
CNP-miR146a Decreases Inflammation in Murine Acute Infectious Lung Injury

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Development of CNP-miR146a
2.2. MRSA Infectious Lung Injury Animal Model
2.3. Proinflammatory Gene Expression Measurement
2.4. Immunohistochemistry for Inflammatory Cell Infiltration
2.5. Statistical Analyses
3. Results
3.1. Intratracheal Treatment with CNP-miR146a
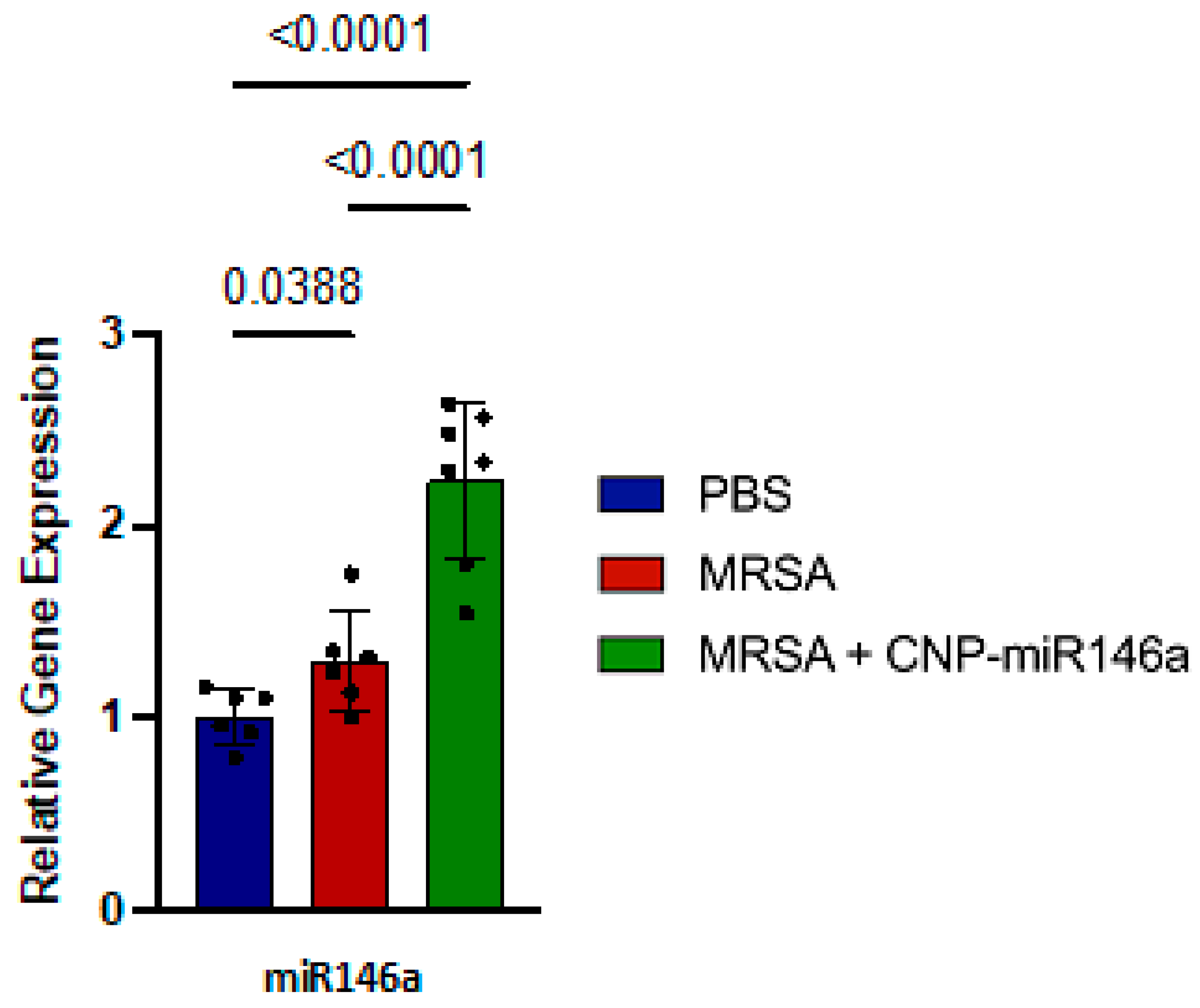
3.2. MiR146a Expression Upregulated in CNP-miR146a Treated Mice
3.3. CNP-miR146a Decreases MRSA-Induced Proinflammatory Gene Expression
3.4. Improved Alveolar–Capillary Barrier Integrity with CNP-miR146a Treatment
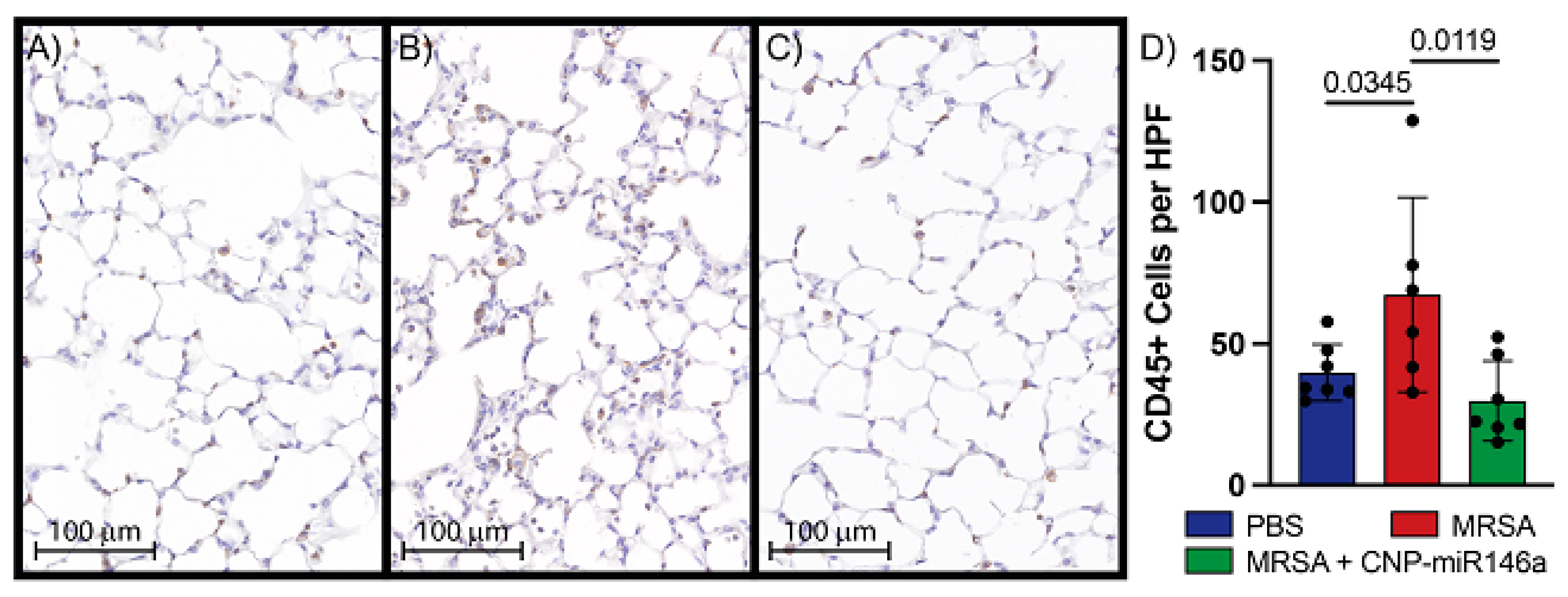
3.5. CNP-miR146a Lowers Inflammatory Cell Infiltrate into the Lung
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Cheung, A.M.; Tansey, C.M.; Tomlinson, G.; Diaz-Granados, N.; Matté, A.; Barr, A.; Mehta, S.; Mazer, C.D.; Guest, C.B.; Stewart, T.E.; et al. Two-year outcomes, health care use, and costs of survivors of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2006, 174, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Khemani, R.G.; Smith, L.S.; Zimmerman, J.J.; Erickson, S. Pediatric acute respiratory distress syndrome: Definition, incidence, and epidemiology: Proceedings from the Pediatric Acute Lung Injury Consensus Conference. Pediatr. Crit. Care Med. 2015, 16 (Suppl. S5), S23–S40. [Google Scholar] [CrossRef]
- Herridge, M.S.; Tansey, C.M.; Matté, A.; Tomlinson, G.; Diaz-Granados, N.; Cooper, A.; Cheung, A.M. Functional disability 5 years after acute respiratory distress syndrome. N. Engl. J. Med. 2011, 364, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, B.B.; Suri, R.; Suchyta, M.R.; Digrande, K.F.; Sherwood, K.D.; Colantuoni, E.; Dinglas, V.D.; Needham, D.M.; Hopkins, R.O. Return to work after critical illness: A systematic review and meta-analysis. Thorax 2020, 75, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Boucher, P.E.; Taplin, J.; Clement, F. The Cost of ARDS: A Systematic Review. Chest 2022, 161, 684–696. [Google Scholar] [CrossRef]
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef]
