
Comparative Stability Study of Polysorbate 20 and Polysorbate 80 Related to Oxidative Degradation

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
Formulation and Storage of the Polysorbate Samples
2.2. Methods
2.2.1. Inductively Coupled Plasma-Mass Spectrometry (ICP-MS)
2.2.2. pH Measurements
2.2.3. Fluorescence Micelle Assay (FMA)
2.2.4. Ferrous Oxidation with Xylenol Orange (FOX) Assay
2.2.5. Gas Chromatography (GC)
2.2.6. Reversed Phase-Ultra High Performance Liquid Chromatography-Mass Spectrometry (RP-UPLC-MS)
2.2.7. Oxygen Measurements
3. Results
3.1. FMA Analysis and Reactive Oxygen Species
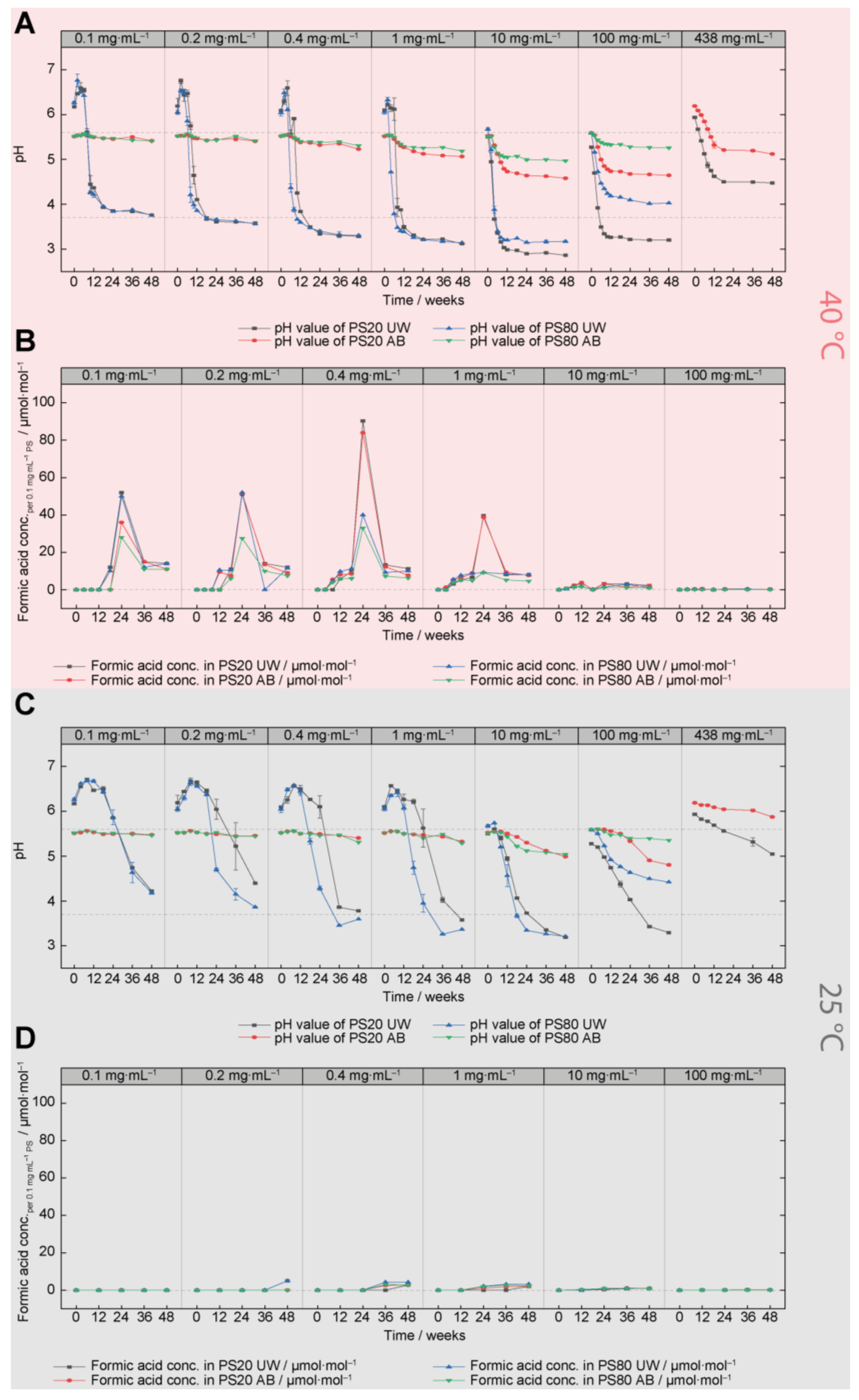
