
A Species-Specific Anti-Human P2X7 Monoclonal Antibody Reduces Graft-versus-Host Disease in Humanised Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Purification and Conjugation of the Anti-hP2X7 and Isotype Control mAbs
2.3. P2X7 Expression by Immunolabelling and Flow Cytometry
2.4. ATP-Induced YO-PRO-12+ Dye Uptake Assay
2.5. Human Peripheral Blood Mononuclear Cell Isolation
2.6. Human Peripheral Blood Mononuclear Cell In Vitro Culture
2.7. Humanised Mouse Model of Graft-Versus-Host Disease
2.8. Histological Analyses
2.9. Immunophenotyping by Flow Cytometry
2.10. Serum Cytokine Analyses
2.11. Data Presentation and Statistical Analyses
3. Results
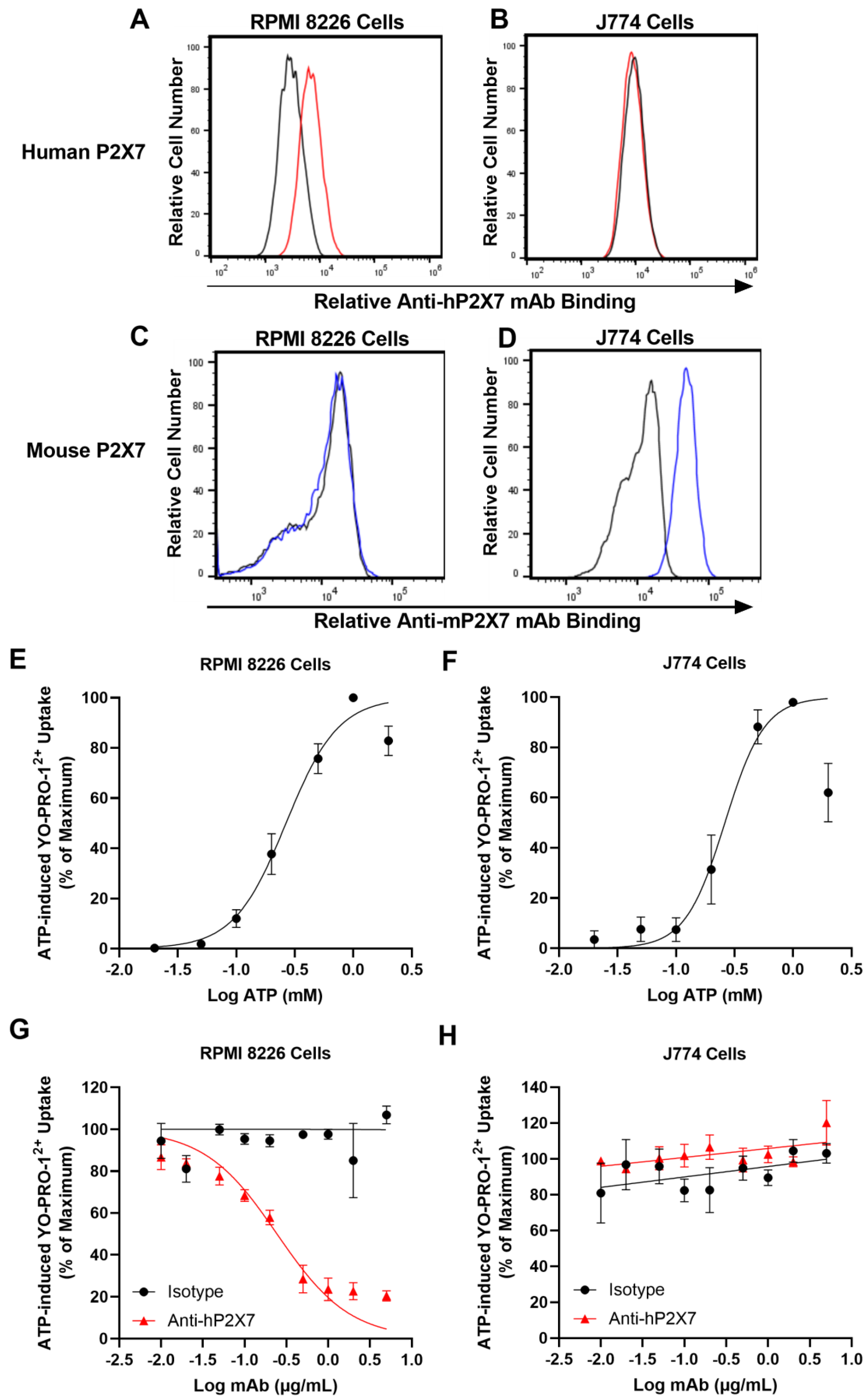
3.1. The Anti-hP2X7 mAb Binds and Blocks Human but Not Mouse P2X7 In Vitro
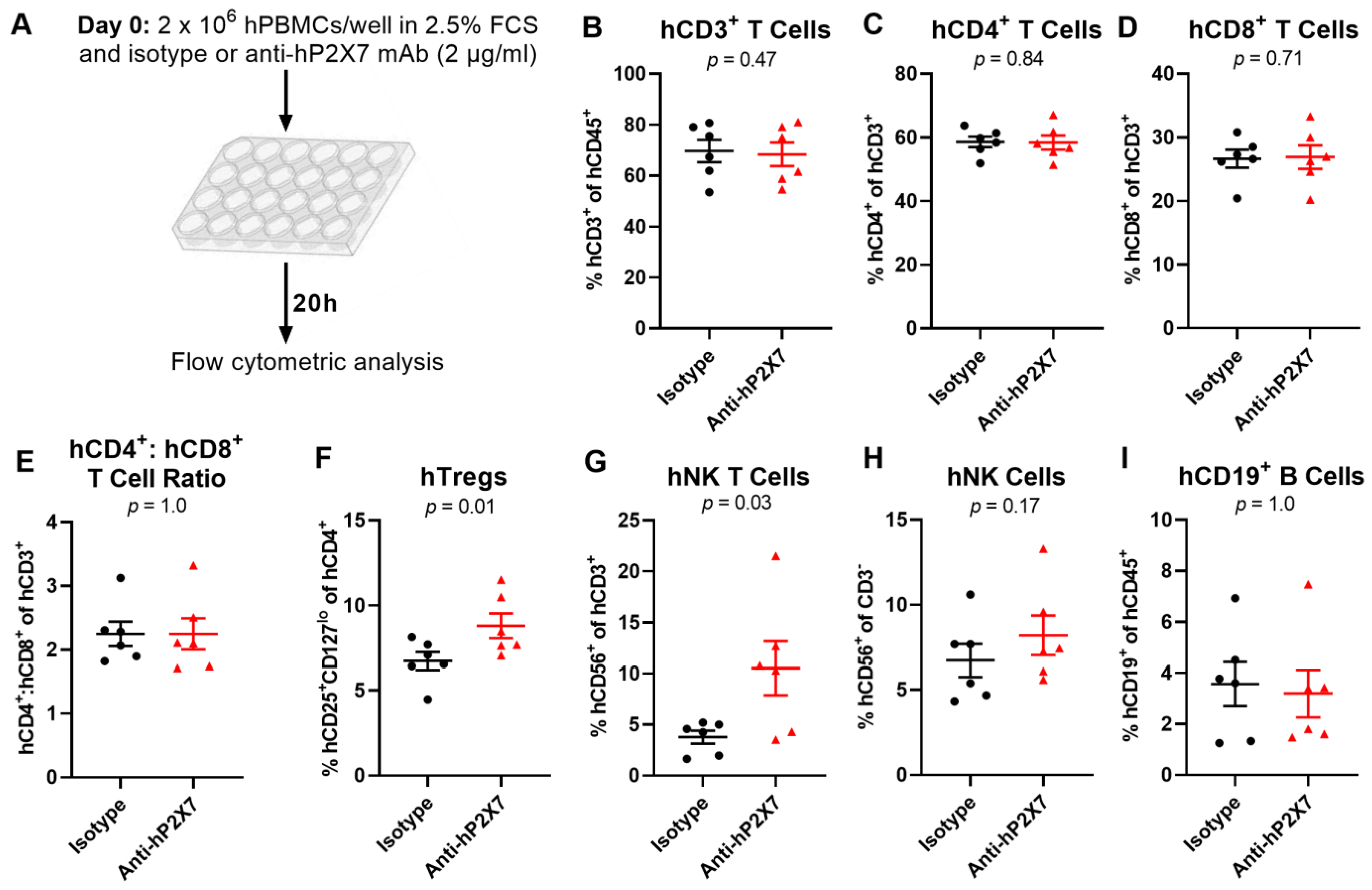
3.2. The Anti-hP2X7 mAb Prevents the Loss of hTregs and hNK T Cells In Vitro
3.3. The Anti-hP2X7 mAb Does Not Affect Early GVHD Development in Humanised Mice
3.4. The Anti-hP2X7 mAb Increases Proportions of Splenic hTregs and hNK Cells at Day 21
3.5. The Anti-hP2X7 mAb Does Not Affect Human Cytokine Concentrations in Sera at Day 21
3.6. The Anti-hP2X7 mAb Reduces Clinical GVHD in Humanised Mice
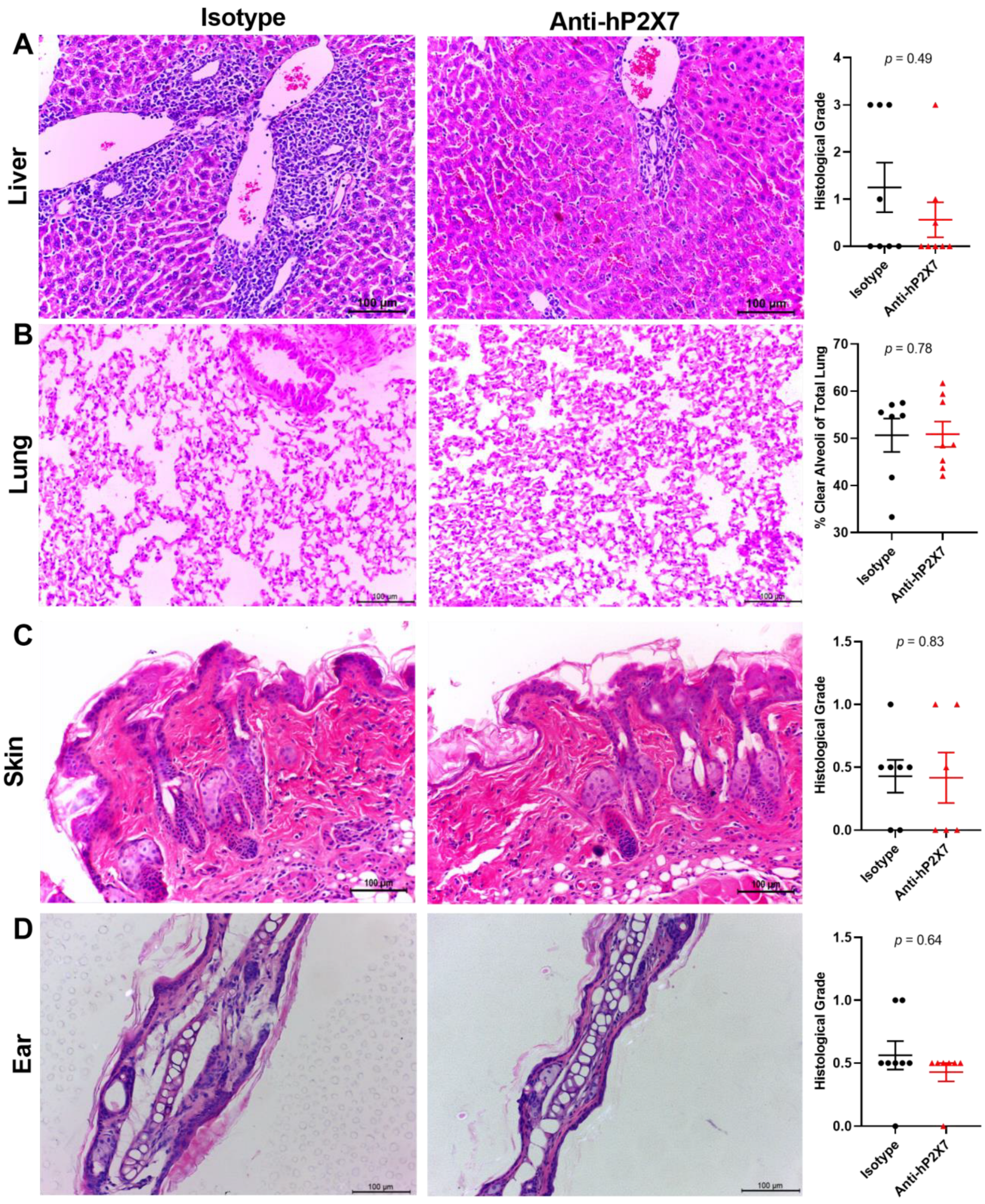
3.7. The Anti-hP2X7 mAb Reduces Histological GVHD in the Liver and Lung at Endpoint
3.8. The Anti-hP2X7 mAb Altered Cell Proportions in the Spleen and Liver of Humanised Mice at Endpoint
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kekre, N.; Antin, J.H. Hematopoietic stem cell transplantation donor sources in the 21st century: Choosing the ideal donor when a perfect match does not exist. Blood 2014, 124, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Holtan, S.G.; Yu, J.; Choe, H.K.; Paranagama, D.; Tang, J.; Naim, A.; Galvin, J.; Joachim Deeg, H. Disease progression, treatments, hospitalization, and clinical outcomes in acute GVHD: A multicenter chart review. Bone Marrow Transplant. 2022, 57, 1581–1585. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Levy, R.; Chao, N.J. Pathophysiologic mechanisms of acute graft-vs.-host disease. Biol. Blood Marrow Transplant. 1999, 5, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.R.; Koyama, M. Cytokines and costimulation in acute graft-versus-host disease. Blood 2020, 136, 418–428. [Google Scholar] [CrossRef]
