Abstract
Abiraterone acetate (AbirAc) is the most used steroidal therapeutic agent for treatment of prostate cancer. The mainly hydrophobic molecular surface of AbirAc results in its poor solubility and plays an important role for retention of abiraterone in the cavity of the receptor formed by peptide chains and heme fragments. In order to evaluate the hydrolytic stability of AbirAc, to modify its solubility by formation of new solid forms and to model bonding of this medication with the heme, a series of d-metal complexes with AbirAc was obtained. AbirAc remains stable in water, acetonitrile, tetrahydrofuran, and ethanol, and readily interacts with dications as a terminal ligand to create discrete complexes, including [FePC(AbirAc)2] and [ZnTPP(AbirAc)] (H2PC = phthalocyanine and H2TPP = 5,10,15,20-tetraphenylporphyrine) models for ligand–receptor bonding. In reactions with silver(I) nitrate, AbirAc acts as a bridge ligand. Energies of chemical bonding between AbirAc and these cations vary from 97 to 235 kJ mol−1 and exceed those between metal atoms and water molecules. This can be indicative of the ability of abiraterone to replace solvent molecules in the coordination sphere of biometals in living cells, although the model [ZnTPP] complex remains stable in CDCl3, CD2Cl2, and 1,1,2,2-tetrachloroethane-d2 solvents and decomposes in polar dimethylsulfoxide-d6 and methanol-d4 solvents, as follows from the 1H DOSY spectra. Dynamics of its behavior in 1,1,2,2-tetrachloroethane-d2 were studied by ROESY and NMR spectra.
1. Introduction
The number of medications based on active pharmaceutical ingredients (API) in the form of salts and complexes has increased in recent decades, both in the total number and in the number of cations used [1,2,3]. This growth is related to ability of cations to tune physical chemical properties of an API, such as solubility or tabletability [4,5,6,7,8], and increased selectivity of their supramolecular binding with biological macromolecules and/or bioactivity [9,10], as well as to the existence of metallopharmaceuticals [11,12,13,14], metallodrugs, and bioMOFs with controlled release of API [15,16,17,18,19,20]. The Orange Book of Approved Drug Products with Therapeutic Equivalence Evaluations [21] contains information about salts of sodium, potassium, calcium, magnesium, iron, platinum, silver, and zinc. Other medications contain bismuth [22], lithium [23], gold [24], and other metal ions. The majority of these salts contain biometals, either present in the human body in relatively large amounts (sodium, potassium, calcium), or known as essential trace metals found in metalloproteins and enzymes (magnesium, zinc, iron).
The complexes of an API with the latter elements as well as with the other biometals present in enzymes (copper, manganese, cobalt, etc.) can also be used as model systems for analysis of chemical bonding between the API and cofactors in living systems [25]. These complexes give valuable information about the number of API molecules able to interact with the cation, the stereochemistry of its coordination environment, and the functional groups involved in chemical bonding [26]. Using density functional (DFT) calculations, data about charge density redistribution and energies of API–metal bonding can be obtained as well [27,28]. Finally, such complexes can be used in the first stage of DFT simulation of metal–ligand interactions in the biological environment [29].
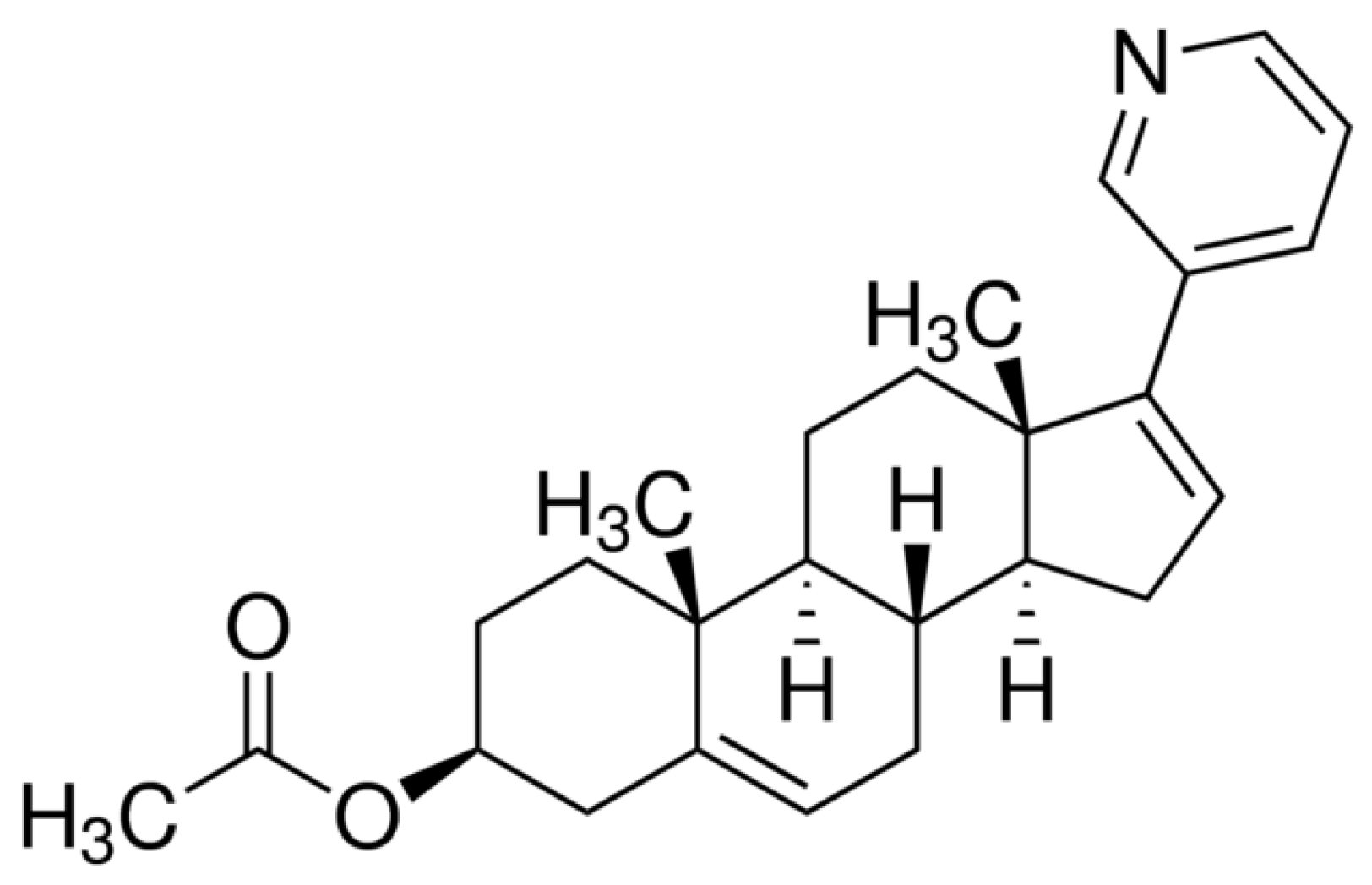
Abiraterone acetate (AbirAc, Scheme 1), commercially available as Zytiga and Yonsa, is the most used API for the treatment of prostate cancer. Abiraterone acetate is known to be hydrolyzed by pancreatic cholesterol esterase to afford pure abiraterone, AbirOH, which inhibits the biosynthesis of androgens [30]. Abiraterone in its stable crystalline form is not druggable due to its extremely low solubility in aqueous media (<0.5 μg/mL) [31]. Abiraterone acetate is even less soluble in water so Zytiga contains it in micronized form [32]. Moreover, it represents the largest food effect of all marketed drugs dependent on the fat content of the food [33]. Thus, attempts to find more soluble and less food-dependent forms of abiraterone are still ongoing. The search for novel solid forms of AbirAc with enhanced solubility included co-crystallization of AbirAc with some co-formers [34,35] and formation of (HAbirOH)Hal · H2O salts (Hal = Cl– [36]; Br– [37]). Based on numerous X-ray diffraction data of ligand (AbirOH): receptor (human cytochrome P450 17A1 or CYP17A1) complexes, the most specific feature of their binding is coordination between the iron atom of the heme fragment and the nitrogen atom NPy of the pyridine-3-yl ring of abiraterone [38,39,40,41,42]. The steroid moiety of abiraterone forms numerous hydrophobic interactions, while the hydroxyl group can take part in H-bonding. The AbirAc is a neutral ditopic molecule, potentially able to act as a terminal ligand or a linker to bioMOFs. Hence, we attested its potential to obtain metallopharmaceuticals based on such biometals as Fe(II), Co(II), Cu(II), Ni(II), and Zn(II), as well as two 5d transition metals, Ag(I) and Cd(II).
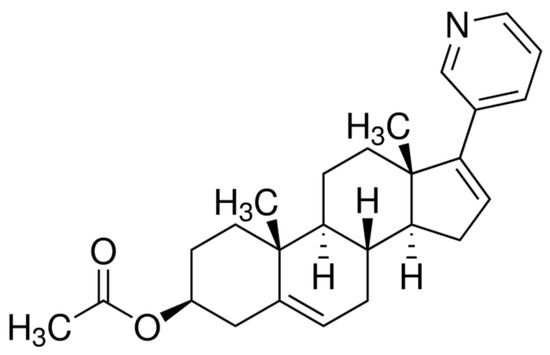
Scheme 1.
Molecular structure of AbirAc.
