
Development of Novel ROCK Inhibitors via 3D-QSAR and Molecular Docking Studies: A Framework for Multi-Target Drug Design

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
- (1)
- To identify key structural features of ROCK1/ROCK2 inhibitors using computer-aided drug design methods, especially ligand-based approaches such as 3D-Quantitative Structure-Activity Relationship (3D-QSAR) and structure-based methods such as molecular docking;
- (2)
- To develop novel ROCK inhibitors and synthesize compounds with the best-predicted activity based on the computational models;
- (3)
- To evaluate the activity of the synthesized ROCK inhibitors using enzyme and cancer cell assays to assess their efficacy and potential as therapeutic agents;
- (4)
- To propose future perspectives on multi-target HDAC/ROCK drug design based on the structure of novel developed ROCK inhibitors.
2. Materials and Methods
2.1. Design of Novel ROCK Inhibitors
2.1.1. Preparation of Compounds for Molecular Docking and 3D-QSAR Study
2.1.2. Molecular Docking
2.1.3. Development of 3D-QSAR (ROCK1) and 3D-QSAR (ROCK2) Models
2.1.4. Selection of Training and Test Set Compounds
2.1.5. Internal and External Validation of Model
2.1.6. Applicability Domain
2.2. Synthesis—General Procedures
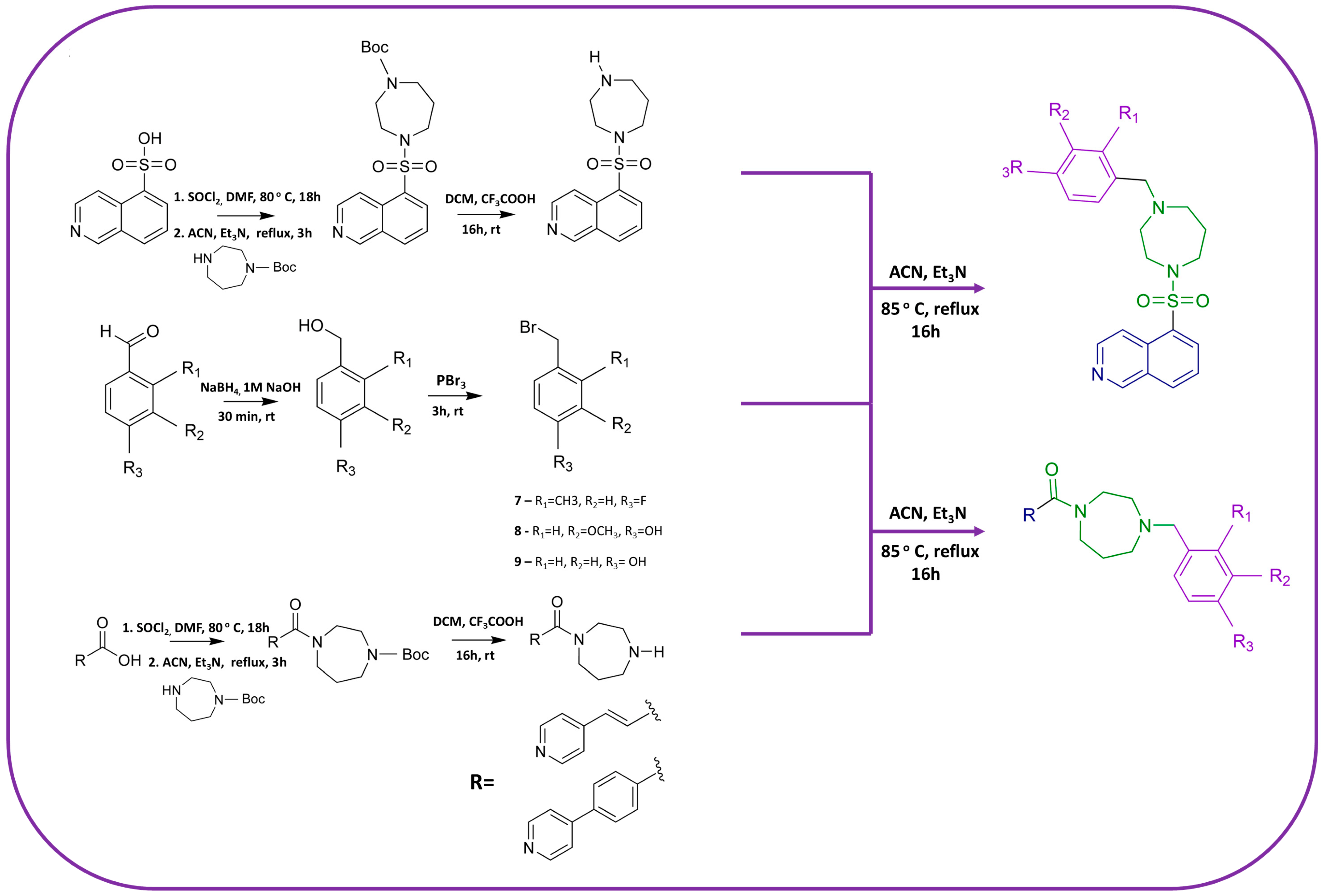
2.2.1. Synthesis of Benzyl Bromide Derivatives—General Procedure A
2.2.2. Synthesis of Homopiperazine Derivatives—General Procedure B
