
Targeting DNA Damage Repair and Immune Checkpoint Proteins for Optimizing the Treatment of Endometrial Cancer


Abstract
:1. Introduction
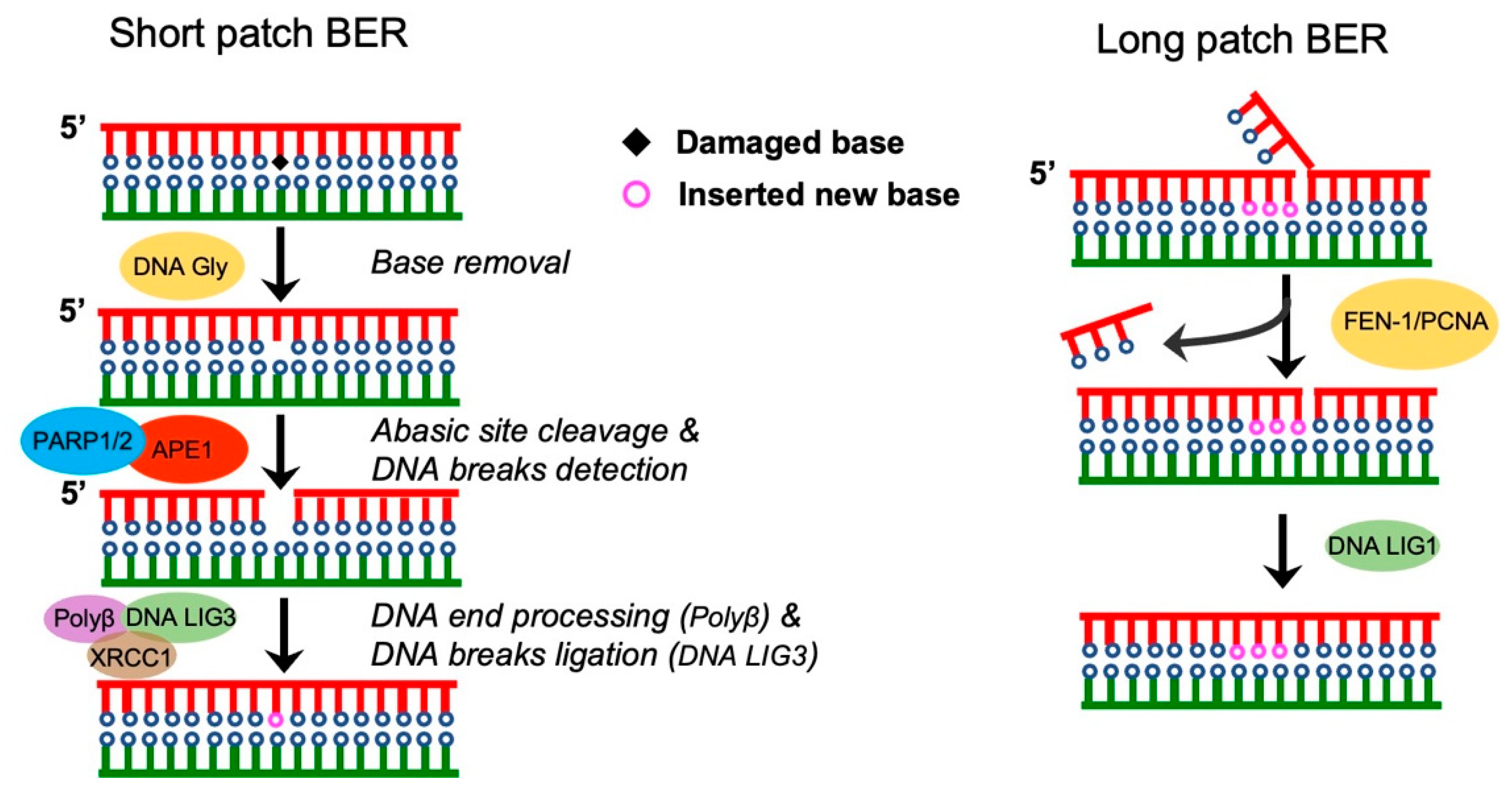
2. Single-Strand Break Repair
Base Excision Repair (BER)
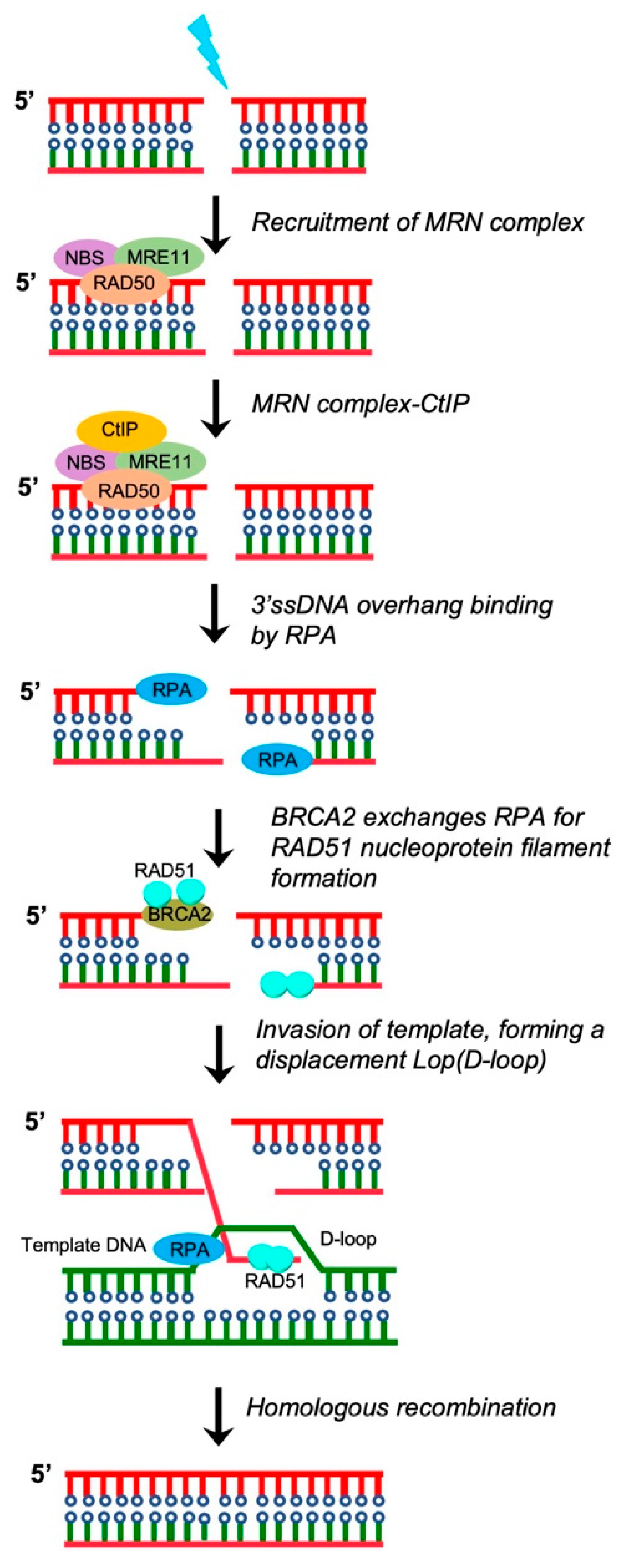
3. DSB Repair
3.1. Homologous Recombination (HR) Repair
3.2. Classical Non-Homologous End Joining Repair (c-NHEJ)
3.3. Alternative Non-Homologous End Joining (A-NHEJ)
4. Single-Strand Annealing (SSA)
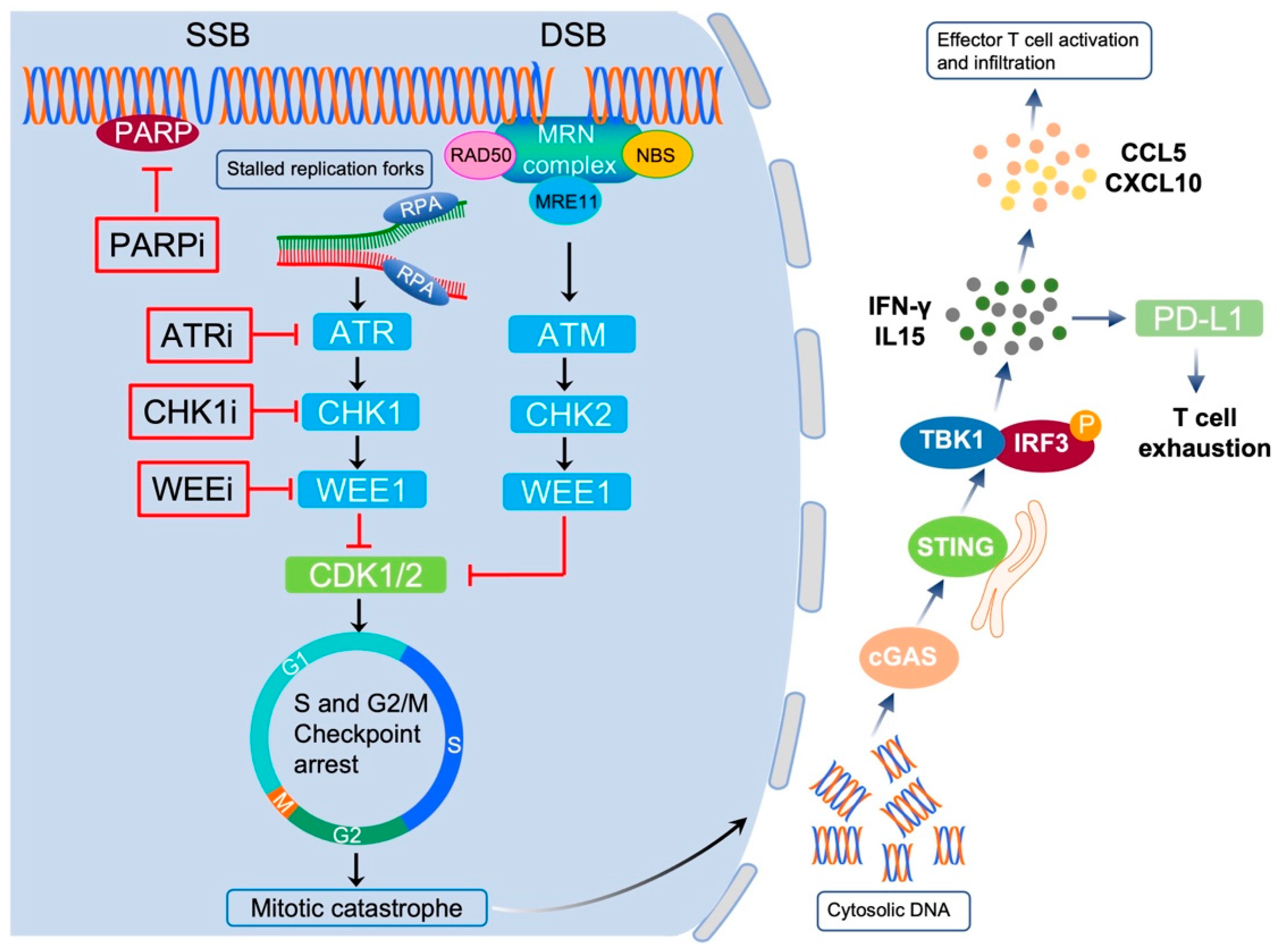
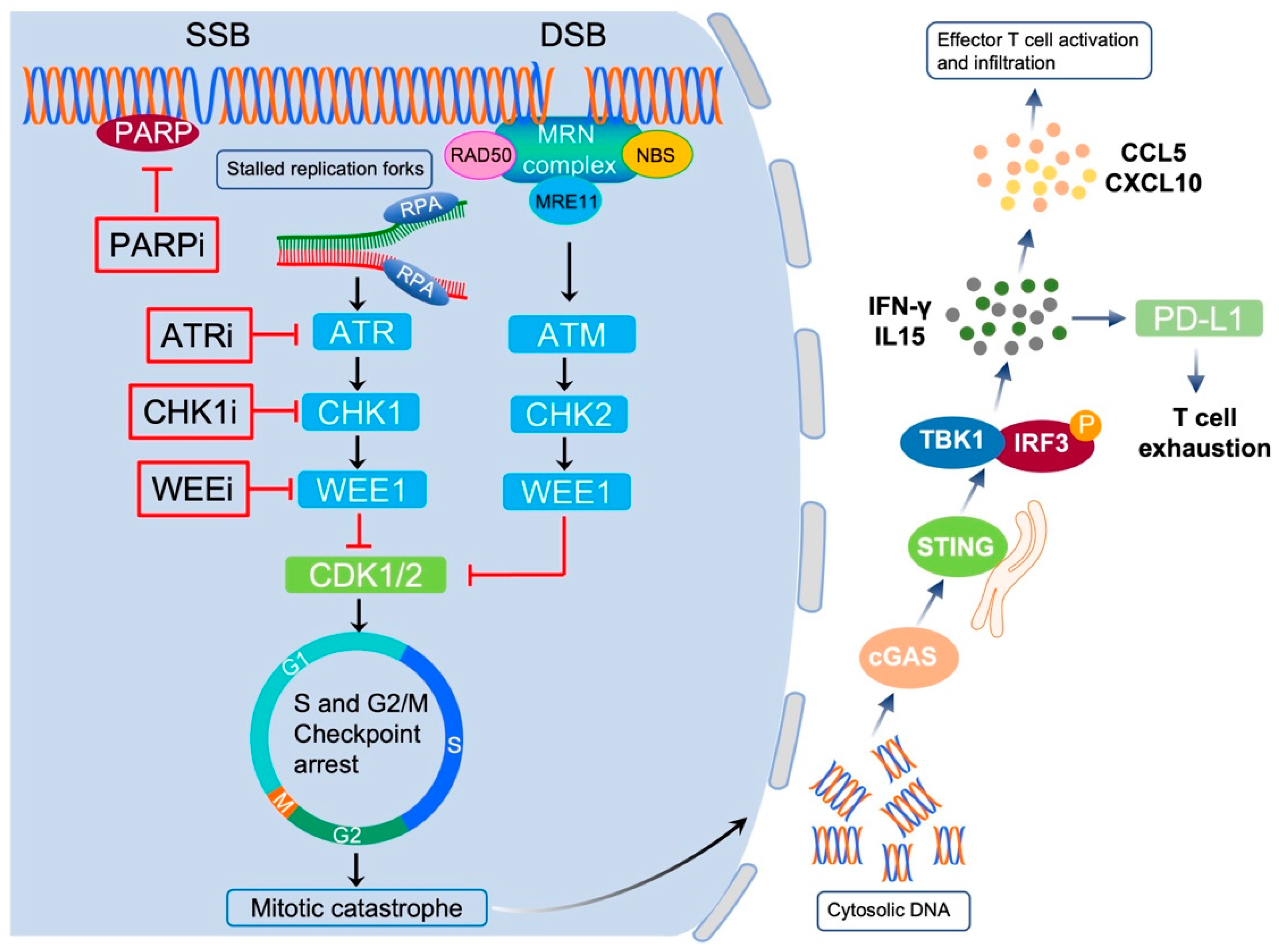
5. Targeting DNA Damage Repair Proteins in EC
5.1. Targeting PARP in EC
5.2. Targeting ATR-CHK1 Signaling in EC
5.3. Targeting WEE1 in EC
5.4. Ongoing Clinical Trials
6. DDR Correlates with Cancer Cell Immunogenicity
7. Exploiting the Interplay between DDR and Immunity for Cancer Therapy
7.1. Pembrolizumab
7.2. Nivolumab
7.3. Dostarlimab
7.4. Durvalumab
7.5. Other Anti-PD-L1 Antibodies
7.6. Immunotherapy in Combination with Chemotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune Checkpoint Inhibitor | Patient Number | Response Biomarker | Phase | Status | Reference |
|---|---|---|---|---|---|
| Pembrolizumab | 75 | POLE-mutation and MSI | Completed | Completed | [98] |
| Pembrolizumab | 47 | MSI or MMR-D | Phase I | Recruiting | [99] |
| Pembrolizumab | 25 | MSI-high | Phase II | Active, not yet recruiting | [101] |
| pembrolizumab | 24 | MMR-D | Phase II | Active, not yet recruiting | [102] |
| Nivolumab | 2 | POLE and MSH6 mutation | unknown | Unknown | [103] |
| Nivolumab | 2 | MSI-high | Phase II | Unknown | [104] |
| Nivolumab | 13 | MMR-D | Phase II | Active, not yet recruiting | [105] |
| Dostarlimab | 104 | MMR-D | Phase I | Recruiting | [106] |
| Dostarlimab | 75 | MMR-D | Phase I | Recruiting | [107] |
| Durvalumab | 71 | MMR-D | Completed | Completed | [109] |
| Atezolizumab | 15 | MMR-D | Completed | Completed | [111] |
| Avelumab | 33 | MMR-D | Phase II | Active, not yet recruiting | [112] |
8. Immunotherapy in Combination with PARP Inhibitors
9. Conclusions
10. Future Directions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial cancer. Lancet 2022, 399, 1412–1428. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; MacKay, H.; Ray-Coquard, I.; Levine, D.A.; Westin, S.N.; Aoki, D.; Oaknin, A. Endometrial cancer. Nat. Rev. Dis. Primers 2021, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Matias-Guiu, X.; Prat, J. Molecular pathology of endometrial carcinoma. Histopathology 2013, 62, 111–123. [Google Scholar] [CrossRef]
- Secord, A.A.; Havrilesky, L.J.; Bae-Jump, V.; Chin, J.; Calingaert, B.; Bland, A.; Rutledge, T.; Berchuck, A.; Clarkepearson, D.; Gehrig, P. The role of multi-modality adjuvant chemotherapy and radiation in women with advanced stage endometrial cancer. Gynecol. Oncol. 2007, 107, 285–291. [Google Scholar]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Daikoku, T.; Hirota, Y.; Tranguch, S.; Joshi, A.R.; DeMayo, F.J.; Lydon, J.P.; Ellenson, L.H.; Dey, S.K. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008, 68, 5619–5627. [Google Scholar] [CrossRef]
