
Targeted Delivery of Celastrol by GA-Modified Liposomal Calcium Carbonate Nanoparticles to Enhance Antitumor Efficacy Against Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Preparation and Characterization of CEL-Loaded GA/LCC Core-Shell Nanoparticles
2.4. In Vitro Drug Release Assay
2.5. In Vitro Cellular Uptake
2.6. Determination of Intracellular ROS Concentration
2.7. Rhodamine 123 Staining
2.8. In Vitro Cytotoxicity and Apoptosis Assay
2.9. In Vitro Cell Migration and Invasion Tests
2.10. Inhibitory Effect on the Tumor Spheroids
2.11. In Vitro Western Blotting Studies
2.12. Statistical Analysis
3. Results and Discussion
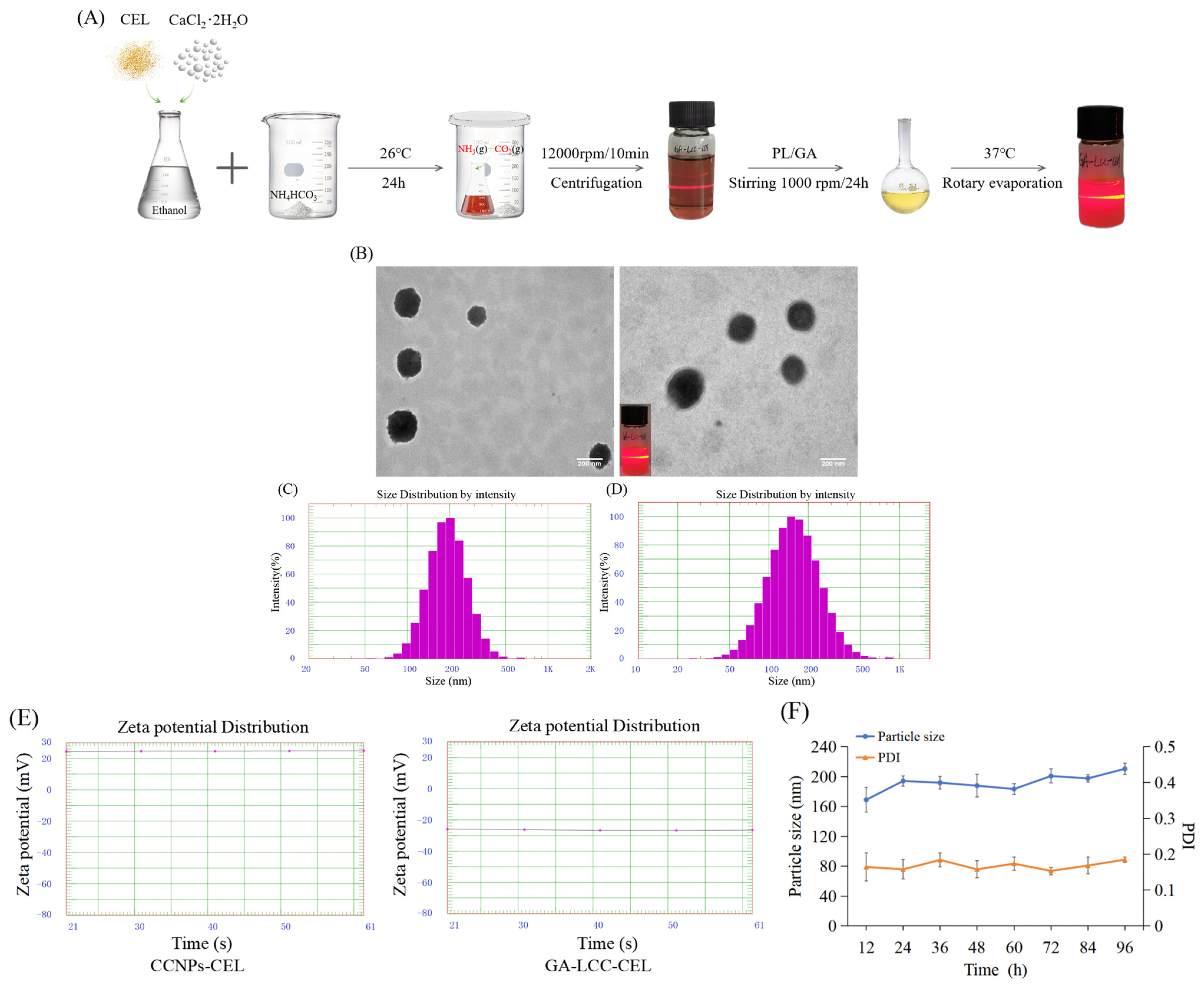
3.1. Preparation and Characterization of CEL-Loaded GA/LCC Core-Shell Nanoparticles
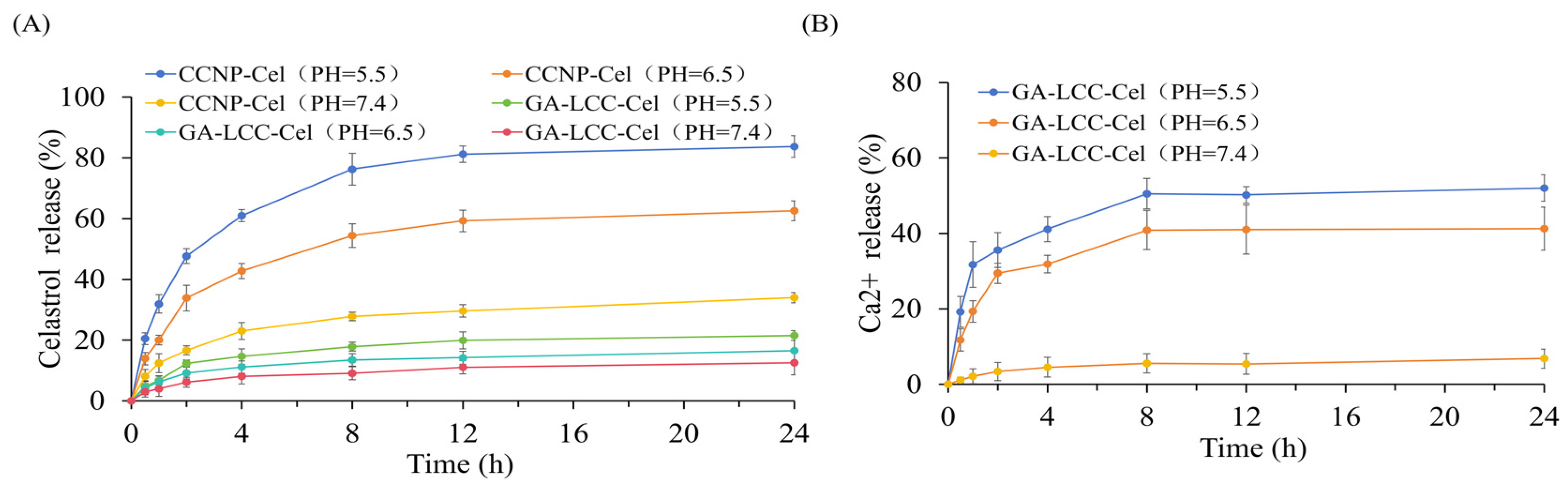
3.2. In Vitro Drug Release Assay
3.3. In Vitro Cellular Uptake
3.4. In Vitro Mitochondrial Dysfunction
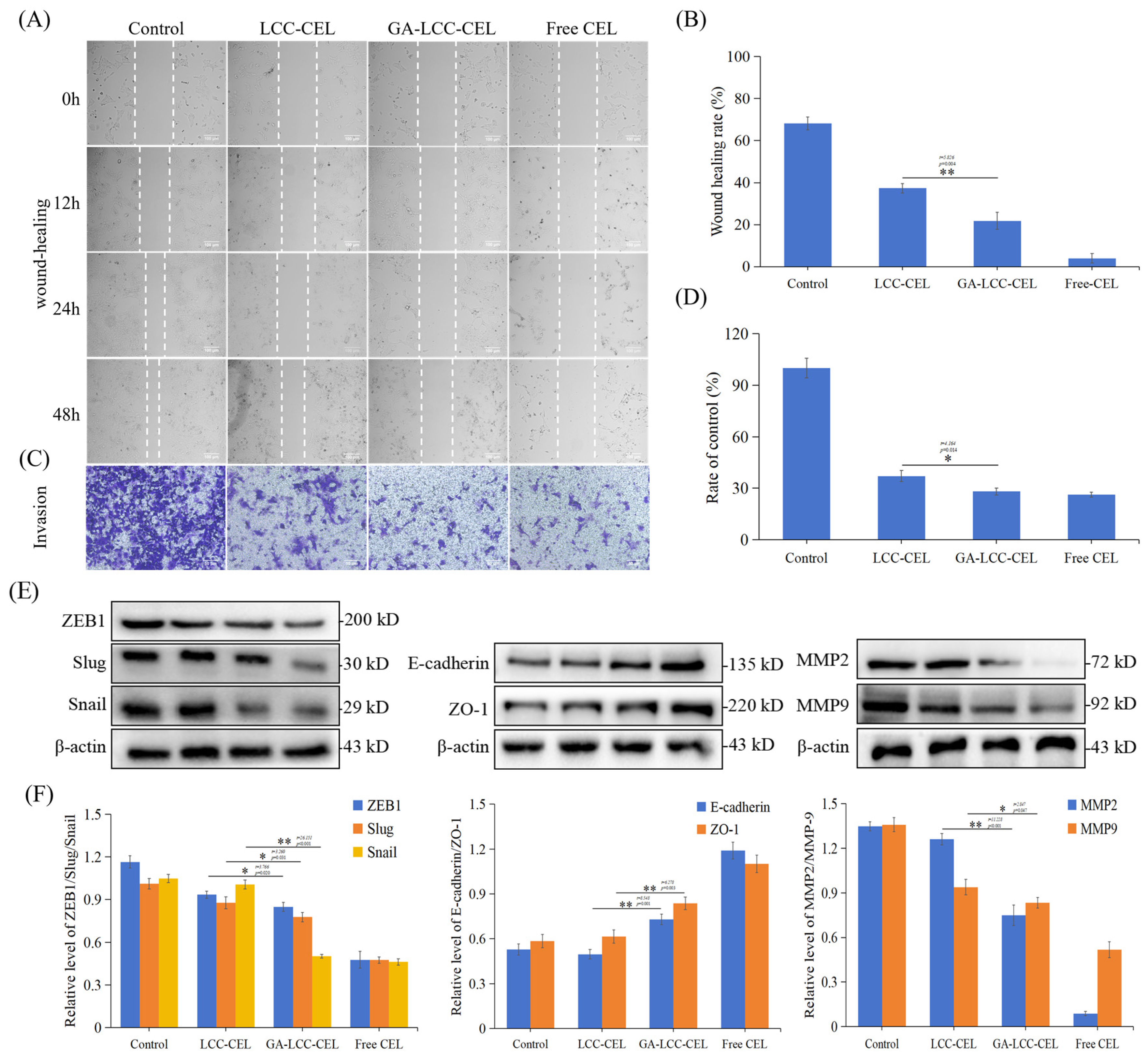
3.5. In Vitro Cell Migration and Invasion Tests
3.6. Inhibitory Effect on the Tumor Spheroids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quijia, C.R.; Enríquez, A.Q.; Zappia, C.D.; Peroni, R.N.; Chorilli, M. Administration of Inhibitory Molecules through Nanoparticles in Breast Cancer Therapy. Curr. Med. Chem. 2024, 31, 726–761. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ge, J.; Chen, Y.; Liu, T.; Chen, L.; Liu, C.; Ma, D.; Cai, Y.; Xu, Y.; Shao, Z.; et al. Combined Single-Cell and Spatial Transcriptomics Reveal the Metabolic Evolvement of Breast Cancer during Early Dissemination. Adv. Sci. 2023, 10, e2205395. [Google Scholar] [CrossRef] [PubMed]
