Abstract
Artemisinin is a natural compound extracted from Artemisia species belonging to the Asteraceae family. Currently, artemisinin and its derivatives are considered among the most significant small-molecule antimalarial drugs. Artemisinin and its derivatives have also been shown to possess selective anticancer properties, however, there are several limitations and gaps in knowledge that retard their repurposing as effective anticancer agents. Hybridization resulting from a covalent combination of artemisinin with one or more active pharmacophores has emerged as a promising approach to overcome several issues. The variety of hybridization partners allows improvement in artemisinin activity by tuning the ability of conjugated artemisinin to interact with various molecule targets involved in multiple biological pathways. This review highlights the current scenario of artemisinin-derived hybrids with potential anticancer activity. The synthetic approaches to achieve the corresponding hybrids and the structure–activity relationships are discussed to facilitate further rational design of more effective candidates.
1. Introduction
Cancer is considered to be one of the most life-threatening diseases throughout the world nowadays. World Health Organization estimated 10 million people worldwide to have died from cancer in 2020 [1]. Although chemotherapy is the conventional treatment for cancer, chemotherapeutic drugs frequently confer relevant levels of toxicity, are subject to drug resistance and lack bioavailability that limit their therapeutic potential and lower the rate of success in cancer [2,3,4,5]. To address these limitations, in recent decades significant efforts have been directed towards the development of combination therapies which involve the use of different drugs (Figure 1A) with diverse molecular mechanisms that can cooperatively affect and prevent cancer cells from developing drug resistance [6]. However, dose-limiting toxicities and drug–drug interactions can in turn limit the use of combination therapies. Hybrid molecules as multitarget anticancer agents have been envisaged as a valuable strategy to overcome the drawbacks of combination therapies as well as drug resistance [6,7].
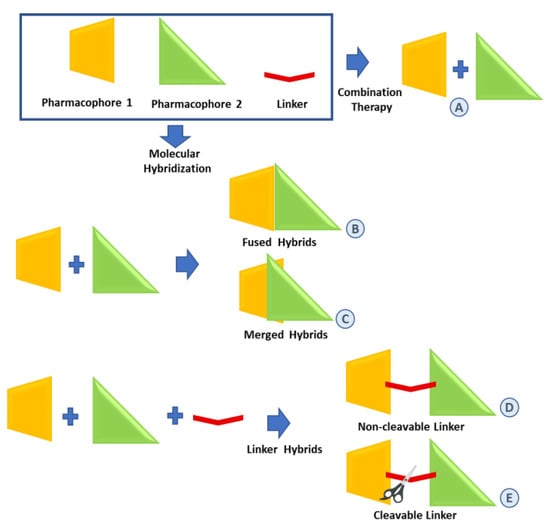
Figure 1.
Strategies for the combination of pharmacophores. (A): combination therapy; (B–E): molecular hybridization approaches.
Molecular hybridization is a modern strategy used in drug discovery for rational drug design [8]. Based on the covalent combination of pharmacophoric moieties of different bioactive substances, molecular hybridization produces new hybrid compounds that can be more than the simple sum of the parent pharmacophores [7,8]. In general, the concept of hybridizing various organic molecular motifs with different pharmacological activities can result in compounds with modified selectivity profile, different and/or dual modes of action, reduced undesired side effects, ability to overcome multidrug resistance and improved safety profile [9].
Typically, hybrid molecules containing two or more pharmacophoric units can be classified depending on the manner in which they are covalently linked. In the case of directly linked hybrids (fused hybrids in Figure 1B), the two molecular units are fused without using any linkers, that is, each molecule provides a functional group to build a link normally resulting in an enzymatically hydrolyzable ester, carbamate or amide. Merged hybrids (Figure 1C) result from the overlapping of structural motifs of both pharmacophores which may or may not retain the original functional activities. Linker hybrids are characterized by the presence of well-identifiable linkers classified as non-cleavable and cleavable. A non-cleavable linker (Figure 1D) leads to a hybrid that can be able to retain the biological activity and the affinity for the biological target of the different units by maintaining its structure or even convey a novel biological action. A cleavable linker (Figure 1E) is designed to release the parent agents with independent action under physiological or enzymatic conditions.
Over recent decades, molecular hybridization has been extensively used for the preparation of novel drug candidates with improved biological properties [9]. The relevance of a molecular hybridization strategy in cancer chemotherapy is evident by the number of hybrid compounds approved or under clinical trials [10,11].
Natural plant extracts represent a very important source of bioactive compounds for pharmacotherapy, especially for cancer and infectious diseases [12,13]. From the chemical point of view, numerous bioactive natural products from plants, such as taxoides deriving from the Pacific yew tree [14,15,16,17,18,19], are very interesting, due to the presence of reactive centers that enable the formation of covalent bonds with other molecules to give hybrid drugs. However, developing bioactive natural products into drugs has remained challenging, therefore, the use of natural compounds in the preparation of novel hybrid molecules has been considered an interesting strategy to empower their biological properties or to expand their biological space [20].
Artemisia annua (Figure 2), a plant belonging to the Asteraceae family, has been safely used in traditional Chinese medicine over the centuries to treat a variety of fevers and respiratory tract infections. Artemisinin (1) (Figure 2), the bioactive component of Artemisia annua, and its semisynthetic derivatives, collectively called artemisinins, are very interesting compounds best known as antimalarial agents.
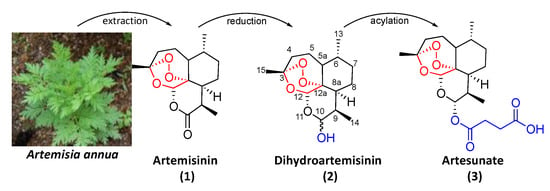
Figure 2.
Picture of Artemisia annua and molecular structures of artemisinin (1), dihydroartemisinin (2) and artesunate (3).
Dr. Youyou Tu’s research team discovered the effectiveness of Artemisia annua extracts in the inhibition of the parasite Plasmodium falciparum in the early 1970s [21]. In the next decades, artemisinin (1) and its derivatives were developed into drugs and became the gold standard for malaria treatment. More recent studies showed that artemisinins also exhibit interesting anticancer activities in vitro and in vivo against a wide range of cancer types [22,23,24,25,26], for instance, breast [27,28], ovarian [29], lung [30] and prostate [31,32].
Although the anticancer mechanism of artemisinin has not yet been fully elucidated [33,34], it has been reported that artemisinin exerts anticancer activity in the micromolar range mainly through apoptosis involving multiple targets and affecting multiple signaling pathways [23,25,26,35,36]. Non-apoptotic cell death mechanisms involved in anticancer activity of artemisinin, including oncosis-like cell death, autophagy and ferroptosis, have also been disclosed [34,35].
Some artemisinin derivatives have been identified as having potential for repurposing in anticancer therapy [23,26], [although several unfavorable properties including poor stability and water solubility and limited bioavailability prevent a large pharmacological application of artemisinin and its derivatives. To overcome artemisinin’s short half-life, and the consequent need for multiple doses each day, the World Health Organization has recommended artemisinin-based combination therapies (ACTs) as first-line treatment for uncomplicated malaria, by combining an artemisinin derivative and a slow-acting antimalarial drug [35,36,37,38,39]. Many studies have also been successfully devoted to the combination of artemisinins with chemotherapeutics or phytochemicals for cancer treatment [25,26,35,40,41]. Indeed, the synergistic effect of dihydroartemisinin (2) (Figure 2) in cancer combination therapies underlies some clinical trials involving dihydroartemisinin as a sensitizer agent [42].
On the other hand, molecular hybridization of an artemisinin moiety with other anticancer pharmacophores could be a valuable strategy not only to overcome short half-life, poor solubility and limited bioavailability but to provide combination therapies in a single multifunctional agent with enhanced activity and selectivity and reduced side effects and drug resistance compared to conventional treatments [6,9]. Over recent decades, numerous artemisinin-derived hybrids have been reported and evaluated for anticancer activity.
Considering our experience in design and synthesis of molecular hybrids for pharmacological applications [19,43,44,45,46], we decided to present herein a timely overview of the latest advances in the field of artemisinin-based hybrids for cancer disease. In particular, this review focuses on the chemical structure and biological evaluation of the most promising hybrids of artemisinin semisynthetic derivatives such as dihydroartemisinin (2) and artesunate (3) as well as their derivatives and highlights the strategies and the synthetic approaches to achieve the corresponding hybrids.
2. Chemical Features of Artemisinin and Selected Artemisinin Derivatives
Artemisinin (1) is a sesquiterpene lactone characterized by an endoperoxide bridge, located in the 1,2,4-trioxane ring (Figure 2). The endoperoxide moiety of artemisinin (1) has been shown to be pharmacologically relevant for both antimalarial and anticancer activities [26,33,47]. However, the lack of the endoperoxide moiety does not completely suppress anticancer activity [48].
Dihydroartemisinin (2) (Figure 2), a semisynthetic derivative of artemisinin (1), preserves the 1,2,4-trioxane ring whereas the C-10 carbonyl function of the lactone ring is modified into a lactol group by reduction with mild hydride-reducing agents [49]. The presence of the hemiacetal moiety improves the water solubility with respect to the parent artemisinin (1) and offers the chance for further chemical modifications. Dihydroartemisinin (2) presents enhanced antimalarial activity with respect to artemisinin (1) [26,50].
Artesunate (3) (Figure 2) is a more hydrophilic derivative of artemisinin (1) obtained by acylation of dihydroartemisinin by treatment with succinic anhydride under basic conditions [51]. Artesunate (3) shows improved solubility, absorption and pharmacokinetics with respect to artemisinin (1) and it is hydrolyzed under physiological conditions within minutes to its active metabolite dihydroartemisinin (2) [52].
3. Artemisinin Hybrids Based on Natural and Synthetic Pharmacophores
In the following section, we report selected examples of artemisinin-based hybrids in which artemisinin derivatives have been conjugated with biologically active natural compounds such as bile acids, steroids, alkaloids, terpenes, flavonoids, coumarins and cinnamic acids. Hybrids of artemisinin derivatives with the synthetic anticancer tamoxifen and sulfasalazine drugs are also reported. Table 1 briefly summarizes the hybrids discussed below, underlining the improvement in biological activities achieved through the hybridization.

Table 1.
Summary of included Artemisinin hybrids.
3.1. Artemisinin–Bile Acid Hybrids
Bile acids are endogenous natural compounds with pharmacologically relevant activities also known to exert growth inhibition of several cancer cell lines through, among others, apoptosis, membrane alterations, modulation of nuclear receptors and oxidative stress [91]. However, the low cytotoxicity (IC50 > 100 μM) prevents their application as anticancer drugs and encourages their use in developing novel anticancer hybrids [91,92]. Indeed, bile acids present different available positions suitable for chemical modifications and are able to improve bioavailability [93], oral adsorption, cellular selectivity and site-specific delivery thanks to their amphiphilic properties and stability in dynamic pH variations, as well as the targeting of specific receptors [94,95].
Recently, our research group explored convergent synthetic approaches to dihydroartemisinin–bile acid hybrids through both condensation reactions and click chemistry (Scheme 1A,B) [44]. Simple condensation reactions mediated by EDCI between dihydroartemisinin (2) and the appropriate bile acids 4–7 as well as 3-azido bile acid derivatives 8–11 revealed a successful synthetic strategy leading to the corresponding dihydroartemisinin–bile acid fused hybrids 12–19 in good yield. Hybrids 12–19 resulted from a direct conjugation of the dihydroartemisinin (2) lactol group with the C-24 carboxylic position of bile acids through a biologically labeled ester bond (Scheme 1A). Moreover, propargyl-dihydroartemisinin 20 was conjugated to 3α-azide derivative 21 via click chemistry to obtain 22 by the formation of a triazole linker which is stable under enzymatic and physiological conditions (Scheme 1B). Compound 20 was reacted with bile acid azide 21 in the presence of CuI to obtain the desired hybrid 22 in 28% yield. Though the click reaction is considered a high-yield one, in the present case a low yield was reported, probably due to the sensitivity of dihydroartemisinin (2) to the redox conditions employed in click chemistry. The linker can also play a role in the biological activity of the hybrids. In particular, the triazole ring is known to improve pharmacological, pharmacokinetic and physiochemical profiles of bioactive compounds, and it therefore can be somewhat considered a pharmacophore itself [96].
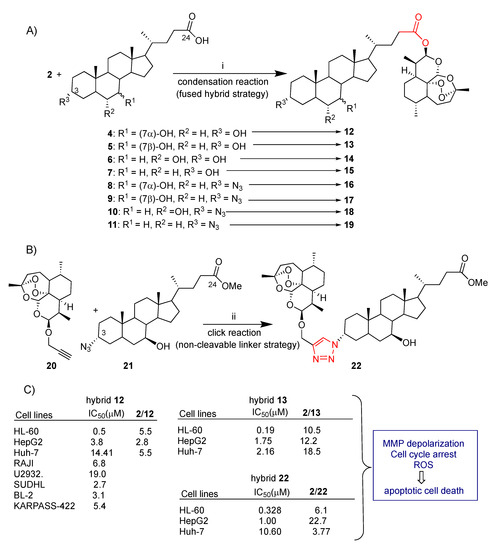
Scheme 1.
(A) Synthesis of dihydroartemisinin–bile acid hybrids 12–19 via condensation reaction: (i) EDC, DMAP, DMF, 25 °C, 18 h, 25–60% yield; (B) synthesis of dihydroartemisinin–bile acid hybrid 22 via click reaction: (ii) CuI 0.1 eq, r.t., 18 h, 28% yield; (C) selected biological data.
Dihydroartemisinin–bile acid hybrids 12–19 and 22 were tested for their in vitro cyctotoxicity against HL-60 leukemia cells. All hybrids showed enhanced cytotoxicity with respect to unconjugated dihydroartemisinin (2) with a dihydroartemisinin (2)/hybrid ratio ranging between 3 and 10.5 [44]. Further insights on mechanism were found for the most potent hybrid 13, an ursodeoxycholic bile acid derivative, (dihydroartemisin (2)/13 = 10.5) (Scheme 1C). Flow cytometry and Western blot analyses revealed that hybrid 13 induced apoptosis in HL-60 cells but no clear ROS production was detected in HL-60 cells treated with dihydroartemisinin (2) or hybrid 13 [44]. Chenodeoxycholic hybrid 12 was also tested in vitro against a selection of diffuse large B-cell lymphomas such as RAJI, U2932, SUDHL-4, KARPAS-422 and BL-2 cancer cell lines [53]. Hybrid 12 showed a significant concentration-dependent antiproliferative effect against all the diffuse large B-cell lymphomas tested (Scheme 1C). The mechanism investigation was limited to flow cytometry analysis with Annexin V-FITC staining that showed a highly predominant dose/time-dependent apoptotic mechanism of antiproliferative activity with a high early apoptosis percentage in SUDHL-4, KARPAS-422 and BL-2 cell lines [53].
Dihydroartemisinin (2) is known to significantly inhibit hepatocellular carcinoma cell growth in vitro and in vivo [97,98,99] but the poor stability limits its utilization. Therefore, dihydroartemisinin (2) conjugation with bile acids, also able to target hepatocyte transporters, may be desirable.