- Parsons, P.E.; Eisner, M.D.; Thompson, B.T.; Matthay, M.A.; Ancukiewicz, M.; Bernard, G.R. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit. Care Med. 2005, 33, 1–6; discussion 230–232. [Google Scholar] [CrossRef]
- Fielding-Singh, V.; Matthay, M.A.; Calfee, C.S. Beyond Low Tidal Volume Ventilation: Treatment Adjuncts for Severe Respiratory Failure in Acute Respiratory Distress Syndrome. Crit. Care Med. 2018, 46, 1820–1831. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; Van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Schwartz, M.D.; Moore, E.E.; Moore, F.A.; Shenkar, R.; Moine, P.; Haenel, J.B.; Abraham, E. Nuclear factor-kappa B is activated in alveolar macrophages from patients with acute respiratory distress syndrome. Crit. Care Med. 1996, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Nepal, N.; Rogers, S.; Manne, N.D.; Arvapalli, R.; Rice, K.M.; Blough, E.R. Inhibition of MAP kinase/NF-kB mediated signaling and attenuation of lipopolysaccharide induced severe sepsis by cerium oxide nanoparticles. Biomaterials 2015, 59, 160–171. [Google Scholar] [CrossRef]
- D'Alessio, F.R. Mouse Models of Acute Lung Injury and ARDS. Methods Mol. Biol. 2018, 1809, 341–350. [Google Scholar] [PubMed]
- Kulkarni, H.S.; Lee, J.S.; Bastarache, J.A.; Kuebler, W.M.; Downey, G.P.; Albaiceta, G.M.; Altemeier, W.A.; Artigas, A.; Bates, J.H.T.; Calfee, C.S.; et al. Update on the Features and Measurements of Experimental Acute Lung Injury in Animals: An Official American Thoracic Society Workshop Report. Am. J. Respir. Cell Mol. Biol. 2022, 66, e1–e14. [Google Scholar] [CrossRef]
- Vishnupriya, M.; Naveenkumar, M.; Manjima, K.; Sooryasree, N.V.; Saranya, T.; Ramya, S.; Winster, S.H.; Paulpandi, M.; Balachandar, V.; Arul, N. Post-COVID pulmonary fibrosis: Therapeutic efficacy using with mesenchymal stem cells-How the lung heals. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2748–2751. [Google Scholar] [PubMed]
- Jarczak, D.; Nierhaus, A. Cytokine Storm-Definition, Causes, and Implications. Int. J. Mol. Sci. 2022, 23, 11740. [Google Scholar] [CrossRef]