2. Materials and Methods
2.1. General Procedures
All reagents and solvents were purchased from Sigma Aldrich (Merck LLC, Moscow, Russia) and used as supplied (analytical grade of purity). See Supporting Information for synthetic details and spectra. 1H and 13C NMR spectra were recorded on a 300 MHz (300.1, 75.5 MHz, respectively) and 400 MHz (400.1, 100.6 MHz, respectively) Bruker Avance spectrometers (Bruker BioSpin GMBH, Rheinstetten, Germany) in CDCl3, CD2Cl2, TCE-d2 (1,1,2,2-tetrachloroethane), DMSO-d6, and methanol-d4 solutions using 0.05% Me4Si as the external or internal standard. Determinations of structures and stereochemistry of obtained compounds and assignments of 1H, 13C signals were conducted with the aid of 2D COSY, TOCSY, NOESY, ROESY, edited-HSQC, and HMBC spectra, as well as using diffusion-ordered NMR studies (DOSY-LED). The powder X-ray diffraction patterns of all the samples were obtained in reflection mode. The measurements were performed with a Bruker D8 Advance diffractometer (Bragg-Brentano geometry, Bruker AXS, Inc., Madison, WI, USA) equipped with motorized slits and a LynxEye 1D position-sensitive detector (CuKa, Ni-filter). The measurement range was 2Ω = 3–60°. Rietveld full-profile X-ray analysis of the patterns of crystalline substances was conducted using TOPAS 4.2 software [43] and is provided on Figures S1–S9 (Supporting Information). The background, profile, preferred orientation, scale factor(s), and unit cell parameters were refined. The preferred orientation was taken into account with the spherical harmonics approach [44].
2.2. Crystallographic Details
The intensities of reflections for 1, 2, 4–6, and 11–14 were measured with a Bruker APEX DUO diffractometer using MoKα (λ = 0.71073 Å) radiation (Bruker AXS, Inc., Madison, WI, USA). X-ray diffraction data for 3, 7–10 were collected by the “Belok/XSA” beamline of the Kurchatov Synchrotron Radiation Source [45,46]. Diffraction patterns were collected using a 1-axis MarDTB goniometer equipped with a Rayonix SX165 CCD 2D positional sensitive CCD detector (λ = 0.745 Å, φ-scanning in 1.0° steps) in direct geometry with the detector plane perpendicular to the beam. For each data set, ~120 diffraction frames were collected at 100 K. The data were indexed and integrated by XDS (ver. 2023) software suite [47]. Crystal structures were solved by the dual-space algorithm [48] and refined by the full-matrix least squares against F2. Non-hydrogen atoms were refined in anisotropic approximation except disordered solvent molecules in 4. Hydrogen atoms were included in the refinement by the riding model with Uiso(H) = 1.5 Ueq(X) for methyl and hydroxo groups, and with 1.2 Ueq(X) for the other atoms. Contribution of highly disordered solvent molecules to the structure factors of 14 was taken into account using solvent mask procedure of OLEX2 [49]. Complex 6 is a twin which was integrated using CELL_NOW and TWINABS algorithms, and refined using the BASF/HKLF 5 combination of instructions. All calculations were conducted using the SHELXTL ver. 2018/3 [50] and OLEX2 [49] program packages.
Experimental details and the results of these refinements are listed in Tables S1 and S2 (Supporting Information). Crystallographic information files are available from the Cambridge Crystallographic Data Center upon request (https://ccdc.cam.ac.uk/structure, (accessed on 22 August 2023) deposition numbers are 2215031–2215044).
2.3. DFT Calculations
Preliminary optimization of molecular geometry of complexes 1, 2, 5, 7–10, 12, and 13 was carried out using the Q-Chem program (PBE/6-31G(d)) [51]. Thereafter, the final optimization, frequencies calculations, and electron density calculation were performed with the Orca 5.1 program [52] and PBE0/def2-TZVP method/basis set combination. The calculated electron density function for the complexes studied was analyzed with the QTAIM approach using Multiwfn software, ver. 3.8 [53]. Coordinates of atoms for optimized complexes are given as Supporting Information.
3. Results
3.1. Synthesis
Our studies were initiated by investigating the reaction between abiraterone acetate and transition metal salts (Table 1). Previously, AbirAc was found to be soluble in ethanol (EtOH; at r.t. mole fraction solubility S × 105 = 1106.45(2)), tetrahydrofuran (THF; S × 105 > 10,000), acetonitrile (CH3CN; S × 105 = 209.76(7)), and acetone (S × 105 = 1806.39(6)) [31]. We attested these solvents as well as diethyl ether (Et2O) and dichloromethane (CH2Cl2) for complexation reactions. The majority of salts are insoluble in Et2O, CH2Cl2, and acetone (denoted with a down arrow in Table 1). Despite CoCl2·2H2O, Fe(acac)3, and Fe(AcO)2 (acac– = acetylacetonate; AcO– = acetate) formation with AbirAc transparent solutions, no complexes precipitated from these mixtures (denoted with a dash). During interaction of CuCl2·2H2O and AbirAc, only powders precipitated which could not be characterized using single crystal XRD. A total of 14 new complexes were obtained and characterized by 1H NMR and powder and single-crystal X-ray structural analysis. A total of 13 of these complexes contained abiraterone acetate coordinated by a metal atom. No hydrolytic elimination was observed in reaction conditions applied despite the slightly acidic nature of some solutions.

Table 1.
Reaction mixtures attested for complex formation.
3.2. Molecular and Crystal Structures
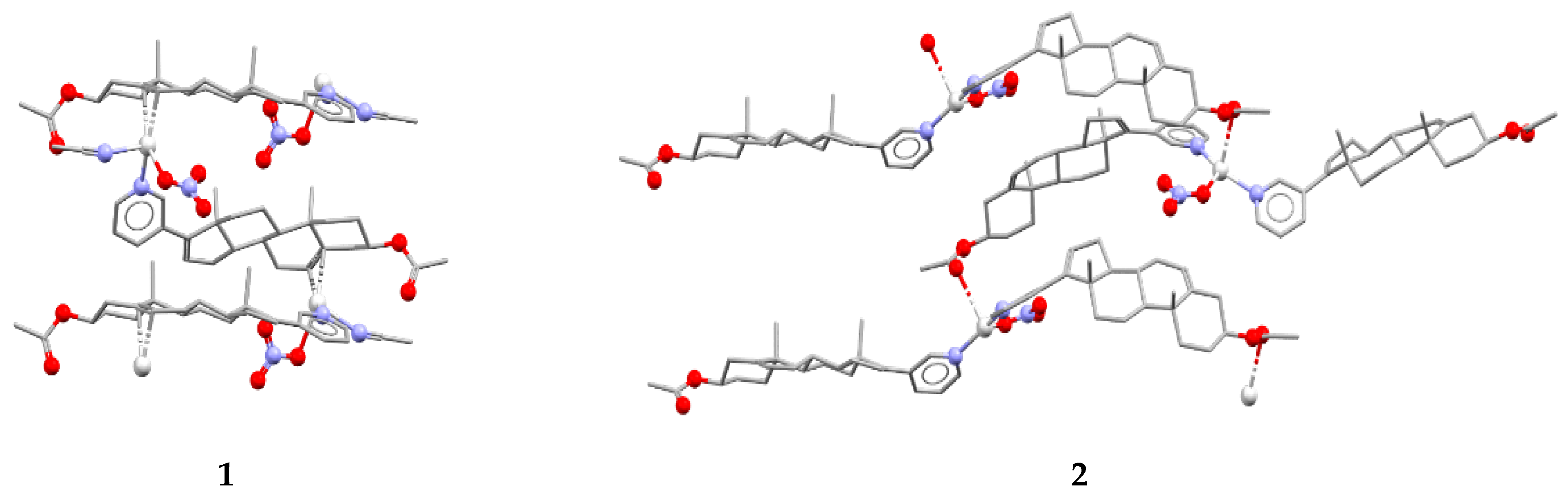
In reactions with silver(I) atoms in acetonitrile and ethanol, AbirAc acts as a linker to form 1D coordination polymers [Ag(AbirAc)(CH3CN)]NO3 (1) and [Ag(AbirAc)2]NO3 (2), respectively (Figure 1). In both cases, a Ag–NPy bond as long as 2.157(6)–2.265(3) Å occurs. In addition, in (1), a double bond of steroid fragment of AbirAc is coordinated by a silver(I) atom through a π-bond (r(Ag–C) = 2.363(4)–2.475(4) Å); and in (2), one of two symmetrically independent AbirAc molecules forms an Ag–O bond with a carbonyl fragment (r(Ag–O) = 2.857(7) Å), while another AbirAc is terminal. In (1), a silver(I) atom beside an AbirAc coordinates a terminal nitrate-anion and an acetonitrile molecule to form a pseudo-trigonal pyramidal AgN2OC2 coordination polyhedron with a metal atom 0.19(1) Å shifted from the plane formed by two carbon atoms, an oxygen atom, and a nitrogen atom to the nitrogen atom of the acetonitrile molecule. In (2), a silver(I) atom is 0.10(1) Å shifted from the base of an AgN2O2 trigonal pyramid to an apical carbonyl oxygen atom.
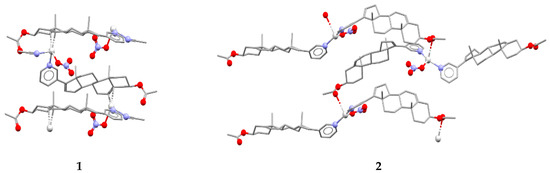
Figure 1.