- Urick, M.E.; Rudd, M.L.; Godwin, A.K.; Sgroi, D.; Merino, M.; Bell, D.W. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011, 71, 4061–4067. [Google Scholar] [CrossRef]
- Rudd, M.L.; Price, J.C.; Fogoros, S.; Godwin, A.K.; Sgroi, D.C.; Merino, M.J.; Bell, D.W. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin. Cancer Res. 2011, 17, 1331–1340. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.C.; Senz, J.; Tone, A.A.; Yang, W.; Prentice, L.M.; Tse, K.; Zeng, T.; et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Gartside, M.; Powell, M.A.; Wellens, C.L.; Gao, F.; Mutch, D.G.; Goodfellow, P.J.; Pollock, P.M. FGFR2 point mutations in 466 endometrioid endometrial tumors: Relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS ONE 2012, 7, e30801. [Google Scholar] [CrossRef]
- Zighelboim, I.; Goodfellow, P.J.; Gao, F.; Gibb, R.K.; Powell, M.A.; Rader, J.S.; Mutch, D.G. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J. Clin. Oncol. 2007, 25, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Wu, R.-C.; Guan, B.; Wu, G.; Zhang, J.; Wang, Y.; Song, L.; Yuan, X.; Wei, L.; Roden, R.B.; et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J. Natl. Cancer Inst. 2012, 104, 1503–1513. [Google Scholar] [CrossRef]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; O’Neil, N.J.; Price, J.C.; Zhang, S.; England, B.M.; Godwin, A.K.; et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310–1315. [Google Scholar] [CrossRef]
- Urick, M.E.; Bell, D.W. In vitro effects of FBXW7 mutation in serous endometrial cancer: Increased levels of potentially druggable proteins and sensitivity to SI-2 and dinaciclib. Mol. Carcinog. 2018, 57, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Urick, M.E.; Bell, D.W. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 2019, 19, 510–521. [Google Scholar] [CrossRef]
- Hong, B.; Le Gallo, M.; Bell, D.W. The mutational landscape of endometrial cancer. Curr. Opin. Genet. Dev. 2015, 30, 25–31. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Bonazzi, V.F.; Kondrashova, O.; Smith, D.; Nones, K.; Sengal, A.T.; Ju, R.; Packer, L.M.; Koufariotis, L.T.; Kazakoff, S.H.; Davidson, A.L.; et al. Patient-derived xenograft models capture genomic heterogeneity in endometrial cancer. Genome Med. 2022, 14, 3. [Google Scholar] [CrossRef]
- Miyasaka, A.; Oda, K.; Ikeda, Y.; Wada-Hiraike, O.; Kashiyama, T.; Enomoto, A.; Hosoya, N.; Koso, T.; Fukuda, T.; Inaba, K.; et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer 2014, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.M.; Paik, D.Y.; Rosales, M.A.; Yep, B.; Cheng, D.; Witte, O.N.; Kayadibi, H.; Ryan, C.M.; Jung, M.E.; Faull, K.; et al. Low levels of circulating estrogen sensitize PTEN-null endometrial tumors to PARP inhibition in vivo. Mol. Cancer Ther. 2013, 12, 2917–2928. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, M.M.; Auguste, A.; van Wijk, L.M.; Schouten, P.C.; Meijers, M.; ter Haar, N.T.; Smit, V.T.; Nout, R.A.; Glaire, M.A.; Church, D.N.; et al. Frequent Homologous Recombination Deficiency in High-grade Endometrial Carcinomas. Clin. Cancer Res. 2019, 25, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef]
