoma | NCT03651206 (Phase 2/3) | Active; not recruiting |
| Pembrolizumab (anti-PD-1 mAb) | Olaparib (PARP inhibitor) | BRCA non-mutated advanced epithelial ovarian cancer | NCT03740165 (Phase 3) | Active; not recruiting |
| Pembrolizumab (anti-PD-1 mAb) | Olaparib (PARP inhibitor) | Unresectable, locally advanced NSCLC | NCT04380636 (Phase 3) | Recruiting |
| Pembrolizumab (anti-PD-1 mAb) | Olaparib (PARP inhibitor) | SCLC | NCT04624204 (Phase 3) | Recruiting |
| Combination with Indoleamine 2,3-dioxygenase-1 (IDO1) inhibitors | ||||
| Nivolumab (anti-PD-1 mAb) | Epacadostat (IDO1 inhibitor) | metastatic NSCLC | NCT03348904 (Phase 3) | Terminated (study halted prematurely and will not resume) |
| Pembrolizumab (anti-PD-1 mAb) | Epacadostat (IDO1 inhibitor) | urothelial cancer | NCT03361865 (Phase 3) | Completed; results not yet published |
| Pembrolizumab (anti-PD-1 mAb) | Epacadostat (IDO1 inhibitor) | metastatic RCC | NCT03260894 (Phase 3) | Active; not recruiting |
| Pembrolizumab (anit-PD-1 mAb) | Epacadostat (IDO1 inhibitor) | various solid cancers | NCT02178722 (Phase 1/2) | Completed; the combination was well tolerated and had encouraging anti-tumor activity in multiple advanced solid tumors. Objective responses in 12 (55%) of 22 patients with melanoma and other solid tumors [72] |
| Combination with various other non-oncology drugs | ||||
| Pembrolizumab (anti-PD-1 mAb) | COX inhibitor (aspirin or celecoxib) | MSI-H/dMMR or high TMB colorectal cancer | NCT03638297 (Phase 2) | Recruiting |
| Nivolumab (anti-PD-1 mAb) | COX-2 inhibitor (celecoxib) | Advanced “cold” solid cancers | NCT03864575 (Phase 2) | Not yet recruiting |
| Pembrolizumab or Nivolumab (anti-PD-1 mAb) | Antidiabetic drug (metformin or rosiglitazone) | Solid cancers | NCT04114136 (Phase 2) | Recruiting |
| Nivolumab (anti-PD-1 mAb) | Antidiabetic drug (metformin) | Stage III–IV NSCLC that cannot be removed by surgery | NCT03048500 (Phase 2) | Active; not recruiting |
| Nivolumab (anti-PD-1 mAb) | Antihypertensive drug—ARB (losartan) | Localized pancreatic cancer | NCT03563248 (Phase 2) | Active; not recruiting |
| Anti-PD-1 mAbs | Antidiabetic drug (metformin) | SCLC | NCT03994744 (Phase 2) | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
To, K.K.W.; Cho, W.C. Drug Repurposing to Circumvent Immune Checkpoint Inhibitor Resistance in Cancer Immunotherapy. Pharmaceutics 2023, 15, 2166. https://doi.org/10.3390/pharmaceutics15082166
To KKW, Cho WC. Drug Repurposing to Circumvent Immune Checkpoint Inhibitor Resistance in Cancer Immunotherapy. Pharmaceutics. 2023; 15(8):2166. https://doi.org/10.3390/pharmaceutics15082166
Chicago/Turabian StyleTo, Kenneth K. W., and William C. Cho. 2023. "Drug Repurposing to Circumvent Immune Checkpoint Inhibitor Resistance in Cancer Immunotherapy" Pharmaceutics 15, no. 8: 2166. https://doi.org/10.3390/pharmaceutics15082166
APA StyleTo, K. K. W., & Cho, W. C. (2023). Drug Repurposing to Circumvent Immune Checkpoint Inhibitor Resistance in Cancer Immunotherapy. Pharmaceutics, 15(8), 2166. https://doi.org/10.3390/pharmaceutics15082166