Almost all hybrids among 12–19 and 22 showed significant enhanced cytotoxic activity with respect to dihydroartemisinin (2) alone towards both HepG2 and Huh-7 liver cancer cell lines [44,45]. In particular, fused hybrid 13 was found to be 12.2- and 18.5-fold more potent than unconjugated dihydroartemisinin (2) in HepG2 and Huh-7 cells, respectively [44,45]. On the other hand, click hybrid 22, linked at C-3 of ursodeoxycholic bile acid, showed higher selectivity towards HepG2 cells with respect to Huh-7 cells, being more potent than unconjugated dihydroartemisinin (2) by 22.7 and 3.77 times, respectively [44,45,46]. The improved stability in cell culture medium of hybrid 13 and 22 with respect to parent dihydroartemisinin (2), as a result of the conjugation, can in part account for the marked improved cytotoxicity. Indeed, HPLC-MS/MS studies demonstrated that both hybrids are completely stable in cell culture medium after 24 h whereas unconjugated dihydroartemisinin (2) decomposed up to 70% after 6 h [45,46]. A mechanism insight for hybrids 13 and 22 in hepatocellular carcinoma cells has been also reported [45,46]. Mechanism studies showed that hybrid 13, similarly to dihydroartemisinin (2), induced G0/G1 arrest in both HepG2 and Huh-7 cells, elevated ROS levels in HepG2 but not in Huh-7 cells and caused depolarization of mitochondrial membrane potential (MMP) in both HepG2 and Huh-7 cells [45]. These effects may ultimately contribute to apoptosis induction. Similarly, click hybrid 22 induced G0/G1 arrest, ROS and mitochondrial membrane potential loss which may activate autophagy and in turn lead to apoptosis [46]. Thus, both hybrids may be potential drug candidates for hepatocellular carcinoma treatment.
Recently, Zou et al. reported [54] the synthesis and the biological evaluation of a library of dihydroartemisinin–bile acid and dihydroartemisinin–steroid hybrids conjugated by condensation of dihydroartemisinin (2) and selected bile acids towards a broad panel of cancer cell lines including liquid and solid tumors. In particular, fused hybrids 23a,b, 24 and 25 depicted in Scheme 2 were obtained by direct condensation mediated by BF3.Et2O of dihydroartemisinin (2) 10-OH with 3-OH of the appropriate bile acid. The condensation under the reaction conditions employed was found to be highly regio- and stereoselective. Hybrids 23a,b, 24 and 25 displayed a marked enhanced anticancer activity compared to dihydroartemisinin (2) against PC3 (prostatic small cell carcinoma), HeLa (cervical cancer) and 786-O (renal cell carcinoma) cell lines and a higher cytotoxic activity with respect to reference drug paclitaxel in NCI-H446 and A549 (lung cancer) and OV-90 (ovarian cancer) cell lines (Scheme 2). In particular, fused hybrid 25, obtained by conjugation of dihydroartemisinin (2) with ursodeoxycholic bile acid, was found to be the most active of the series, being 86.5- and 48-fold more potent than reference drug paclitaxel in OV-90 and A549 cell lines, respectively (Scheme 2). The pharmacokinetic profile of hybrid 25 was also obtained by using male Sprague Dawley rats. Hybrid 25 displayed acceptable maximum concentration and oral exposure. Moreover, a short half-life of 0.8 h, typically associated with artemisinin compounds, and good plasma clearance were also reported.
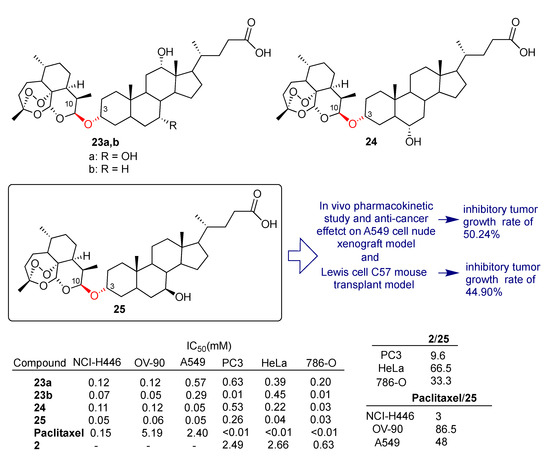
Scheme 2.
Structures of dihydroartemisinin–bile acid hybrids 23a,b–25 and selected biological data.
The A549 cell nude mouse xenograft model and Lewis cell C57 mouse transplant model were used to study the in vivo antitumor effect of hybrid 25. The A549 cell xenograft model showed that hybrid 25 significantly reduced tumor growth, with an inhibitory rate of 50.24%. In the Lewis cell C57 mouse transplant model, a 44.90% inhibitory rate was observed at a dosage of 12 mg/kg, which was significantly higher than that of the paclitaxel group (14.91% at a dosage of 10 mg/kg). It was also demonstrated by flow cytometry analysis that hybrid 25 significantly increased the concentration of CD3 (p < 0.05) and decreased the concentration of CD19 (p < 0.01). A FOXP3 experiment showed that hybrid 25 significantly increased the concentration of Tregs (p < 0.01). The overall data indicated that the conjugation of dihydroartemisinin (2) with ursodeoxycholic bile acid may have some relevance in immunotherapy.
Letis et al. reported [55] the synthesis of a series of artemisinin–cholic bile acid hybrids through condensation reactions mediated by EDCI. The synthesis of the most active hybrid 28 prepared by conjugation of deoxoartemisinin 26 to 3α-NH2 cholic acid derivative 27 is depicted in Scheme 3. The newly prepared hybrids were tested towards sensitive CCRF-CEM and multidrug-resistant CEM/ADR5000 lymphoblastic leukemia cells. Improved cytotoxic activity compared to artemisinin (1) itself was found in all cases [55]. The combination of several semisynthetic dihydroartemisinin derivatives with different cholic bile acid derivatives allowed insights into the structure–activity relationships.
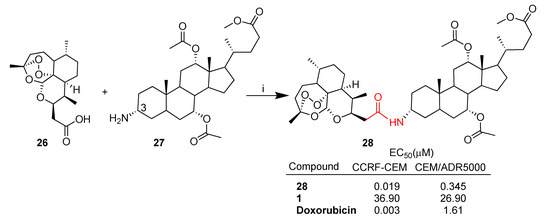
Scheme 3.
Synthesis of hybrid 28 and selected biological data. Reaction conditions: (i) EDC, DMAP, DCM, 24 h, 86% yield.
Compound 28 resulted the most potent hybrid of the series in both CCRF-CEM and CEM/ADR5000 cell lines with an IC50 value of 0.019 μM and 0.345 μM, respectively (Scheme 3). Hybrid 28 showed significant cytoselectivity towards sensitive CCRF-CEM, being 18-fold more potent than in multidrug-resistant CEM/ADR500 cancer cells. Moreover, hybrid 28 was found to be 4.6-fold more potent than reference drug doxorubicin (IC50 = 1.61 μM) and far more active than unconjugated artemisinin (1) (IC50 = 26.90 μM) in multidrug-resistant CEM/ADR5000 cancer cells (Scheme 3). The structure–activity relationship study also highlights some key points. Particularly, the 3α-orientation of the cholic acid skeleton and the incorporation of the artemisinin-derived carboxylic acid 26, forming a two-carbon chain linker between the artemisinin and the cholic acid moieties, contribute to the enhancement of the cytotoxic activity of the amide hybrid 28.
The chemical hybridization of drug compounds with bile acid skeletons appears very attractive since bile acids may improve the bioavailability of parent compounds [95]; nevertheless, so far, in vivo experiments are limited to dihydroartemisinin–ursodeoxycholic acid 25 reported by Zou et al. in 2021 [54].
3.2. Artemisinin–Quinoline and Quinazoline Hybrids
Quinoline and quinazoline alkaloids are two important classes of N-heterocyclic compounds with a wide range of pharmaceutical properties including antibacterial, antiviral, anticancer and antiparasitic effects [100,101,102,103,104,105]. Integration of a quinoline moiety can improve physical and chemical properties as well as pharmacological behavior, therefore, the quinoline ring represents an attractive scaffold in drug design. Quinoline derivatives have been vastly explored as partners in artemisinin combination therapy. Recently, Hermann et al. reported [56] the synthesis of a small library of artesunate–quinoline hybrids obtained through condensation reactions mediated by EDCI and DMAP between artesunate (3) and the series of chloroquinoline triazole derivatives 29–34 (Scheme 4). The newly synthesized hybrids 35–40 were tested against several leukemia cell lines including K562, HL-60 and MOLT-4 (Scheme 4). The best improvement of % growth inhibition compared to the parent quinoline derivative and artesunate (3) was shown by hybrids 36–38. Notably, hybrids 36–38 showed comparable anticancer activity towards K562 cells with respect to the antileukemia reference drug doxorubicin (Scheme 4).
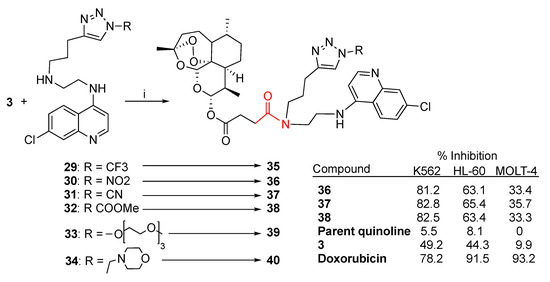
Scheme 4.
Synthesis of artesunate–quinoline hybrids and selected biological data. Reaction conditions: (i) EDC, DMAP, DCM, 0 °C to r.t., overnight, 33–69% yield.
It is worth noting that hybrids 36–38 also showed a marked inhibitory activity against SARS-CoV-2 with EC50 = 13–19 μM [56]. This result should encourage the design of further hybrids with combined anticancer and antiviral properties.
Yao et al. reported [59] the synthesis and biological evaluation of dihydroartemisinin–quinoline hybrid 43 (Scheme 5). Hybrid 43 was prepared by condensation of dihydroartemisinin–benzaldehyde derivative 41 and 4-quinolylhydrazine 42 and tested against the MCF-7 breast cancer cell line (Scheme 5). Hybrid 43 showed significantly higher anticancer activity than that of reference drugs 5-fluorouracil and paclitaxel with an IC50 ≈ 15 μM (Scheme 5). A mechanism insight on cell death revealed that hybrid 43 inactivates the c-Jun N-terminal kinase signaling pathway, leading to the induction of autophagy and apoptosis with NO generation restricted [60].
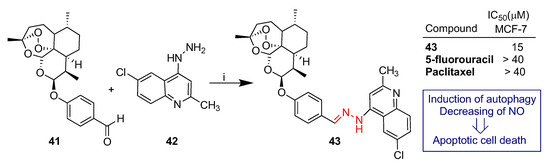
Scheme 5.
Synthesis of artemisinin–quinoline hybrids 43 and selected biological data. Reaction conditions: (i) MeOH, r.t., 7 h.
Deoxoartemisinin derivative 44 was conjugated with 3-aminoquinoline 45 through a condensation reaction (Scheme 6) [57]. The resulting hybrid 46 obtained in high yield was tested in vitro against several cancer cell lines including HSC-2 and Ca.9-22 oral squamous carcinoma cells [57]. Hybrid 46 showed significantly higher cytotoxic activity with respect to parent artemisinin (1) as well as to standard drugs such as cisplatin and paclitaxel (Scheme 6).
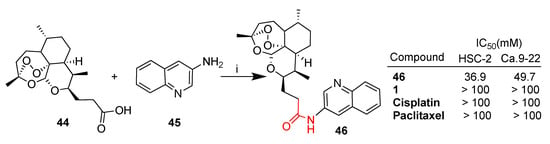
Scheme 6.
Synthesis of quinoline hybrid 46 and selected biological data. Reaction conditions: (i) EDC, DMAP, DMF, r.t., 3 h, 89% yield.
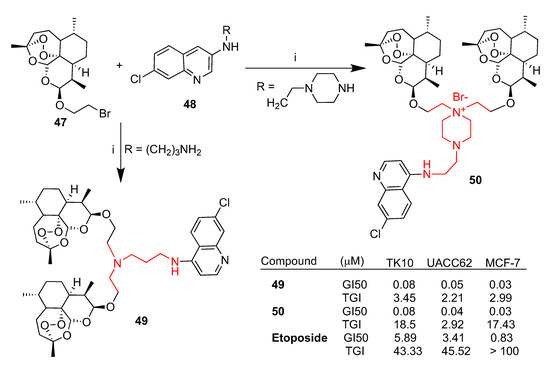
Quinoline–bis(dihydroartemisinin) hybrids 49 and 50 [61] were prepared as shown in Scheme 7 and tested in vitro against TK10 (renal), UACC62 (melanoma) and MCF7 (breast) cancer cell lines [62]. The cytotoxic activity was expressed as 50% growth inhibition (GI50) and drug concentration resulting in total growth inhibition (TGI). The anticancer agent etoposide was used as a reference standard drug. Both quinoline–bis(dihydroartemisinin) hybrids 49 and 50 showed anticancer activity against all cancer cell lines tested with GI50 values in the range of 0.03–0.08 μM and TGI values in the range of 2.21–18.50 μM. Both hybrids were found to be much more potent than the reference drug etoposide (GI50 = 0.83 and 5.89 μM, respectively; TGI = 43.33 and >100 μM, respectively) (Scheme 7). Pharmacokinetic studies were also performed in order to support further desirable in vivo studies. Maximum concentrations of 279 ng/mL and 210 ng/mL in tumors were reached within 16.7 and 20.0 min for hybrids 49 and 50, respectively, and both hybrids showed a faster blood clearance compared to dihydroartemisinin (2) (3.06 and 4.78 L/min/kg vs. 1.19 L/min/kg for 2). The overall data indicated that both hybrids 49 and 50 can be considered as lead compounds for further in vivo investigations.
Scheme 7.
Synthesis of dihydroartemisinin–quinoline hybrids 49 and 50 and selected biological data. Reaction conditions (i): DMF, 90–110 °C, 6–8 h.
The synthesis of a small library of artesunate–indoloquinoline hybrids prepared by a condensation reaction between artesunate (3) and the appropriate indoloquinoline derivative in the presence of EDCI and 1-hydroxybenzotriazole was reported [58].
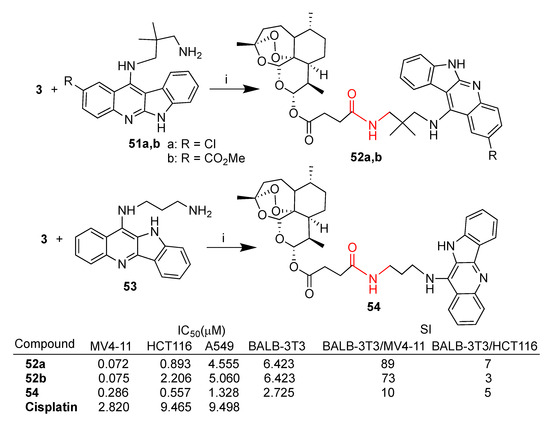
All hybrids were tested in vitro against a selection of cancer cell lines such as leukemia (MV4-11), lung (A549) and colon cancer (HCT-116) as well as healthy cells (BALB-3T3) [58]. The synthesis of selected hybrids 52a,b and 54 is reported in Scheme 8. Hybrids 52a,b and 54 exhibited promising activity against MV4-11 and HCT-116, being significantly more potent than the reference drug cisplatin and showing a good selectivity index with respect to healthy cells (BALB-3T3) (Scheme 8). In the case of the A549 cancer cell line, the hybrids were still more potent than cisplatin but less selective, having IC50 comparable to that reported for the healthy cell line BALB-3T3. Hybrids 52a,b showed the best activity and selectivity profile against leukemia MV4-11 cells with IC50 of 0.072 and 0.075 μM and selectivity index, with respect to the healthy cell line BALB-3T3, of 89 and 73, respectively (Scheme 8).
Scheme 8.
Synthesis of artesunate–indoloquinoline hybrids 52a,b and 54 and selected biological data. Reaction conditions: (i) DCM, EDC, OHBt, r.t., 6 h, 40–50% yield.