2.2.3. Synthesis of Novel Developed ROCK Inhibitors—General Procedure C
2.3. ROCK Enzyme Assays
2.4. Biological Evaluation
2.4.1. Cell Viability (MTT) Assay
2.4.2. Cell Cycle Analysis
2.4.3. Apoptosis Assay
2.4.4. Detection of Mitochondrial Membrane Potential
2.4.5. Evaluation of ROS Production
2.4.6. Generation and Analysis of Tumor Spheroids
2.4.7. Wound Healing Assay
2.4.8. Transwell Migration and Invasion Assays
3. Results and Discussion
3.1. Design of Novel ROCK Inhibitors
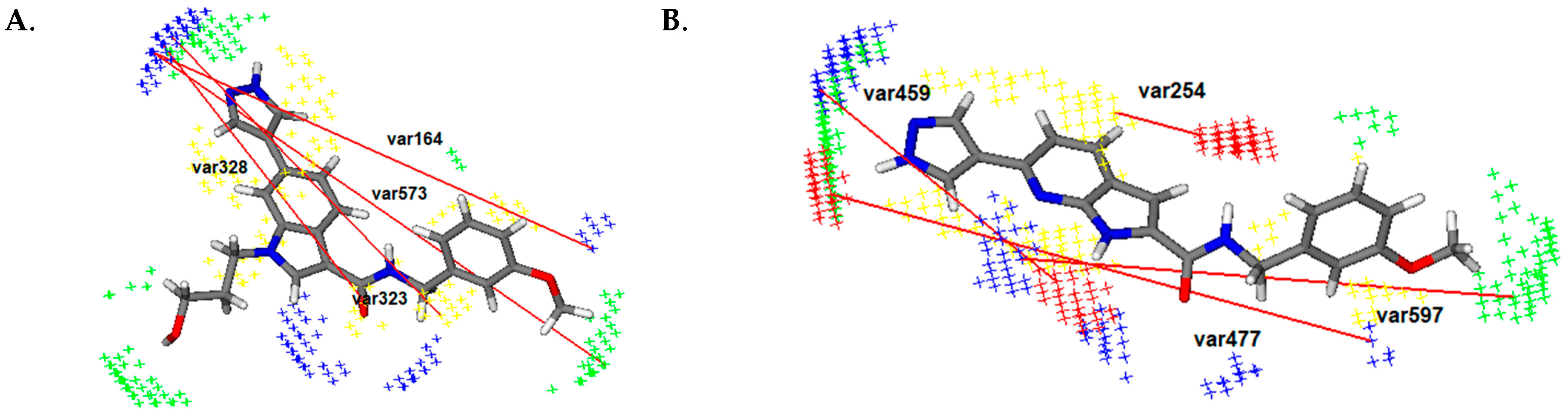
3.1.1. Results of 3D-QSAR Study for ROCK1 and ROCK2
- (1)
- Heterocycle with nitrogen atom as hydrogen-accepting group due to positive impact of var323 (DRY-N1: 13.2–13.6 Å) (ROCK1), var328 (DRY-N1: 15.2–15.6 Å) (ROCK1), var254 (DRY-O: 2.4–2.8 Å) (ROCK2), var459 (O-N1: 10.0–10.4 Å) (ROCK2);
- (2)
- Linker with hydrogen accepting and hydrogen-donating group (amide group), which is shown by var459 (O-N1: 10.0–10.4 Å) (ROCK2);
- (3)
- Phenyl ring with hydrogen bond accepting groups as substituents in meta or para position due to the influence of var573 (N1-TIP: 20.4–20.8 Å) (ROCK1), var383 (DRY-TIP: 14.0–14.4 Å) (ROCK1), var164 (N1-N1: 19.2–19.6 Å) (ROCK1) and var597 (N1-TIP: 15.60–16.0 Å) (ROCK2), var477 (O-N1: 17.2–17.6 Å) (ROCK2).
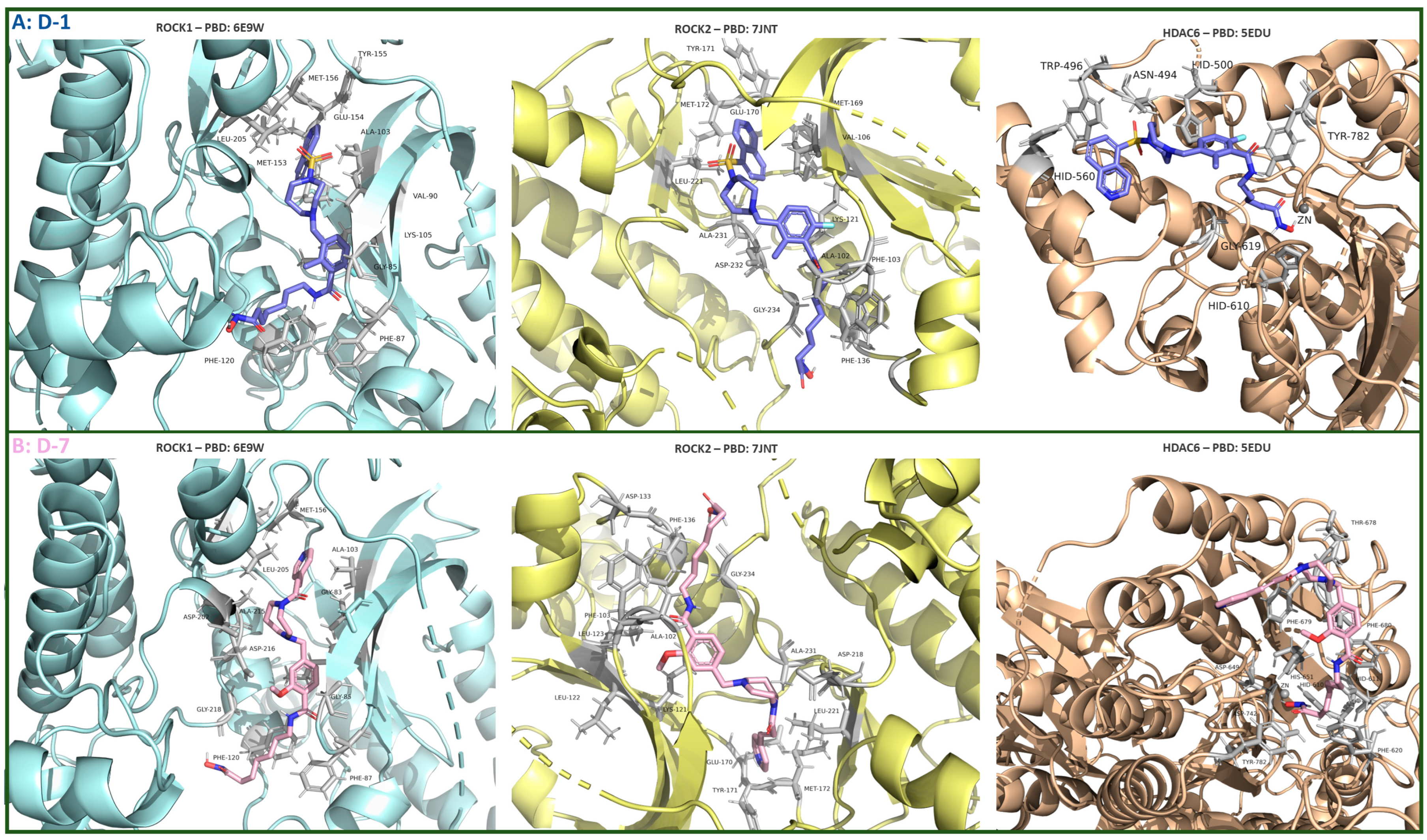
3.1.2. Molecular Docking Study
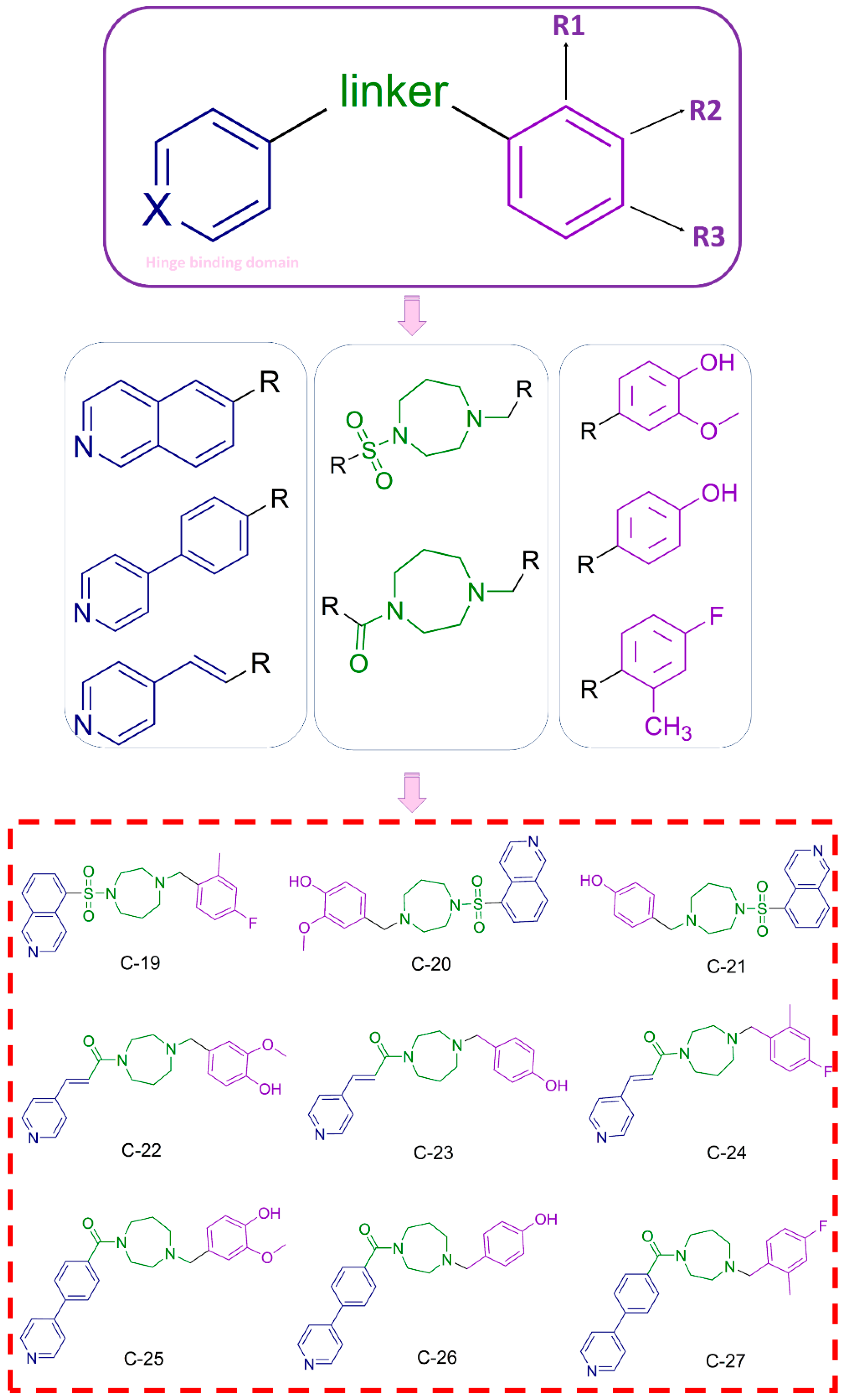
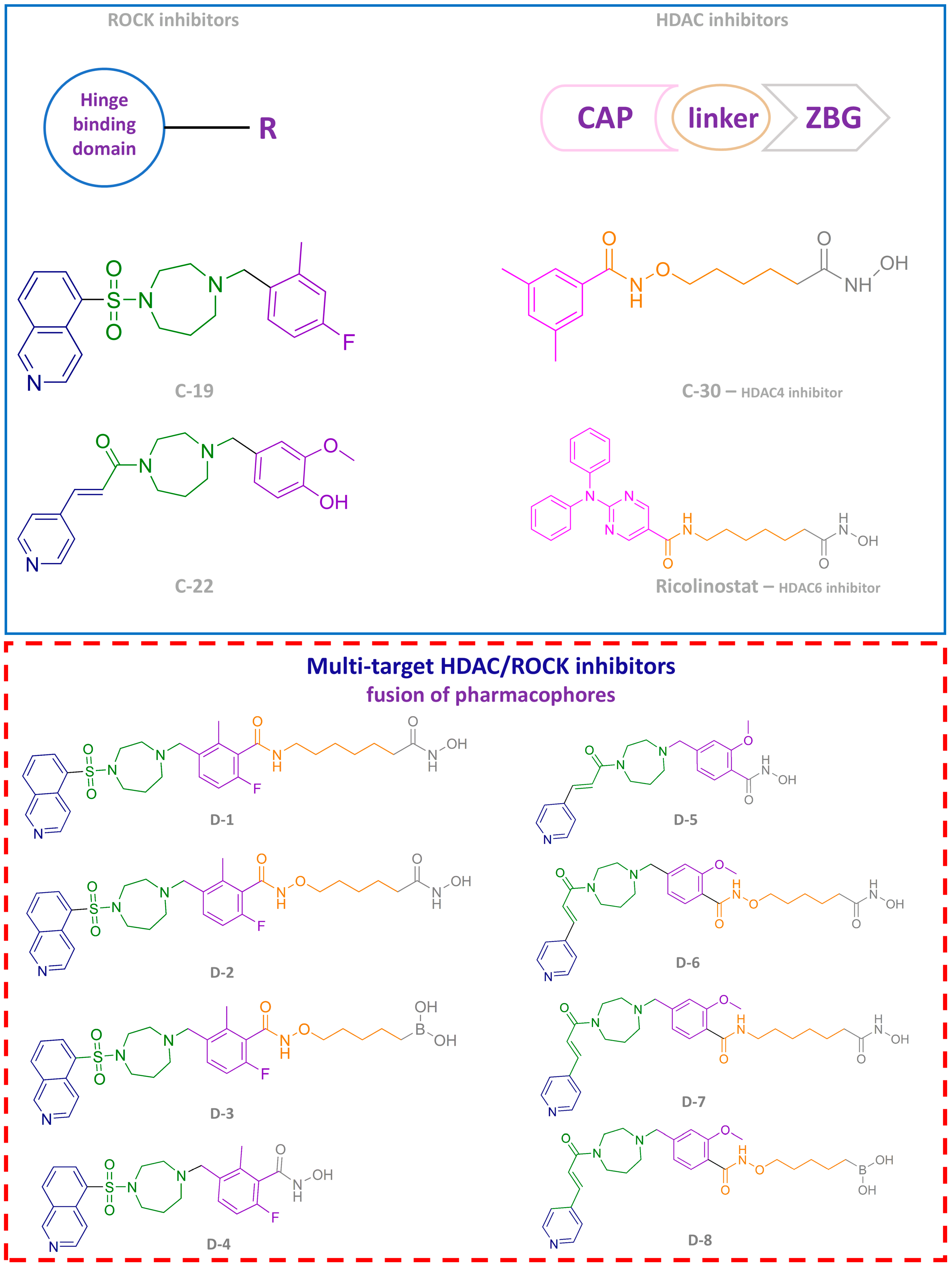
3.1.3. Design and Synthesis of Novel Potential ROCK Inhibitors