- Lin, D.I.; Fine, A.; Danziger, N.A.; Huang, R.S.; Mata, D.A.; Decker, B.; Killian, J.K.; Ramkissoon, S.H.; Lechpammer, M.; Janovitz, T.; et al. Molecular analysis of endometrial serous carcinoma reveals distinct clinicopathologic and genomic subgroups. Gynecol. Oncol. 2022, 164, 558–565. [Google Scholar] [CrossRef]
- Mukherjee, A.; Patterson, A.L.; George, J.W.; Carpenter, T.J.; Madaj, Z.B.; Hostetter, G.; Risinger, J.I.; Teixeira, J.M. Nuclear PTEN Localization Contributes to DNA Damage Response in Endometrial Adenocarcinoma and Could Have a Diagnostic Benefit for Therapeutic Management of the Disease. Mol. Cancer Ther. 2018, 17, 1995–2003. [Google Scholar] [CrossRef]
- Takeuchi, M.; Tanikawa, M.; Nagasaka, K.; Oda, K.; Kawata, Y.; Oki, S.; Agapiti, C.; Sone, K.; Miyagawa, Y.; Hiraike, H.; et al. Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells. Cancers 2019, 11, 1913. [Google Scholar] [CrossRef]
- Lieber, M.R.; Karanjawala, Z.E. Ageing, repetitive genomes and DNA damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 69–75. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA single-strand break repair and human genetic disease. Trends Cell Biol 2022, 32, 733–745. [Google Scholar] [CrossRef]
- Lebedeva, N.A.; Rechkunova, N.I.; Endutkin, A.V.; Lavrik, O.I. Apurinic/Apyrimidinic Endonuclease 1 and Tyrosyl-DNA Phosphodiesterase 1 Prevent Suicidal Covalent DNA-Protein Crosslink at Apurinic/Apyrimidinic Site. Front. Cell Dev. Biol. 2020, 8, 617301. [Google Scholar] [CrossRef]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef]
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996, 271, 9573–9578. [Google Scholar] [CrossRef]
- Palles, C.; West, H.D.; Chew, E.; Galavotti, S.; Flensburg, C.; Grolleman, J.E.; Jansen, E.A.; Curley, H.; Chegwidden, L.; Arbe-Barnes, E.H.; et al. Germline MBD4 deficiency causes a multi-tumor predisposition syndrome. Am. J. Hum. Genet. 2022, 109, 953–960. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e886. [Google Scholar] [CrossRef]
- Long, K.; Gu, L.; Li, L.; Zhang, Z.; Li, E.; Zhang, Y.; He, L.; Pan, F.; Guo, Z.; Hu, Z. Small-molecule inhibition of APE1 induces apoptosis, pyroptosis, and necroptosis in non-small cell lung cancer. Cell Death Dis. 2021, 12, 503. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. 2014, 26, 243–249. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- Gordhandas, S.; Rios-Doria, E.; Cadoo, K.A.; Catchings, A.; Maio, A.; Kemel, Y.; Sheehan, M.; Ranganathan, M.; Green, D.; Aryamvally, A.; et al. Comprehensive analysis of germline drivers in endometrial cancer. J. Natl. Cancer Inst. 2023, 115, 560–569. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Watanabe, G.; Lieber, M.R. Dynamics of the Artemis and DNA-PKcs Complex in the Repair of Double-Strand Breaks. J. Mol. Biol. 2022, 434, 167858. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Kurosawa, A.; Adachi, N. Mutations in XRCC4 cause primordial dwarfism without causing immunodeficiency. J. Hum. Genet. 2016, 61, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Tomkinson, A.E.; Yasui, A. Translocation of XRCC1 and DNA ligase IIIalpha from centrosomes to chromosomes in response to DNA damage in mitotic human cells. Nucleic Acids Res. 2005, 33, 422–429. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Laan, R.L.; Suresh, A.; Wilson, T.E. DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J. Biol. Chem. 2005, 280, 29030–29037. [Google Scholar] [CrossRef]
- Daley, J.M.; Palmbos, P.L.; Wu, D.; Wilson, T.E. Nonhomologous end joining in yeast. Annu. Rev. Genet. 2005, 39, 431–451. [Google Scholar] [CrossRef]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Manthey, G.M.; Bailis, A.M. RAD59 is required for efficient repair of simultaneous double-strand breaks resulting in translocations in Saccharomyces cerevisiae. DNA Repair 2008, 7, 788–800. [Google Scholar] [CrossRef]
- Dasari, S.K.; Joseph, R.; Umamaheswaran, S.; Mangala, L.S.; Bayraktar, E.; Rodriguez-Aguayo, C.; Wu, Y.; Nguyen, N.; Powell, R.T.; Sobieski, M.; et al. Combination of EphA2- and Wee1-Targeted Therapies in Endometrial Cancer. Int. J. Mol. Sci. 2023, 24, 3915. [Google Scholar] [CrossRef]
- Teng, P.N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Mhawech-Fauceglia, P.; Wang, D.; Kim, G.; Sharifian, M.; Chen, X.; Liu, Q.; Lin, Y.G.; Liu, S.; Pejovic, T. Expression of DNA repair proteins in endometrial cancer preicts disease outcome. Gynecol. Oncol. 2014, 132, 593–598. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Philip, C.A.; Laskov, I.; Beauchamp, M.C.; Marques, M.; Amin, O.; Bitharas, J.; Kessous, R.; Kogan, L.; Baloch, T.; Gotlieb, W.H.; et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 2017, 17, 638. [Google Scholar] [CrossRef]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2018, 37, 341–351. [Google Scholar] [CrossRef]
- Gockley, A.A.; Kolin, D.L.; Awtrey, C.S.; Lindeman, N.I.; Matulonis, U.A.; Konstantinopoulos, P.A. Durable response in a woman with recurrent low-grade endometrioid endometrial cancer and a germline BRCA2 mutation treated with a PARP inhibitor. Gynecol. Oncol. 2018, 150, 219–226. [Google Scholar] [CrossRef]
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.C.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 Recovery as a Resistance Mechanism After Exceptional Response to Poly (ADP-ribose) Polymerase Inhibition. J. Clin. Oncol. 2017, 35, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Yang, L.; Li, F.Y.; Zhang, F.; Ding, L.L.; Yang, J.; Lu, J.; Wang, N.N.; Wang, Y. Research Progress of PARP Inhibitor Monotherapy and Combination Therapy for Endometrial Cancer. Curr. Drug Targets 2022, 23, 145–155. [Google Scholar] [CrossRef]