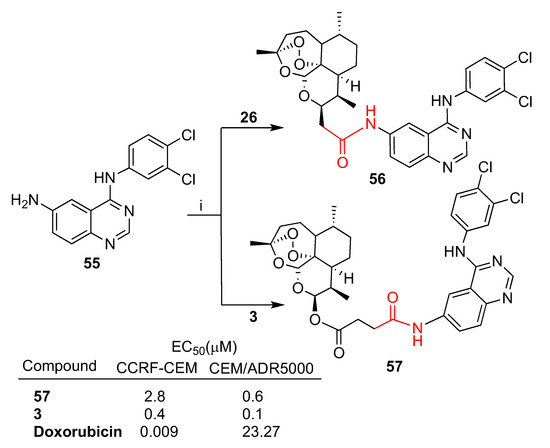
Fröhlich et al. reported [63] the synthesis of hybrids 56 and 57 by conjugation of quinazoline 55 with deoxoartemisinin 26 and artesunate (3), respectively (Scheme 9). Both hybrids were tested for their in vitro cytotoxic activity towards sensitive wild-type CCRF-CEM and multidrug-resistant P-glycoprotein-overexpressing CEM/ADR5000 leukemia cells. Deoxoartemisinin–quinazoline hybrid 56 showed low antileukemia activity towards both cell lines tested. In contrast, artesunate–quinazoline hybrid 57 exhibited an antileukemia effect with EC50 values of 2.8 μM in CCRF-CEM and 0.6 μM in CEM/ADR5000, similar to that of parent artesunate (3). Hybrid 57’s activity was found to be 45 times higher than that of reference drug doxorubicin (EC50 = 23.27 μM) in multidrug-resistant CEM/ADR5000 cells (Scheme 9).
Scheme 9.
Synthesis of artemisinin–quinazoline hybrids 56 and 57 and selected biological data. Reaction conditions: (i) DMF, EDC, DIPEA, OHBt, r.t., 1–3 days, 68–64% yield.
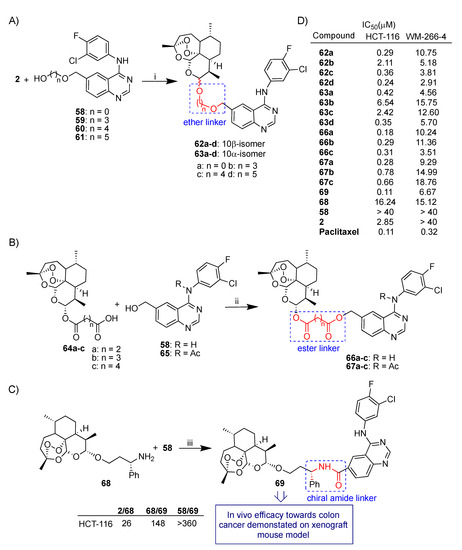
Wang et al. recently reported a wide study on the synthesis and anticancer activity of several series of artemisinin–quinazoline hybrids [64] featuring ether (Scheme 10A) or ester (Scheme 10B) flexible linkers of different lengths besides a more rigid amide chiral linker (Scheme 10C). As depicted in Scheme 10A, dihydroartemisinin (2) was conjugated through condensation reactions with quinazoline derivatives 58–61 to obtain 10β-anomer hybrids 62a–d and 10α-anomer hybrids 63a–d. Hybrids 62a–d and 63a–d were tested in vitro against two cancer cell lines, HCT-116 (colon) and WM-266-4 (melanoma). Interestingly, in most cases 10β-anomer hybrids 62a–d showed higher antiproliferative activity compared to 10α-anomers hybrids 63a–d in both cancer cell lines tested, indicating a possible role of the stereochemistry of dihydroartemisinin (2) C-10 anomeric carbon in the biological activity. On the contrary, a clear correlation between the linker length and the bioactivity could not be assessed. The 10β-anomer 62d with the longest linker alkyl chain was the most active hybrid, being ca. 12- and >14-fold more potent than parent dihydroartemisinin (2) towards HCT-116 and WM-266-4 cancer cell lines, respectively (Scheme 10D). Condensation reactions of artesunate homologues 64a–c with quinazoline 58 as well as its corresponding N-acetylated quinazoline 65 led to hybrids 66a–c and 67a–c, respectively (Scheme 10B). A slight influence of the length of the linkers on the bioactivity can be observed (Scheme 10D). Moreover, N-hydrogen hybrids 66a–c were found to be more potent than the N-acetylated homologues 67a–c towards both cancer cell lines tested.
Scheme 10.
(A–C): synthesis of artemisinin–quinazoline hybrids; (D) selected biological data. Reaction conditions: (i) BF3.Et2O, DCM, EDC, r.t., overnight, 19–33% yield; (ii) Et3N, DCM, EDC, r.t., 63–77% yield; (iii) EDC, DMAP, DPEA, DCM, 40 °C, overnight, 42% yield.
A condensation reaction of amino dihydroartemisinin derivative 68 with quinazoline 58 led to hybrid 69 characterized by an (S)-alkylamide linker (Scheme 10C) [64]. Hybrid 69 showed the best cytotoxic activity towards the HCT-116 colon cancer cell line of all hybrids tested with IC50 = 0.11 μM, comparable to that of reference drug paclitaxel (Scheme 10D) [64].
Notably, molecular hybridization was particularly efficient in enhancing the cytotoxic activity in HCT-116. Thus, hybrid 69 was 148-fold more potent than parent dihydroartemisinin derivative 68 (and in turn 26-fold more potent than dihydroartemisinin (2) itself) as well as > 360-fold more potent than unconjugated quinazoline 58 (Scheme 10D).
Hybrid 69 was further studied for in vivo anticolon cancer activity. A study on xenograft mice disclosed that hybrid 69 actually delayed tumor growth after 18 days of treatment, without affecting the weight of mice compared with the control group [64]. These results highlight hybrid 69 as a very interesting structural lead compound for the development of new anticancer drugs.
3.3. Artemisinin–Nitrogen Mustard Hybrid
Nitrogen mustard compounds are DNA-alkylating agents characterized by antiproliferative potency. Even though several types of nitrogen mustard derivatives such as chlorambucil, melphalan and cyclophosphamide are used in clinical applications, their non-specific DNA alkylation as well as the high dose required to reach effective plasma concentrations increase the risk of drug toxicity and drug resistance [106]. Therefore, the conjugation of nitrogen mustards with bioactive natural molecules characterized by low toxicity can lead to improved antitumor effect and selectivity and reduced toxicity. In recent decades, many nitrogen mustard hybrids have been reported and nitrogen mustard molecular hybridization has turned out to be an effective strategy to obtain anticancer agents with increased activity, reduced toxicity and improved physicochemical properties [106].
Recently, Dai et al. reported [65] a novel class of nitrogen mustard hybrids obtained by conjugation with artemisinin derivatives.
In particular, the ring contracted artemisinin derivative 70 and dihydroartemisinin (2) were conjugated with a series of nitrogen mustards, among other compounds 71a,b, through condensation reactions mediated by DMAP and EDCI (Scheme 11). All novel hybrids were tested in vitro against a selection of cancer cell lines including leukemia CCRF-CEM, HL-60 and K562 as well as healthy liver L02 cells [65]. Among others, hybrids 72a,b and 73 depicted in Scheme 11 were found to be the most promising compounds, displaying significantly higher activity with respect to the reference drug chloroambucil and good selectivity compared to healthy liver L02 cells. In particular, dihydroartemisinin-derived hybrid 73 displayed the best selectivity towards CCRF-CEM cell lines, being ca. 14-fold more active than chloroambucil and ca. 44-fold less toxic than in L02 healthy cells. Further investigation on the cell death mechanism suggested that hybrid 73 promoted both early and late apoptosis. Moreover, Western blot assay revealed that hybrid 73 downregulated the expression of the antiapoptotic protein Bcl-2, while the expression of procaspase-3 and cleaved caspase-3 did not show any significant changes. Additionally, hybrid 73 significantly improved the activity of glutathione peroxidase to reduce total ROS levels, therefore inhibiting cell proliferation. The overall data highlight hybrid 73 as a lead compound for possible clinical applications.
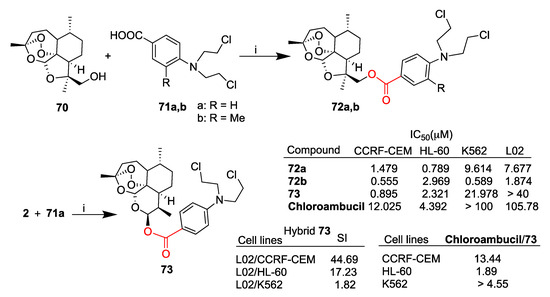
Scheme 11.
Synthesis of artemisinin–nitrogen mustard hybrids and selected biological data. Reaction conditions: (i) DMAP, EDC, DCM, r.t., overnight, 30–60% yield.
Li et al. reported [66] the synthesis of artemisinin-derived hybrids obtained by conjugation of several chemotherapeutic alkylating agents including, among others, chloroambucil, melphalan and flutamide. In particular, molecular hybridization via the condensation reaction of artesunate (3) with melphalan methyl ester 74 led to melphalan-derived hybrid 75 that showed marked enhanced activity against A2780 and OVCAR3 ovarian cancer cell lines with respect to the unconjugated dihydroartemisinin (2) and melphalan drug (Scheme 12). Further mechanism studies showed that hybrid 75 led to S-phase arrest, apoptosis and inhibition of migration. Moreover, hybrid 75 was found to modulate the expression of proteins involved in cell cycle progression, apoptosis and epithelial–mesenchymal transition. In vivo studies in mice highlighted that hybrid 75 can inhibit growth, intraperitoneal dissemination and metastasis of ovarian cancer cells without observable toxic effects. Therefore, hybrid 75 can be considered a promising lead compound for the treatment of ovarian cancer.
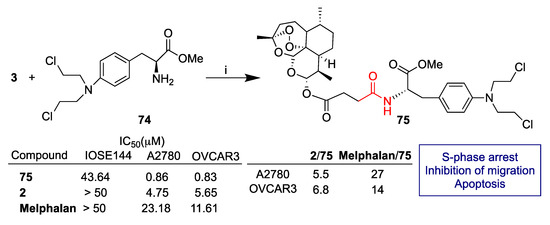
Scheme 12.
Synthesis of artesunate hybrid 75 and selected biological data. Reaction conditions: (i) DMAP, EDC, DCM, r.t., overnight, 88% yield.
3.4. Artemisinin–Tyrosol Hybrids
Tyrosol (76, Scheme 13) is a simple phenolic component of olive oil with well-established antioxidant and anti-inflammatory properties. Although some evidence of antiproliferative activity has been reported in the literature, tyrosol 76 is mainly employed in non-malignant diseases such as diabetes and cardiovascular disorders [107].
Scheme 13.
Synthesis of artemisinin–tyrosol hybrids and selected biological data. Reaction conditions: (i) K2CO3, DMF, 90 °C, overnight, 50% yield; (ii) DMAP, DCC, DCM, r.t., overnight, 53% yield; (iii) Et2O, BF3.Et2O, 0 °C, overnight, 48%; (iv) PPh3, DIAD, DMF, r.t., overnight, 60% yield.
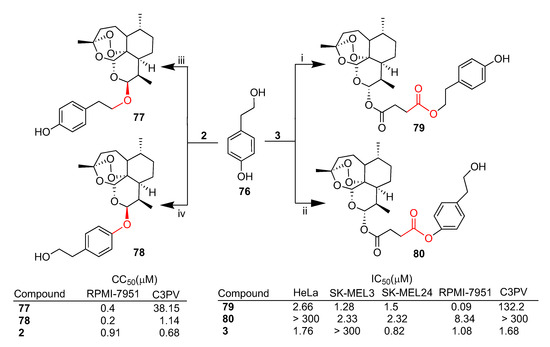
Botta et al. recently reported [67,68] the synthesis of a series of artemisinin–tyrosol hybrids and their in vitro biological evaluation against a panel of cancer cell lines, mainly melanoma. As depicted in Scheme 13, both dihydroartemisinin (2) and artesunate (3) were conjugated with tyrosol 76 in a stereocontrolled manner [68]. Furthermore, depending on the conditions employed, a different regioselectivity in the linkage of tyrosol (76) could be achieved. Hybrids 77 and 79 were obtained by conjugation of dihydroartemisinin (2) and artesunate (3), respectively, with the primary alcohol of tyrosol 76. On the other hand, the conjugation of dihydroartemisinin (2) and artesunate (3) with the phenolic moiety of tyrosol 76 led to hybrids 78 and 80, respectively.
Hybrids 79 and 80 were tested against a panel of cancer cell lines such as HeLa (cervical cancer) and SK-MEL3, SK-MEL24 and RPMI-7951 (metastatic melanoma) as well as normal fibroblast C3PV cells. Both hybrids displayed greater cytoselectivity towards melanoma cancer cell lines with respect to both HeLa and normal fibroblasts. In particular, hybrid 79 showed the best cytotoxicity against metastatic RPMI-7951 melanoma cells with IC50 = 0.09 μM, being 12-fold more potent than the parent unconjugated artesunate (3) (IC50 = 1.08 μM) (Scheme 13). Moreover, hybrid 79 was found to be much less toxic in normal fibroblast C3PV cells (C3PV/RPMI-7951 = 1467) [67]. Interestingly, hybrid 79 showed higher cytotoxic activity compared to hybrid 80 in all cell lines tested, indicating that the regioselective linkage of the alcohol moiety of tyrosol 76 can somewhat influence the hybrid cytotoxicity.
On the contrary, dihydroartemisinin-derived hybrids 77 and 78 showed similar cytotoxicity in the RPMI-7951 melanoma cell line independently of the regioselective linkage of the alcohol moiety of tyrosol 76 (Scheme 13). Hybrids 77 and 78 displayed CC50 values towards RPMI-7951 in the same order of magnitude as that of dihydroartemisinin (2). However, both hybrids 77 and 78 showed a better safety profile as compared to parent dihydroartemisinin (2), being 5.7- and 95-fold less toxic in C3PV normal cells than in RPMI-7951 cancer cells, respectively (Scheme 13) [68].
3.5. Artemisinin–Camptothecin Hybrids
Camptothecin (81, Scheme 14) is a natural alkaloid first isolated form the Chinese tree Camptotheca acuminata. Camptothecin has been reported to exhibit remarkable anticancer activity against leukemia and several solid tumors including colon and hepatic cancers through inhibition of topoisomerase I [108]. However, camptothecin’s negligible water solubility and poor stability limit its clinical use and have prompted researchers to seek molecular hybridization [109] and prodrug strategies [110].
Scheme 14.
Synthesis of artesunate–camptothecin hybrids and selected biological data. Reaction conditions: (i) EDC, DMAP, DMF, 40–64% yield.
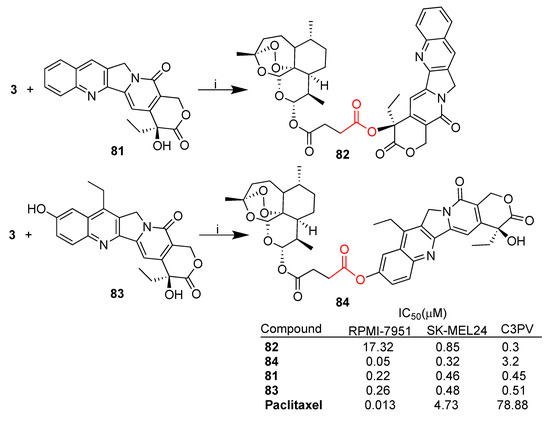
Recently, Botta et al. reported [69] the synthesis of artesunate–camptothecin hybrids 82 and 84 obtained by the condensation reaction between artesunate (3) and camptothecin 81 as well as 7-ethyl-10-hydroxy-camptothecin 83, the active form of the anticancer drug irinotecan (SN 38) (Scheme 14). Hybrids 82 and 84 were tested in vitro against a selection of melanoma cell lines (RPMI7951 and SK-MEL24) as well as towards normal fibroblast C3PV cells using camptothecins 81 and 83 and paclitaxel as reference drugs [69].
Noteworthy, hybrid 84 showed higher antimelanoma activity towards RPMI-7951 cells than camptothecins and activity similar to that of paclitaxel, as well as a good selectivity index with respect to normal fibroblasts (C3PV/RPMI-7951 = 64).
Both hybrids showed high inhibitory activity against human DNA topoisomerase 1 (hTop1p) in both in vivo and in vitro experiments. In particular, hybrid 84 was active at 3 μM in vivo and at 300 nM in vitro. On the contrary, hybrid 82 was more effective in vivo than in vitro [69].