3.2. Acidification of PS20 and PS80 Formulations (pH Analysis and Gas Chromatography)
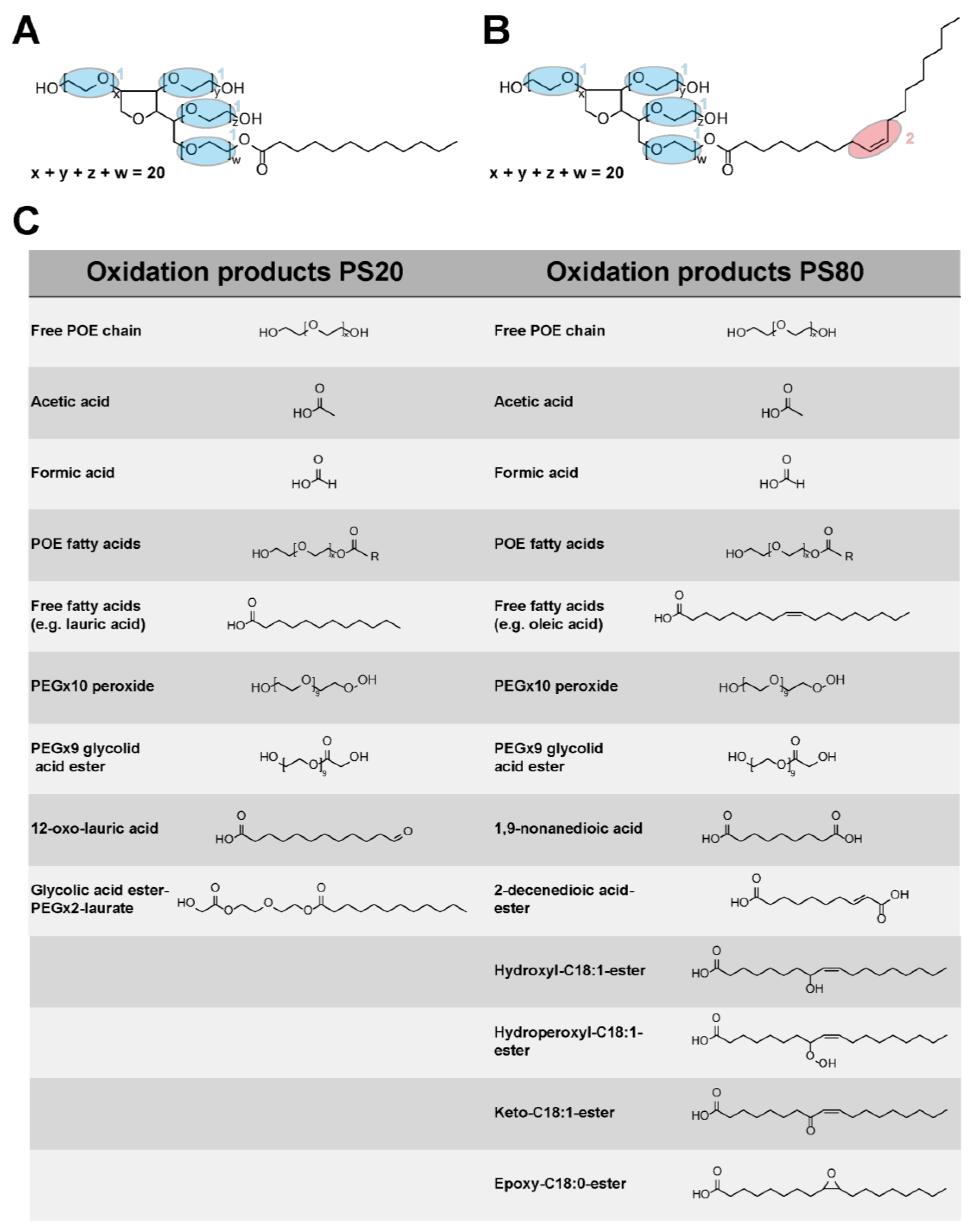
3.3. Mass Spectrometry Analysis of PS20 and PS80
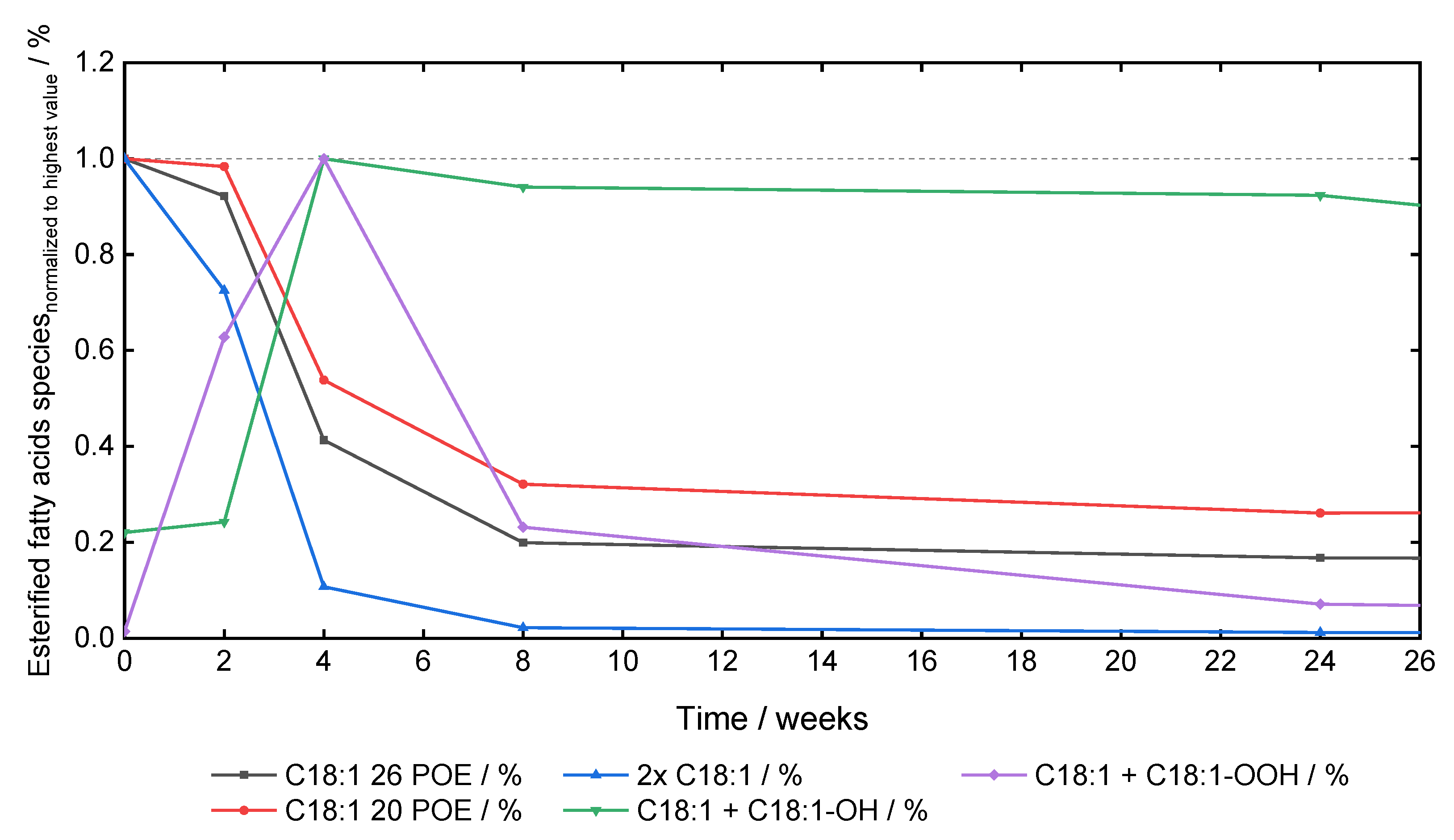
3.4. Oxidation Markers of PS80
4. Discussion
4.1. Oxidation as the Root Cause for PS20 and PS80 Degradation
4.2. Concentration-Dependent Oxidative Degradation of PS20 and PS80
4.3. PS20 vs. PS80 Oxidation
4.4. Oxidation Marker and Fatty Acid Dependent Oxidation of Polysorbates
5. Conclusions
- -
- PS80 is more prone to oxidation than PS20 as it possesses an earlier onset of oxidation, independent of whether it is formulated in water or acetate buffer;
- -
- At 5 °C, PS20 showed no oxidative degradation in water and acetate buffer up to 48 weeks, whereas PS80 oxidation started after 36 weeks;
- -
- Lower temperatures (less oxidative stress) revealed larger differences between PS20 and PS80 oxidation;
- -
- Delayed oxidation was monitored for PS20 formulated in water and high pH values were discussed as a potential origin;
- -
- Oxidation is dependent on the initial PS concentration, most likely due to differences in radical generation and/or due to limiting oxygen availability;
- -
- Metal ions such as iron in the presence of oxygen are most likely the root cause of oxidation.