- Garnett, C.; Apperley, J.F.; Pavlů, J. Treatment and management of graft-versus-host disease: Improving response and survival. Ther. Adv. Hematol. 2013, 4, 366–378. [Google Scholar] [CrossRef]
- Gu, B.J.; Zhang, W.Y.; Bendall, L.J.; Chessell, I.P.; Buell, G.N.; Wiley, J.S. Expression of P2X(7) purinoceptors on human lymphocytes and monocytes: Evidence for nonfunctional P2X(7) receptors. Am. J. Physiol. Cell Physiol. 2000, 279, C1189–C1197. [Google Scholar] [CrossRef]
- Ferrari, D.; La Sala, A.; Chiozzi, P.; Morelli, A.; Falzoni, S.; Girolomoni, G.; Idzko, M.; Dichmann, S.; Norgauer, J.; Di Virgilio, F. The P2 purinergic receptors of human dendritic cells: Identification and coupling to cytokine release. FASEB J. 2000, 14, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, F.D.; Sarti, A.C.; Coutinho-Silva, R. Purinergic signaling, DAMPs, and inflammation. Am. J. Physiol. Cell Physiol. 2020, 318, C832–C835. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 receptor in infection and inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef]
- Sluyter, R.; Cuthbertson, P.; Elhage, A.; Sligar, C.; Watson, D. Purinergic signalling in graft-versus-host disease. Curr. Opin. Pharmacol. 2023, 68, 102346. [Google Scholar] [CrossRef]
- Cuthbertson, P.; Geraghty, N.J.; Adhikary, S.R.; Bird, K.M.; Fuller, S.J.; Watson, D.; Sluyter, R. Purinergic signalling in allogeneic haematopoietic stem cell transplantation and graft-versus-host disease. Int. J. Mol. Sci. 2021, 22, 8343. [Google Scholar] [CrossRef] [PubMed]
- Hubert, S.; Rissiek, B.; Klages, K.; Huehn, J.; Sparwasser, T.; Haag, F.; Koch-Nolte, F.; Boyer, O.; Seman, M.; Adriouch, S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J. Exp. Med. 2010, 207, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Story, M.E.; Hao, X.; Sumpter, T.L.; Mathers, A.R. P2X7 receptor expression and signaling on dendritic cells and CD4(+) T cells is not required but can enhance Th17 differentiation. Front. Cell Dev. Biol. 2022, 10, 687659. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Gaugler, B.; Lamarthee, B.; Mohty, M. Translational opportunities for targeting the Th17 axis in acute graft-vs.-host disease. Mucosal Immunol. 2016, 9, 299–308. [Google Scholar] [CrossRef]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Ganesan, J.; Muller, T.; Durr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Juttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef]
- Koehn, B.H.; Saha, A.; McDonald-Hyman, C.; Loschi, M.; Thangavelu, G.; Ma, L.; Zaiken, M.; Dysthe, J.; Krepps, W.; Panthera, J.; et al. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GVHD. Blood 2019, 134, 1670–1682. [Google Scholar] [CrossRef]
- Zhong, X.; Zhu, F.; Qiao, J.; Zhao, K.; Zhu, S.; Zeng, L.; Chen, X.; Xu, K. The impact of P2X7 receptor antagonist, brilliant blue G on graft-versus-host disease in mice after allogeneic hematopoietic stem cell transplantation. Cell. Immunol. 2016, 310, 71–77. [Google Scholar] [CrossRef]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef]
- Geraghty, N.J.; Belfiore, L.; Ly, D.; Adhikary, S.R.; Fuller, S.J.; Varikatt, W.; Sanderson-Smith, M.L.; Sluyter, V.; Alexander, S.I.; Sluyter, R.; et al. The P2X7 receptor antagonist Brilliant Blue G reduces serum human interferon-gamma in a humanized mouse model of graft-versus-host disease. Clin. Exp. Immunol. 2017, 190, 79–95. [Google Scholar] [CrossRef]
- Cuthbertson, P.; Geraghty, N.J.; Adhikary, S.R.; Casolin, S.; Watson, D.; Sluyter, R. P2X7 receptor antagonism increases regulatory T cells and reduces clinical and histological graft-versus-host disease in a humanised mouse model. Clin. Sci. 2021, 135, 495–513. [Google Scholar] [CrossRef]
- Geraghty, N.J.; Watson, D.; Sluyter, R. Long-term treatment with the P2X7 receptor antagonist Brilliant Blue G reduces liver inflammation in a humanized mouse model of graft-versus-host disease. Cell. Immunol. 2019, 336, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Amiya, T.; Nochi, T.; Fujisawa, K.; Haraguchi, T.; Iba, H.; Tsutsui, H.; Sato, S.; Nakajima, S.; Iijima, H.; et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat. Commun. 2012, 3, 1034. [Google Scholar] [CrossRef] [PubMed]