Fragment of 1D coordination polymers in 1 and 2.
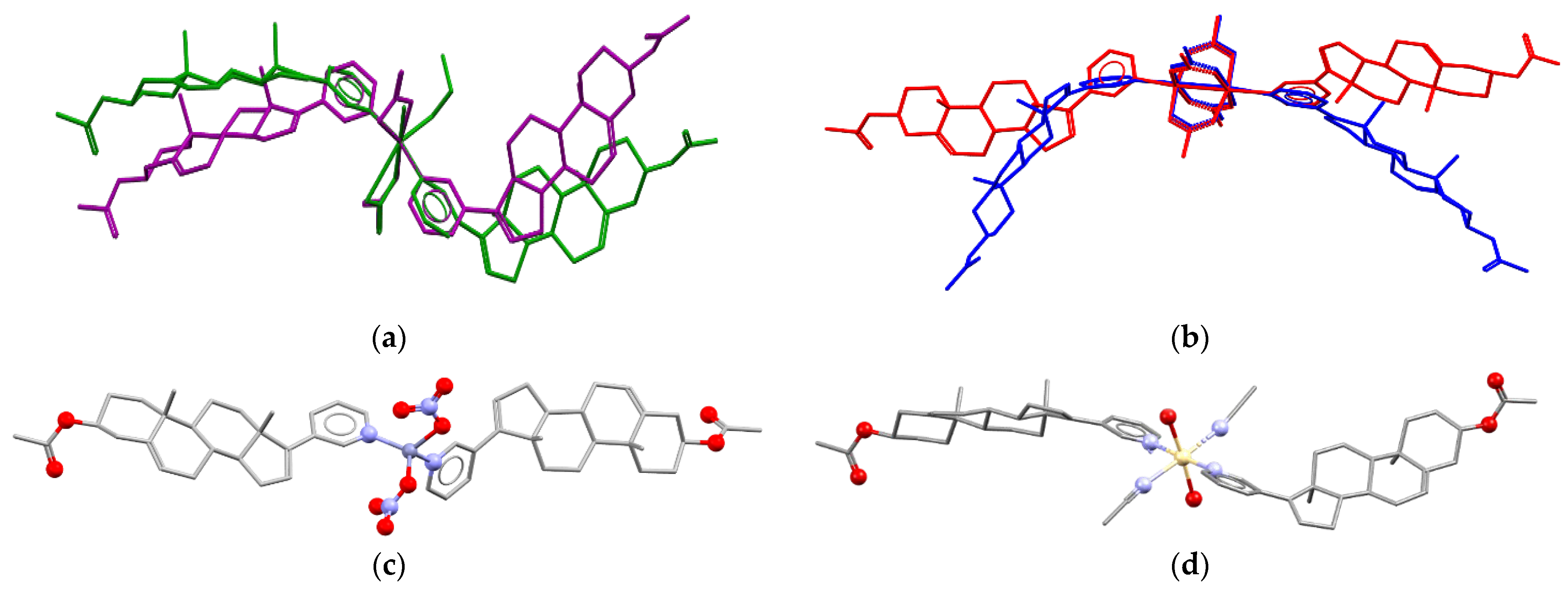
The molar ratio Ag(I):AbirAc in the reaction mixtures was the same; however, it varies in complexes (1) and (2). In copper(II)-containing mixtures, the molar ratio of reagents affected the Cu(II):AbirAc ratio in resulting complexes. For Cu(NO3)2:AbirAc = 1:1 solutions, isostructural complexes (3) and (4) were obtained where Cu(II):AbirAc ratio is equal to 1:2. In solid (3), [Cu(AbirAc)2(NO3)2] and [Cu(AbirAc)2(NO3)(H2O)(CH3CN)]+ complexes co-crystallize in 1:1 ratio, while in (4), [Cu(AbirAc)2(NO3)(H2O)(EtOH)]+ and [Cu(AbirAc)2(NO3)(H2O)2]+ complexes were found to have a ratio of 0.33:0.67 (Figure 2a). When the Cu(OAc)2:AbirAc ratio was taken as 2:1 in an attempt to obtain an AbirAc bridge between copper(II) atoms, complexes [Cu2(AbirAc)2(AcO)4] (5) and [Cu2(AbirAc)2(AcO)4]·2THF (6) based on binuclear “lantern” [Cu2(AcO)4] units precipitated as green plates (Figure 2b). For all these complexes, Cu(II)–NPy bond lengths were equal to 1.979(5)–2.198(6) Å.
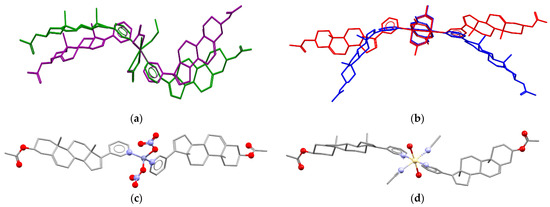
Figure 2.
(a) Conformations of complexes [Cu(AbirAc)2(NO3)2] (violet) in solid 3 and [Cu(AbirAc)2(NO3)(H2O)(EtOH)]+ (green) in solid 4 with overlaid CuN2(NO3) fragments. (b) Conformations of [Cu2(AbirAc)2(AcO)4] complexes in 5 and 6 with overlaid [Cu2(AcO)4] units. Molecular structures of (c) 9 and (d) 10. H atoms are omitted.
The reaction of Co(II), Ni(II), Zn(II) and Cd(II) nitrates with AbirAc in a 1:1 ratio in acetonitrile affords isostructural [M(AbirAc)2(CH3CN)2(H2O)2](NO3)2 (M = Co(II), 7; M = Ni(II), 8 and M = Cd(II), 10) and [Zn(AbirAc)2(NO3)2] (9) with terminal AbirAc, anions and solvent molecules, and metal atoms in, respectively, octahedral and tetrahedral environments (Figure 2c,d). Unfortunately, the only iron(II) complex with AbirAc was obtained by reaction of FeBr3 with AbirAc in acidic media and had the composition (AbirAcH)[FeBr4] (11). With the absence of HBr, single crystals of (HAbirOH)Br · H2O and colloid Fe(OH)3 precipitated from the same FeBr3:AbirAc = 1:1 mixture, and the structure of this salt has been published elsewhere [37]. The recrystallization of AbirAc from 2M and concentrated 48% HBr affords only the starting AbirAc. Thus, the presence of iron(III) salt abiraterone acetate can be hydrolyzed to AbirOH or HAbirOH+.
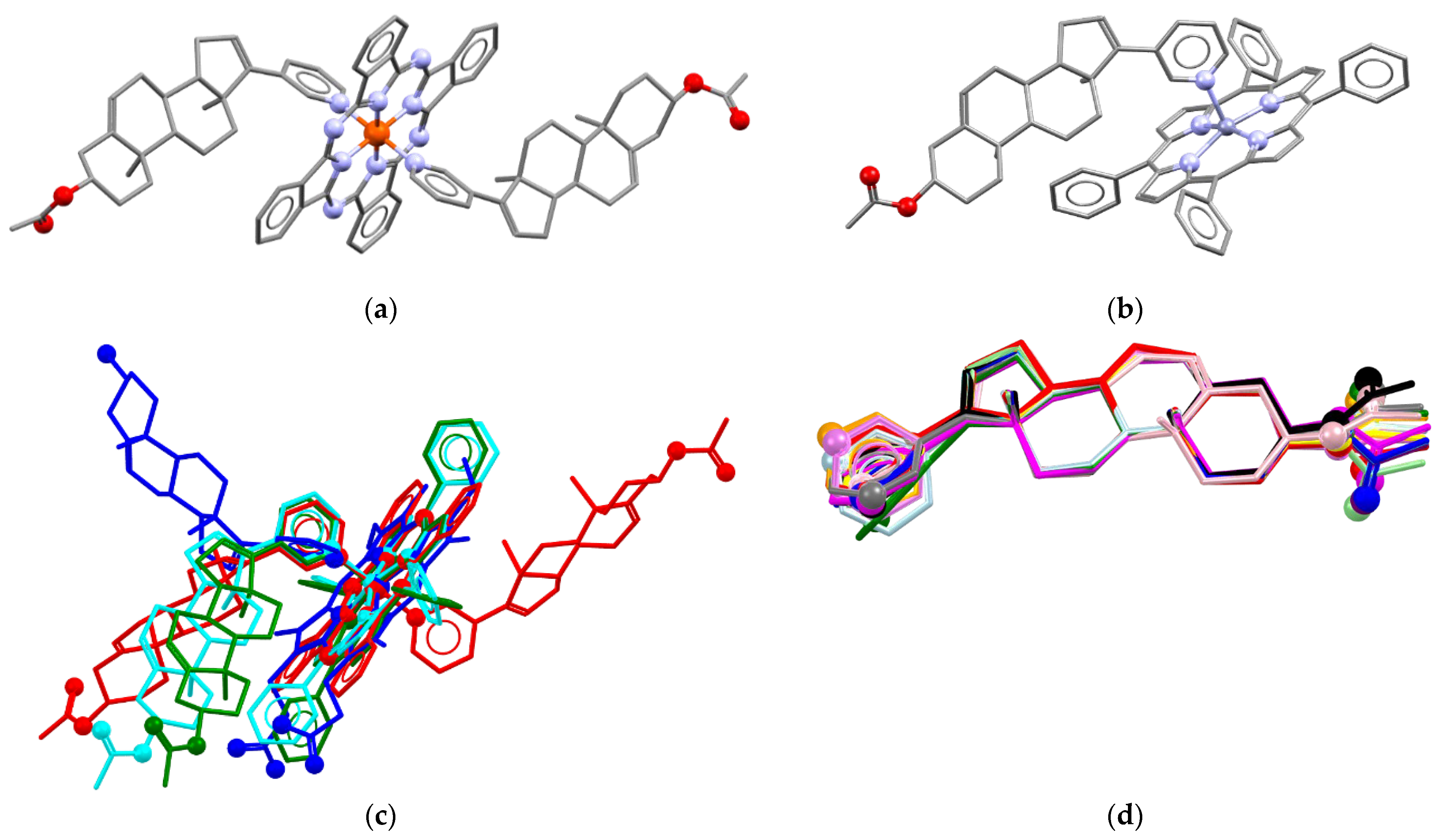
In order to obtain an iron(II) complex with AbirAc, iron(II) phthalocyaninate (FePC) was also attested as complexing agent. The 2:1 mixture in acetonitrile afforded violet needle single crystals which were characterized by means of X-ray diffraction. The complex has the composition [FePC(AbirAc)2] (12), where two AbirAc molecules interact with the iron(II) atom through the nitrogen atoms of their heterocycles, and the metal atom realizes the octahedral coordination (Figure 3a). The AbirAc ligand with methyl groups directed from the PC ring is situated ca. 57° to the [FePC] plane, and the angle between the AbirAc involved in C-H…PC intramolecular bonding and the [FePC] plane is equal to 40°. Thus, not only Fe-N bond lengths, but also the mutual orientation of the iron(II) phthalocyaninate plane and steroid fragment in the complex closely resemble those observed in AbirOH:receptor complexes where the corresponding angle is close to 60° (Figure 3c). Finally, zinc(II) tetraphenylporphyrinate was allowed to react with AbirAc. Both reagents are soluble in alcohols and CH2Cl2 [31,54,55]. Dark-violet needles of the [ZnTPP(AbirAc)] (13) and [ZnTPP(AbirAc)]·CH3OH (14) compositions precipitated from ethanol and methanol, respectively. Zinc(II) in 13 and 14 is 0.306(1) and 0.228(4) Å shifted from the base of a square pyramid formed by TPP nitrogen atoms with a pyridyl nitrogen atom in the vertex (Figure 3b). Methyl groups and ring C of the steroid core of AbirAc take part in H…H bonding with a phenyl ring of TPP to form the angle between the AbirAc and the TPP plane close to that in 12. The presence of intramolecular bonding between the AbirAc molecule and the TPP fragment in the solution was additionally confirmed by 1H NMR spectroscopy (see below).