3.2. Enzyme Assays
3.3. Biological Evaluation
3.3.1. Cytotoxic Effect of Newly Designed and Synthesized ROCK Inhibitors
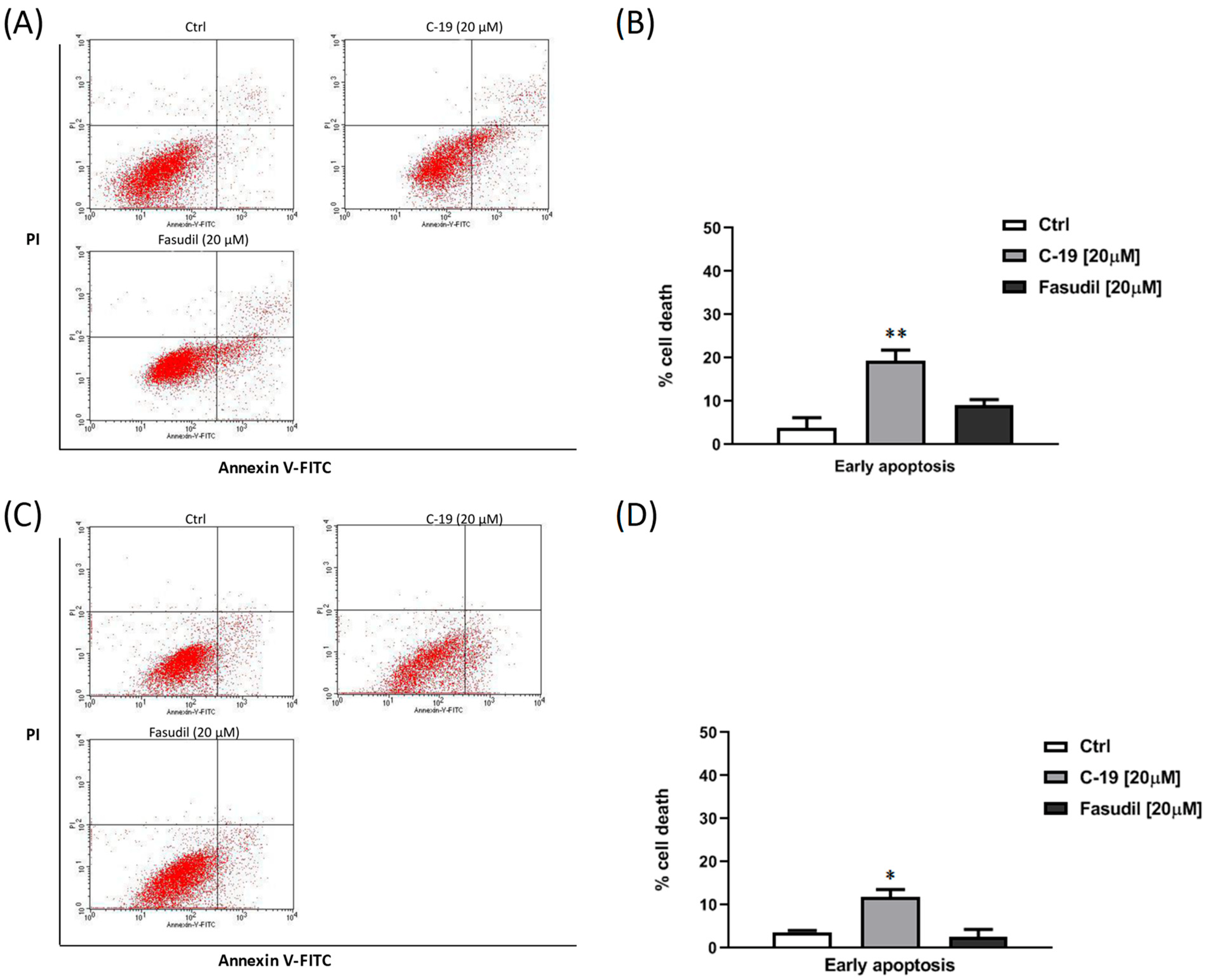
3.3.2. Newly Designed and Synthesized ROCK Inhibitor C-19 Induces Early Apoptosis in Mia PaCa-2 and Panc-1 Cells
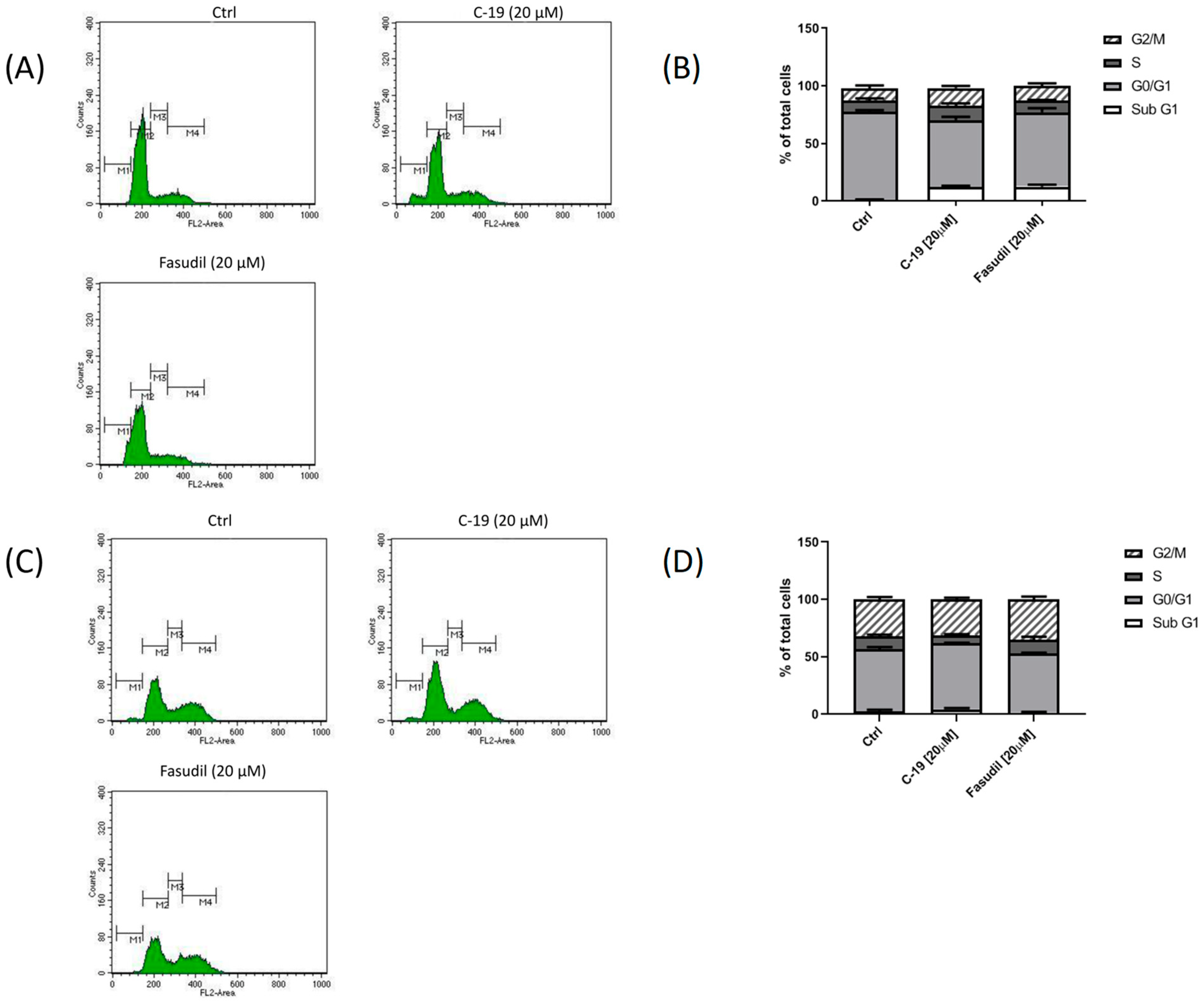
3.3.3. Newly Designed and Synthesized ROCK Inhibitor C-19 Induces Changes in Cell Cycle Phase Distribution in Mia PaCa-2 and Panc-1 Cells
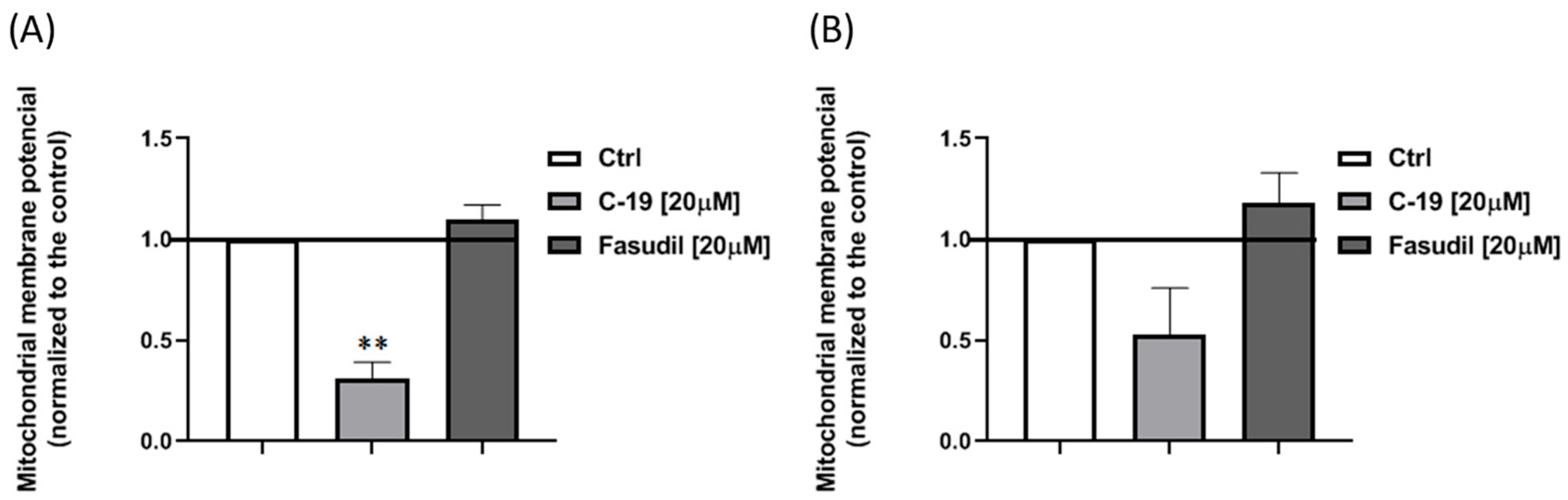
3.3.4. Newly Designed and Synthesized ROCK Inhibitor C-19 Induces a Decrease of Mitochondrial Membrane Potential in Mia Paca-2 Cells
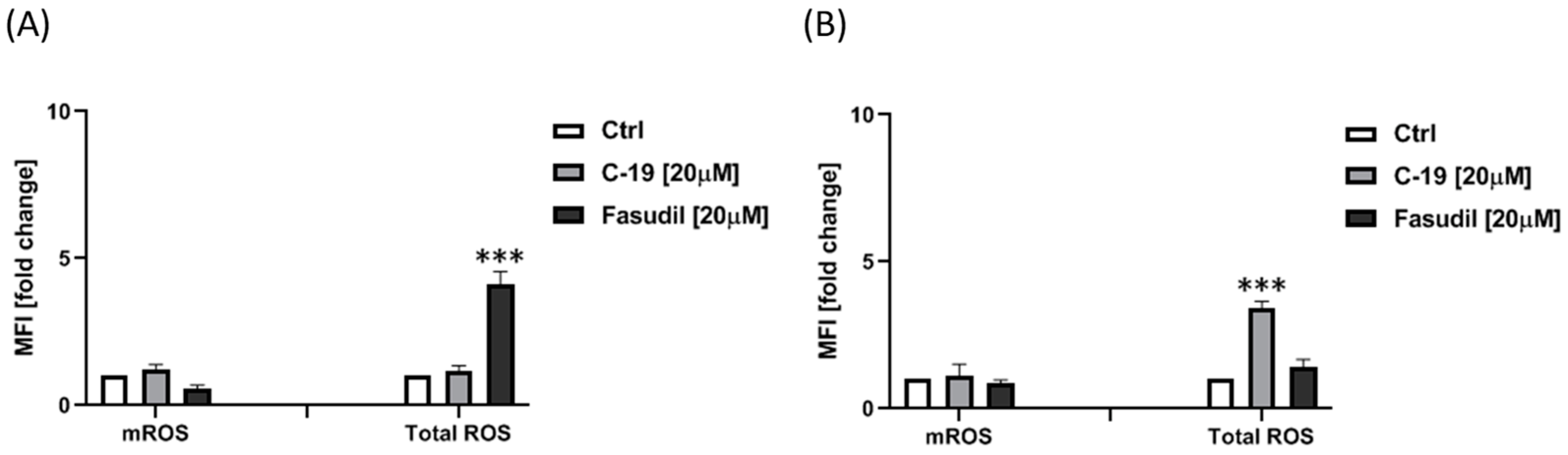
3.3.5. Newly Designed and Synthesized ROCK Inhibitor C-19 Induces an Increase of Total ROS in Panc-1 Cells
3.3.6. Analyses of the Efficacy of C-19 in a 3D Cancer Model
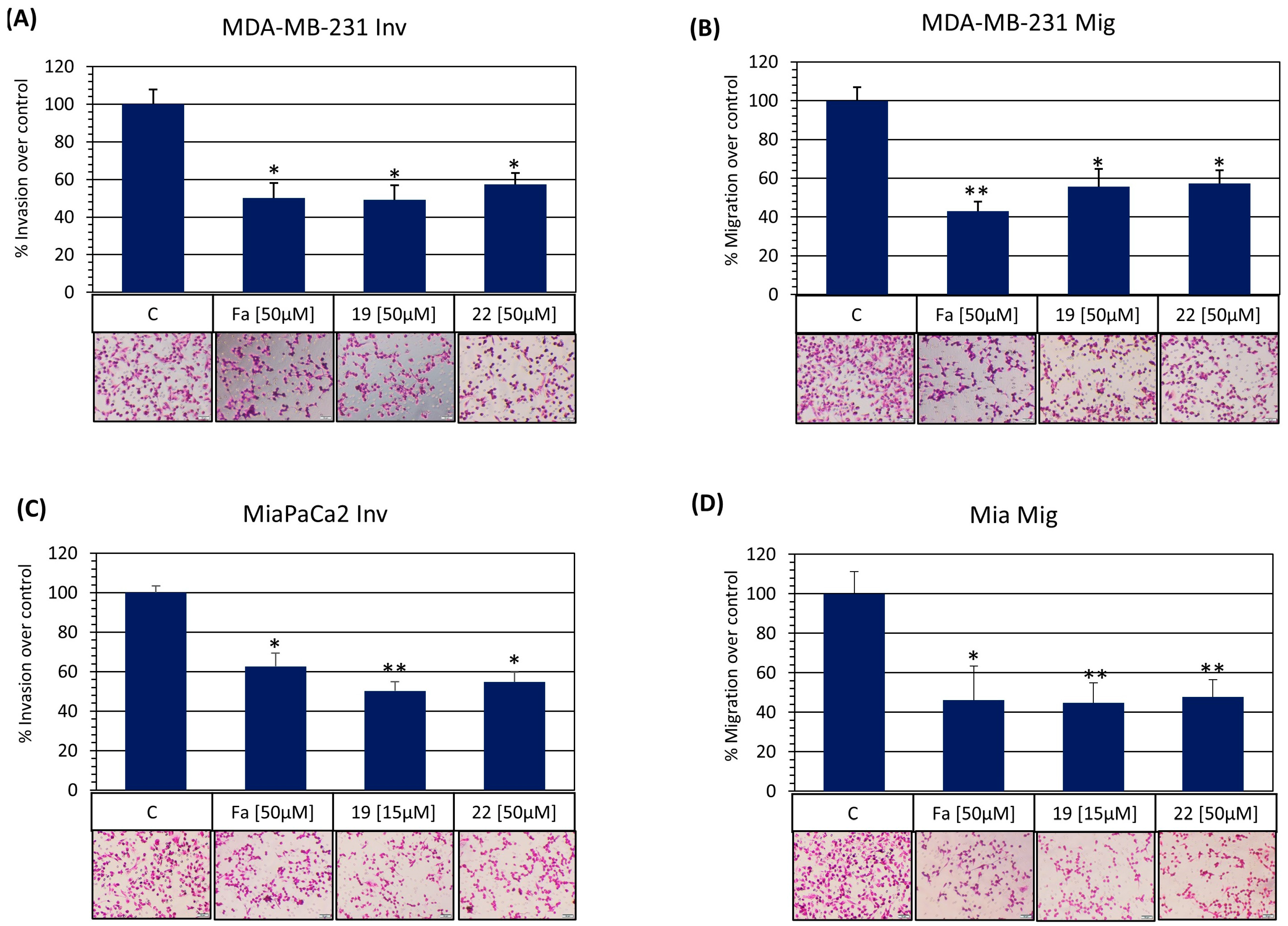
3.3.7. Synthetized ROCK Inhibitors Inhibit Cell Migration and Invasion In Vitro
3.3.8. Future Perspectives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Structural Characterization of Synthesized Compounds
References
- Morgan-Fisher, M.; Wewer, U.M.; Yoneda, A. Regulation of ROCK Activity in Cancer. J. Histochem. Cytochem. 2013, 61, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Skarp, K.-P.; Vartiainen, M.K. Actin on DNA—An Ancient and Dynamic Relationship. Cytoskeleton 2010, 67, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and Actin Dynamics in Membrane Protrusions and Vesicle Trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