- Akbarali, H.I.; Muchhala, K.H.; Jessup, D.K.; Cheatham, S. Chemotherapy induced gastrointestinal toxicities. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2022; Volume 155, pp. 131–166. [Google Scholar] [CrossRef]
- Gupta, M.; Chandan, K.; Sarwat, M. Natural products and their derivatives as immune check point inhibitors: Targeting cytokine/chemokine signalling in cancer. Semin. Cancer Biol. 2022, 86, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, K.; Zhang, X.; Zhou, Y.; Li, Z.; Shang, F. Celastrol Ameliorates Inflammation in Human Retinal Pigment Epithelial Cells by Suppressing NF-κB Signaling. J. Ocul. Pharmacol. Ther. 2019, 35, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Fan, R.; Wang, L.; He, W.; Ge, H.; Chen, H.; Xu, W.; Zhang, J.; Xu, W.; Feng, Y.; et al. Synthesis and biological evaluation of celastrol derivatives as potent antitumor agents with STAT3 inhibition. J. Enzym. Inhib. Med. Chem. 2021, 37, 236–251. [Google Scholar] [CrossRef]
- He, Q.-W.; Feng, J.-H.; Hu, X.-L.; Long, H.; Huang, X.-F.; Jiang, Z.-Z.; Zhang, X.-Q.; Ye, W.-C.; Wang, H. Synthesis and Biological Evaluation of Celastrol Derivatives as Potential Immunosuppressive Agents. J. Nat. Prod. 2020, 83, 2578–2586. [Google Scholar] [CrossRef]
- Nakayama, T.; Okimura, K.; Shen, J.; Guh, Y.-J.; Tamai, T.K.; Shimada, A.; Minou, S.; Okushi, Y.; Shimmura, T.; Furukawa, Y.; et al. Seasonal changes in NRF2 antioxidant pathway regulates winter depression-like behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 9594–9603. [Google Scholar] [CrossRef]