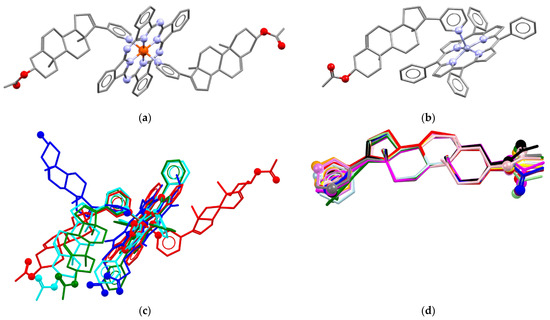
Figure 3.
Molecular structures of (a) 12 and (b) 13. (c) Comparison of molecular conformations in [FePC(AbirAc)2] (red), [ZnTPP(AbirAc)2] (13, cyan; 14, green) with [FePP(AbirOH)] (H2PP = protoporphyrine) in PDB 3ruk [38] (blue) with overlaid N4 equatorial planes. (d) Molecular conformations of AbirAc in 1 (red), 2 (orange), 3 (yellow), 4 (light-green), 5 (green), 6 (light-blue), 7 (cyan), 8 (blue), 9 (violet), 10 (purple), 11 (magenta), 12 (pink), 13 (grey), and 14 (black). Overlaid carbon atoms belong to olefin and methyl groups. Non-carbon atoms are depicted as spheres. H atoms are omitted.
Thus, abiraterone acetate can form complexes with transition metals through the nitrogen atom of the pyridine ring. Silver(I) atoms are able also to coordinate through acetate or olefin groups. Conformation of the AbirAc group in the studied solids is very rigid with only the pyridine ring and acetate fragments freely rotating along single bonds (Figure 3d).
Despite the presence of bulky AbirAc, coordinated water molecules as well as solvate methanol and pyridinium proton are able to take part in H-bonding. Parameters of H-bonds are listed in Table 2. In 3, 4, 7, 8, and 10, the H-bonds occur between water molecules and uncoordinated nitrate anions. Solid 11 contains N1—H1···O2 bonds between pyridinium and acetate groups. In crystals of 14, H-bonding between methanol and acetate groups can be seen. However, in all complexes but 11, analysis of H-bonding and H-bonded architectures is prohibited by strong disorder of the nitrate anions, solvent, and AbirAc molecules.
Table 2.
H-bond parameters (Å, °) for solid 3, 4, 7, 8, 10, 11, and 14.
3.3. Bond Energies
In general, EtOH, THF, and CH3CN solvent molecules used in the synthesis are able to act as ligands towards metal ions, and in solutions an excess of these molecules towards reagents is always present. Thus, the fact that AbirAc was coordinated by cations upon crystallization from these solvents can be indicative that the energy of the M–NPy bond is similar or lower than that of M–NSolv and M–OSolv bonds. In order to check this assumption PBE0/Def2-TZVP calculations of isolated complexes 1–2, 5, 7–10, 12, and 13 were performed. Analysis of charge distribution within the Bader ‘Atoms in Molecules’ approach [56] revealed all expected bond critical points (bcp), including Ag-C in 1 and Ag-O=C in 2. Bond energies estimated using the empirical correlation proposed by Espinosa, Mollins, and Lecomte (EML) [57], although having significant limitations [58], can nevertheless be used for semi-qualitative evaluation of the strength of intermolecular interactions (Table 3). Particularly, Eint estimated for Fe–PC and Zn–TPP binding using this correlation (−865.1 and −561.6 kJ mol−1) are close to calculated values (−946.5 and −629.1 kJ mol−1) given in Ref. [59]. The value of E(M-NPy) of −233.7…−235.5 and −160.1…−162.8 kJ mol−1 in 7 and 12 can also be compared with that of −248.3 estimated by Rodgers, Stanley, and Amunugama [60] and that of −139.9 kJ mol−1 calculated for [FePP(Py)2] by Liao and Scheiner [61].
Table 3.
Energies of coordination bonds (kJ mol−1) in selected complexes a.
Despite having a different charge, Eint for the bond between silver(I) and AbirAc is close to that for copper(II), cadmium(II), and zinc(II) atoms. Eint(M–NPy) for the other transition metals decreases in the range of Ni(II) (c.a. –150 kJ mol−1)-Fe(II) (c.a. –160 kJ mol−1)-Co(II) (c.a. –235 kJ mol−1). For heteroligand complexes 7, 8, and 10, Eint values fall from M–Oaqua to M–NCH3CN and to M–NPy. The latter values are also lower than Eint for M–ONO3 bonds; thus, it is not surprising that we obtained the target complexes from metal nitrates, and that the majority of complexes realized the 2:1 ratio of AbirAc and cation.
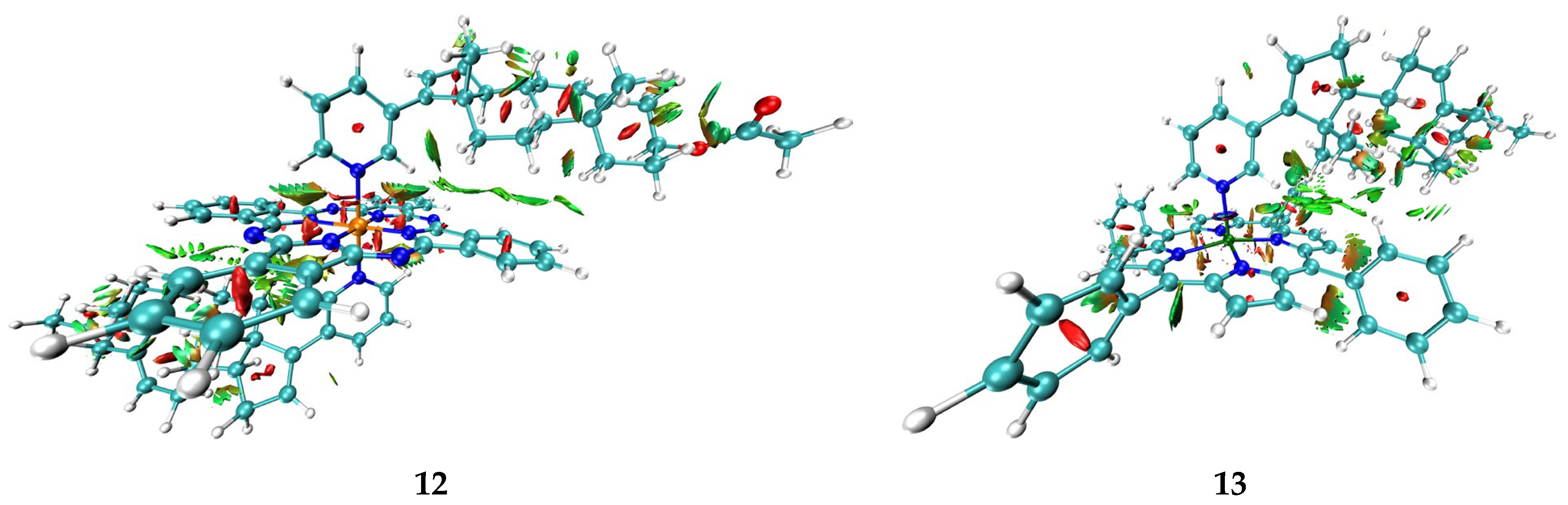
Taking into account data about the connectivity of biometals in proteins and enzymes, as well as the abovementioned regularities in Eint for metal–solvent and metal–abiraterone bonds, one can assume that AbirOH in the absence of steric hindrances can potentially replace water molecules in the coordination sphere of not only iron porphyrin but also of other biometals. At the same time, as the energy of interactions with heme iron is higher than that with other transition metals except for cobalt(II), it remains the main target of AbirOH in living cells. In addition, the abiraterone complexes with heme porphyrin as compared with ions connected with water and amino acids only are probably additionally stabilized with intramolecular bonding. Particularly, for complexes 12–14 few other bcp corresponding to intramolecular AbirAc…PC and AbirAc…TPP interactions were found. In Figure 4, intramolecular bonding in this complex is visualized by means of the NCI (non-covalent interaction) method [62]. This method relies on two functions: dimensionless reduced density gradient (RDG, s(r)) and signλ2ρ(r), which is a product of λ2 eigenvector by electron density at a given point. NCI analysis has shown that phthalocyanine and porphyrin moieties are bonded with abiraterone by a number of C-H…π interactions and C-H…N weak bonds (Figure 4). The isosurfaces of the RDG function that correspond to the former type of interactions can be described as shapeless fuzzy regions where the values of the signλ2ρ(r) function are close to zero. Such character of bonding corresponds to van der Waals interactions. In the case of C-H…N weak bonds, the isosurfaces of RDG function are partly characterized by positive values of thesignλ2ρ(r) function, which is indicative to some extent of steric repulsion between the pyridine fragment of the abiraterone moiety and the phthalocyanine or porphyrin systems.