- Buell, G.; Chessell, I.P.; Michel, A.D.; Collo, G.; Salazzo, M.; Herren, S.; Gretener, D.; Grahames, C.; Kaur, R.; Kosco-Vilbois, M.H.; et al. Blockade of human P2X7 receptor function with a monoclonal antibody. Blood 1998, 92, 3521–3528. [Google Scholar] [CrossRef]
- Elhage, A.; Sligar, C.; Cuthbertson, P.; Watson, D.; Sluyter, R. Insights into mechanisms of graft-versus-host disease through humanised mouse models. Biosci. Rep. 2022, 42, BSR20211986. [Google Scholar] [CrossRef] [PubMed]
- Elhage, A.; Turner, R.J.; Cuthbertson, P.; Watson, D.; Sluyter, R. Preparation of the murine anti-human P2X7 receptor monoclonal antibody (clone L4). In The P2X7 Receptor: Methods and Protocols; Nicke, A., Ed.; Springer: New York, NY, USA, 2022; pp. 77–98. [Google Scholar] [CrossRef]
- Cuthbertson, P.; Elhage, A.; Al-Rifai, D.; Sophocleous, R.A.; Turner, R.J.; Aboelela, A.; Majed, H.; Bujaroski, R.S.; Jalilian, I.; Kelso, M.J.; et al. 6-furopyridine hexamethylene amiloride is a non-selective P2X7 receptor antagonist. Biomolecules 2022, 12, 1309. [Google Scholar] [CrossRef]
- Watson, D.; Adhikary, S.R.; Cuthbertson, P.; Geraghty, N.J.; Bird, K.M.; Elhage, A.; Sligar, C.; Sluyter, R. Humanized mouse model to study the P2X7 receptor in graft-versus-host disease. In The P2X7 Receptor: Methods and Protocols; Nicke, A., Ed.; Springer: New York, NY, USA, 2022; pp. 315–340. [Google Scholar] [CrossRef]
- Turner, R.J.; Geraghty, N.J.; Williams, J.G.; Ly, D.; Brungs, D.; Carolan, M.G.; Guy, T.V.; Watson, D.; de Leon, J.F.; Sluyter, R. Comparison of peripheral blood mononuclear cell isolation techniques and the impact of cryopreservation on human lymphocytes expressing CD39 and CD73. Purinergic Signal. 2020, 16, 389–401. [Google Scholar] [CrossRef]
- Cuthbertson, P.; Adhikary, S.R.; Geraghty, N.J.; Guy, T.V.; Hadjiashrafi, A.; Fuller, S.J.; Ly, D.; Watson, D.; Sluyter, R. Increased P2X7 expression in the gastrointestinal tract and skin in a humanised mouse model of graft-versus-host disease. Clin. Sci. 2020, 134, 207–223. [Google Scholar] [CrossRef]