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bee, J.S.; Randolph, T.W.; Carpenter, J.F.; Bishop, S.M.; Dimitrova, M.N. Effects of Surfaces and Leachables on the Stability of Biopharmaceuticals. J. Pharm. Sci. 2011, 100, 4158–4170. [Google Scholar] [CrossRef]
- Wang, W. Instability, Stabilization, and Formulation of Liquid Protein Pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef] [PubMed]
- Frokjaer, S.; Otzen, D.E. Protein Drug Stability: A Formulation Challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Brovč, E.V.; Mravljak, J.; Šink, R.; Pajk, S. Rational Design to Biologics Development: The Polysorbates Point of View. Int. J. Pharm. 2020, 581, 119285. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Hawe, A.; Jiskoot, W. Pharmaceutical Biotechnology: Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2019; pp. 83–103. [Google Scholar]
- Daugherty, A.L.; Mrsny, R.J. Formulation and Delivery Issues for Monoclonal Antibody Therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef] [PubMed]
- Kishore, R.S.K.; Kiese, S.; Fischer, S.; Pappenberger, A.; Grauschopf, U.; Mahler, H.-C. The Degradation of Polysorbates 20 and 80 and Its Potential Impact on the Stability of Biotherapeutics. Pharm. Res. 2011, 28, 1194–1210. [Google Scholar] [CrossRef]
- Kerwin, B.A. Polysorbates 20 and 80 Used in the Formulation of Protein Biotherapeutics: Structure and Degradation Pathways. J. Pharm. Sci. 2008, 97, 2924–2935. [Google Scholar] [CrossRef]
- Donbrow, M.; Azaz, E.; Pillersdorf, A. Autoxidation of Polysorbates. J. Pharm. Sci. 1978, 67, 1676–1681. [Google Scholar] [CrossRef]
- Dwivedi, M.; Blech, M.; Presser, I.; Garidel, P. Polysorbate Degradation in Biotherapeutic Formulations: Identification and Discussion of Current Root Causes. Int. J. Pharm. 2018, 552, 422–436. [Google Scholar] [CrossRef]
- Hewitt, D.; Alvarez, M.; Robinson, K.; Ji, J.; Wang, Y.J.; Kao, Y.-H.; Zhang, T. Mixed-Mode and Reversed-Phase Liquid Chromatography–Tandem Mass Spectrometry Methodologies to Study Composition and Base Hydrolysis of Polysorbate 20 and 80. J. Chromatogr. A 2011, 1218, 2138–2145. [Google Scholar] [CrossRef]
- Bates, T.R.; Nightingale, C.H.; Dixon, E. Kinetics of Hydrolysis of Polyoxyethylene(20)Sorbitan Fatty Acid Ester Surfactants. J. Pharm. Pharmacol. 1973, 25, 470–477. [Google Scholar] [CrossRef] [PubMed]
- McShan, A.C.; Kei, P.; Ji, J.A.; Kim, D.C.; Wang, Y.J. Hydrolysis of Polysorbate 20 and 80 by a Range of Carboxylester Hydrolases. PDA J. Pharm. Sci. Technol. 2016, 70, 332–345. [Google Scholar] [CrossRef]
- Chiu, J.; Valente, K.N.; Levy, N.E.; Min, L.; Lenhoff, A.M.; Lee, K.H. Knockout of a Difficult-To-Remove CHO Host Cell Protein, Lipoprotein Lipase, for Improved Polysorbate Stability in Monoclonal Antibody Formulations. Biotechnol. Bioeng. 2017, 114, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.; Tomlinson, A.; Yuk, I.H.; Kufer, R.; Spensberger, B.; Falkenstein, R.; Shen, A.; Li, H.; Duan, D.; Liu, W.; et al. Identification and Characterization of Polysorbate-Degrading Enzymes in a Monoclonal Antibody Formulation. J. Pharm. Sci. 2021, 110, 3558–3567. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Salamat-Miller, N.; Salinas, P.A.; Taylor, K.D.; Basu, S.K. Residual Host Cell Protein Promotes Polysorbate 20 Degradation in a Sulfatase Drug Product Leading to Free Fatty Acid Particles. J. Pharm. Sci. 2016, 105, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Kovner, D.; Yuk, I.H.; Shen, A.; Li, H.; Graf, T.; Gupta, S.; Liu, W.; Tomlinson, A. Characterization of Recombinantly-Expressed Hydrolytic Enzymes from Chinese Hamster Ovary Cells: Identification of Host Cell Proteins That Degrade Polysorbate. J. Pharm. Sci. 2023, 112, 1351–1363. [Google Scholar] [CrossRef]