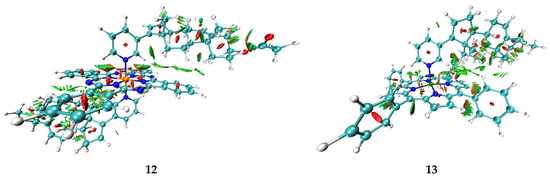
Figure 4.
Isosurfaces of the RDG function (s(r) = 0.3) of 12 and 13, colored according to λ2 values (−0.02–0.04 a.u.).
3.4. Structure in Solution
Finally, peculiarities of chemical bonding of AbirAc and zinc(II) 5,10,15,20-tetraphenylporphyrinate in solutions and the potential for drug release of a complex were studied in detail, with the example of complex 13, using NMR spectroscopy known to be sensitive to molecular dynamics in solution (Figures S10–S52, Supporting Information). The complex and coordination of pyridine nitrogen by zinc(II) atoms were found to remain stable in solutions of low-polar solvents, for example, in organochlorides. In polar solvents such as DMSO, AbirAc fully splits away from the coordination sphere of zinc(II) and is replaced by solvent molecules which are present in solution in large numbers and ensure equilibrium during AbirAc release. The dynamics of drug release in polar solvents could not be estimated due to the speed of the process. The existence a of stable [ZnTPP(AbirAc)] complex, as well as its decomposition into two separate AbirAc and [ZnTPP] complexes, was confirmed by a series of independent NMR techniques.
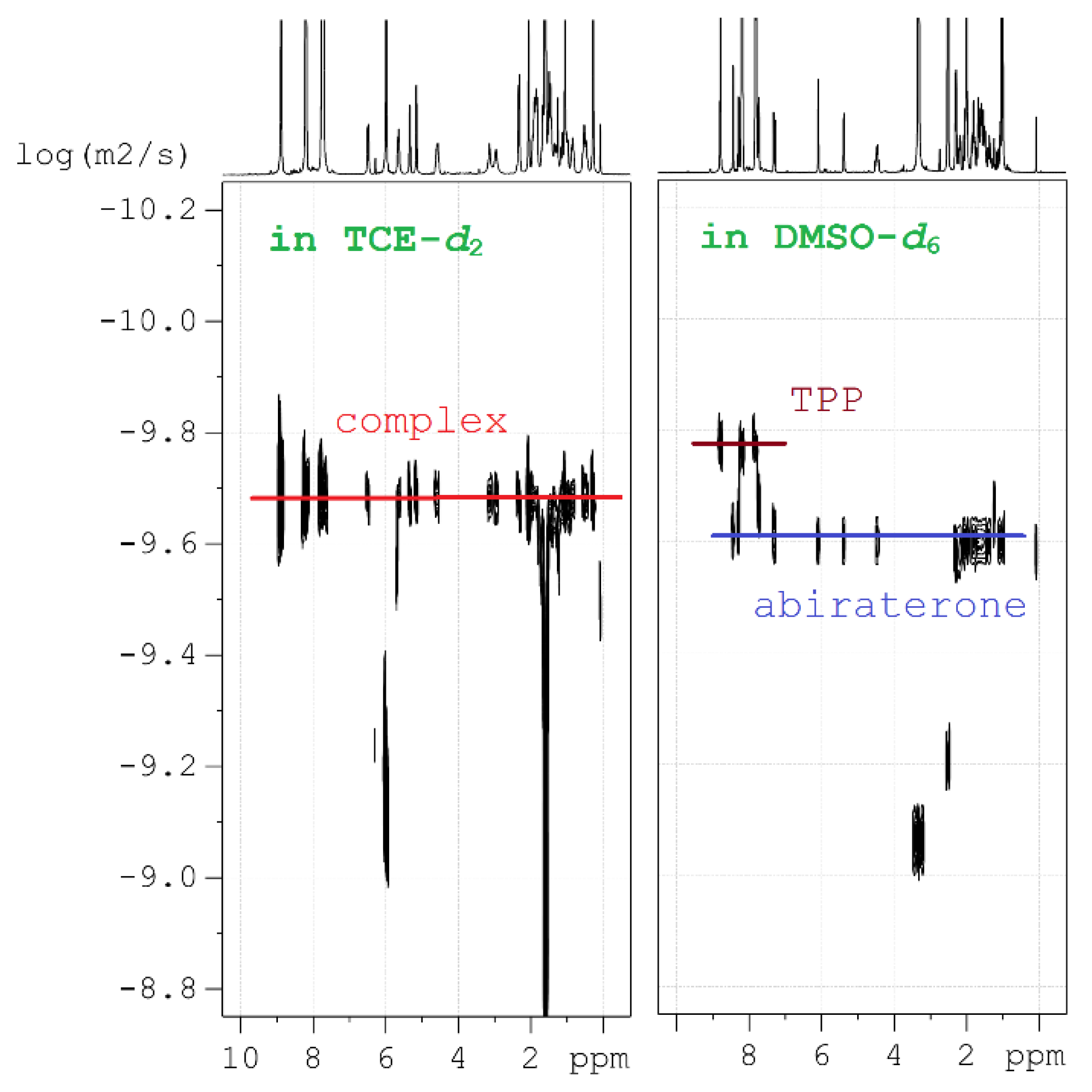
(1) Diffusion-ordered NMR spectroscopy (DOSY) was applied to estimate the dynamics of molecular motion in the solutions and to estimate their molecular mass (MW). This method is well suited to analyze chemical bonding, because in low-polar solvents the [ZnTPP(AbirAc)] complex moves as a whole and has one diffusion coefficient (Figure 5). In polar solvents, AbirAc and [ZnTPP] act as two independent molecules with different diffusion coefficients because of varying size and mass (Figure 5). For complex 14, the diffusion coefficient was compared with those of its individual components sensitive to MW increase and diffusion-slowing accompanying the complex formation (see ESI).
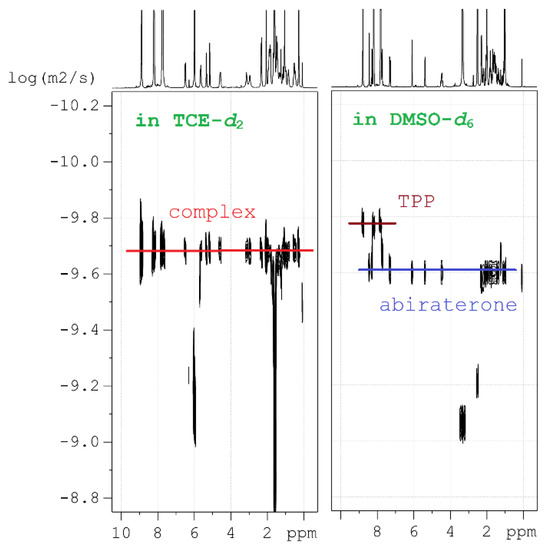
Figure 5.
Diffusion NMR studies: representation of two fundamentally different 2D 1H DOSY spectra in 1,1,2,2-tetrachloroethane-d2 (TCE-d2) and DMSO-d6, one of which contains complex 14, and the other containing free molecules of [ZnTPP] and AbirAc.