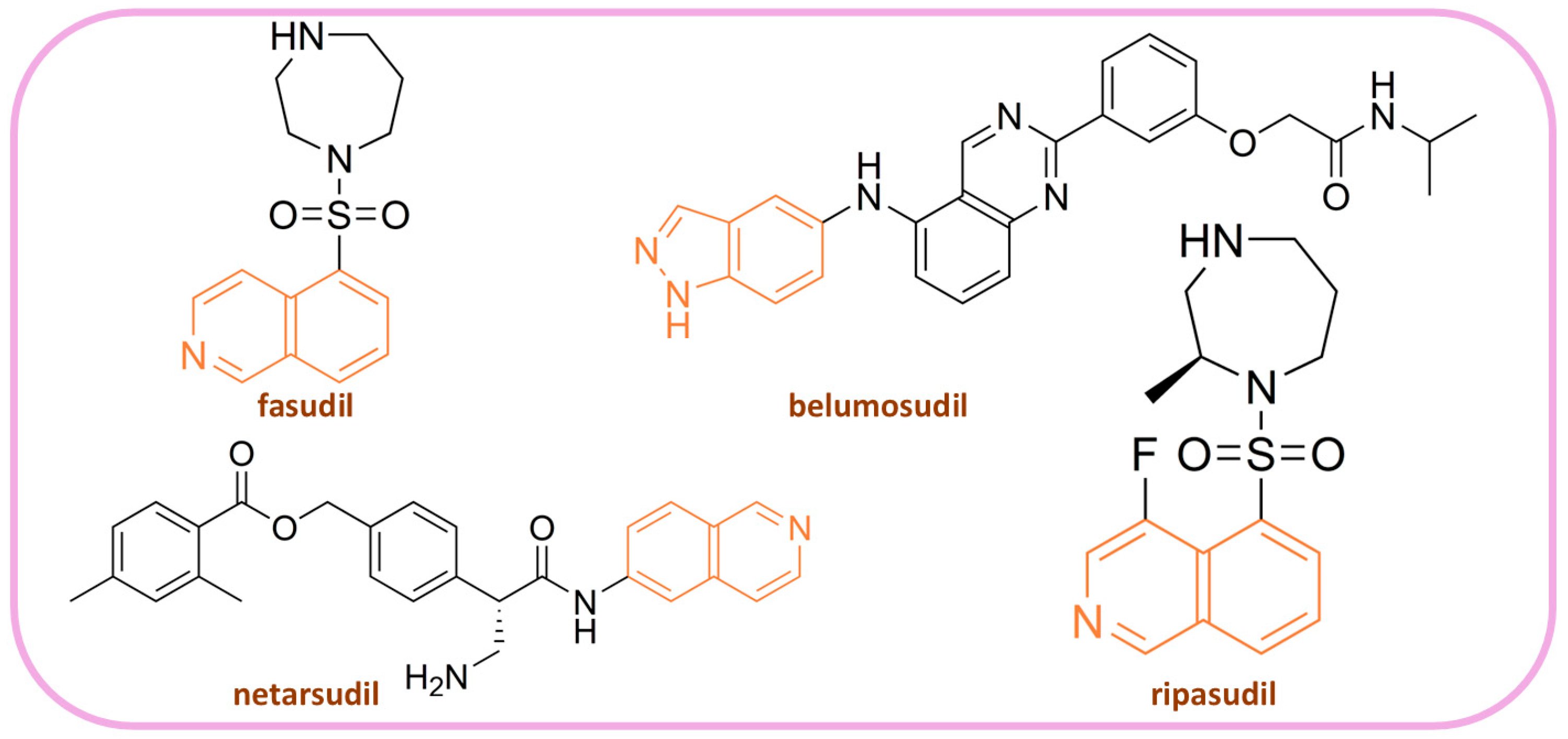
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel Insights into the Roles of Rho Kinase in Cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef]
- Lee, M.-H.; Kundu, J.K.; Chae, J.-I.; Shim, J.-H. Targeting ROCK/LIMK/Cofilin Signaling Pathway in Cancer. Arch. Pharm. Res. 2019, 42, 481–491. [Google Scholar] [CrossRef]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef]
- Julian, L.; Olson, M.F. Rho-Associated Coiled-Coil Containing Kinases (ROCK): Structure, Regulation, and Functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and Activation of Myosin by Rho-Associated Kinase (Rho-Kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the Actin Cytoskeleton Through Protein Kinases ROCK and LIM-Kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Barcelo, J.; Samain, R.; Sanz-Moreno, V. Preclinical to Clinical Utility of ROCK Inhibitors in Cancer. Trends Cancer 2023, 9, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wu, X.; Surma, M.; Vemula, S.; Zhang, L.; Yang, Y.; Kapur, R.; Wei, L. Distinct Roles for ROCK1 and ROCK2 in the Regulation of Cell Detachment. Cell Death Dis. 2013, 4, e483. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.A.; Badoual, M.; Asmussen, H.; Patel, H.; Whitmore, L.; Horwitz, A.R. ROCK1 and 2 Differentially Regulate Actomyosin Organization to Drive Cell and Synaptic Polarity. J. Cell Biol. 2015, 210, 225–242. [Google Scholar] [CrossRef]