- Yang, X.; Wu, F.; Li, L.; Lynch, E.C.; Xie, L.; Zhao, Y.; Fang, K.; Li, J.; Luo, J.; Xu, L.; et al. Celastrol alleviates metabolic disturbance in high-fat diet-induced obese mice through increasing energy expenditure by ameliorating metabolic inflammation. Phytother. Res. 2020, 35, 297–310. [Google Scholar] [CrossRef]
- Corson, T.W.; Crews, C.M. Molecular Understanding and Modern Application of Traditional Medicines: Triumphs and Trials. Cell 2007, 130, 769–774. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, H.; Ding, C.; Jiang, D.; Zhao, Z.; Li, Y.; Ding, X.; Gao, J.; Zhou, H.; Luo, C.; et al. Celastrol suppresses colorectal cancer via covalent targeting peroxiredoxin 1. Signal Transduct. Target. Ther. 2023, 8, 51. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, X.; Zhao, P.; Zhao, H.; Gao, W.; Wang, L. Celastrol Suppresses Glioma Vasculogenic Mimicry Formation and Angiogenesis by Blocking the PI3K/Akt/mTOR Signaling Pathway. Front. Pharmacol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, Z.; Wang, X.; Zhang, T.; Hu, Y.; Wang, D.; Sun, H.; Zhang, L.; Zhu, Y. Therapeutic effect of multifunctional celastrol nanoparticles with mitochondrial alkaline drug release in breast cancer. Mater. Today Adv. 2022, 17, 100328. [Google Scholar] [CrossRef]
- Xu, L.-N.; Zhao, N.; Chen, J.-Y.; Ye, P.-P.; Nan, X.-W.; Zhou, H.-H.; Jiang, Q.-W.; Yang, Y.; Huang, J.-R.; Yuan, M.-L.; et al. Celastrol Inhibits the Growth of Ovarian Cancer Cells in vitro and in vivo. Front. Oncol. 2019, 9, 2. [Google Scholar] [CrossRef]