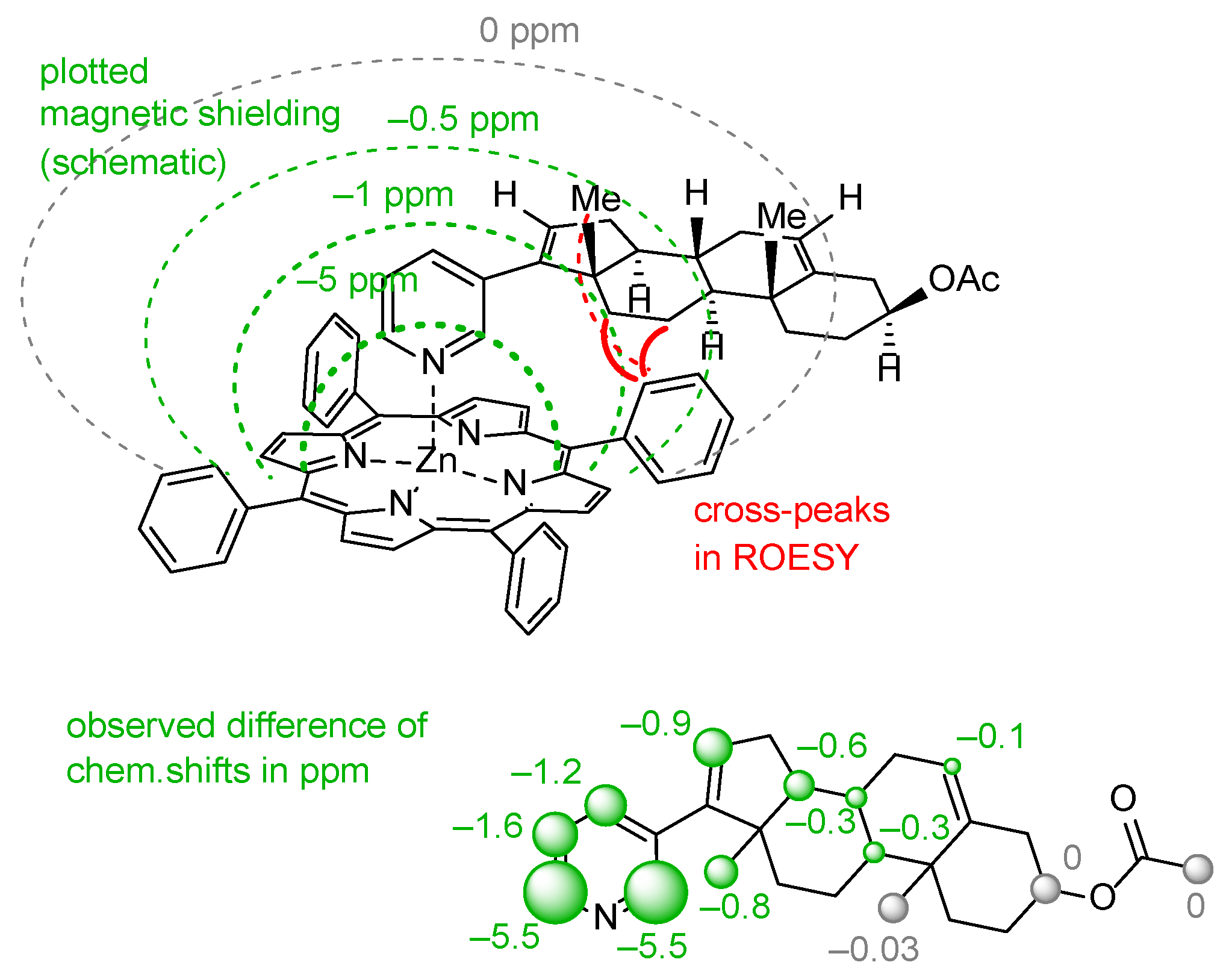
(2) Another method is analysis of NOE interactions between protons of abiraterone acetate and tetraphenylporphyrinate which can be found only for the [ZnTPP(AcirAc)] complex. Taking the high molecular mass of the complex as well as the relatively free rotation of abiraterone acetate along the Zn-N coordination bond, ROESY spectra should be analyzed, while intensities of NOE interactions are rather low (Figure 6).
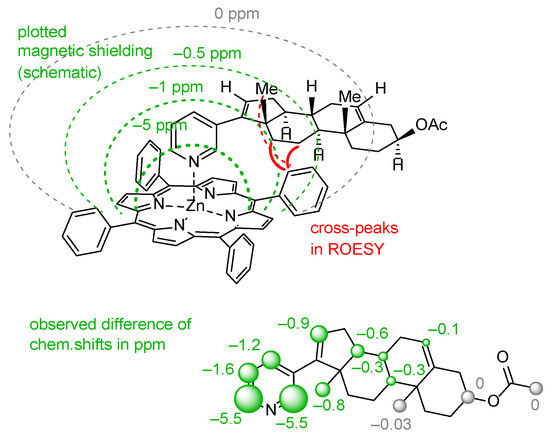
Figure 6.
Schematic representation of the magnetic shielding of aromatic π-system in [ZnTPP] framework in complex 14, and its impact on the chemical shifts of signals in the abiraterone molecule.
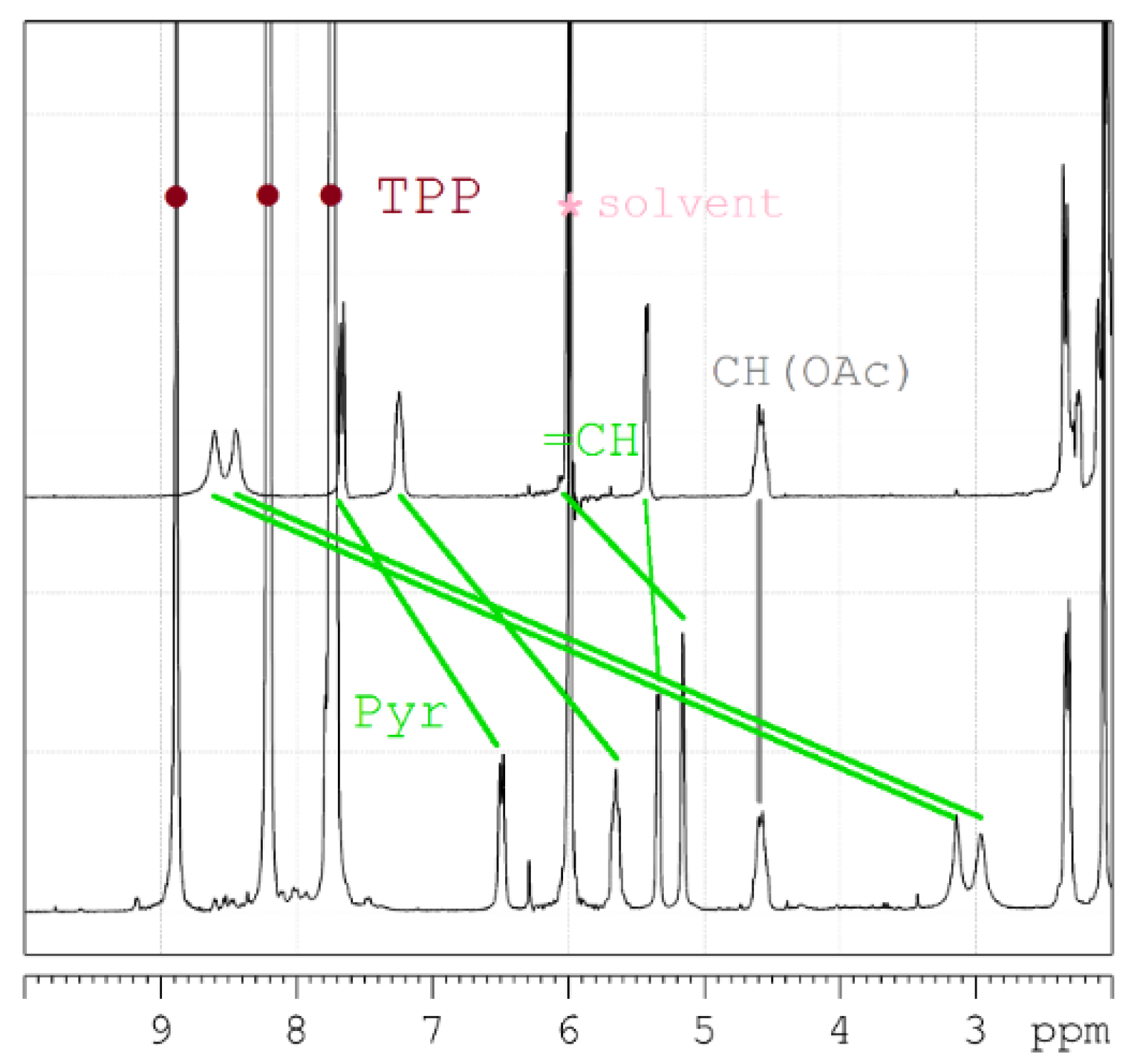
(3) The third, less common, approach is analysis of variation in chemical shifts of signals in 1H NMR spectra due to the effect of strong ring electronic currents in the extended π-system of porphyrin and four phenyl rings when the AbirAc molecule partially enters the anisotropy cone of the [ZnTPP] upon coordination with zinc(II) [63]. The values of the magnetic shielding for the closest to TPP protons achieve as much as −5.5 ppm and gradually decrease to zero for the farthest atoms (Figure 6 and Figure 7). At the same time the signals corresponding to the phenyl and porphyrin protons remain unchanged upon coordination of AbirAc. This approach gives the most detailed structural information about the mutual disposition of AbirAc and [ZnTPP] in solution, because it allows detection of the closest protons for these two molecules, while the ring currents of the aromatic π-system have almost no effect on the other side of AbirAc. In solution, disposition of AbirAc towards the [ZnTPP] differs from that in a solid state. The molecule is nearly 90° shifted and free rotation dynamic is hindered (Figure 7).