- Westin, S.N.; Labrie, M.; Litton, J.K.; Blucher, A.; Fang, Y.; Vellano, C.P.; Marszalek, J.R.; Feng, N.; Ma, X.; Creason, A.; et al. Phase Ib Dose Expansion and Translational Analyses of Olaparib in Combination with Capivasertib in Recurrent Endometrial, Triple-Negative Breast, and Ovarian Cancer. Clin. Cancer Res. 2021, 27, 6354–6365. [Google Scholar] [CrossRef]
- Yadav, G.; Roque, D.M.; Bellone, S.; Manavella, D.D.; Hartwich, T.M.; Zipponi, M.; Harold, J.; Tymon-Rosario, J.; Mutlu, L.; Altwerger, G.; et al. Synergistic activity of neratinib in combination with olaparib in uterine serous carcinoma overexpressing HER2/neu. Gynecol. Oncol. 2022, 166, 351–357. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, P.; Wang, L.; Chen, S.; Basappa, B.; Zhu, T.; Lobie, P.E.; Pandey, V. Inhibition of BAD-Ser99 phosphorylation synergizes with PARP inhibition to ablate PTEN-deficient endometrial carcinoma. Cell Death Dis. 2022, 13, 558. [Google Scholar] [CrossRef]
- Giovannini, S.; Weller, M.C.; Hanzlikova, H.; Shiota, T.; Takeda, S.; Jiricny, J. ATAD5 deficiency alters DNA damage metabolism and sensitizes cells to PARP inhibition. Nucleic Acids Res. 2020, 48, 4928–4939. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef]
- Manic, G.; Sistigu, A.; Corradi, F.; Musella, M.; De Maria, R.; Vitale, I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin. Cancer Biol. 2018, 53, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting replication stress in cancer therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef]
- Nakayama, K.; Rahman, M.T.; Rahman, M.; Nakamura, K.; Ishikawa, M.; Katagiri, H.; Sato, E.; Ishibashi, T.; Iida, K.; Ishikawa, N.; et al. CCNE1 amplification is associated with aggressive potential in endometrioid endometrial carcinomas. Int. J. Oncol. 2016, 48, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; George, E.; Kinose, Y.; Kim, H.; Shah, J.B.; Peake, J.D.; Ferman, B.; Medvedev, S.; Murtha, T.; Barger, C.J.; et al. CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models. Cell Rep. Med. 2021, 2, 100394. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Lindqvist, A.; Rodriguez-Bravo, V.; Medema, R.H. The decision to enter mitosis: Feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009, 185, 193–202. [Google Scholar] [CrossRef]
- Meng, X.; Bi, J.; Li, Y.; Yang, S.; Zhang, Y.; Li, M.; Liu, H.; Li, Y.; Mcdonald, M.E.; Thiel, K.W.; et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers 2018, 10, 149. [Google Scholar] [CrossRef]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef]
- Reislander, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Marin-Jimenez, J.A.; Garcia-Mulero, S.; Matias-Guiu, X.; Piulats, J.M. Facts and Hopes in Immunotherapy of Endometrial Cancer. Clin. Cancer Res. 2022, 28, 4849–4860. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Q.; Zhu, Y.; Zhang, S.; Peng, Q.; Strickland, A.L.; Zheng, W.; Zhou, F. PD-L1 Expression in Endometrial Serous Carcinoma and Its Prognostic Significance. Cancer Manag. Res. 2021, 13, 9157–9165. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.-J.; Berton-Rigaud, D.; Elez, E.; Pishvaian, M.J.; Rugo, H.S.; Puzanov, I.; Mehnert, J.M.; Aung, K.L.; Lopez, J.; et al. Safety and Antitumor Activity of Pembrolizumab in Advanced Programmed Death Ligand 1-Positive Endometrial Cancer: Results From the KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 2535–2541. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Maio, M.; Ascierto, P.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.; Bariani, G.; Acosta, A.D.J.; Doi, T.; Longo, F.; et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: Updated analysis from the phase II KEYNOTE-158 study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Casak, S.J.; Marcus, L.; Fashoyin-Aje, L.; Mushti, S.L.; Cheng, J.; Shen, Y.L.; Pierce, W.F.; Her, L.; Goldberg, K.B.; Theoret, M.R.; et al. FDA Approval Summary: Pembrolizumab for the First-line Treatment of Patients with MSI-H/dMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021, 27, 4680–4684. [Google Scholar] [CrossRef]
- Bellone, S.; Roque, D.M.; Siegel, E.R.; Buza, N.; Hui, P.; Bonazzoli, E.; Guglielmi, A.; Zammataro, L.; Nagarkatti, N.; Zaidi, S.; et al. A phase 2 evaluation of pembrolizumab for recurrent Lynch-like versus sporadic endometrial cancers with microsatellite instability. Cancer 2022, 128, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.D.; Michaels, T.; Bellone, S.; Hartwich, T.M.; Bonazzoli, E.; Iwasaki, A.; Song, E.; Santin, A.D. Distinct Mechanisms of Mismatch-Repair Deficiency Delineate Two Modes of Response to Anti-PD-1 Immunotherapy in Endometrial Carcinoma. Cancer Discov. 2023, 13, 312–331. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Bellone, S.; Buza, N.; Choi, J.; Schwartz, P.E.; Schlessinger, J.; Lifton, R.P. Regression of Chemotherapy-Resistant Polymerase epsilon (POLE) Ultra-Mutated and MSH6 Hyper-Mutated Endometrial Tumors with Nivolumab. Clin. Cancer Res. 2016, 22, 5682–5687. [Google Scholar] [CrossRef]
- Tamura, K.; Hasegawa, K.; Katsumata, N.; Matsumoto, K.; Mukai, H.; Takahashi, S.; Nomura, H.; Minami, H. Efficacy and safety of nivolumab in Japanese patients with uterine cervical cancer, uterine corpus cancer, or soft tissue sarcoma: Multicenter, open-label phase 2 trial. Cancer Sci. 2019, 110, 2894–2904. [Google Scholar] [CrossRef]
- Azad, N.S.; Gray, R.J.; Overman, M.J.; Schoenfeld, J.D.; Mitchell, E.P.; Zwiebel, J.A.; Sharon, E.; Streicher, H.; Li, S.; McShane, L.M.; et al. Nivolumab Is Effective in Mismatch Repair-Deficient Noncolorectal Cancers: Results From Arm Z1D-A Subprotocol of the NCI-MATCH (EAY131) Study. J. Clin. Oncol. 2020, 38, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients with Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Brown, J.; Mathews, C.; Press, J.; Sabatier, R.; O’Malley, D.M.; Samouelian, V.; Boni, V.; et al. Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) or proficient/stable (MMRp/MSS) endometrial cancer: Interim results from GARNET-a phase I, single-arm study. J. Immunother. Cancer 2022, 10, e003777. [Google Scholar] [PubMed]