- Yin, L.; Morishige, K.; Takahashi, T.; Hashimoto, K.; Ogata, S.; Tsutsumi, S.; Takata, K.; Ohta, T.; Kawagoe, J.; Takahashi, K.; et al. Fasudil Inhibits Vascular Endothelial Growth Factor–Induced Angiogenesis in Vitro and in Vivo. Mol. Cancer Ther. 2007, 6, 1517–1525. [Google Scholar] [CrossRef]
- Ying, H.; Biroc, S.L.; Li, W.; Alicke, B.; Xuan, J.-A.; Pagila, R.; Ohashi, Y.; Okada, T.; Kamata, Y.; Dinter, H. The Rho Kinase Inhibitor Fasudil Inhibits Tumor Progression in Human and Rat Tumor Models. Mol. Cancer Ther. 2006, 5, 2158–2164. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Hoff, D.D.V.; Han, H. Inhibition of ROCK1 Kinase Modulates Both Tumor Cells and Stromal Fibroblasts in Pancreatic Cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar] [CrossRef]
- Vennin, C.; Rath, N.; Pajic, M.; Olson, M.F.; Timpson, P. Targeting ROCK Activity to Disrupt and Prime Pancreatic Cancer for Chemotherapy. Small GTPases 2020, 11, 45–52. [Google Scholar] [CrossRef]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; del Monte-Nieto, G.; Conway, J.R.W.; et al. Transient Tissue Priming via ROCK Inhibition Uncouples Pancreatic Cancer Progression, Sensitivity to Chemotherapy, and Metastasis. Sci. Transl. Med. 2017, 9, eaai8504. [Google Scholar] [CrossRef]