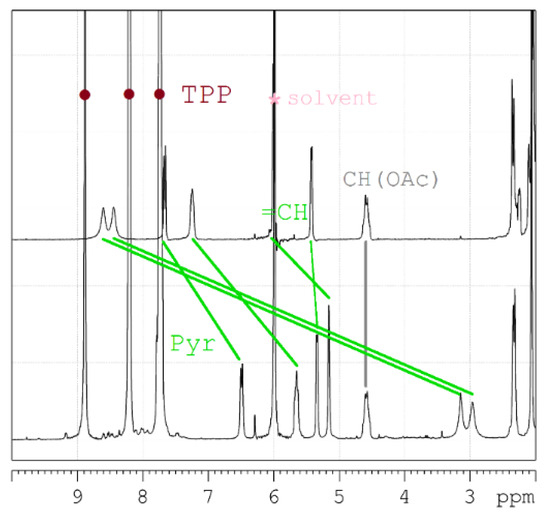
Figure 7.
Downfield region of the 1H NMR spectra to demonstrate clearly the shift of the abiraterone acetate signals due to coordination with [ZnTPP] (AbirAc = top; [ZnTPP(AbirAc)] = bottom; solvent = 1,1,2,2-tetrachloroethane-d2; assignment = TOCSY, HSQC, HMBC).
4. Conclusions
In summary, self-assembly of abiraterone acetate with transition metal salts was studied, and its first complexes with transition metals were obtained. Energies of coordination bonds for nine complexes were estimated from the theoretical charge density of isolated molecules. The bond between a metal ion and the nitrogen atom of the 3-pyridyl ring of abiraterone acetate is stronger than bonds with solvent (acetonitrile or water) molecules and anions (nitrate or acetate) for a given cation. Thus, it is not surprising that abiraterone acetate readily replaces solvent molecules in the coordination sphere of metal ions by the nitrogen atom of the pyridyl ring upon crystallization. The strength of M–N(pyridyl) bond complexes increases in the following manner: E(AgI–N)≈E(CuII–N) ≈ E(CdII–N) ≈ E(ZnII–N) < E(NiII–N) < E(FeII–N) < E(CoII–N). The bonding in phthalocyaninates and porphyrinates can be additionally supported by weak intramolecular interactions between the steroid fragment of abiraterone and the π-system of the macrocycle. Nevertheless, a zinc(II) porphyrinate-containing complex with abiraterone acetate remains stable only in low-polar solvents, while drug release was observed in polar solvents which makes this family of compounds prospective as metallopharmaceuticals.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pharmaceutics15092180/s1: Synthetic details and spectral data; Figures S1–S9: Rietveld refinement of powder XRD patterns; Figures S10–S52: NMR spectra; Tables S1 and S2: Crystallographic data and structure refinement details for 1–14; OUT files for complexes 1, 2, 5, 7–10, 12, and 13. Crystallographic information files are available from the Cambridge Crystallographic Data Center upon request (https://ccdc.cam.ac.uk/structure, accessed on 22 August 2023; deposition numbers are 2215031–2215044).
Author Contributions
Conceptualization, A.K.; formal analysis, A.K.; investigation, P.B., P.D. and A.K.; writing—original draft preparation, A.V. and R.N.; writing—review and editing, A.V. and A.K.; visualization, A.V. and R.N.; funding acquisition, A.K. The manuscript was written through contributions of all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Russian Science Foundation, grant No. 20-13-00241.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Compounds, spectra, and XRD data are available from authors.
Acknowledgments
X-ray diffraction studies were supported by the Ministry of Science and Higher Education of the Russian Federation (Contract No. 075-03-2023-642) and performed using the equipment of the Center for Molecular Studies of INEOS RAS.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Paulekuhn, G.S.; Dressman, J.B.; Saal, C. Trends in Active Pharmaceutical Ingredient Salt Selection Based on Analysis of the Orange Book Database. J. Med. Chem. 2007, 50, 6665–6672. [Google Scholar] [CrossRef] [PubMed]
- Berge, S.M.; Bighley, L.D.; Monkhouse, D.C. Pharmaceutical Salts. J. Pharm. Sci. 1977, 66, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G.; Stahl, P.H. Pharmaceutical Salts, Properties, Selection, and Use. A Handbook; Helvetica Chimica Acta/Wiley-VCH: Weinheim, Germany, 2001; ISBN 978-3-906390-26-0. [Google Scholar]
- Fini, A.; Fazio, G.; Hervás, M.-J.F.; Holgado, M.A.; Rabasco, A.M. Factors Governing the Dissolution of Diclofenac Salts. Eur. J. Pharm. Sci. 1996, 4, 231–238. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Maini, L.; Prosperi, S.; Gobetto, R.; Chierotti, M.R. From Unexpected Reactions to a New Family of Ionic Co-Crystals: The Case of Barbituric Acid with Alkali Bromides and Caesium Iodide. Chem. Commun. 2010, 46, 7715–7717. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Grepioni, F.; Lampronti, G.I.; Maini, L.; Turrina, A. Ionic Co-Crystals of Organic Molecules with Metal Halides: A New Prospect in the Solid Formulation of Active Pharmaceutical Ingredients. Cryst. Growth Des. 2011, 11, 5621–5627. [Google Scholar] [CrossRef]
- Grepioni, F.; Wouters, J.; Braga, D.; Nanna, S.; Fours, B.; Coquerel, G.; Longfils, G.; Rome, S.; Aerts, L.; Quéré, L. Ionic Co-Crystals of Racetams: Solid-State Properties Enhancement of Neutral Active Pharmaceutical Ingredients via Addition of Mg2+ and Ca2+ Chlorides. CrystEngComm 2014, 16, 5887–5896. [Google Scholar] [CrossRef]
- Surov, A.O.; Drozd, K.V.; Ramazanova, A.G.; Churakov, A.V.; Vologzhanina, A.V.; Kulikova, E.S.; Perlovich, G.L. Polymorphism of Carbamazepine Pharmaceutical Cocrystal: Structural Analysis and Solubility Performance. Pharmaceutics 2023, 15, 1747. [Google Scholar] [CrossRef]
- Meggers, E. Targeting Proteins with Metal Complexes. Chem. Commun. 2009, 1001–1010. [Google Scholar] [CrossRef]
- Blanck, S.; Maksimoska, J.; Baumeister, J.; Harms, K.; Marmorstein, R.; Meggers, E. The Art of Filling Protein Pockets Efficiently with Octahedral Metal Complexes. Angew. Chem. Int. Ed. 2012, 51, 5244–5246. [Google Scholar] [CrossRef]
- Delori, A.; Friščić, T.; Jones, W. The Role of Mechanochemistry and Supramolecular Design in the Development of Pharmaceutical Materials. CrystEngComm 2012, 14, 2350–2362. [Google Scholar] [CrossRef]
- Ma, Z.; Moulton, B. Supramolecular Medicinal Chemistry: Mixed-Ligand Coordination Complexes. Mol. Pharm. 2007, 4, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Grepioni, F.; André, V.; Duarte, M.T. Drug-Containing Coordination and Hydrogen Bonding Networks Obtained Mechanochemically. CrystEngComm 2009, 11, 2618–2621. [Google Scholar] [CrossRef]
- Chow, E.H.H.; Strobridge, F.C.; Friščić, T. Mechanochemistry of Magnesium Oxide Revisited: Facile Derivatisation of Pharmaceuticals Using Coordination and Supramolecular Chemistry. Chem. Commun. 2010, 46, 6368–6370. [Google Scholar] [CrossRef]
- McKinlay, A.C.; Morris, R.E.; Horcajada, P.; Férey, G.; Gref, R.; Couvreur, P.; Serre, C. BioMOFs: Metal–Organic Frameworks for Biological and Medical Applications. Angew. Chem. Int. Ed. 2010, 49, 6260–6266. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Serre, C.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Metal–Organic Frameworks as Efficient Materials for Drug Delivery. Angew. Chem. 2006, 118, 6120–6124. [Google Scholar] [CrossRef]
- Keskin, S.; Kızılel, S. Biomedical Applications of Metal Organic Frameworks. Ind. Eng. Chem. Res. 2011, 50, 1799–1812. [Google Scholar] [CrossRef]
- An, J.; Geib, S.J.; Rosi, N.L. Cation-Triggered Drug Release from a Porous Zinc–Adeninate Metal–Organic Framework. J. Am. Chem. Soc. 2009, 131, 8376–8377. [Google Scholar] [CrossRef]
- Kremer, L.E.; McLeod, A.I.; Aitken, J.B.; Levina, A.; Lay, P.A. Vanadium(V) and -(IV) Complexes of Anionic Polysaccharides: Controlled Release Pharmaceutical Formulations and Models of Vanadium Biotransformation Products. J. Inorg. Biochem. 2015, 147, 227–234. [Google Scholar] [CrossRef]
- Babarao, R.; Jiang, J. Unraveling the Energetics and Dynamics of Ibuprofen in Mesoporous Metal–Organic Frameworks. J. Phys. Chem. C 2009, 113, 18287–18291. [Google Scholar] [CrossRef]
- Orange Book. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book (accessed on 22 August 2023).