The overall data indicate the effectiveness of molecular hybridization since hybrid 84 displayed significant enhanced cytotoxicity and cytoselectivity towards cancer cells with respect to normal ones. Moreover, the inhibitory activity of parent camptothecin against hTop1p is preserved in both hybrids 82 and 84.
3.6. Artemisinin–Thymoquinone Hybrids
Thymoquinone is a monoterpene obtained from Nigella sativa’s black seed oil [111]. Thymoquinone has received much attention for its wide-ranging pharmacological properties including antioxidant, anti-inflammatory, antidiabetic, antihistaminic, antimicrobial, anticonvulsant and anticancer effects [111]. In the last decade, many studies have been reported on the anticancer effects of thymoquinone on a broad range of cancer types including lung, breast, prostate, gastric, colon, bladder and bone as well as on the use of thymoquinone in combination anticancer therapy [112]. In particular, thymoquinone was reported to exhibit activity against colon cancer by triggering apoptosis through different mechanisms including downregulation of STAT3 activation by reducing JAK2 and c-Src activity, p53- and p21-dependent mechanisms as well as G1-phase cell cycle arrest [112].
Tsogoeva’s research group reported extensive studies on the design, synthesis and biological activity of artemisinin–thymoquinone hybrids [70,71,72]. The synthesis of a selection of artemisinin–thymoquinone hybrids is depicted in Scheme 15. Newly synthesized hybrids 86a–c, 88b,c and 91 were evaluated for anticancer activity in HCT-116 and HT-29 colon cancer cell lines [70] and/or sensitive wild-type CCRF-CEM and multidrug-resistant CEM/ADR5000 leukemia cells [71]. Artesunate–thymoquinone hybrids 86a–c were obtained in good yield by the condensation reaction of artesunate (3) and thimoquinone derivatives 85a–c (Scheme 15A).
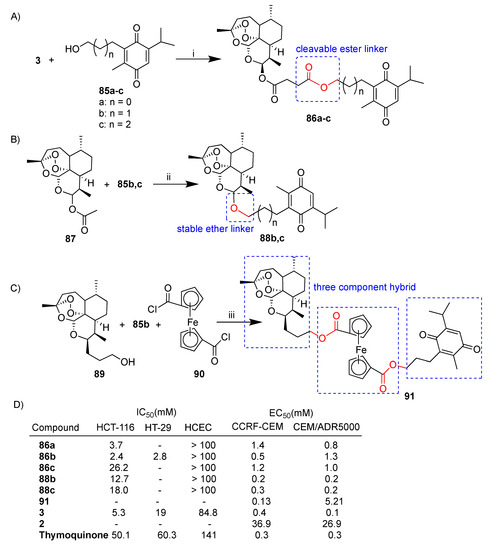
Scheme 15.
(A–C): synthesis of artemisinin–thymoquinone hybrids. (D): selected biological data. Reaction conditions: (i) DDC, DMAP, DCM, r.t., overnight, 80–86% yield; (ii) TMSOTf, CHCl3, 0 °C, 1 h, 32–39% yield; (iii) DCM, DMAP, r.t., overnight, 26% yield.
Hybrids 86a–c characterized by labile ester linkers of different lengths were tested on colon cancer and leukemia cells as well as healthy HCEC cells. Notably, the conjugation led in all cases to hybrid compounds being less toxic towards normal cells than parent artesunate (3) (Scheme 15D). Among all, hybrid 86b showed the best anticancer activity against HCT116 cells, being ca. 2- and 21-fold more potent than artesunate (3) and thymoquinone, respectively. Similar results were obtained in the case of the HT-29 colon cancer cell line. Also, in the case of leukemia cell lines, hybrid 86b was the most active of the series with significant cytoselectivity towards sensitive CCRF-CEM. Instead, hybrids 86a–c were found to be around 10-fold less potent than parent artesunate (3) in the multidrug-resistant CEM/ADR5000 cell line (Scheme 15D) [71]. A mechanism insight indicated that hybrid 86b led to an increase in ROS and induced an elevated level of the DNA-damage marker g-H2AX in colon cancer cells [70].
Dihydroartemisinin acetate 87 was reacted with thymoquinone derivatives 85b,c in order to obtain hybrids 88b,c characterized by stable ether linkers of different lengths (Scheme 15B) [70]. Both hybrids 88b,c exhibited similar activity to that of artesunate derivatives 85b,c and thymoquinone towards leukemia cell lines [71] whereas they were found to be 3–4-fold less potent than artesunate (3) alone in the HCT-116 colon cancer cell line (Scheme 15D) [70].
Hybrid 91, a deoxoartemisinin-derived hybrid containing two subunits of ferrocene and thymoquinone prepared as reported in Scheme 15C, was tested against both leukemia CCRF-CEM and CEM/ADR5000 cell lines [72]. Hybrid 91 showed marked cytoselectivity towards sensitive CCRF-CEM with respect to multidrug-resistant CEM/ADR5000 (CCRF-CEM/CEM/ADR5000 = 39) (Scheme 15D) [72]. Most of the artemisinin–thymoquinone hybrids reported showed interesting cytoselectivity which is an important aspect for drug development and one of the main goals of molecular hybridization.
3.7. Artemisinin–Chalcone Hybrids
Chalcones are a class of compounds consisting of two aryl rings linked by an α,β-unsaturated ketone moiety and characterized by the common chemical scaffold 1,3-diaryl-2-propen-1-one [113]. Chalcone moieties are common substructures in numerous natural products, including vegetables, fruits, teas, and other plants, belonging to the flavonoid family. Chalcone derivatives are versatile as pharmaceutically active compounds with broad interesting biological activities including anticancer [114], anti-HIV, antimalarial, antioxidant, anti-inflammatory and antiallergic activities [113]. Several chalcone-derived compounds have been approved for clinical use, for instance, metochalcone and sofalcone used as a choleretic drug and antiulcer and mucoprotective drug, respectively [115]. Chalcones have been considered as privileged scaffolds for molecular hybridization to improve biological activities, solubility and/or oral bioavailability as well as to overcome drug resistance. Many chalcone-derived hybrids have been reported in the literature for development of novel anticancer agents with multiple mechanisms of action [116], including artemisinin–chalcone hybrids.
Gau et al. reported a library of dihydroartemisinin–chalcone hybrids conjugated via ether linkers [117]. The hybrids have been evaluated in vitro for their cytotoxicity against a panel of cancer cell lines such as HL-60 (leukemia), MIA PaCa-2 (pancreatic cancer), PC-3 (prostate cancer), HepG2 (hepatocellular carcinoma) and LS-180 (colon cancer) [113]. Hybrids 93a,b, obtained by conjugation of dihydroartemisin derivative 47 and chalcone derivative 92a,b, showed the best activity in HL-60 leukemia cells (Scheme 16). Indeed, hybrids 93a,b (IC50 = 0.3 and 0.4 μM, respectively) were found to be 5- and 7-fold more potent than dihydroartemisinin (2) (IC50 = 2 μM), respectively, and showed comparable cytotoxicity to that of reference drug doxorubicin (IC50 = 0.3 μM) (Scheme 16).
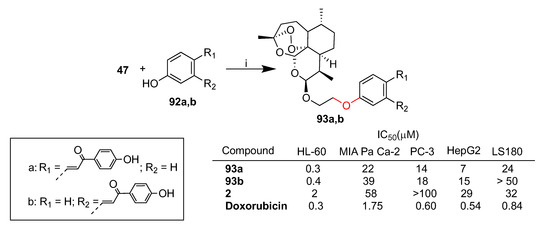
Scheme 16.
Synthesis of dihydroartemisinin–chalcone hybrids and selected biological data. Reaction conditions: (i) K2CO3, DMF, DCM, 60 °C, 30 min, 55% yield.
A mechanism insight in HL-60 cells was also reported. Both hybrids 93a,b were found to be significantly more effective in inducing apoptotic subG0 population and in triggering mitochondrial membrane potential loss than dihydroartemisinin (2).
Smit et al. reported [75] a library of dihydroartemisinin–chalcone hybrids conjugated via ester linkage that have been evaluated in vitro for their cytotoxicity against TK-10 (renal cancer), UACC-62 (melanoma) and MCF-7 (breast cancer) as well as healthy WI-38 cell lines. The synthesis of selected hybrids 95a–d, prepared by condensation of dihydroartemisinin (2) and a series of chalcone derivatives 94a–d, is depicted in Scheme 17. Hybrids 95a–c were found to be highly cytoselective towards cancer cell lines with respect to normal cells. Hybrid 95a was the most potent of the series against all cancer cell lines tested, showing comparable or higher cytotoxic activity with respect to reference drug parthenolide (Scheme 17).
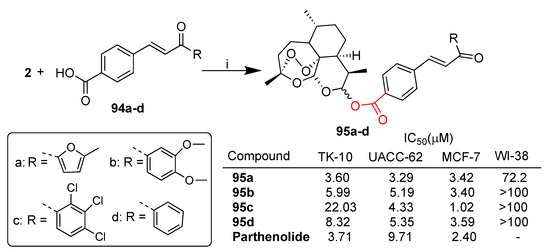
Scheme 17.
Synthesis of dihydroartemisinin–chalcone hybrids and selected biological data. Reaction conditions: (i) DCI, DCM, r.t., 24 h, 32–61% yield.
The structure–activity relationship revealed that the presence of a heterocycle instead of a phenyl ring on the chalcone moiety improved the activity as well as the introduction of electron-withdrawing groups into the phenyl ring.
Xie et al. reported [73,74] two series of dihydroartemisinin–chalcone hybrids prepared by conjugation through ether linkers of α- and β-C10-aminodihydroartemisinin anomers 97 and 98, respectively, with chalcone derivatives 96a–l (Scheme 18). Hybrids 99a–l and 100a–l were tested in vitro against the following five cancer cell lines: HT-29 (colon), A549 and A-460 (lung), MDA-MB-231 (breast), HeLa (cervical).
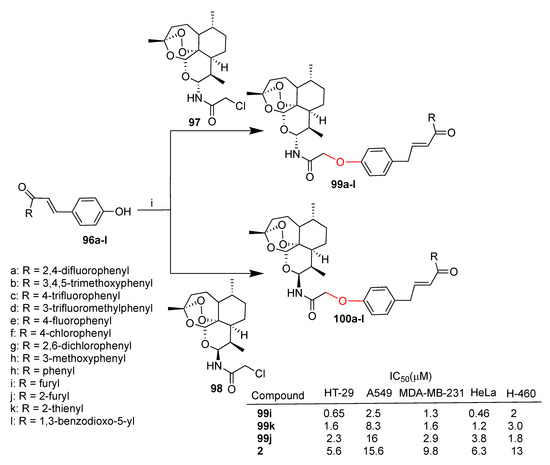
Scheme 18.
Synthesis of dihydroartemisinin–chalcone hybrids and selected biological data. Reaction conditions: (i) K2CO3, DMF, NaI, 60 °C, 5 h, 70–90% yield.
The numerousness of the reported hybrids allowed the establishment of the structure–activity relationship. Particularly, the anomeric configuration at the C-10 position of the dihydroartemisinin moiety does not affect the activity of hybrids. Almost all hybrids of 99a–l were found to be from 2.6- to 40-fold more active than the parent dihydroartemisinin (2) in all cancer cell lines tested, proving the effectiveness of the conjugation with a chalcone moiety. Moreover, the presence of a chalcone moiety also improved the cytoselectivity towards HT-29 and HeLa cell lines with respect to MDA-MB-231, A549 and H460 cell lines. By comparing hybrids 99i–k it can be evidenced that the bioisosteric replacement of phenyl (99i) with a thiophene (99k) and furan (99j) rings significantly decreased the activity against the five tumor cell lines tested (Scheme 18).
3.8. Artemisinin–Coumarin Hybrids
Coumarin and its derivatives are a family of benzopyrones largely distributed in nature, being present in the seeds, fruits, flowers or roots of various plant species [118,119]. Coumarins are characterized by a two-ring backbone consisting of a phenyl group fused with an α-pyrone ring. Coumarin’s potent biological activity is related to its conjugated electronic system, which is able to react with different molecules including enzymes and receptors of living organisms. Coumarin and its derivatives have been recognized as interesting anticancer candidates due to their ability to induce cell differentiation and apoptosis [118,119,120].
Guo et al. reported the synthesis of several series of dihydroartemisinin–coumarin hybrids via click chemistry [76,77,78] together with some artemisinin–coumarin hybrids via ether linkers [78].
The synthesis of a selection of hybrids such as click hybrids 103a–e and the homologues 104a–e as well as homologue hybrids 108a–e and 109a–e is reported in Scheme 19A.
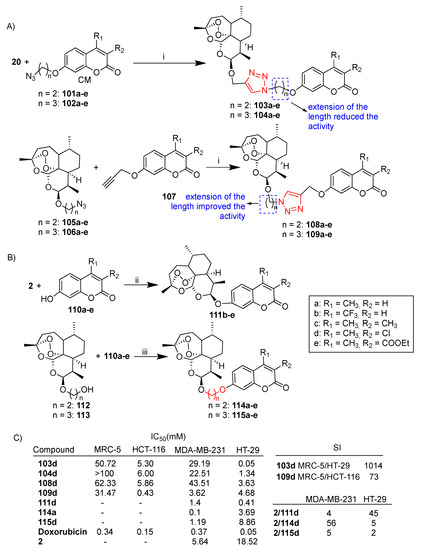
Scheme 19.
(A,B) Synthesis of dihydroartemisinin–coumarin hybrids; (C) selected biological data. Reaction conditions: (i) CuSO4, DMF, sodium ascorbate, r.t., 8–10 h, 20–40% yield; (ii) (CF3CO2)2, Et3N, DCM, 0–5 °C, 18 h, 12–17% yield; (iii) K2CO2, DMF, 60 °C, 5 h, 38–60% yield.
All hybrids were tested against selected cancer cell lines HCT116 and HT-29 (colon), MDA-MB-231 (breast) and normal MRC-5 fibroblast cells. By comparing hybrids 103a–e, 104a–e, 108a–e and 109a–e, it can be noted that the extension of the length of the alkyl chain between the triazole ring and the coumarin moiety decreased the activity (hybrid activity 103 > 104) whereas the extension of the length of the alkyl linker between the dihydroartemisinin and triazole moiety increased the activity (hybrid activity 108 < 109) (Scheme 19C).
The best activity was exhibited by 3-chlorol-4-methyl-coumarin-derived hybrids belonging to the series d, suggesting that the different electronic effect of substituents at the C-3 and C-4 skeleton of the coumarin moiety could play a positive role in the cytotoxicity of hybrids. In particular, hybrid 103d was the most potent with IC50 = 0.05 μM, comparable to that of reference drug doxorubicin. Moreover, hybrid 103d showed the best cytoselectivity towards HT-29 cancer cells with respect to normal MRC-5 cells (MRC-5/HT-29 = 1014) (Scheme 19C). Further studies in HT-29 colon cancer cells showed that hybrid 103d inhibited proliferation, arrested the cell cycle progression, induced both apoptosis and ferroptosis and inhibited migration [78].
The synthesis of dihydroartemisin-derived fused hybrids 111b–e as well as of hybrids 114a–e and 115a–e conjugated through ether linkers is depicted in Scheme 19B. All hybrids were evaluated in vitro against MDA-MB-231 and HT-29 cancer cells. Among hybrids 111b–e, 3-chloro-4-methyl-coumarin-derived hybrid 111d showed the best activity towards the HT-29 colon cancer cell line, being 45-fold more potent than unconjugated dihydroartemisinin (2) (Scheme 19C). Among hybrids 114a–e and 115a–e, hybrid 114a showed the best activity towards MDA-MB-231, being 56-fold more active than dihydroartemisinin (2) (Scheme 19C) [78].