- Niemiec, S.M.; Hilton, S.A.; Wallbank, A.; Azeltine, M.; Louiselle, A.E.; Elajaili, H.; Allawzi, A.; Xu, J.; Mattson, C.; Dewberry, L.C.; et al. Cerium oxide nanoparticle delivery of microRNA-146a for local treatment of acute lung injury. Nanomedicine 2021, 34, 102388. [Google Scholar] [CrossRef]
- Niemiec, S.M.; Hilton, S.A.; Wallbank, A.; Louiselle, A.E.; Elajaili, H.; Hu, J.; Liechty, K.W. Lung function improves after delayed treatment with CNP-miR146a following acute lung injury. Nanomedicine 2022, 40, 102498. [Google Scholar] [CrossRef]
- Wallbank, A.M.; Vaughn, A.E.; Niemiec, S.; Bilodeaux, J.; Lehmann, T.; Knudsen, L.; Smith, B.J. CNP-miR146a improves outcomes in a two-hit acute- and ventilator-induced lung injury model. Nanomedicine 2023, 50, 102679. [Google Scholar] [CrossRef]
- Heckert, E.G.; Karakoti, A.S.; Seal, S.; Self, W.T. The role of cerium redox state in the SOD mimetic activity of nanoceria. Biomaterials 2008, 29, 2705–2709. [Google Scholar] [CrossRef]
- Kolanthai, E.; Fu, Y.; Kumar, U.; Babu, B.; Venkatesan, A.K.; Liechty, K.W.; Seal, S. Nanoparticle mediated RNA delivery for wound healing. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2022, 14, e1741. [Google Scholar]
- Li, L.; Chen, X.P.; Li, Y.J. MicroRNA-146a and human disease1. Scand. J. Immunol. 2010, 71, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sig. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Rubinstein, E.; Kollef, M.H.; Nathwani, D. Pneumonia caused by methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2008, 46 (Suppl. S5), S378–S385. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Brunsvold, M.E.; Reddy, R.C.; Hyzy, R.C. Community-acquired methicillin-resistant Staphylococcus aureus pneumonia and ARDS: 1-year follow-up. Chest 2009, 136, 1407–1412. [Google Scholar] [CrossRef]
- Hayashida, A.; Bartlett, A.H.; Foster, T.J.; Park, P.W. Staphylococcus aureus beta-toxin induces lung injury through syndecan-1. Am. J. Pathol. 2009, 174, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Zemans, R.L.; Matthay, M.A. What drives neutrophils to the alveoli in ARDS? Thorax 2017, 72, 1–3. [Google Scholar] [CrossRef]