- Meng, F.; Su, Y.; Xu, B. Rho-associated Protein Kinase-dependent Moesin Phosphorylation Is Required for PD-L1 Stabilization in Breast Cancer. Mol. Oncol. 2020, 14, 2701–2712. [Google Scholar] [CrossRef]
- Djokovic, N.; Djuric, A.; Ruzic, D.; Srdic-Rajic, T.; Nikolic, K. Correlating Basal Gene Expression across Chemical Sensitivity Data to Screen for Novel Synergistic Interactors of HDAC Inhibitors in Pancreatic Carcinoma. Pharmaceuticals 2023, 16, 294. [Google Scholar] [CrossRef] [PubMed]
- Iskit, S.; Lieftink, C.; Halonen, P.; Shahrabi, A.; Possik, P.A.; Beijersbergen, R.L.; Peeper, D.S. Integrated in Vivo Genetic and Pharmacologic Screening Identifies Co-Inhibition of EGRF and ROCK as a Potential Treatment Regimen for Triple-Negative Breast Cancer. Oncotarget 2016, 7, 42859–42872. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.; Kumar, R.; Papadatos-Pastos, D.; Mateo, J.; Brown, J.S.; Garces, A.H.I.; Ruddle, R.; Decordova, S.; Jueliger, S.; Ferraldeschi, R.; et al. First-in-Human Study of AT13148, a Dual ROCK-AKT Inhibitor in Patients with Solid Tumors. Clin. Cancer Res. 2020, 26, 4777–4784. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone Deacetylase 4 Controls Chondrocyte Hypertrophy during Skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.-L.; Chua, K.; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Mol. Cell Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef]
- Kaur, S.; Rajoria, P.; Chopra, M. HDAC6: A Unique HDAC Family Member as a Cancer Target. Cell Oncol. 2022, 45, 779–829. [Google Scholar] [CrossRef]