- Farrell, A.W.; Gadeock, S.; Pupovac, A.; Wang, B.; Jalilian, I.; Ranson, M.; Sluyter, R. P2X7 receptor activation induces cell death and CD23 shedding in human RPMI 8226 multiple myeloma cells. Biochim. Biophys. Acta 2010, 1800, 1173–1182. [Google Scholar] [CrossRef]
- Coutinho-Silva, R.; Ojcius, D.M.; Górecki, D.C.; Persechini, P.M.; Bisaggio, R.C.; Mendes, A.N.; Marks, J.; Burnstock, G.; Dunn, P.M. Multiple P2X and P2Y receptor subtypes in mouse J774, spleen and peritoneal macrophages. Biochem. Pharmacol. 2005, 69, 641–655. [Google Scholar] [CrossRef]
- Sligar, C.; Cuthbertson, P.; Miles, N.A.; Adhikary, S.R.; Elhage, A.; Zhang, G.; Alexander, S.I.; Sluyter, R.; Watson, D. Tocilizumab increases regulatory T cells, reduces natural killer cells and delays graft-versus-host disease development in humanized mice treated with post-transplant cyclophosphamide. Immunol. Cell Biol. 2023, 101, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Donnelly-Roberts, D.L.; Namovic, M.T.; Han, P.; Jarvis, M.F. Mammalian P2X7 receptor pharmacology: Comparison of recombinant mouse, rat and human P2X7 receptors. Br. J. Pharmacol. 2009, 157, 1203–1214. [Google Scholar] [CrossRef]
- Adhikary, S.R.; Geraghty, N.J.; Cuthbertson, P.; Sluyter, R.; Watson, D. Altered donor P2X7 activity in human leukocytes correlates with P2RX7 genotype but does not affect the development of graft-versus-host disease in humanised mice. Purinergic Signal. 2019, 15, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Pupovac, A.; Geraghty, N.J.; Watson, D.; Sluyter, R. Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol. Cell Biol. 2015, 93, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, N.J.; Elhage, A.; Cuthbertson, P.; Watson, D.; Sluyter, R. The P2X7 Receptor Antagonist AZ10606120 Does Not Alter Graft-Versus-Host Disease Development and Increases Serum Human Interferon-γ in a Humanized Mouse Model. OBM Transplant. 2022, 6, 166. [Google Scholar] [CrossRef]