- Markham, A. Dostarlimab: First Approval. Drugs 2021, 81, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Antill, Y.; Kok, P.-S.; Robledo, K.; Yip, S.; Cummins, M.; Smith, D.; Spurdle, A.; Barnes, E.; Lee, Y.C.; Friedlander, M.; et al. Clinical activity of durvalumab for patients with advanced mismatch repair-deficient and repair-proficient endometrial cancer. A nonrandomized phase 2 clinical trial. J. Immunother. Cancer 2021, 9, e002255. [Google Scholar] [CrossRef]
- Post, C.; Westermann, A.; Boere, I.; Witteveen, P.; Ottevanger, P.; Sonke, G.; Lalisang, R.; Putter, H.; Kranenbarg, E.M.-K.; Braak, J.; et al. Efficacy and safety of durvalumab with olaparib in metastatic or recurrent endometrial cancer (phase II DOMEC trial). Gynecol. Oncol. 2022, 165, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Gordon, M.; Veneris, J.; Braiteh, F.; Balmanoukian, A.; Eder, J.P.; Oaknin, A.; Hamilton, E.; Wang, Y.; Sarkar, I.; et al. Safety, clinical activity and biomarker assessments of atezolizumab from a Phase I study in advanced/recurrent ovarian and uterine cancers. Gynecol. Oncol. 2019, 154, 314–322. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Luo, W.; Liu, J.F.; Gulhan, D.C.; Krasner, C.; Ishizuka, J.J.; Gockley, A.A.; Buss, M.; Growdon, W.B.; Crowe, H.; et al. Phase II Study of Avelumab in Patients with Mismatch Repair Deficient and Mismatch Repair Proficient Recurrent/Persistent Endometrial Cancer. J. Clin. Oncol. 2019, 37, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Barber, E.L.; Chen, S.; Pineda, M.J.; Robertson, S.E.; Hill, E.K.; Teoh, D.; Schilder, J.; O’Shea, K.L.; Kocherginsky, M.; Zhang, B.; et al. Clinical and Biological Activity of Chemoimmunotherapy in Advanced Endometrial Adenocarcinoma: A Phase II Trial of the Big Ten Cancer Research Consortium. Cancer Res. Commun. 2022, 2, 1293–1303. [Google Scholar] [CrossRef]
- Eskander, R.N.; Sill, M.W.; Beffa, L.; Moore, R.G.; Hope, J.M.; Musa, F.B.; Mannel, R.; Shahin, M.S.; Cantuaria, G.H.; Girda, E.; et al. Pembrolizumab plus Chemotherapy in Advanced Endometrial Cancer. N. Engl. J. Med. 2023, 388, 2159–2170. [Google Scholar] [CrossRef]
- Goodman, A.M.; Sokol, E.S.; Frampton, G.M.; Lippman, S.M.; Kurzrock, R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol. Res. 2019, 7, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Zighelboim, I.; Schmidt, A.P.; Gao, F.; Thaker, P.H.; Powell, M.A.; Rader, J.S.; Gibb, R.K.; Mutch, D.G.; Goodfellow, P.J. ATR mutation in endometrioid endometrial cancer is associated with poor clinical outcomes. J. Clin. Oncol. 2009, 27, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, W.; Zhang, L.; Sridhara, R.; Spillman, D.; Mathai, J.P.; Scott, B.; Golding, S.J.; Coory, M.; et al. FDA Approval Summary: Pembrolizumab plus Lenvatinib for Endometrial Carcinoma, a Collaborative International Review under Project Orbis. Clin. Cancer Res. 2020, 26, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.; Huang, Y.; Cao, K.; Liu, T.; Shen, H.; Cui, J.; Li, B.; Cai, J.; Gao, F.; et al. Long non-coding RNA ANRIL promotes homologous recombination-mediated DNA repair by maintaining ATR protein stability to enhance cancer resistance. Mol. Cancer 2021, 20, 94. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Gockley, A.A.; Xiong, N.; Krasner, C.; Horowitz, N.; Campos, S.; Wright, A.A.; Liu, J.F.; Shea, M.; Yeku, O.; et al. Evaluation of Treatment with Talazoparib and Avelumab in Patients with Recurrent Mismatch Repair Proficient Endometrial Cancer. JAMA Oncol. 2022, 8, 1317–1322. [Google Scholar] [CrossRef]

| Inhibitor | Combination with | Phase | Status | NCT Number |
|---|---|---|---|---|
| Olaparib | Monotherapy | Phase I | Not yet recruiting | NCT05320757 |
| Olaparib | CYH33 | Phase I | Recruiting | NCT04586335 |
| Niraparib | Monotherapy | Phase II | Recruiting | NCT04716686 |
| Rucaparib | Nivolumab | Phase I and II | Terminated | NCT03572478 |
| Niraparib | TSR-042 | Phase II | Active | NCT03016338 |
| Olaparib | Carboplatin | Phase I | Completed | NCT01237067 |
| Olaparib | Selumetinib | Phase II | Recruiting | NCT05554328 |
| AZD5305 | Paclitaxel or Carboplatin or T-Dxd or Dato-Dxd or Camizestrant | Phase I and II | Recruiting | NCT04644068 |
| Olaparib | AZD2014 or AZD5363 | Phase I and II | Active | NCT02208375 |
| Rucaparib | Bevacizumab | Phase II | Active | NCT03476798 |
| Niraparib | Copanlisib | Phase I | Active | NCT03586661 |
| Niraparib | Dostarlimab | Phase II | Not yet recruiting | NCT05870761 |
| Olaparib or AZD6738 | AZD6738 or Durvalumab | Phase II | Recruiting | NCT03682289 |
| Olaparib | DS-8201a | Phase I | Recruiting | NCT04585958 |
| BBI-355 | Monotherapy | Phase I | Recruiting | NCT05827614 |
| BAY1895344 | Chemotherapy | Phase I | Recruiting | NCT04491942 |
| ART0380 | Monotherapy | Phase II | Not yet recruiting | NCT05798611 |
| AZD1775 | Radiotherapy and chemotherapy | Phase I | Active | NCT03345784 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, X.; Sun, C.; Cheng, J.; Hong, B. Targeting DNA Damage Repair and Immune Checkpoint Proteins for Optimizing the Treatment of Endometrial Cancer. Pharmaceutics 2023, 15, 2241. https://doi.org/10.3390/pharmaceutics15092241
Bian X, Sun C, Cheng J, Hong B. Targeting DNA Damage Repair and Immune Checkpoint Proteins for Optimizing the Treatment of Endometrial Cancer. Pharmaceutics. 2023; 15(9):2241. https://doi.org/10.3390/pharmaceutics15092241
Chicago/Turabian StyleBian, Xing, Chuanbo Sun, Jin Cheng, and Bo Hong. 2023. "Targeting DNA Damage Repair and Immune Checkpoint Proteins for Optimizing the Treatment of Endometrial Cancer" Pharmaceutics 15, no. 9: 2241. https://doi.org/10.3390/pharmaceutics15092241
APA StyleBian, X., Sun, C., Cheng, J., & Hong, B. (2023). Targeting DNA Damage Repair and Immune Checkpoint Proteins for Optimizing the Treatment of Endometrial Cancer. Pharmaceutics, 15(9), 2241. https://doi.org/10.3390/pharmaceutics15092241