- Svensson Grape, E.; Rooth, V.; Nero, M.; Willhammar, T.; Inge, A.K. Structure of the Active Pharmaceutical Ingredient Bismuth Subsalicylate. Nat. Commun. 2022, 13, 1984. [Google Scholar] [CrossRef]
- Lestari, M.; Lusi, M. A Mixed Molecular Salt of Lithium and Sodium Breaks the Hume-Rothery Rules for Solid Solutions. Chem. Commun. 2019, 55, 2297–2300. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R&D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Palermo, G.; Spinello, A.; Saha, A.; Magistrato, A. Frontiers of Metal-Coordinating Drug Design. Expert Opin. Drug Discov. 2021, 16, 497–511. [Google Scholar] [CrossRef]
- Palermo, G.; Magistrato, A.; Riedel, T.; von Erlach, T.; Davey, C.A.; Dyson, P.J.; Rothlisberger, U. Fighting Cancer with Transition Metal Complexes: From Naked DNA to Protein and Chromatin Targeting Strategies. ChemMedChem 2016, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, S.; Hansen, C.H.; Petrunak, E.M.; Scott, E.E.; Styrishave, B.; Jørgensen, F.S.; Olsen, L. Promising Tools in Prostate Cancer Research: Selective Non-Steroidal Cytochrome P450 17A1 Inhibitors. Sci. Rep. 2016, 6, 29468. [Google Scholar] [CrossRef]
- Bonomo, S.; Jørgensen, F.S.; Olsen, L. Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J. Chem. Inf. Model. 2017, 57, 1123–1133. [Google Scholar] [CrossRef]
- Vidossich, P.; Magistrato, A. QM/MM Molecular Dynamics Studies of Metal Binding Proteins. Biomolecules 2014, 4, 616–645. [Google Scholar] [CrossRef]
- Sakai, Y.; Fukami, T.; Nagaoka, M.; Hirosawa, K.; Ichida, H.; Sato, R.; Suzuki, K.; Nakano, M.; Nakajima, M. Arylacetamide Deacetylase as a Determinant of the Hydrolysis and Activation of Abiraterone Acetate in Mice and Humans. Life Sci. 2021, 284, 119896. [Google Scholar] [CrossRef]
- Solymosi, T.; Tóth, F.; Orosz, J.; Basa-Dénes, O.; Angi, R.; Jordán, T.; Ötvös, Z.; Glavinas, H. Solubility Measurements at 296 and 310 K and Physicochemical Characterization of Abiraterone and Abiraterone Acetate. J. Chem. Eng. Data 2018, 63, 4453–4458. [Google Scholar] [CrossRef]
- Acharya, M.; Gonzalez, M.; Mannens, G.; De Vries, R.; Lopez, C.; Griffin, T.; Tran, N. A Phase I, Open-Label, Single-Dose, Mass Balance Study of 14C-Labeled Abiraterone Acetate in Healthy Male Subjects. Xenobiotica 2013, 43, 379–389. [Google Scholar] [CrossRef]
- Geboers, S.; Stappaerts, J.; Mols, R.; Snoeys, J.; Tack, J.; Annaert, P.; Augustijns, P. The Effect of Food on the Intraluminal Behavior of Abiraterone Acetate in Man. J. Pharm. Sci. 2016, 105, 2974–2981. [Google Scholar] [CrossRef] [PubMed]
- Chennuru, R.; Devarapalli, R.; Rengaraj, P.; Srinivas, P.L.; Dey, S.; Reddy, C.M. Improving Solubility of Poorly Soluble Abiraterone Acetate by Cocrystal Design Aided by In Silico Screening. Cryst. Growth Des. 2020, 20, 5018–5030. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, Y.; Xia, M.; Dai, W.; Zhu, B.; Mei, X. Improving the Dissolution Behaviors and Bioavailability of Abiraterone Acetate via Multicomponent Crystal Forms. Int. J. Pharm. 2022, 614, 121460. [Google Scholar] [CrossRef] [PubMed]
- Silveira, R.G.; Cunha, B.N.; Tenório, J.C.; Alves de Aguiar, D.V.; da Cruz Souza, P.; Vaz, B.G.; Ellena, J.; Batista, A.A.; Martins, F.T. A Simple Alternative to Prodrug: The Hydrochloride Salt Monohydrate of the Prostate Anticancer Drug Abiraterone. J. Mol. Struct. 2019, 1190, 165–170. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Buikin, P.A.; Dorovatovskii, P.V.; Vologzhanina, A.V. Synthesis, NoSpherA2 Refinement, and Noncovalent Bonding of Abiraterone Bromide Monohydrate. Struct. Chem. 2023, in press. [Google Scholar] [CrossRef]
- DeVore, N.M.; Scott, E.E. Structures of Cytochrome P450 17A1 with Prostate Cancer Drugs Abiraterone and TOK-001. Nature 2012, 482, 116–119. [Google Scholar] [CrossRef]
- Petrunak, E.M.; DeVore, N.M.; Porubsky, P.R.; Scott, E.E. Structures of Human Steroidogenic Cytochrome P450 17A1 with Substrates. J. Biol. Chem. 2014, 289, 32952–32964. [Google Scholar] [CrossRef]
- Pallan, P.S.; Nagy, L.D.; Lei, L.; Gonzalez, E.; Kramlinger, V.M.; Azumaya, C.M.; Wawrzak, Z.; Waterman, M.R.; Guengerich, F.P.; Egli, M. Structural and Kinetic Basis of Steroid 17α,20-Lyase Activity in Teleost Fish Cytochrome P450 17A1 and Its Absence in Cytochrome P450 17A2. J. Biol. Chem. 2015, 290, 3248–3268. [Google Scholar] [CrossRef]
- Gonzalez, E.; Johnson, K.M.; Pallan, P.S.; Phan, T.T.N.; Zhang, W.; Lei, L.; Wawrzak, Z.; Yoshimoto, F.K.; Egli, M.; Guengerich, F.P. Inherent Steroid 17α,20-Lyase Activity in Defunct Cytochrome P450 17A Enzymes. J. Biol. Chem. 2018, 293, 541–556. [Google Scholar] [CrossRef]
- Petrunak, E.M.; Bart, A.G.; Peng, H.-M.; Auchus, R.J.; Scott, E.E. Human Cytochrome P450 17A1 Structures with Metabolites of Prostate Cancer Drug Abiraterone Reveal Substrate-Binding Plasticity and a Second Binding Site. J. Biol. Chem. 2023, 299, 102999. [Google Scholar] [CrossRef]
- Available online: https://topas.awh.durham.ac.uk/doku.php?id=manual (accessed on 22 August 2023).
- Järvinen, M. Application of Symmetrized Harmonics Expansion to Correction of the Preferred Orientation Effect. J. Appl. Cryst. 1993, 26, 525–531. [Google Scholar] [CrossRef]
- Svetogorov, R.D.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA Diffraction Beamline for Studying Crystalline Samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Lazarenko, V.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Burlov, A.S.; Koshchienko, Y.V.; Vlasenko, V.G.; Khrustalev, V.N. High-Throughput Small-Molecule Crystallography at the ‘Belok’ Beamline of the Kurchatov Synchrotron Radiation Source: Transition Metal Complexes with Azomethine Ligands as a Case Study. Crystals 2017, 7, 325. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Cryst. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Epifanovsky, E.; Gilbert, A.T.B.; Feng, X.; Lee, J.; Mao, Y.; Mardirossian, N.; Pokhilko, P.; White, A.F.; Coons, M.P.; Dempwolff, A.L.; et al. Software for the Frontiers of Quantum Chemistry: An Overview of Developments in the Q-Chem 5 Package. J. Chem. Phys. 2021, 155, 084801. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Compt. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. The Porphyrin Handbook Volume 3 (The Porphyrin Handbook); Academic Press: Cambridge, MA, USA, 1999. [Google Scholar]
- Zheng, W.; Shan, N.; Yu, L.; Wang, X. UV–Visible, Fluorescence and EPR Properties of Porphyrins and Metalloporphyrins. Dye. Pigment. 2008, 77, 153–157. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Oxford University Press: Oxford, UK, 1994; ISBN 978-0-19-855865-1. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Liao, M.-S.; Scheiner, S. Comparative Study of Metal-Porphyrins, -Porphyrazines, and -Phthalocyanines. J. Compt. Chem. 2002, 23, 1391–1403. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, M.T.; Stanley, J.R.; Amunugama, R. Periodic Trends in the Binding of Metal Ions to Pyridine Studied by Threshold Collision-Induced Dissociation and Density Functional Theory. J. Am. Chem. Soc. 2000, 122, 10969–10978. [Google Scholar] [CrossRef]
- Liao, M.-S.; Scheiner, S. Electronic Structure and Bonding in Unligated and Ligated FeII Porphyrins. J. Chem. Phys. 2002, 116, 3635–3645. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Lo Presti, L. Non-Covalent Interaction via the Reduced Density Gradient: Independent Atom Model vs Experimental Multipolar Electron Densities. Compt. Theor. Chem. 2012, 998, 148–163. [Google Scholar] [CrossRef]
- Rickhaus, M.; Jirasek, M.; Tejerina, L.; Gotfredsen, H.; Peeks, M.D.; Haver, R.; Jiang, H.-W.; Claridge, T.D.W.; Anderson, H.L. Global Aromaticity at the Nanoscale. Nat. Chem. 2020, 12, 236–241. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).