- Sluyter, R.; Adriouch, S.; Fuller, S.J.; Nicke, A.; Sophocleous, R.A.; Watson, D. Animal Models for the Investigation of P2X7 Receptors. Int. J. Mol. Sci. 2023, 24, 8225. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Mielke, S.; Ahmadzadeh, M.; Kilical, Y.; Savani, B.N.; Zeilah, J.; Keyvanfar, K.; Montero, A.; Hensel, N.; Kurlander, R.; et al. High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood 2006, 108, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Takahashi, T.; Hiruma, K.; Kanda, Y.; Tanaka, Y.; Ogawa, S.; Chiba, S.; Miura, O.; Sakamaki, H.; Hirai, H. Recovery of Vα24+ NKT cells after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004, 34, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, S.; Zou, Y.; Yan, X.; Wu, H.; Zhou, M.; Sun, Y.C.; Zhang, Y.; Zhu, H.; Xu, K.; et al. NK cell predicts the severity of acute graft-versus-host disease in patients after allogeneic stem cell transplantation using antithymocyte globulin (ATG) in pretreatment scheme. BMC Immunol. 2019, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, P.; Janin, A.; Peffault de Latour, R.; Leboeuf, C.; Desveaux, A.; Keyvanfar, K.; Robin, M.; Clave, E.; Douay, C.; Quinquenel, A.; et al. Th17/Treg ratio in human graft-versus-host disease. Blood 2010, 116, 1165–1171. [Google Scholar] [CrossRef]
- Bayegi, S.N.; Hamidieh, A.A.; Behfar, M.; Saghazadeh, A.; Bozorgmehr, M.; Karamlou, Y.; Shekarabi, M.; Tajik, N.; Delbandi, A.A.; Zavareh, F.T.; et al. T helper 17 and regulatory T-cell profile and graft-versus-host disease after allogeneic hematopoietic stem cell transplantation in pediatric patients with beta-thalassemia. Transpl. Immunol. 2023, 77, 101803. [Google Scholar] [CrossRef]
- Hippen, K.L.; Merkel, S.C.; Schirm, D.K.; Nelson, C.; Tennis, N.C.; Riley, J.L.; June, C.H.; Miller, J.S.; Wagner, J.E.; Blazar, B.R. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant. 2011, 11, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Coman, T.; Rossignol, J.; D’Aveni, M.; Fabiani, B.; Dussiot, M.; Rignault, R.; Babdor, J.; Bouillé, M.; Herbelin, A.; Coté, F.; et al. Human CD4- invariant NKT lymphocytes regulate graft versus host disease. Oncoimmunology 2018, 7, e1470735. [Google Scholar] [CrossRef] [PubMed]