3.9. Artemisinin–Tamoxifen Hybrids
Tamoxifen belongs to a class of anticancer drugs that are selective estrogen receptor modulators extensively used in hormone-dependent breast cancer therapy over recent decades [121,122]. Chronic tamoxifen use can increase the risk of uterine cancer and drug resistance. For these reasons, hybridization of tamoxifen with other anticancer agents may be effective to overcome adverse effects and drug resistance from tamoxifen usage alone.
Frohlich et al. recently reported [79] the synthesis of some artemisinin–tamoxifen hybrids as well as their biological evaluation against two cancer cell lines, PC-3 (prostate) and MCF-7 (breast) [79]. In particular, both artesunate (3) and deoxoartemisinin derivative 26 were reacted with two metabolites of tamoxifen, 2-hydroxytamoxifen 116 (afimoxifene) and (Z)-N-desmethyl-tamoxifen 119, to give the corresponding hybrids 117, 118 and 120, 121, respectively (Scheme 20). Hybrids 117 and 118 presented ester linkers, whereas hybrids 120 and 121 presented amide linkers (Scheme 20). All hybrids displayed higher activity than dihydroartemisinin (2) and artesunate (3) against both PC-3 and MCF-7. Hybrid 121 was found to be the most potent of the series with EC50 = 2.74 and 3.91 μM in PC-3 and MCF-7 cells, respectively. Moreover, hybrid 121 showed marked improved activity compared to dihydroartemisinin (2), being 93- and 5-fold more potent in PC-3 and MCF-7, respectively.
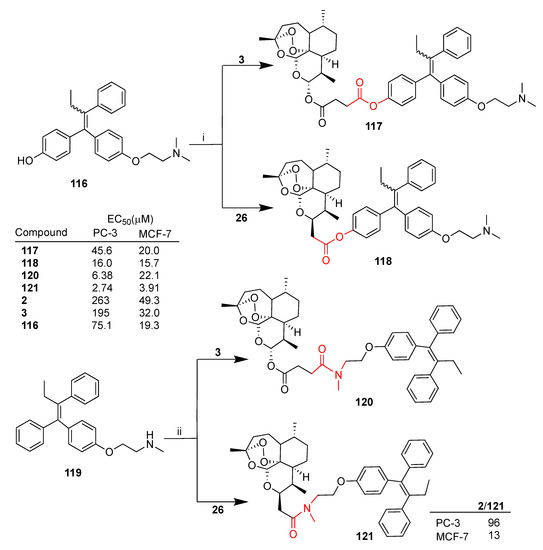
Scheme 20.
Synthesis of artemisinin–tamoxifen hybrids and selected biological data. Reaction conditions: (i) DMAP, THF, DCC, r.t., 80% yield; (ii) DMAP, THF, EDC, TEA, r.t., 68–75% yield.
3.10. Artemisinin–Steroid Hybrids
Steroids are natural organic bioactive compounds present in animals as well as in plants and fungi. Steroids are considered important scaffolds in medicinal chemistry due to their molecular structure as well as to their ability to interact with various biological targets in different pathways [123].
Frohlich et al. reported [80] a study on the synthesis of a series of artemisinin–estrogen hybrids and their in vitro evaluation against a selection of breast (MCF-7, MDA-MB-231, MDA-MB-361 and T47D) and cervical (HeLa, SiHa and C33A) cancer cell lines [80]. In particular, estradiol amine derivative 122 and estrone derivative 125 were conjugated with both artesunate (3) and deoxoartemisinin derivative 26 to give the corresponding hybrids with amide linkers 123, 124 (Scheme 21A) and with ester linkers 126, 127 (Scheme 21B). Moreover, estradiol 122 was reacted with dihydroartemisinin alkyne derivative 20 via click reaction to give hybrid 128 characterized by a 1,2,3-triazole linker (Scheme 21C). Almost all hybrids displayed improved growth inhibition than parent artemisinins and estrogen derivatives. In particular, hybrid 127 showed the best activity against breast cancer cell lines with submicromolar EC50 values ranging from 0.18 and 0.93 μM and EC50 = 0.15 μM in the C33A cervical cancer line with (Scheme 21D). On the other hand, hybrid 124 showed interesting activity towards all cancer cell lines tested (Scheme 21D).
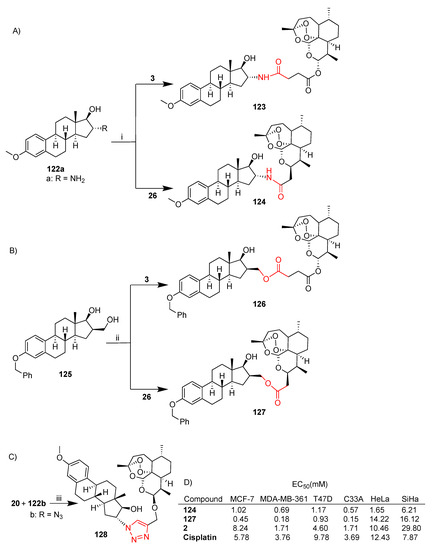
Scheme 21.
(A–C): synthesis of artemisinin–estrogen hybrids; (D) selected biological data. Reaction conditions: (i) EDC, DCM, ACN, r.t., overnight, 45–80% yield; (ii) DCC, DMAP, DCM, r.t., overnight 55–95% yield; (iii) CuSO4, DCM/H2O, sodium ascorbate, r.t., overnight, 42% yield.
More recently, the same research group reported a library of 16 artesunate–estrogen hybrids, prepared via condensation reactions, that were evaluated in vitro on prostate PC-3 and breast MCF-7 cancer cell lines [79]. Among others, estrogen derivatives bearing a sulfamate moiety were used as building blocks. Sulfamate-based estrogen analogues have been reported to display a potential as multitargeting agents for hormone-independent diseases [124]. As reported in Scheme 22, the new hybrids 130 and 132a,b were synthesized through a multistep synthesis involving the condensation reaction between artesunate (3) and estrogen derivatives 129 and 131a,b followed by the introduction of the sulfamate moiety and deprotection. All hybrids are characterized by ester linkers. Hybrids 132 were obtained as pure anomers α (132a) and β (132b) [79].
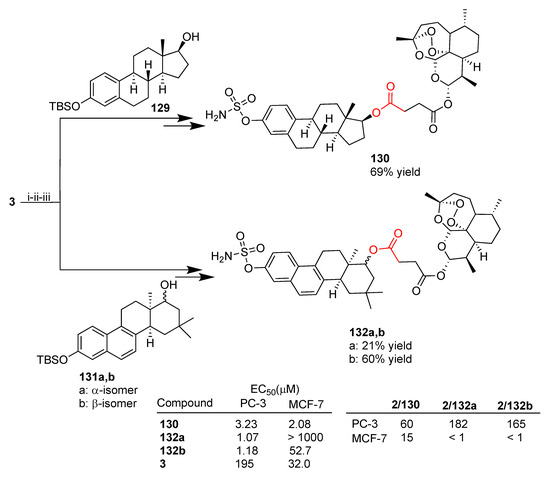
Scheme 22.
Synthesis of artemisinin–estrogen hybrids and selected biological data. Reaction conditions: (i) EDC, DMAP, THF, r.t.; (ii) TBAF, THF, r.t., 2 h; (iii) NH2SO2Cl, DMA, r.t., overnight.
All hybrids tested were found to be more potent than unconjugated artesunate (3) towards PC-3 prostate cancer cells and most of them were found to be more potent towards MCF-7 breast cancer cells as a result of the effectiveness of molecular hybridization. In particular, hybrid 130 was the most active of the whole series towards MCF-7 breast cancer cells, being 15-fold more potent than unconjugated artesunate (3). Hybrids 132a,b showed highly marked cytoselectivity towards PC-3 prostate cancer cells with 182- and 165-fold enhanced activity, respectively, compared to parent artesunate (3) (Scheme 22).
3.11. Artemisinin–Cinnamic Acid Hybrids
Cinnamic acid is a phytochemical isolated from cinnamon bark known for its well-established antimicrobial activity [125]. Cinnamic acid was also reported to have potential anticancer activity in several tumors, therefore it can be considered an interesting scaffold for molecular hybridization [126]
Xu et al. reported [81] the synthesis of a series of dihydroartemisinin–cinnamic acid hybrids through condensation of a series of cinnamic acids substituted on the benzene ring with dihydroartemisinin derivative 133 (Scheme 23). Disubstituted dihydroartemisinin hybrids prepared by conjugation of suitable cinnamic acid derivatives 134a–c to both C-10 and C-7 hydroxyl groups of dihydroartemisinin derivative 133 have also been reported (Scheme 23) [81]. All hybrids were tested in vitro against four cancer cell lines (PC-3 prostate, SGC-7921 gastric, A549 lung and MDA-MB-435s melanoma) as well as normal L02 cells. Monosubstituted dihydroartemisinin hybrids showed in all cases poor activity whereas the corresponding disubstituted dihydroartemisinin hybrids displayed interesting anticancer activity [81]. In Scheme 23, a selection of disubstituted dihydroartemisinin hybrids 135a–c are reported. In particular, compound 135a was the most active of the whole series in the A549 cancer cell line with IC50 = 0.2 μM and was 34-fold more potent than reference drug 5-fluorouracil and much less cytotoxic in normal L02 cells (L02/A549 = 401). Molecular mechanism studies in the A549 cancer cell line indicated that hybrid 135a induced apoptosis in a dose-dependent manner. In the same paper [81], it was also reported that both ROS and ferrous ions were involved in the cytotoxicity and apoptosis induced by hybrid 135a. Therefore, cell lines containing a higher level of ferrous as well as endogenous oxidative stress would be more susceptible to anticancer activity of hybrid 135a.
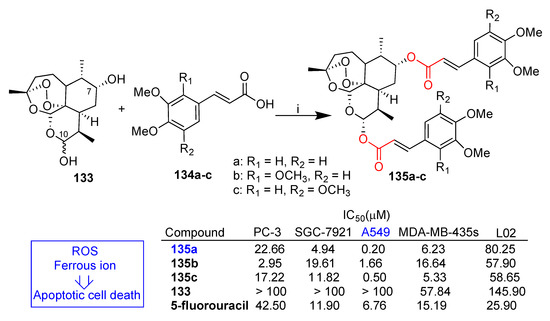
Scheme 23.
Synthesis of artemisinin–cinnamic acid hybrids and selected biological data. Reaction conditions: (i) EDC, DMAP, DCM, r.t., 12 h, ca. 50% yield.
3.12. Artemisinin–Acridine Hybrids
Acridine derivatives are an important class of heterocycles characterized by a broad spectrum of pharmaceutical properties including anti-inflammatory, antimicrobial, antitubercular, antiparasitic, antimalarial, antiviral as well as anticancer [127]. The variety of pathways involved in acridine anticancer activity, such as inhibition of cell proliferation, apoptosis, cell cycle arrest, retarding migration, invasion and metastasis, encourages acridine’s use as appealing scaffolds for rational design of novel anticancer drug candidates [127,128].
Jones et al. reported [82] the synthesis of several hybrids obtained by reaction between deoxoartemisin 26 and the appropriate diaminoacridine 136a–c and 138. The resulting hybrids 137a–c, characterized by amide linkers of different lengths, and hybrid 139, with a piperazine ring linker, were tested in vitro against a panel of cancer cell lines (leukemia HL-60, colon cancer HT29-AK, breast MDA-MB-231 and MCF-7) in comparison to parent dihydroartemisinin (2) and acridine 136a (Scheme 24). All hybrids tested were found to be cytoselective towards leukemia and breast cancer cell lines, displaying significant enhanced activity with respect to parent dihydroartemisinin (2). Compound 137b is the only hybrid with significant activity towards the HT29-AK colon cancer cell line with 1.6-fold enhanced activity with respect to unconjugated dihydroartemisinin (2). This result may be related to the length of the linker that seems to play a role in the cytoselectivity and activity in the series of hybrids 136a–c. Hybrid 136b was further investigated in HL-60 leukemia cells. Flow cytometry analysis demonstrated that this hybrid can promote apoptotic cell death and bind covalently to intraparasitic cellular targets in the presence of iron (II) [82].
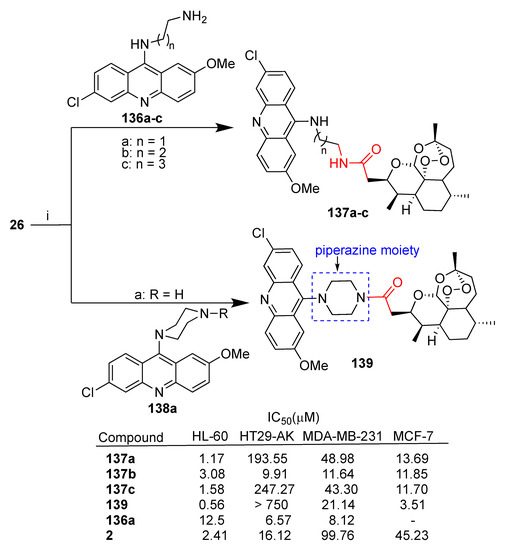
Scheme 24.
Synthesis of artemisinin–acridine hybrids and selected biological data. Reaction conditions: (i) (COCl)2, DCM, r.t., 1.30 h, then NEt3, DCM, r.t., 16 h, 55–74% yield.
On the other hand, hybrid 139 with the piperazine moiety was the most potent against both HL-60 and MCF-7 cell lines with improved activity compared to parent dihydroartemisinin (2) of 6- and 13-fold, respectively (Scheme 24).
Joubert et al. reported [83] the synthesis of dihydroartemisinin–acridine hybrids obtained by reaction of dihydroartemisinin bromo derivative 47 and a series of diaminoacridine derivatives that were tested against HeLa cervical cancer cells. The synthesis of two hybrids containing a piperazine linker is reported in Scheme 25. The hybrids containing the piperazine ring showed higher activity [83]. In particular, hybrid 141 was the most potent of the whole series with comparable activity to that of parent dihydroartemisinin (2). Of note, hybrid 141 with a longer linker displayed 3-fold higher activity than 140, underlining a possible role of the linker length in the bioactivity.
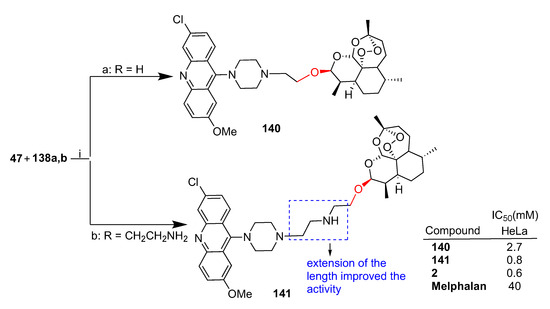
Scheme 25.
Synthesis of artemisinin–acridine hybrids and selected biological data. Reaction conditions: (i) K2CO3, KI, ACN, microwave, 10 min, 45 °C, 64–71% yield.
3.13. Artemisinin–Isatin Hybrids
Isatin, an indole derivative, is a natural alkaloid present in plants of the Isatis genus spread all over the world [129]. Isatin is a synthetic versatile pharmacophore since nitrogen N-1, C-2 and C-3 carbonyl groups as well as any carbon in the benzene ring (Scheme 26) can be modified, leading to a broad spectrum of derivatives with biological properties including, among others, antiviral [130], antimicrobial [131], antibacterial [132] and anticancer [129,133]. Some isatin derivatives have been tested in clinical trials [134] or approved as anticancer drugs [135,136]. Recently, isatin has also attracted attention as a conjugation partner of dihydroartemisin.
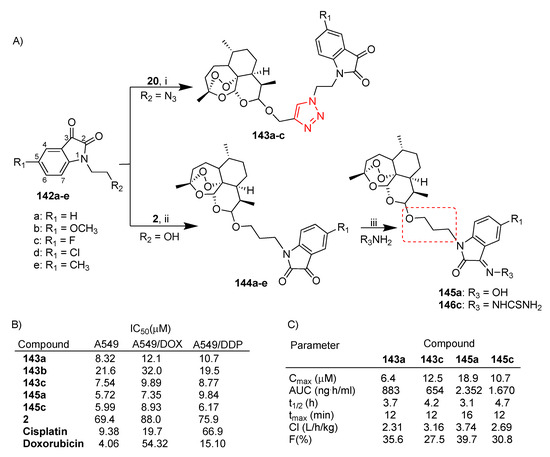
Scheme 26.