- Hall, T.; Sandefur, S.L.; Frye, C.C.; Tuley, T.L.; Huang, L. Polysorbates 20 and 80 Degradation by Group XV Lysosomal Phospholipase A2 Isomer X1 in Monoclonal Antibody Formulations. J. Pharm. Sci. 2016, 105, 1633–1642. [Google Scholar] [CrossRef]
- Roy, I.; Patel, A.; Kumar, V.; Nanda, T.; Assenberg, R.; Wuchner, K.; Amin, K. Polysorbate Degradation and Particle Formation in a High Concentration MAb: Formulation Strategies to Minimize Effect of Enzymatic Polysorbate Degradation. J. Pharm. Sci. 2021, 110, 3313–3323. [Google Scholar] [CrossRef]
- Zhang, S.; Xiao, H.; Li, N. Degradation of Polysorbate 20 by Sialate O-Acetylesterase in Monoclonal Antibody Formulations. J. Pharm. Sci. 2021, 110, 3866–3873. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Li, H.; Richardson, D.D.; Roush, D.J. The Measurement and Control of High-Risk Host Cell Proteins for Polysorbate Degradation in Biologics Formulation. Antib. Ther. 2022, 5, 42–54. [Google Scholar] [CrossRef]
- Wuchner, K.; Yi, L.; Chery, C.; Nikels, F.; Junge, F.; Crotts, G.; Rinaldi, G.; Starkey, J.A.; Bechtold-Peters, K.; Shuman, M.; et al. Industry Perspective on the Use and Characterization of Polysorbates for Biopharmaceutical Products Part 1: Survey Report on Current State and Common Practices for Handling and Control of Polysorbates. J. Pharm. Sci. 2022, 111, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Ha, E.; Wang, W.; Wang, Y.J. Peroxide Formation in Polysorbate 80 and Protein Stability. J. Pharm. Sci. 2002, 91, 2252–2264. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.R.; Wei, Y.; Prajapati, I.; Chakraborty, A.; Peters, B.; Kalonia, C.; Hudak, S.; Choudhary, S.; Esfandiary, R.; Dhar, P.; et al. Comparison of Polysorbate 80 Hydrolysis and Oxidation on the Aggregation of a Monoclonal Antibody. J. Pharm. Sci. 2020, 109, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Kranz, W.; Wuchner, K.; Corradini, E.; Menzen, T.; Hawe, A. Micelle Driven Oxidation Mechansim and Novel Oxidation Markers for Different Grades of Polysorbate 20 and 80. J. Pharm. Sci. 2020, 109, 3064–3077. [Google Scholar] [CrossRef]
- Kranz, W.; Wuchner, K.; Corradini, E.; Berger, M.; Hawe, A. Factors Influencing Polysorbate’s Sensitivity Against Enzymatic Hydrolysis and Oxidative Degradation. J. Pharm. Sci. 2019, 108, 2022–2032. [Google Scholar] [CrossRef]
- Bensaid, F.; Dagallier, C.; Authelin, J.-R.; Audat, H.; Filipe, V.; Narradon, C.; Guibal, P.; Clavier, S.; Wils, P. Mechanistic Understanding of Metal-Catalyzed Oxidation of Polysorbate 80 and Monoclonal Antibody in Biotherapeutic Formulations. Int. J. Pharm. 2022, 615, 121496. [Google Scholar] [CrossRef]
- Yarbrough, M.; Hodge, T.; Menard, D.; Jerome, R.; Ryczek, J.; Moore, D.; Baldus, P.; Warne, N.; Ohtake, S. Edetate Disodium as a Polysorbate Degradation and Monoclonal Antibody Oxidation Stabilizer. J. Pharm. Sci. 2019, 108, 1631–1635. [Google Scholar] [CrossRef]
- Doyle, L.M.; Sharma, A.N.; Gopalrathnam, G.; Huang, L.; Bradley, S. A Mechanistic Understanding of Polysorbate 80 Oxidation in Histidine and Citrate Buffer Systems-Part 2. PDA J. Pharm. Sci. Technol. 2019, 73, 320–330. [Google Scholar] [CrossRef]
- Gopalrathnam, G.; Sharma, A.N.; Dodd, S.W.; Huang, L. Impact of Stainless Steel Exposure on the Oxidation of Polysorbate 80 in Histidine Placebo and Active Monoclonal Antibody Formulation. PDA J. Pharm. Sci. Technol. 2018, 72, 163–175. [Google Scholar] [CrossRef]
- Liu, H.; Jin, Y.; Menon, R.; Laskowich, E.; Bareford, L.; de Vilmorin, P.; Kolwyck, D.; Yeung, B.; Yi, L. Characterization of Polysorbate 80 by Liquid Chromatography-Mass Spectrometry to Understand Its Susceptibility to Degradation and Its Oxidative Degradation Pathway. J. Pharm. Sci. 2022, 111, 323–334. [Google Scholar] [CrossRef]