- Gregoire-Gauthier, J.; Fontaine, F.; Benchimol, L.; Nicoletti, S.; Selleri, S.; Dieng, M.M.; Haddad, E. Role of Natural Killer Cells in Intravenous Immunoglobulin–Induced Graft-versus-Host Disease Inhibition in NOD/LtSz-scidIL2rg−/− (NSG) Mice. Biol. Blood Marrow Transplant. 2015, 21, 821–828. [Google Scholar] [CrossRef]
- Delens, L.; Ehx, G.; Somja, J.; Vrancken, L.; Belle, L.; Seidel, L.; Grégoire, C.; Fransolet, G.; Ritacco, C.; Hannon, M.; et al. In vitro Th17-polarized human CD4+ T cells exacerbate xenogeneic graft-versus-host disease. Biol. Blood Marrow Transplant. 2019, 25, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Kaminskiy, Y.; Kuznetsova, V.; Kudriaeva, A.; Zmievskaya, E.; Bulatov, E. Neglected, yet significant role of FOXP1 in T-cell quiescence, differentiation and exhaustion. Front. Immunol. 2022, 13, 971045. [Google Scholar] [CrossRef]
- Liu, Q.; Kim, C.H. Control of tissue-resident invariant NKT cells by vitamin A metabolites and P2X7-mediated cell death. J. Immunol. 2019, 203, 1189–1197. [Google Scholar] [CrossRef]
- Atarashi, K.; Nishimura, J.; Shima, T.; Umesaki, Y.; Yamamoto, M.; Onoue, M.; Yagita, H.; Ishii, N.; Evans, R.; Honda, K.; et al. ATP drives lamina propria TH17 cell differentiation. Nature 2008, 455, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, J.; Li, R.; Zhu, F.; Xu, W.; Zha, G.; He, G.; Cao, H.; Wang, Y.; Yang, J. ATP/P2X7-NLRP3 axis of dendritic cells participates in the regulation of airway inflammation and hyper-responsiveness in asthma by mediating HMGB1 expression and secretion. Exp. Cell Res. 2018, 366, 1–15. [Google Scholar] [CrossRef]
- Kawamura, H.; Aswad, F.; Minagawa, M.; Govindarajan, S.; Dennert, G. P2X7 receptors regulate NKT cells in autoimmune hepatitis. J. Immunol. 2006, 176, 2152–2160. [Google Scholar] [CrossRef]
- Chessell, I.P.; Michel, A.D.; Humphrey, P.P. Effects of antagonists at the human recombinant P2X7 receptor. Br. J. Pharmacol. 1998, 124, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Peñín-Franch, A.; Lucas-Ruiz, F.; de Torre-Minguela, C.; Alarcón-Vila, C.; Hernández-Caselles, T.; Pelegrín, P. P2X7 receptor activation impairs antitumour activity of natural killer cells. Br. J. Pharmacol. 2023, 180, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef] [PubMed]