(A) Synthesis of dihydroartemisinin–isatin hybrids; (B) selected biological data; (C) pharmacokinetic data: maximum plasma concentration (Cmax), area under curve (AUC), half-life (t1/2), peak time (tmax), clearance rate (Cl), bioavailability (F). Reaction conditions: (i) CuSO4, DMF, 60 °C, 8 h, 28–37% yield; (ii) BF3.Et2O, DCM, 0 °C, overnight, 39–61% yield; (iii) NaHCO3, THF, 60 °C, 12 h, 87 and 26% yield.
Gao et al. reported the synthesis of a large number of dihydroartemisinin–isatin hybrids via both click chemistry [84] and condensation reactions [85] (Scheme 26A). All hybrids were evaluated in vitro for antiproliferative activity in A549 and A549 doxorubicin- and cisplatin-resistant (namely A549/DOX and A549/DDP, respectively) lung cancer cell lines [84,85]. As shown in Scheme 26A, hybrids 143a–c, characterized by the triazole linker, presented different substituents on the C-5 position of the isatin moiety. The SAR indicated that the presence of an electron-withdrawing group such as fluorine (hybrid 143c) improved the activity whereas the electron-donating group methoxy (hybrid 143b) led to a significant decrease in the activity (Scheme 26B). Further substitutions of the carbonyl group at the C-3 position of isatin with a hydroxime, alkoxime, benzyloxime and thiosemicarbazide moiety reduced the activity (Scheme 26) [84]. Click hybrids 143a–c were found to be more active (IC50 = 7.54–32.0 μM) than unconjugated dihydroartemisinin (2) (IC50 = 69.4–88.0 μM) in the drug-sensitive and multidrug-resistant A549 cancer cell lines tested (Scheme 26B). On the other hand, all hybrids displayed no cytotoxicity in normal fibroblast HIH/3T3 cells (IC50 > 100 μM) [84]. It is worth noting that hybrids 143a–c were found to be at least 6-fold more active than cisplatin in A549/DDP cells (Scheme 26B).
Hybrids 144a–e (Scheme 26A), characterized by a three-carbon alkyl spacer, did not show any activity against all A549 cell lines tested; only hybrid 144c displayed antiproliferative activity comparable to that of unconjugated dihydroartemisinin (2) (Scheme 26B). On the other hand, the introduction of a hydroxime and a thiosemicarbazide at the C-3 position of the isatin moiety of hybrids 144a,c led to the corresponding hybrids 145a and 146c with significant enhanced activity with respect to parent dihydroartemisinin (2) (>10-folds) in all cancer cell lines tested (Scheme 26B) [85]. Moreover, hybrids 145a and 146c showed interesting activity in both multidrug-resistant 549/DOX and 549/DDP (Scheme 26B) [85].
Hybrids 143a,c, 145a and 146c were selected for further pharmacokinetic studies in a CD-1 mouse model by single intravenous administration (dose = 30 mg/kg). The results are reported in Scheme 26C.
Cao et al. reported [86,87] a series of dihydroartemisinin–isatin hybrids conjugated through a nucleophilic substitution by reacting a series of tosylate derivatives of dihydroartemisinin (2) with isatin derivatives substituted at C-3 with different groups such as methoxy, fluorine and methyl. The synthesis of the most potent hybrids of the series is reported in Scheme 27. The new hybrids 149a [86] and 149b [87] presented alkyl chains of different lengths and different substituents at the C-5 position of the isatin moiety. Hybrids 149a and 149b were in turn modified at the C-3 position of the isatin moiety with, among others, hydroxime and thiosemicarbazide moieties, leading to hybrids 150a and 150b, respectively. The new hybrids were tested for their antiproliferative activity against breast cancer cell lines MCF-7 and MDA-MB-231 as well as their corresponding multidrug-resistant cell lines. Most of the hybrids were found to be significantly more potent than parent dihydroartemisinin (2) in all cancer cell lines tested and, in particular, hybrids 149a,b and 150a,b were found to be 4- and 5-fold more potent (Scheme 27). Accordingly with previous data reported in the literature [84,85], the nature of the substituents at both C-5 and C-3 positions of isatin influenced the activity, as well as the length of the alkyl chain. It is worth noting that all hybrids were found to be not cytotoxic towards normal MCF-10A breast cells (IC50 > 100 μM).
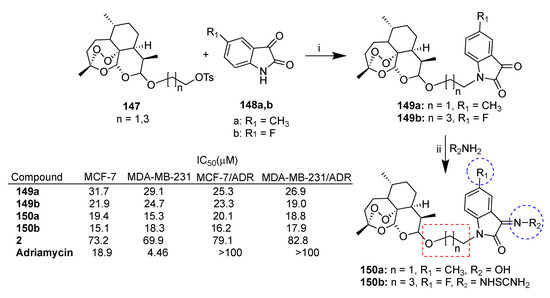
Scheme 27.
Synthesis of dihydroartemisinin–isatin hybrids and selected biological data. Reaction conditions: (i) K2CO3, DMF, r.t., overnight; (ii) AcONa, THF/H2O, 60 °C, 12 h.
Xu et al. reported [88,89] a series of dihydroartemisinin–isatin hybrids conjugated through ester linkers of different lengths. In particular, dihydroartemisinin (2) was reacted with isatin carboxylic acid homologue derivatives 151a–d (Scheme 28) and their C-5 derivatives substituted with fluorine, methyl and methoxy moieties. All hybrids were tested for antiproliferative activity against MCF-7 and triple-negative MDA-MB-231 breast cancer cell lines as well as their corresponding multidrug-resistant MCF-7/ADR and MDA-MB-231/ADR. The synthesis of the most potent hybrids of the series 153a–c is reported in Scheme 28. Hybrid 153a [89], presenting the shortest linker (two-carbon alkyl chain) and the thiosemicarbazide moiety at the C-3 position of isatin, showed high activity and cytoselectivity, being 53- and 46-fold more potent than parent dihydroartemisinin (2) towards triple-negative MDA-MB-231 and MDA-MB-231/ADR breast cell lines and 75- and 55-fold more potent than in MCF-7 and MCF-7/ADR cell lines, respectively (Scheme 28). Analogously, hybrid 153b (R = OBn) [88], with a three-carbon alkyl chain and the benzoxime moiety at the C-3 skeleton of isatin, showed a similar activity profile to that of hybrid 153a (Scheme 28). On the contrary, hybrid 153b (R = OEt) [88], with a three-carbon alkyl chain and the ethoxime moiety at the C-3 position of isatin, was found to be cytoselective towards MCF-7 cells with IC50 = 1.27 μM (MDA-MB-231/MCF-7 = 11.8) (Scheme 28). Hybrid 153c [89], with a four-carbon alkyl chain and the oxime moiety at the C-3 position of isatin, showed interesting activity in all breast cancer cell lines tested (IC50 = 2.85–10.18 μM). In particular, hybrid 153c was found to be 29-fold more active than dihydroartemisinin (2) in multidrug-resistant triple-negative MDA-MB-231/ADR (Scheme 28). The overall data indicated that the conjugation of dihydroartemisinin (2) with isatin through ester bonds could be helpful to overcome drug resistance and to tackle triple-negative breast cancer.
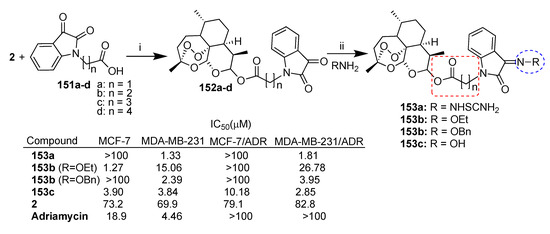
Scheme 28.
Synthesis of dihydroartemisinin-isatin hybrids and selected biological data. Reaction conditions: (i) HATU, DIEA, DMF, r.t., overnight; (ii) AcONa, THF/H2O, 60 °C, 12 h.
3.14. Artemisinin–Sulfasalazine Hybrid
Sulfasalazine (154, Scheme 29) is an anti-inflammatory drug in clinical use for chronic diseases such as ulcerative colitis and Crohn’s disease [137] and rheumatoid arthritis [138]. Sulfasalazine’s anticancer activity has also been reported in glioma [139,140] and other cancers [141,142]. Moreover, it has been employed as a chemosensitizer in radiotherapy [140,143] and chemotherapy [144,145,146,147].
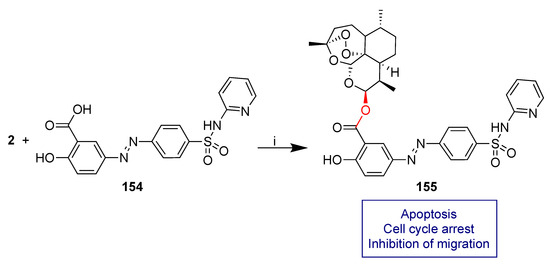
Scheme 29.
Synthesis of dihydroartemisinin–sulfasalazine hybrid. Reaction conditions: (i) DCC, DMAP, ACN, 40 °C, 8 h 21% yield.
Ackermann et al. recently reported a study on the anticancer activity in selected glioma cell lines of dihydroartemisinin–sulfasalazine fused hybrid 155 obtained by condensation reaction of dihydroartemisinin (2) and sulfasalazine 154 (Scheme 29) [90].
Hybrid 155 showed significant improved anticancer activity in U87 and TN22 human glioma cells with respect to both unconjugated dihydroartemisinin (2) and sulfasalazine 154 as well as their 1:1 combination. In particular, treatment of TN22 cells with 5 μM of hybrid 155 led to a 60% reduction of cell viability whereas unconjugated dihydroartemisinin (2) and dihydroartemisinin (2) and sulfasalazine 154 1:1 combinations reduced the cell viability by only 20%. No effect on cell viability was found by treatment with sulfasalazine 154 alone. These data prove a relevant effectiveness of molecular hybridization. A deep mechanism insight demonstrated that the remarkable reduction of cell viability can be attributed to apoptosis and cell cycle arrest. Moreover, hybrid 155 showed significant inhibition of cell migration which is an important property to fight malignancies with invasive growth such as glioma.
4. Conclusions
The research on artemisinin’s anticancer activity has demonstrated that despite challenges, repurposing of artemisinin and its derivatives as anticancer drugs is possible. Indeed, artemisinins have shown anticancer activity against different cancer types by acting through various pathways. Anticancer efficacy of artemisinins has also been proved in in vivo studies with evidence of tumor growth inhibition in xenograft models. Human trials reported so far have demonstrated that some artemisinins are safe and have significant anticancer efficacy. However, artemisinins are characterized by poor pharmacokinetic properties such as low tissue distribution and short half-life that limit their applicability in clinical anticancer therapy. To avoid these limitations, different strategies have been implemented, such as the use of combination therapy and nanoformulations, that can overcome the pharmacokinetic barriers of artemisinins, as well as molecular hybridization. This last option can help not only to overcome the intrinsic limitations of artemisinins but also to improve anticancer activity by lowering the toxicity as a result of enhanced cytoselectivity and to improve the efficacy against multidrug-resistant cancers. The discovery of artemisinin’s anticancer activity is relatively recent, therefore, the research on artemisinin moiety molecular hybridization has been developed mainly over the last 10–15 years. Although many efforts have been made, as demonstrated by the numerousness of the hybrids reported in the literature, the lack of systematic studies on the molecular mechanism of action of hybrids as well as the lack of in vivo studies still limit the development of new artemisinin-based hybrid drugs. Indeed, while many hybrids among the herein reported ones showed interesting improved anticancer activity against selected cancer cell lines, few cases have been extensively studied, for instance, ursodeoxycholic bile acid hybrids 13 and 22 in hepatocellular carcinoma [45,46], cinnamic acid hybrid 135a in lung carcinoma [81] and sulfasalazine hybrid 155 in glioma [90]. Up to now, studies in xenograft models have been restricted to very few cases such as ursodeoxycholic bile acid hybrid 25 in lung cancer [54] and quinazoline hybrid 69 in colon cancer [64]. More in-depth studies into these aspects are required. The eventual development of artemisinin-based hybrids into drugs for cancer chemotherapy can have great relevance.