- Schröter, A.; Peter, S.; Mahler, H.-C.; Zerr, A.; Danton, O.; Chalus, P.; Koulov, A.V.; Huwyler, J.; Jahn, M. Identification of an Oxidizing Leachable from a Clinical Syringe Rubber Stopper. J. Pharm. Sci. 2021, 110, 3410–3417. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, I.; Subelzu, N.; Zhang, Y.; Wu, Y.; Schöneich, C. Near UV and Visible Light Photo-Degradation Mechanisms in Citrate Buffer: One-Electron Reduction of Peptide and Protein Disulfides Promotes Oxidation and Cis/Trans Isomerization of Unsaturated Fatty Acids of Polysorbate 80. J. Pharm. Sci. 2022, 111, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Haber, F.; Weiss, J. The Catalytic Decomposition of Hydrogen Peroxide by Iron Salts. Proc. Royal Soc. Lond. Ser. A Math. Phys. Sci. 1934, 147, 332–351. [Google Scholar] [CrossRef]
- Koppenol, W.H. The Haber-Weiss Cycle—70 Years Later. Redox Rep. 2001, 6, 229–234. [Google Scholar] [CrossRef]
- Fenton, H.J.H. Oxidation of Tartaric Acid in Presence of Iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Rachmilovich-Calis, S.; Masarwa, A.; Meyerstein, N.; Meyerstein, D.; Eldik, R. van New Mechanistic Aspects of the Fenton Reaction. Chem. A Eur. J. 2009, 15, 8303–8309. [Google Scholar] [CrossRef]
- Goldstein, S.; Meyerstein, D.; Czapski, G. The Fenton Reagents. Free Radic Biol. Med. 1993, 15, 435–445. [Google Scholar] [CrossRef]
- Goldstein, S.; Meyerstein, D. Comments on the Mechanism of the “Fenton-Like” Reaction. Acc. Chem. Res. 1999, 32, 547–550. [Google Scholar] [CrossRef]
- Masarwa, M.; Cohen, H.; Meyerstein, D.; Hickman, D.L.; Bakac, A.; Espenson, J.H. Reactions of Low-Valent Transition-Metal Complexes with Hydrogen Peroxide. Are They “Fenton-like” or Not? 1. The Case of Cu+aq and Cr2+aq. J. Am. Chem. Soc. 1988, 110, 4293–4297. [Google Scholar] [CrossRef]
- Luzzatto, E.; Cohen, H.; Stockheim, C.; Wieghardt, K.; Meyerstein, D. Reactions of Low-Valent Transition Metal Complexes with Hydrogen Peroxide. Are They “Fenton-Like” or Not? 4. The Case of Fe(II)L, L = Edta; Hedta and Tcma. Free Radic. Res. 1995, 23, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Bamnolker, H.; Cohen, H.; Meyerstein, D. Reactions of Low-Valent Transition-Metal Complexes with Hydrogen Peroxide. Are They “Fenton-Like” or Not? 3. The Case of Fe(II){N(CH2CO2)3}(H2O)2. Free Radic. Res. Commun. 1991, 15, 231–241. [Google Scholar] [CrossRef]
- Jung, Y.S.; Lim, W.T.; Park, J.Y.; Kim, Y.H. Effect of PH on Fenton and Fenton-like Oxidation. Environ. Technol. 2009, 30, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-R.; Li, X.-Y.; Li, X.-Z.; Li, H.-B. Degradation of Melatonin by UV, UV/H2O2, Fe2+/H2O2 and UV/Fe2+/H2O2 Processes. Sep. Purif. Technol. 2009, 68, 261–266. [Google Scholar] [CrossRef]
- Tang, W.Z.; Huang, C.P. 2,4-Dichlorophenol Oxidation Kinetics by Fenton’s Reagent. Environ. Technol. 1996, 17, 1371–1378. [Google Scholar] [CrossRef]
- Hovorka, S.W.; Schöneich, C. Oxidative Degradation of Pharmaceuticals: Theory, Mechanisms and Inhibition. J. Pharm. Sci. 2001, 90, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Mittag, J.J.; Trutschel, M.-L.; Kruschwitz, H.; Mäder, K.; Buske, J.; Garidel, P. Characterization of Radicals in Polysorbate 80 Using Electron Paramagnetic Resonance (EPR) Spectroscopy and Spin Trapping. Int. J. Pharm. X 2022, 4, 100123. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Detection and Characterisation of Radicals in Biological Materials Using EPR Methodology. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 708–721. [Google Scholar] [CrossRef]
- Finkelstein, E.; Rosen, G.M.; Rauckman, E.J. Spin Trapping. Kinetics of the Reaction of Superoxide and Hydroxyl Radicals with Nitrones. J. Am. Chem. Soc. 1980, 102, 4994–4999. [Google Scholar] [CrossRef]