- Cuttini, E.; Goi, C.; Pellarin, E.; Vida, R.; Brancolini, C. HDAC4 in Cancer: A Multitasking Platform to Drive Not Only Epigenetic Modifications. Front. Mol. Biosci. 2023, 10, 1116660. [Google Scholar] [CrossRef] [PubMed]
- Mertsch, S.; Krämer, O.H. The Interplay between Histone Deacetylases and Rho Kinases Is Important for Cancer and Neurodegeneration. Cytokine Growth Factor Rev. 2017, 37, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Büttner, F.H.; Chen, R.; Hickey, E.; Jakes, S.; Kaplita, P.; Kashem, M.A.; Kerr, S.; Kugler, S.; Paw, Z.; et al. Substituted 2H-Isoquinolin-1-One as Potent Rho-Kinase Inhibitors. Part 1: Hit-to-Lead Account. Bioorganic Med. Chem. Lett. 2010, 20, 3235–3239. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Martin, M.P.; Liu, Y.; Wang, B.; Patel, R.A.; Zhu, J.-Y.; Sun, N.; Pireddu, R.; Lawrence, N.J.; Li, J.; et al. Fragment-Based and Structure-Guided Discovery and Optimization of Rho Kinase Inhibitors. J. Med. Chem. 2012, 55, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Sessions, E.H.; Chowdhury, S.; Yin, Y.; Pocas, J.R.; Grant, W.; Schröter, T.; Lin, L.; Ruiz, C.; Cameron, M.D.; LoGrasso, P.; et al. Discovery and Optimization of Indole and 7-Azaindoles as Rho Kinase (ROCK) Inhibitors (Part-II). Bioorganic Med. Chem. Lett. 2011, 21, 7113–7118. [Google Scholar] [CrossRef]
- ChEMBL Database. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 2 August 2024).