- Danquah, W.; Meyer-Schwesinger, C.; Rissiek, B.; Pinto, C.; Serracant-Prat, A.; Amadi, M.; Iacenda, D.; Knop, J.-H.; Hammel, A.; Bergmann, P.; et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci. Transl. Med. 2016, 8, 366ra162. [Google Scholar] [CrossRef]
- Wilmes, M.; Pinto Espinoza, C.; Ludewig, P.; Stabernack, J.; Liesz, A.; Nicke, A.; Gelderblom, M.; Gerloff, C.; Falzoni, S.; Tolosa, E.; et al. Blocking P2X7 by intracerebroventricular injection of P2X7-specific nanobodies reduces stroke lesions. J. Neuroinflamm. 2022, 19, 256. [Google Scholar] [CrossRef]
- Demeules, M.; Scarpitta, A.; Hardet, R.; Gondé, H.; Abad, C.; Blandin, M.; Menzel, S.; Duan, Y.; Rissiek, B.; Magnus, T.; et al. Evaluation of nanobody-based biologics targeting purinergic checkpoints in tumor models in vivo. Front. Immunol. 2022, 13, 1012534. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Vultaggio-Poma, V.; Falzoni, S.; Giuliani, A.L. The coming of age of the P2X7 receptor in diagnostic medicine. Int. J. Mol. Sci. 2023, 24, 9465. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elhage, A.; Cuthbertson, P.; Sligar, C.; Watson, D.; Sluyter, R. A Species-Specific Anti-Human P2X7 Monoclonal Antibody Reduces Graft-versus-Host Disease in Humanised Mice. Pharmaceutics 2023, 15, 2263. https://doi.org/10.3390/pharmaceutics15092263
Elhage A, Cuthbertson P, Sligar C, Watson D, Sluyter R. A Species-Specific Anti-Human P2X7 Monoclonal Antibody Reduces Graft-versus-Host Disease in Humanised Mice. Pharmaceutics. 2023; 15(9):2263. https://doi.org/10.3390/pharmaceutics15092263
Chicago/Turabian StyleElhage, Amal, Peter Cuthbertson, Chloe Sligar, Debbie Watson, and Ronald Sluyter. 2023. "A Species-Specific Anti-Human P2X7 Monoclonal Antibody Reduces Graft-versus-Host Disease in Humanised Mice" Pharmaceutics 15, no. 9: 2263. https://doi.org/10.3390/pharmaceutics15092263
APA StyleElhage, A., Cuthbertson, P., Sligar, C., Watson, D., & Sluyter, R. (2023). A Species-Specific Anti-Human P2X7 Monoclonal Antibody Reduces Graft-versus-Host Disease in Humanised Mice. Pharmaceutics, 15(9), 2263. https://doi.org/10.3390/pharmaceutics15092263