- Goldstein, S.; Rosen, G.M.; Russo, A.; Samuni, A. Kinetics of Spin Trapping Superoxide, Hydroxyl, and Aliphatic Radicals by Cyclic Nitrones. J. Phys. Chem. A 2004, 108, 6679–6685. [Google Scholar] [CrossRef]
- Buettner, G.R.; Mason, R.P. Spin-Trapping Methods for Detecting Superoxide and Hydroxyl Free Radicals in Vitro and in Vivo. Methods Enzym. 1990, 186, 127–133. [Google Scholar] [CrossRef]
- Brovč, E.V.; Mravljak, J.; Šink, R.; Pajk, S. Degradation of Polysorbates 20 and 80 Catalysed by Histidine Chloride Buffer. Eur. J. Pharm. Biopharm. 2020, 154, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Borisov, O.V.; Ji, J.A.; Wang, Y.J. Oxidative Degradation of Polysorbate Surfactants Studied by Liquid Chromatography–Mass Spectrometry. J. Pharm. Sci. 2015, 104, 1005–1018. [Google Scholar] [CrossRef]
- Hvattum, E.; Yip, W.L.; Grace, D.; Dyrstad, K. Characterization of Polysorbate 80 with Liquid Chromatography Mass Spectrometry and Nuclear Magnetic Resonance Spectroscopy: Specific Determination of Oxidation Products of Thermally Oxidized Polysorbate 80. J. Pharm. Biomed. Anal. 2012, 62, 7–16. [Google Scholar] [CrossRef]
- Yao, J.; Dokuru, D.K.; Noestheden, M.; Park, S.S.; Kerwin, B.A.; Jona, J.; Ostovic, D.; Reid, D.L. A Quantitative Kinetic Study of Polysorbate Autoxidation: The Role of Unsaturated Fatty Acid Ester Substituents. Pharm. Res. 2009, 26, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Lippold, S.; Koshari, S.H.S.; Kopf, R.; Schuller, R.; Buckel, T.; Zarraga, I.E.; Koehn, H. Impact of Mono- and Poly-Ester Fractions on Polysorbate Quantitation Using Mixed-Mode HPLC-CAD/ELSD and the Fluorescence Micelle Assay. J. Pharm. Biomed. 2017, 132, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yadav, S.; Wang, Y.J.; Mozziconacci, O.; Schöneich, C. Dual Effect of Histidine on Polysorbate 20 Stability: Mechanistic Studies. Pharm. Res. 2018, 35, 33. [Google Scholar] [CrossRef]
- Weber, J.; Buske, J.; Mäder, K.; Garidel, P.; Diederichs, T. Oxidation of Polysorbates—An Underestimated Degradation Pathway? Int. J. Pharm. X 2023, 6, 100202. [Google Scholar] [CrossRef]
- Glücklich, N.; Carle, S.; Buske, J.; Mäder, K.; Garidel, P. Assessing the Polysorbate Degradation Fingerprints and Kinetics of Lipases—How the Activity of Polysorbate Degrading Hydrolases Is Influenced by the Assay and Assay Conditions. Eur. J. Pharm. Sci. 2021, 166, 105980. [Google Scholar] [CrossRef]
- Jaeger, J.; Sorensen, K.; Wolff, S.P. Peroxide Accumulation in Detergents. J. Biochem. Bioph. Methods 1994, 29, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, V.; Stenroos, L. Quantitative Analysis of Volatile Fatty Acids in Aqueous Solution by Gas Chromatography. Anal. Chem. 1967, 39, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Evers, D.-H.; Schultz-Fademrecht, T.; Garidel, P.; Buske, J. Development and Validation of a Selective Marker-Based Quantification of Polysorbate 20 in Biopharmaceutical Formulations Using UPLC-QDa Detection. J. Chromatogr. B 2020, 1157, 122287. [Google Scholar] [CrossRef] [PubMed]
- Hipper, E.; Lehmann, F.; Kaiser, W.; Hübner, G.; Buske, J.; Blech, M.; Hinderberger, D.; Garidel, P. Protein Photodegradation in the Visible Range? Insights into Protein Photooxidation with Respect to Protein Concentration. Int. J. Pharm. X 2023, 5, 100155. [Google Scholar] [CrossRef]
- Dwivedi, M.; Buske, J.; Haemmerling, F.; Blech, M.; Garidel, P. Acidic and Alkaline Hydrolysis of Polysorbates under Aqueous Conditions: Towards Understanding Polysorbate Degradation in Biopharmaceutical Formulations. Eur. J. Pharm. Sci. 2020, 144, 105211. [Google Scholar] [CrossRef]
- Zhou, S.; Schöneich, C.; Singh, S.K. Biologics Formulation Factors Affecting Metal Leachables from Stainless Steel. AAPS Pharm. Sci. Technol. 2011, 12, 411–421. [Google Scholar] [CrossRef]