- Marvin. Available online: https://chemaxon.com/marvin (accessed on 2 August 2024).
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Hehre, W.J. Ab Initio Molecular Orbital Theory. Acc. Chem. Res. 1976, 9, 399–406. [Google Scholar] [CrossRef]
- Frisch, M. Gaussian 09, Revision d.01; Gaussian Inc.: Wallingford, CT, USA, 2009; Volume 201. [Google Scholar]
- CambridgeSoft ChemBio3D Ultra. Get the Software Safely and Easily. Available online: https://cambridgesoft-chembio3d-ultra.software.informer.com/13.0/ (accessed on 2 August 2024).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Bank RPD. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 13 November 2023).
- Martínez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef]
- BIOVIA. Available online: https://www.3ds.com/products/biovia (accessed on 2 August 2024).
- Pastor, M.; Cruciani, G.; McLay, I.; Pickett, S.; Clementi, S. GRid-INdependent Descriptors (GRIND): A Novel Class of Alignment-Independent Three-Dimensional Molecular Descriptors. J. Med. Chem. 2000, 43, 3233–3243. [Google Scholar] [CrossRef]
- Artese, A.; Cross, S.; Costa, G.; Distinto, S.; Parrotta, L.; Alcaro, S.; Ortuso, F.; Cruciani, G. Molecular Interaction Fields in Drug Discovery: Recent Advances and Future Perspectives. WIREs Comput. Mol. Sci. 2013, 3, 594–613. [Google Scholar] [CrossRef]
- Baroni, M.; Costantino, G.; Cruciani, G.; Riganelli, D.; Valigi, R.; Clementi, S. Generating Optimal Linear PLS Estimations (GOLPE): An Advanced Chemometric Tool for Handling 3D-QSAR Problems. Quant. Struct. Act. Relat. 1993, 12, 9–20. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of Q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative Studies on Some Metrics for External Validation of QSPR Models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a Simple Approach for Determining Applicability Domain of QSAR Models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Reers, M.; Smiley, S.T.; Mottola-Hartshorn, C.; Chen, A.; Lin, M.; Chen, L.B. [29] Mitochondrial Membrane Potential Monitored by JC-1 Dy. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1995; Volume 260, pp. 406–417. ISBN 978-0-12-182161-6. [Google Scholar]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Badr-Eldin, S.M.; Aldawsari, H.M.; Kotta, S.; Deb, P.K.; Venugopala, K.N. Three-Dimensional In Vitro Cell Culture Models for Efficient Drug Discovery: Progress So Far and Future Prospects. Pharmaceuticals 2022, 15, 926. [Google Scholar] [CrossRef]
- Guan, G.; Cannon, R.D.; Coates, D.E.; Mei, L. Effect of the Rho-Kinase/ROCK Signaling Pathway on Cytoskeleton Components. Genes 2023, 14, 272. [Google Scholar] [CrossRef] [PubMed]
- Molinie, N.; Gautreau, A. Directional Collective Migration in Wound Healing Assays. In Cell Migration: Methods and Protocols; Gautreau, A., Ed.; Springer: New York, NY, USA, 2018; pp. 11–19. ISBN 978-1-4939-7701-7. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Internal Validation Parameters | ||||
| R2 | Q2LOO | RMSEE | ||
| 0.914 | 0.59 | 0.393 | ||
| Criteria | >0.9 | >0.5 | ||
| External Validation Parameters | ||||
| R2pred | RMSEP | rm2 | r/2m | |
| 0.792 | 0.561 | 0.759 | 0.573 | |
| Criteria | >0.6 | ≤2RMSEE | >0.5 | >0.5 |
| Δr2m | (r2 − r/20)/r2 | k’ | ||
| 0.666 | 0.186 | 0.099 | 0.974 | |
| Criteria | >0.5 | <0.2 | <0.1 | 0.85 ≤ k’ ≤ 1.15 |
| Internal Validation Parameters | ||||
| R2 | Q2LOO | RMSEE | ||
| 0.933 | 0.54 | 0.327 | ||
| Criteria | >0.9 | >0.5 | ||
| External Validation Parameters | ||||
| R2pred | RMSEP | rm2 | r/2m | |
| 0.851 | 0.499 | 0.753 | 0.616 | |
| Criteria | >0.6 | ≤2RMSEE | >0.5 | >0.5 |
| Δr2m | (r2 − r/20)/r2 | k’ | ||
| 0.684 | 0.137 | 0.032 | 0.963 | |
| Criteria | >0.5 | <0.2 | <0.1 | 0.85 ≤ k’ ≤ 1.15 |
| Cd | ROCK1 | ROCK2 | ||||
|---|---|---|---|---|---|---|
| pIC50 pred | pIC50 exp | pIC50 exp − pIC50 pred | pIC50 pred | pIC50 exp | pIC50 exp − pIC50 pred | |
| C-19 | 5.76 | 5.37 | −0.39 | 5.61 | 5.21 | −0.40 |
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| MDA-MB-231 | HCC1937 | MiaPaCa-2 | Panc-1 | MRC-5 | |
| C-19 | >50 (n.d) | >50 (n.d) | 19.10 ± 1.89 (4.1) | 35.03 ± 4.43 (2.2) | 78.25 ± 4.22 |
| C-22 | >50 (n.d) | >50 (n.d) | >50 (n.d) | >50 (n.d) | >50 |
| Fasudil | >50 (n.d) | >50 (n.d) | >50 (n.d) | >50 (n.d) | 92.02 ± 3.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beljkas, M.; Petkovic, M.; Vuletic, A.; Djuric, A.; Santibanez, J.F.; Srdic-Rajic, T.; Nikolic, K.; Oljacic, S. Development of Novel ROCK Inhibitors via 3D-QSAR and Molecular Docking Studies: A Framework for Multi-Target Drug Design. Pharmaceutics 2024, 16, 1250. https://doi.org/10.3390/pharmaceutics16101250
Beljkas M, Petkovic M, Vuletic A, Djuric A, Santibanez JF, Srdic-Rajic T, Nikolic K, Oljacic S. Development of Novel ROCK Inhibitors via 3D-QSAR and Molecular Docking Studies: A Framework for Multi-Target Drug Design. Pharmaceutics. 2024; 16(10):1250. https://doi.org/10.3390/pharmaceutics16101250
Chicago/Turabian StyleBeljkas, Milan, Milos Petkovic, Ana Vuletic, Ana Djuric, Juan Francisco Santibanez, Tatjana Srdic-Rajic, Katarina Nikolic, and Slavica Oljacic. 2024. "Development of Novel ROCK Inhibitors via 3D-QSAR and Molecular Docking Studies: A Framework for Multi-Target Drug Design" Pharmaceutics 16, no. 10: 1250. https://doi.org/10.3390/pharmaceutics16101250