- Neal, C.J.; Fox, C.R.; Sakthivel, T.S.; Kumar, U.; Fu, Y.; Drake, C.; Seal, S. Metal-Mediated Nanoscale Cerium Oxide Inactivates Human Coronavirus and Rhinovirus by Surface Disruption. ACS Nano 2021, 15, 14544–14556. [Google Scholar] [CrossRef]
- Neal, C.J.; Sakthivel, T.S.; Fu, Y.; Seal, S. Aging of nanoscale cerium oxide in a peroxide environment: Its influence on the redox, surface, and dispersion character. J. Phys. Chem. 2021, 125, 27323–27334. [Google Scholar] [CrossRef]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; Chambers, R.C. The mercurial nature of neutrophils: Still an enigma in ARDS? Am. J. Physiol. Lung. Cell. Mol. Physiol. 2014, 306, L217–L230. [Google Scholar] [CrossRef] [PubMed]
- Crimi, E.; Slutsky, A.S. Inflammation and the acute respiratory distress syndrome. Best Pract. Res. Clin. Anaesthesiol. 2004, 18, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Sul, C.; Lewis, C.; Dee, N.; Burns, N.; Oshima, K.; Schmidt, E.; Nozik, E. Release of extracellular superoxide dismutase into alveolar fluid protects against acute lung injury and inflammation in Staphylococcus aureus pneumonia. Am. J. Physiol. Lung. Cell Mol. Physiol. 2023, 324, L445–L455. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaughn, A.E.; Lehmann, T.; Sul, C.; Wallbank, A.M.; Lyttle, B.D.; Bardill, J.; Burns, N.; Apte, A.; Nozik, E.S.; Smith, B.; et al. CNP-miR146a Decreases Inflammation in Murine Acute Infectious Lung Injury. Pharmaceutics 2023, 15, 2210. https://doi.org/10.3390/pharmaceutics15092210
Vaughn AE, Lehmann T, Sul C, Wallbank AM, Lyttle BD, Bardill J, Burns N, Apte A, Nozik ES, Smith B, et al. CNP-miR146a Decreases Inflammation in Murine Acute Infectious Lung Injury. Pharmaceutics. 2023; 15(9):2210. https://doi.org/10.3390/pharmaceutics15092210
Chicago/Turabian StyleVaughn, Alyssa E., Tanner Lehmann, Christina Sul, Alison M. Wallbank, Bailey D. Lyttle, James Bardill, Nana Burns, Anisha Apte, Eva S. Nozik, Bradford Smith, and et al. 2023. "CNP-miR146a Decreases Inflammation in Murine Acute Infectious Lung Injury" Pharmaceutics 15, no. 9: 2210. https://doi.org/10.3390/pharmaceutics15092210