- Allmendinger, A.; Lebouc, V.; Bonati, L.; Woehr, A.; Kishore, R.S.K.; Abstiens, K. Glass Leachables as a Nucleation Factor for Free Fatty Acid Particle Formation in Biopharmaceutical Formulations. J. Pharm. Sci. 2021, 110, 785–795. [Google Scholar] [CrossRef]
- Ditter, D.; Nieto, A.; Mahler, H.-C.; Roehl, H.; Wahl, M.; Huwyler, J.; Allmendinger, A. Evaluation of Glass Delamination Risk in Pharmaceutical 10 ML/10R Vials. J. Pharm. Sci. 2018, 107, 624–637. [Google Scholar] [CrossRef]
- Ditter, D.; Mahler, H.-C.; Gohlke, L.; Nieto, A.; Roehl, H.; Huwyler, J.; Wahl, M.; Allmendinger, A. Impact of Vial Washing and Depyrogenation on Surface Properties and Delamination Risk of Glass Vials. Pharm. Res. 2018, 35, 146. [Google Scholar] [CrossRef]
- Gregoritza, K.; Cai, S.K.; Siketanc, M.; Woehr, A.; Lebouc, V.; Kishore, R.S.K.; Nicoulin, V.; Bleher, S.; Allmendinger, A. Metal-Induced Fatty Acid Particle Formation Resulting from Hydrolytic Polysorbate Degradation. J. Pharm. Sci. 2022, 111, 743–751. [Google Scholar] [CrossRef]
- Musakhanian, J.; Rodier, J.-D.; Dave, M. Oxidative Stability in Lipid Formulations: A Review of the Mechanisms, Drivers, and Inhibitors of Oxidation. AAPS Pharm. Sci. Technol. 2022, 23, 151. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, A.; Zarraga, I.E.; Demeule, B. Characterization of Polysorbate Ester Fractions and Implications in Protein Drug Product Stability. Mol. Pharm. 2020, 17, 2345–2353. [Google Scholar] [CrossRef]
- Nayem, J.; Zhang, Z.; Tomlinson, A.; Zarraga, I.E.; Wagner, N.J.; Liu, Y. Micellar Morphology of Polysorbate 20 and 80 and Their Ester Fractions in Solution via Small-Angle Neutron Scattering. J. Pharm. Sci. 2020, 109, 1498–1508. [Google Scholar] [CrossRef]
- Denisov, E.T.; Afanas’ev, I.B. Oxidation and Antioxidants in Organic Chemistry and Biology. Angew. Chem. 2005, 118, 4831–4832. [Google Scholar] [CrossRef]
- Wang, R.L.C.; Kreuzer, H.J.; Grunze, M. Molecular Conformation and Solvation of Oligo(Ethylene Glycol)-Terminated Self-Assembled Monolayers and Their Resistance to Protein Adsorption. J. Phys. Chem. B 1997, 101, 9767–9773. [Google Scholar] [CrossRef]
- Tasaki, K. Poly(Oxyethylene)−Water Interactions: A Molecular Dynamics Study. J. Am. Chem. Soc. 1996, 118, 8459–8469. [Google Scholar] [CrossRef]
- Braun, H.-G.; Meyer, E. Structure Formation of Ultrathin PEO Films at Solid Interfaces—Complex Pattern Formation by Dewetting and Crystallization. Int. J. Mol. Sci. 2013, 14, 3254–3264. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | PS20 RM /ng⋅mL−1 | 100 mg⋅mL−1 PS20 UW after Storage of 48 Weeks at 40 °C /ng⋅mL−1 | PS80 RM /ng⋅mL−1 | 100 mg⋅mL−1 PS80 UW after Storage of 48 Weeks at 40 °C /ng⋅mL−1 | 1 M Acetate Buffer /ng⋅mL−1 |
|---|---|---|---|---|---|
| Al | <250 | 1233.5 | <250 | 668.2 | <250 |
| B | <250 | 1100.5 | <250 | 572.2 | 2330.5 |
| Cu | <10 | 1.9 | <10 | 12.4 | <10 |
| Fe | 141.8 | 44.1 | 102.2 | 23.9 | <100 |
| Mo | 13.4 | 7.4 | 13.8 | 7.4 | 12.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozuch, B.; Weber, J.; Buske, J.; Mäder, K.; Garidel, P.; Diederichs, T. Comparative Stability Study of Polysorbate 20 and Polysorbate 80 Related to Oxidative Degradation. Pharmaceutics 2023, 15, 2332. https://doi.org/10.3390/pharmaceutics15092332
Kozuch B, Weber J, Buske J, Mäder K, Garidel P, Diederichs T. Comparative Stability Study of Polysorbate 20 and Polysorbate 80 Related to Oxidative Degradation. Pharmaceutics. 2023; 15(9):2332. https://doi.org/10.3390/pharmaceutics15092332
Chicago/Turabian StyleKozuch, Benedykt, Johanna Weber, Julia Buske, Karsten Mäder, Patrick Garidel, and Tim Diederichs. 2023. "Comparative Stability Study of Polysorbate 20 and Polysorbate 80 Related to Oxidative Degradation" Pharmaceutics 15, no. 9: 2332. https://doi.org/10.3390/pharmaceutics15092332