Author Contributions
Conceptualization and writing—original draft preparation: M.L.N. and D.P.; review and editing: E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Penny, L.K.; Wallace, H.M. The challenges for cancer chemoprevention. Chem. Soc. Rev. 2015, 44, 8836–8847. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Rahman, T. The difficulties in cancer treatment. Ecancermedicalscience 2012, 6, ed16. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Gediya, L.K.; Njar, V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009, 4, 1099–1111. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Danuello, A.; da Bolzani, V.S.; Barreiro, E.J.; Manssour, C.A. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- Shalini; Kumar, V. Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural Products in Drug Discovery and Development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Cordell, G.A. Biodiversity and drug discovery—A symbiotic relationship. Phytochemistry 2000, 55, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Strobl, J.S.; Bane, S.; Schilling, J.K.; McCracken, M.; Chatterjee, S.K.; Rahim-Bata, R.; Kingston, D.G.I. Design, Synthesis, and Bioactivities of Steroid-Linked Taxol Analogues as Potential Targeted Drugs for Prostate and Breast Cancer. J. Nat. Prod. 2004, 67, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wittman, M.D.; Kadow, J.F.; Vyas, D.M.; Lee, F.L.; Rose, W.C.; Long, B.H.; Johnston, K. Synthesis and Antitumor Activity of Novel paclitaxel ± Chlorambucil Hybrids. Bioorg. Med. Chem. Lett. 2001, 11, 811–814. [Google Scholar] [CrossRef]
- Kingston, D.G.I.; Snyder, J.P. The quest for a simple bioactive analog of paclitaxel as a potential anticancer agent. Acc. Chem. Res. 2014, 47, 2682–2691. [Google Scholar] [CrossRef]
- Daniel, J.; Montaleytang, M.; Nagarajan, S.; Picard, S.; Clermont, G.; Lazar, A.N.; Dumas, N.; Correard, F.; Braguer, D.; Blanchard-Desce, M.; et al. Hydrophilic Fluorescent Nanoprodrug of paclitaxel for Glioblastoma Chemotherapy. ACS Omega 2019, 4, 18342–18354. [Google Scholar] [CrossRef]
- Roussi, F.; Quoc, A.N.; Thoret, S.; Guéritte, F.; Guénard, D. The design and synthesis of new steroidal compounds as potential mimics of taxoids. Eur. J. Org. Chem. 2005, 2005, 3952–3961. [Google Scholar] [CrossRef]
- Melloni, E.; Marchesi, E.; Preti, L.; Casciano, F.; Rimondi, E.; Romani, A.; Secchiero, P.; Navacchia, M.L.; Perrone, D. Synthesis and Biological Investigation of Bile Acid-Paclitaxel Hybrids. Molecules 2022, 27, 471. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem.-Int. Ed. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, D.; Goswami, A.; Saikia, P.P.; Barua, N.C.; Rao, P.G. Artemisinin and its derivatives: A novel class of anti-malarial and anti-cancer agents. Chem. Soc. Rev. 2010, 39, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Woon, C.Y.-N.; Liu, C.-G.; Cheng, J.-T.; You, M.; Sethi, G.; Wong, A.L.-A.; Ho, P.C.-L.; Zhang, D.; Ong, P.; et al. Repurposing Artemisinin and its Derivatives as Anticancer Drugs: A Chance or Challenge? Front. Pharmacol. 2021, 12, 828856. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.C.; Singh, N.P.; Sasaki, T. Development of artemisinin compounds for cancer treatment. Investig. New Drugs 2013, 31, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.K.; Xu, C.; Kalesh, K.A.; He, Y.; Lin, Q.; Wong, W.S.F.; Shen, H.M.; Wang, J. Artemisinin as an anticancer drug: Recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017, 37, 1492–1517. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, X.; Chen, W.; Chen, Y.; Zhang, Q.; Mo, S.; Lu, J. Dihydroartemisinin: A potential natural anticancer drug. Int. J. Biol. Sci. 2021, 17, 603–622. [Google Scholar] [CrossRef]
- Wen, L.; Liu, L.; Wen, L.; Yu, T.; Wei, F. Artesunate promotes G2/M cell cycle arrest in MCF7 breast cancer cells through ATM activation. Breast Cancer 2018, 25, 681–686. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhan, X.; Wang, L.; Ho, R.J.Y.; Sasaki, T. pH-responsive artemisinin dimer in lipid nanoparticles are effective against human breast cancer in a xenograft model. J. Pharm. Sci. 2015, 104, 1815–1824. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Chen, K.; Ba, Q.; Zhang, X.; Li, J.; Wang, J.; Wang, H.; Liu, H. Structural optimization and biological evaluation for novel artemisinin derivatives against liver and ovarian cancers. Eur. J. Med. Chem. 2021, 211, 113000. [Google Scholar] [CrossRef]
- Zhou, C.; Pan, W.; Wang, X.P.; Chen, T.S. Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. J. Cell. Physiol. 2012, 227, 3778–3786. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.; Zhang, J.; He, A.; Wang, Y.L.; Han, K.; Su, Y.; Yin, J.; Lv, X.; Hu, H. Artesunate suppresses the viability and mobility of prostate cancer cells through UCA1, the sponge of miR-184. Oncotarget 2017, 8, 18260–18270. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zou, W.Q.; Du, S.J.; Wu, M.J.; Xiang, T.X.; Luo, Z.G. Mechanism of dihydroartemisinin-induced apoptosis in prostate cancer PC3 cells: An iTRAQ-based proteomic analysis. Life Sci. 2016, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, B. Biological Actions of Artemisinin: Insights from Medicinal Chemistry Studies. Molecules 2010, 15, 1378–1397. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Giordo, R.; Pintus, G.; Mohammed, S.A.; Orhan, I.E.; Fokou, P.V.T.; Sharopov, F.; Adetunji, C.O.; Gulsunoglu-Konuskan, Z.; Ydyrys, A.; et al. Medicinal and mechanistic overview of artemisinin in the treatment of human diseases. Biomed. Pharmacother. 2023, 163, 114866. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, H.; Mu, L.; Yang, X. Artemisinins as Anticancer Drugs: Novel Therapeutic Approaches, Molecular Mechanisms, and Clinical Trials. Front. Pharmacol. 2020, 11, 529881. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Barton, V.E.; Ward, S.A. The molecular mechanism of action of artemisinin-The debate continues. Molecules 2010, 15, 1705–1721. [Google Scholar] [CrossRef]
- World Health Organization. WHO Calls for an Immediate Halt to Provision of Single-Drug Artemisinin Malaria Pills; World Health Organization: Washington, DC, USA, 2006; p. 2006. [Google Scholar]
- World Health Organization. WHO Resolution WHA6018 Malaria, including proposal for establishment of World Malaria Day In Sixtieth World Health Assembly, Geneva, Resolutions and Decisions, Annexes; World Health Organization: Geneva, Switzerland, 2007; (WHA60/2007/REC/1). [Google Scholar]
- Nosten, F.; White, N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007, 77, 181–192. [Google Scholar] [CrossRef]
- Tsamesidis, I.; Reybier, K.; Marchetti, G.; Pau, M.C.; Virdis, P.; Fozza, C.; Nepveu, F.; Low, P.S.; Turrini, F.M.; Pantaleo, A. Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes. Antioxidants 2020, 9, 753–774. [Google Scholar] [CrossRef]
- Chien, H.D.; Pantaleo, A.; Kesely, K.R.; Noomuna, P.; Putt, K.S.; Tuan, T.A.; Low, P.S.; Turrini, F.M. Imatinib augments standard malaria combination therapy without added toxicity. J. Exp. Med. 2021, 218, e20210724. [Google Scholar] [CrossRef]
- Li, Q.; Ma, Q.; Cheng, J.; Zhou, X.; Pu, W.; Zhong, X.; Guo, X. Dihydroartemisinin as a sensitizing agent in cancer therapies. OncoTargets Ther. 2021, 14, 2563–2573. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Mari, L.; Chinaglia, N.; Gallerani, E.; Gavioli, R.; Capobianco, M.L.; Perrone, D. Rational design of nucleoside-bile acid conjugates incorporating a triazole moiety for anticancer evaluation and SAR exploration. Molecules 2017, 22, 1710. [Google Scholar] [CrossRef]
- Marchesi, E.; Chinaglia, N.; Capobianco, M.L.; Marchetti, P.; Huang, T.E.; Weng, H.C.; Guh, J.H.; Hsu, L.C.; Perrone, D.; Navacchia, M.L. Dihydroartemisinin–Bile Acid Hybridization as an Effective Approach to Enhance Dihydroartemisinin Anticancer Activity. ChemMedChem 2019, 14, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.E.; Deng, Y.N.; Hsu, J.L.; Leu, W.J.; Marchesi, E.; Capobianco, M.L.; Marchetti, P.; Navacchia, M.L.; Guh, J.H.; Perrone, D.; et al. Evaluation of the Anticancer Activity of a Bile Acid-Dihydroartemisinin Hybrid Ursodeoxycholic-Dihydroartemisinin in Hepatocellular Carcinoma Cells. Front. Pharmacol. 2020, 11, 599067. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.F.; Kung, F.L.; Huang, T.E.; Deng, Y.N.; Guh, J.H.; Marchetti, P.; Marchesi, E.; Perrone, D.; Navacchia, M.L.; Hsu, L.C. Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells. Molecules 2023, 28, 2358. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its derivatives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 2012, 247597. [Google Scholar] [CrossRef] [PubMed]
- Galal, A.M.; Ross, S.A.; ElSohly, M.A.; Hala, N.; ElSohly, H.N.; . El-Feraly, F.S.; Ahmed, M.S.; McPhail, E.T. Deoxyartemisinin derivatives from photooxygenation of anhydrodeoxydihydroartemisinin and their cytotoxic evaluation. J. Nat. Prod. 2002, 65, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, P.; Chen, Y.-X.; Li, L.-Q.; Gai, D.-S.; Wang, Y.-P.Z. Synthesis of some derivatives of artemisinine. Chin. Sci. Bull. (Repr. Kexue Tongbao) 1979, 24, 667–669. [Google Scholar]
- Posner, G.H.; Paik, I.H.; Chang, W.; Borstnik, K.; Sinishtaj, S.; Rosenthal, A.S.; Shapiro, T.A. Malaria-infected mice are cured by a single dose of novel artemisinin derivatives. J. Med. Chem. 2007, 50, 2516–2519. [Google Scholar] [CrossRef]
- Presser, A.; Feichtinger, A.; Buzzi, S. A simplified and scalable synthesis of artesunate. Monatshefte Fur Chem. 2017, 148, 63–68. [Google Scholar] [CrossRef]
- Li, Q.G.; Peggins, J.O.; Fleckenstein, L.L.; Masonic, K.; Heiffer, M.H.; Brewer, T.G. The pharmacokinetics and bioavailability of dihydroartemisinin, arteether, artemether, artesunic acid and artelinic acid in rats. J. Pharm. Pharmacol. 1998, 50, 173–182. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Perrone, D. Bile Acid Conjugates with Anticancer Activity: Most Recent Research. Molecules 2021, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Liu, C.; Li, C.; Fu, R.; Xu, W.; Bian, H.; Dong, X.; Zhao, X.; Xu, Z.; Zhang, J.; et al. Study on the structure-activity relationship of dihydroartemisinin derivatives: Discovery, synthesis, and biological evaluation of dihydroartemisinin-bile acid conjugates as potential anticancer agents. Eur. J. Med. Chem. 2021, 225, 113754. [Google Scholar] [CrossRef] [PubMed]
- Letis, A.S.; Seo, E.J.; Nikolaropoulos, S.S.; Efferth, T.; Giannis, A.; Fousteris, M.A. Synthesis and cytotoxic activity of new artemisinin hybrid molecules against human leukemia cells. Bioorg. Med. Chem. 2017, 25, 3357–3367. [Google Scholar] [CrossRef]
- Herrmann, L.; Yaremenko, I.A.; Çapcı, A.; Struwe, J.; Tailor, D.; Dheeraj, A.; Hodek, J.; Belyakova, Y.Y.; Radulov, P.S.; Weber, J.; et al. Synthesis and in vitro Study of Artemisinin/Synthetic Peroxide-Based Hybrid Compounds against SARS-CoV-2 and Cancer. ChemMedChem 2022, 17, e202200005. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.; Kim, M.; Chung, W.Y.; Park, K.K.; Jung, M. Discovery of artemisinin-glycolipid hybrids as anti-oral cancer agents. Chem. Pharm. Bull. 2011, 59, 1471–1475. [Google Scholar] [CrossRef]
- Wang, L.; Switalska, M.; Wang, N.; Du, Z.J.; Fukumoto, Y.; Diep, N.K.; Kiguchi, R.; Nokami, J.; Wietrzyk, J.; Inokuchi, T. Design, synthesis, and biological evaluation of artemisinin-indoloquinoline hybrids as potent antiproliferative agents. Molecules 2014, 19, 19021–19035. [Google Scholar] [CrossRef]
- Yao, G.; Chen, H.; Chen, L.; Ge, M.; Yang, J.; Liu, W.; Xia, M.; Hayashi, T.; Guo, C.; Ikejima, T. Autophagy promotes apoptosis induction through repressed nitric oxide generation in the treatment of human breast cancer MCF-7 cells with L-A03, a dihydroartemisinin derivative. Med. Chem. Res. 2017, 26, 1427–1436. [Google Scholar] [CrossRef]
- Yao, G.D.; Ge, M.Y.; Li, D.Q.; Chen, L.; Hayashi, T.; Tashiro, S.I.; Onodera, S.; Guo, C.; Song, S.J.; Ikejima, T. L-A03, a dihydroartemisinin derivative, promotes apoptotic cell death of human breast cancer MCF-7 cells by targeting c-Jun N-terminal kinase. Biomed. Pharmacother. 2018, 105, 320–325. [Google Scholar] [CrossRef]
- Lombard, M.C.; N’Da, D.D.; Breytenbach, J.C.; Smith, P.J.; Lategan, C.A. Artemisinin-quinoline hybrid-dimers: Synthesis and in vitro antiplasmodial activity. Bioorg. Med. Chem. Lett. 2010, 20, 6975–6977. [Google Scholar] [CrossRef]
- Lombard, M.C.; N’Da, D.D.; Breytenbach, J.C.; Kolesnikova, N.I.; Tran Van Ba, C.; Wein, S.; Norman, J.; Denti, P.; Vial, H.; Wiesner, L. Antimalarial and anticancer activities of artemisinin-quinoline hybrid-dimers and pharmacokinetic properties in mice. Eur. J. Pharm. Sci. 2012, 47, 834–841. [Google Scholar] [CrossRef]
- Fröhlich, T.; Reiter, C.; Ibrahim, M.M.; Beutel, J.; Hutterer, C.; Zeitträger, I.; Bahsi, H.; Leidenberger, M.; Friedrich, O.; Kappes, B.; et al. Synthesis of Novel Hybrids of Quinazoline and Artemisinin with High Activities against Plasmodium falciparum, Human Cytomegalovirus, and Leukemia Cells. ACS Omega 2017, 2, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Kong, L.; Liu, H.; Zhang, Y.; Zhang, L.; Liu, X.; Yuan, F.; Li, Y.; Zuo, Z. Design and synthesis of novel artemisinin derivatives with potent activities against colorectal cancer in vitro and in vivo. Eur. J. Med. Chem. 2019, 182, 111665. [Google Scholar] [CrossRef]
- Dai, T.; Lin, L.; Chen, H.; Lu, W.; Yang, X.; Yang, L.; Liu, Y.; Cui, J.; Sun, D. Novel nitrogen mustard-artemisinin hybrids with potent anti-leukemia action through DNA damage and activation of GPx. Eur. J. Med. Chem. 2022, 244, 114783. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Liu, Y.; Zhang, X.; Chen, T.; Chen, K.; Ba, Q.; Li, J.; Liu, H.; Wang, H. Preclinical Efficacy and Safety Assessment of Artemisinin-Chemotherapeutic Agent Conjugates for Ovarian Cancer. EBioMedicine 2016, 14, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Botta, L.; Filippi, S.; Bizzarri, B.M.; Zippilli, C.; Meschini, R.; Pogni, R.; Baratto, M.C.; Villanova, L.; Saladino, R. Synthesis and Evaluation of Artemisinin-Based Hybrid and Dimer Derivatives as Antimelanoma Agents. ACS Omega 2020, 5, 243–251. [Google Scholar] [CrossRef]
- Botta, L.; Cesarini, S.; Zippilli, C.; Filippi, S.; Bizzarri, B.M.; Baratto, M.C.; Pogni, R.; Saladino, R. Stereoselective Access to Antimelanoma Agents by Hybridization and Dimerization of Dihydroartemisinin and Artesunic acid. ChemMedChem 2021, 16, 2270–2277. [Google Scholar] [CrossRef]
- Botta, L.; Filippi, S.; Zippilli, C.; Cesarini, S.; Bizzarri, B.M.; Cirigliano, A.; Rinaldi, T.; Paiardini, A.; Fiorucci, D.; Saladino, R.; et al. Artemisinin Derivatives with Antimelanoma Activity Show Inhibitory Effect against Human DNA Topoisomerase 1. ACS Med. Chem. Lett. 2020, 11, 1035–1040. [Google Scholar] [CrossRef]
- Fröhlich, T.; Ndreshkjana, B.; Muenzner, J.K.; Reiter, C.; Hofmeister, E.; Mederer, S.; Fatfat, M.; El-Baba, C.; Gali-Muhtasib, H.; Schneider-Stock, R.; et al. Synthesis of Novel Hybrids of Thymoquinone and Artemisinin with High Activity and Selectivity Against Colon Cancer. ChemMedChem 2017, 12, 226–234. [Google Scholar] [CrossRef]
- Fröhlich, T.; Reiter, C.; Saeed, M.E.M.; Hutterer, C.; Hahn, F.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Marschall, M.; Efferth, T.; et al. Synthesis of Thymoquinone-Artemisinin Hybrids: New Potent Antileukemia, Antiviral, and Antimalarial Agents. ACS Med. Chem. Lett. 2018, 9, 534–539. [Google Scholar] [CrossRef]
- Çapcı Karagöz, A.; Reiter, C.; Seo, E.J.; Gruber, L.; Hahn, F.; Leidenberger, M.; Klein, V.; Hampel, F.; Friedrich, O.; Marschall, M.; et al. Access to new highly potent antileukemia, antiviral and antimalarial agents via hybridization of natural products (homo)egonol, thymoquinone and artemisinin. Bioorg. Med. Chem. 2018, 26, 3610–3618. [Google Scholar] [CrossRef]
- Xie, L.; Zhai, X.; Ren, L.; Meng, H.; Liu, C.; Zhu, W.; Zhao, Y. Design, synthesis and antitumor activity of novel artemisinin derivatives using hybrid approach. Chem. Pharm. Bull. 2011, 59, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhai, X.; Liu, C.; Li, P.; Li, Y.; Guo, G.; Gong, P. Anti-tumor activity of new artemisinin-chalcone hybrids. Arch. Pharm. 2011, 344, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Smit, F.J.; Van Biljon, R.A.; Birkholtz, L.M.; N’da, D.D. Synthesis and in vitro biological evaluation of dihydroartemisinyl-chalcone esters. Eur. J. Med. Chem. 2015, 90, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liang, Z.; Xu, H.; Mou, Y.; Guo, C. Design, synthesis and cytotoxicity of novel dihydroartemisinin-coumarin hybrids via click chemistry. Molecules 2016, 21, 758. [Google Scholar] [CrossRef]
- Yu, H.; Hou, Z.; Yang, X.; Mou, Y.; Guo, C. Design, Synthesis, and Mechanism of Dihydroartemisinin–Coumarin Hybrids as Potential Anti-Neuroinflammatory AgentsNo Title. Molecules 2019, 24, 1672. [Google Scholar] [CrossRef]
- Yu, H.; Hou, Z.; Tian, Y.; Mou, Y.; Guo, C. Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents. Eur. J. Med. Chem. 2018, 151, 434–449. [Google Scholar] [CrossRef]
- Fröhlich, T.; Mai, C.; Bogautdinov, R.P.; Morozkina, S.N.; Shavva, A.G.; Friedrich, O.; Gilbert, D.F.; Tsogoeva, S.B. Synthesis of Tamoxifen-Artemisinin and Estrogen-Artemisinin Hybrids Highly Potent Against Breast and Prostate Cancer. ChemMedChem 2020, 15, 1473–1479. [Google Scholar] [CrossRef]
- Fröhlich, T.; Kiss, A.; Wölfling, J.; Mernyák, E.; Kulmány, Á.E.; Minorics, R.; Zupkó, I.; Leidenberger, M.; Friedrich, O.; Kappes, B.; et al. Synthesis of Artemisinin-Estrogen Hybrids Highly Active against HCMV, P. falciparum, and Cervical and Breast Cancer. ACS Med. Chem. Lett. 2018, 9, 1128–1133. [Google Scholar] [CrossRef]
- Xu, C.C.; Deng, T.; Fan, M.L.; Lv, W.B.; Liu, J.H.; Yu, B.Y. Synthesis and in vitro antitumor evaluation of dihydroartemisinin-cinnamic acid ester derivatives. Eur. J. Med. Chem. 2016, 107, 192–203. [Google Scholar] [CrossRef]
- Jones, M.; Mercer, A.E.; Stocks, P.A.; La Pensée, L.J.I.; Cosstick, R.; Kevin Park, B.K.; Kennedy, M.E.; Piantanida, I.; Ward, S.A.; Davies, J.; et al. Antitumour and antimalarial activity of artemisinin–acridine hybrids. Bioorg. Med. Chem. Lett. 2009, 19, 2033–2037. [Google Scholar] [CrossRef]
- Joubert, J.P.; Smit, F.J.; du Plessis, L.; Smith, P.J.; David, D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin–acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27. [Google Scholar] [CrossRef]
- Hou, H.; Qu, B.; Su, C.; Hou, G.; Gao, F. Design, Synthesis and Anti-Lung Cancer Evaluation of 1, 2, 3-Triazole Tethered Dihydroartemisinin-Isatin Hybrids. Front. Pharmacol. 2021, 12, 801580. [Google Scholar] [CrossRef]
- Dong, M.; Zheng, G.; Gao, F.; Li, M.; Zhong, C. Three-Carbon Linked Dihydroartemisinin-Isatin Hybrids: Design, Synthesis and Their Antiproliferative Anticancer Activity. Front. Pharmacol. 2022, 13, 834317. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, H.; Xu, J.; Gao, F.; Cao, W. The Anti-Breast Cancer Activity of Dihydroartemisinin-5-methylisatin Hybrids Tethered via Different Carbon Spacers. Molecules 2022, 27, 7994. [Google Scholar] [CrossRef]
- Ding, F.; Chen, X.; Cao, W.; Dong, T.; Wang, P. The anti-breast cancer potential of dihydroartemisinin-isatin hybrids with hydrogen bond donors at C-3 position of isatin moiety. Fitoterapia 2023, 165, 105426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, X.; Tang, M.; Liu, X.; Deng, J.; Zhou, W.; Xu, Z. Design, synthesis and anti-breast cancer properties of butyric ester tethered dihydroartemisinin-isatin hybrids. Med. Chem. Res. 2023, 32, 705–712. [Google Scholar] [CrossRef]
- Liu, S.; Wang, S.; Xu, D.; Pan, B.; Chen, L.; Zhao, S.; Xu, Z.; Zhou, W. Novel ester tethered dihydroartemisinin-3-(oxime/thiosemicarbazide)isatin hybrids as potential anti-breast cancer agents: Synthesis, in vitro cytotoxicity and structure–activity relationship. Drug Dev. Res. 2023, 2040, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.; Çapcı, A.; Buchfelder, M.; Tsogoeva, S.B.; Savaskan, N. Chemical hybridization of sulfasalazine and dihydroartemisinin promotes brain tumor cell death. Sci. Rep. 2021, 11, 20766. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Wang, Q.-H.; Molina-Molina, E.; Baccetto, R.L.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acid and Cancer: Direct and environmental-dependent effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef]
- Goossens, J.-F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and Ursodeoxycholic Acid Conjugation: Design of a New Prodrug Potentially Able To Bypass the Active Efflux Transport Systems of the Central Nervous System. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef]
- Faustino, C.; Serafim, C.; Rijo, P.; Reis, C.P. Bile acids and bile acid derivatives: Use in drug delivery systems and as therapeutic agents. Expert Opin. Drug Deliv. 2016, 13, 1133–1148. [Google Scholar] [CrossRef]
- Pavlovic, N.; Golocorbin-Kon, S.; Danic, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic. Profiles Front. Pharmacol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Luo, Z.; Xiang, T.; Wang, K.; Li, J.; Wang, P. Dihydroartemisinin Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in HepG2 Human Hepatoma Cells. Tumori J. 2011, 97, 771–780. [Google Scholar] [CrossRef]
- Im, E.; Yeo, C.; Lee, H.J.; Lee, E.O. Dihydroartemisinin induced caspase-dependent apoptosis through inhibiting the specificity protein 1 pathway in hepatocellular carcinoma SK-Hep-1 cells. Life Sci. 2018, 192, 286–292. [Google Scholar] [CrossRef]
- Qin, G.; Zhao, C.B.; Zhang, L.; Liu, H.; Quan, Y.; Chai, L.; Wu, S.; Wang, X.; Chen, T. Dihydroartemisinin induces apoptosis preferentially via a Bim-mediated intrinsic pathway in hepatocarcinoma cells. Apoptosis 2015, 20, 1072–1086. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin. Drug Discov. 2017, 12, 583–597. [Google Scholar] [CrossRef]
- Nanda, A.K.; Ganguli, S.; Chakraborty, R. Antibacterial activity of some 3-(arylideneamino)-2-phenylquinazoline-4(3H)-ones: Synthesis and preliminary QSAR studies. Molecules 2007, 12, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.P.; Wang, X.Y.; Song, B.A.; Yang, S.; Yan, K.; Xu, G.F.; Bhadury, P.S.; Liu, F.; Jin, L.H.; Hu, D.Y. Synthesis, antiviral and antifungal bioactivity of 2-cyano-acrylate derivatives containing phosphonyl moieties. Molecules 2007, 12, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, Y.; Wang, M.; Liu, Q.; Lei, X.; Wu, M.; Guo, S.; Yi, D.; Li, Q.; Ma, L.; et al. Quinoline and Quinazoline Derivatives Inhibit Viral RNA Synthesis by SARS-CoV-2 RdRp. ACS Infect. Dis. 2021, 7, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, Z.; Jin, L.; Song, B.; Liu, G.; Chen, J.; Chen, Z.; Hu, D.; Xue, W.; Xu, R. Synthesis and bioactivity of 4-alkyl(aryl)thioquinazoline derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 2193–2196. [Google Scholar] [CrossRef]
- Chen, Y.; Jia, Y.; Song, W.; Zhang, L. Therapeutic Potential of Nitrogen Mustard Based Hybrid Molecules. Front. Pharmacol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Yu, E.; Yue, H.; Li, Q. Enhanced liver targeting of camptothecin via conjugation with deoxycholic acid. Molecules 2019, 24, 1179. [Google Scholar] [CrossRef]
- Liu, Y.-Q.; Li, W.-Q.; Morris-Natschke, S.L.; Qian, K.; Yang, L.; Zhu, G.-X.; Wu, X.-B.; Chen, A.-L.; Zhang, S.-Y.; Song, Z.-L.; et al. Perspectives on Biologically Active Camptothecin Derivatives. Med. Res. Rev. 2015, 35, 753–789. [Google Scholar] [CrossRef]
- Li, X.; Zhao, T.; Cheng, D.; Chu, C.; Tong, S.; Yan, J.; Li, Q.Y. Synthesis and biological activity of some bile acid-based camptothecin analogues. Molecules 2014, 19, 3761–3776. [Google Scholar] [CrossRef]
- Tan, X.; Zhou, H.; Wang, C.; Liu, X.; Yang, X.; Liu, W. GSH-responsive camptothecin prodrug-based hybrid micellar nanoparticles enable antitumor chemo-immunotherapy by PD-L1 knockdown. Nano Res. 2023, 16, 834–848. [Google Scholar] [CrossRef]
- Tabassum, S.; Norhayati Rosli, N.; Ichwan, S.J.A.; Mishra, P. Thymoquinone and its pharmacological perspective: A review. Pharmacol. Res.-Mod. Chin. Med. 2021, 1, 100020. [Google Scholar] [CrossRef]
- Sarkar, C.; Jamaddar, S.; Islam, T.; Mondal, M.; Islam, M.T.; Mubarak, M.S. Therapeutic perspectives of the black cumin component thymoquinone: A review. Food Funct. 2021, 12, 6167–6213. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 11306. [Google Scholar] [CrossRef]
- Bahare, S.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, A.; Iqbal, A.; Docea1, O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Gaur, R.; Pathania, A.S.; Malik, F.A.; Bhakuni, R.S.; Verma, R.K. Synthesis of a series of novel dihydroartemisinin monomers and dimers containing chalcone as a linker and their anticancer activity. Eur. J. Med. Chem. 2016, 122, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Cruz-Martins, N.; Lόpez-Jornet, P.; Pons-Fuster Lopez, E.; Harun, N.; Yeskaliyeva, B.; Beyatli, A.; Sytar, O.; Shaheen, S.; Sharopov, F.; et al. Natural Coumarins: Exploring the Pharmacological Complexity and Underlying Molecular Mechanisms. Oxid. Med. Cell. Longev. 2021, 2021, 6492346. [Google Scholar] [CrossRef] [PubMed]
- Akkol, E.K.; Gen, Y.; Büsra Karpuz, B.; Sobarzo-Sanchez, E.; Capasso, R. Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Lu, W.J.; Desta, Z.; Flockhart, D.A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 131, 473–481. [Google Scholar] [CrossRef]
- Ekholm, M.; Bendahl, P.O.; Fernö, M.; Nordenskjöld, B.; Stål, O.; Rydén, L. Effects of adjuvant tamoxifen over three decades on breast cancer–free and distant recurrence–free interval among premenopausal women with oestrogen receptor–positive breast cancer randomised in the Swedish SBII:2pre trial. Eur. J. Cancer 2019, 110, 53–61. [Google Scholar] [CrossRef]
- Dey, P.; Kundu, A.; Chakraborty, H.J.; Kar, B.; Choi, W.S.; Lee, B.M.; Bhakta, T.; Atanasov, A.G.; Kim, H.S. Therapeutic value of steroidal alkaloids in cancer: Current trends and future perspectives. Int. J. Cancer 2019, 145, 1731–1744. [Google Scholar] [CrossRef]
- Potter, B.V.L. Steroid sulphatase inhibition via aryl sulphamates: Clinical progress, mechanism and future prospects. J. Mol. Endocrinol. 2018, 61, T233–T252. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Liu, L.; Hudgins, W.R.; Shack, S.; Yin, M.Q.; Samid, D. Cinnamic acid: A natural product with potential use in cancer intervention. Int. J. Cancer 1995, 62, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent developments in the synthesis and biological activity of acridine/acridone analogues. RSC Adv. 2017, 7, 15776–15804. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, X. Current Scenario of Acridine Hybrids with Anticancer Potential. Curr. Top. Med. Chem. 2021, 21, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Ferraz de Paiva, R.E.; Vieira, E.G.; Rodrigues da Silva, D.; Wegermann, C.A.; Costa Ferreira, A.M. Anticancer Compounds Based on Isatin-Derivatives: Strategies to Ameliorate Selectivity and Efficiency. Front. Mol. Biosci. 2021, 7, 627272. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Chen, Q.; Yang, G.F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Bharathi Dileepan, A.G.; Daniel Prakash, T.; Ganesh Kumar, A.; Shameela Rajam, P.; Violet Dhayabaran, V.; Rajaram, R. Isatin based macrocyclic Schiff base ligands as novel candidates for antimicrobial and antioxidant drug design: In vitro DNA binding and biological studies. J. Photochem. Photobiol. B Biol. 2018, 183, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Guo, H. Isatin derivatives and their anti-bacterial activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A. Oxindole inhibitors of cyclin-dependent kinase as anti-tumor agents. In Inhibitors of Cyclin-Dependent Kinases as Anti-Tumor Agents; Smith, P.J., Yue, E.W., Eds.; CRC Press: Boca Raton, FL, USA; Taylor& Francis Group: Abingdon, UK, 2007; pp. 265–281. [Google Scholar]
- Prakash, C.R.; Theivendren, P.; Raja, S. Indolin-2-Ones in Clinical Trials as Potential Kinase Inhibitors: A Review. Pharmacol. Pharm. 2012, 3, 62–71. [Google Scholar] [CrossRef]
- Roth, G.J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, S.L.; Kaiser, R. Nintedanib: From discovery to the clinic. J. Med. Chem. 2015, 58, 1053–1063. [Google Scholar] [CrossRef]
- Izzedine, H.; Buhaescu, I.; Rixe, O.; Deray, G. Sunitinib malate. Cancer Chemother. Pharmacol. 2007, 60, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Harbord, M.; Eliakim, R.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnár, T.; Raine, T.; Sebastian, S.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 2: Current management. J. Crohn’s Colitis 2017, 11, 769–784. [Google Scholar] [CrossRef]
- Sehm, T.; Fan, Z.; Ghoochani, A.; Rauh, M.; Engelhorn, T.; Minakaki, G.; Dörfler, A.; Klucken, J.; Buchfelder, M.; Eyüpoglu, I.Y.; et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget 2016, 7, 36021–36033. [Google Scholar] [CrossRef]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Førde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.H.; Wang, J.; et al. Drug repurposing: Sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x-c cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef]
- Lo, M.; Ling, V.; Low, C.; Wang, Y.Z.; Gout, P.W. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr. Oncol. 2010, 17, 9–16. [Google Scholar] [CrossRef]
- Nagane, M.; Kanai, E.; Shibata, Y.; Shimizu, T.; Yoshioka, C.; Maruo, T.; Yamashita, T. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE 2018, 13, e0195151. [Google Scholar] [CrossRef]
- Awasthi, S.; Sharma, R.; Singhal, S.S.; Herzog, N.K.; Chaubey, M.; Awasthi, Y.C. Modulation of cisplatin cytotoxicity by sulphasalazine. Br. J. Cancer 1994, 70, 190–194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, M.Z.; Chen, G.; Wang, P.; Lu, W.H.; Zhu, C.F.; Song, M.; Yang, J.; Wen, S.; Xu, R.H.; Hu, Y.; et al. Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Narang, V.S.; Pauletti, G.M.; Gout, P.W.; Buckley, D.J.; Buckley, A.R. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: Enhancement of growth-inhibitory activity of doxorubicin. Chemotherapy 2007, 53, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Müerköster, S.; Arlt, A.; Witt, M.; Gehrz, A.; Haye, S.; March, C.; Grohmann, F.; Wegehenkel, K.; Kalthoff, H.; Fölsch, U.R.; et al. Usage of the NF-κB inhibitor sulfasalazine as sensitizing agent in combined chemotherapy of pancreatic cancer. Int. J. Cancer 2003, 104, 469–476. [Google Scholar] [CrossRef] [PubMed]

































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).