- Lim, H.Y.; Ong, P.S.; Wang, L.; Goel, A.; Ding, L.; Wong, A.L.-A.; Ho, P.C.-L.; Sethi, G.; Xiang, X.; Goh, B.C. Celastrol in cancer therapy: Recent developments, challenges and prospects. Cancer Lett. 2021, 521, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Wagh, P.R.; Desai, P.; Prabhu, S.; Wang, J. Nanotechnology-Based Celastrol Formulations and Their Therapeutic Applications. Front. Pharmacol. 2021, 12, 673209. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.; Teja, B.V.; Pandey, G.; Mittapelly, N.; Trivedi, R.; Mishra, P. An insight into functionalized calcium based inorganic nanomaterials in biomedicine: Trends and transitions. Colloids Surf. B Biointerfaces 2015, 133, 120–139. [Google Scholar] [CrossRef]
- Martínez-Parra, L.; Piñol-Cancer, M.; Sanchez-Cano, C.; Miguel-Coello, A.B.; Di Silvio, D.; Gomez, A.M.; Uriel, C.; Plaza-García, S.; Gallego, M.; Pazos, R.; et al. A Comparative Study of Ultrasmall Calcium Carbonate Nanoparticles for Targeting and Imaging Atherosclerotic Plaque. ACS Nano 2023, 17, 13811–13825. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, S.; Song, H.; Yu, T.; Zheng, X.; Chu, Q. CaCO3 nanoparticles incorporated with KAE to enable amplified calcium overload cancer therapy. Biomaterials 2021, 277, 121080. [Google Scholar] [CrossRef]
- Zhu, T.; Dittrich, M. Carbonate Precipitation through Microbial Activities in Natural Environment, and Their Potential in Biotechnology: A Review. Front. Bioeng. Biotechnol. 2016, 4, 4. [Google Scholar] [CrossRef]
- Wei, K.; Zhang, J.; Li, X.; Shi, P.; Fu, P. High density lipoprotein coated calcium carbonate nanoparticle for chemotherapy of breast cancer. J. Biomater. Appl. 2019, 34, 178–187. [Google Scholar] [CrossRef]
- Ding, Y.; Brand, E.; Wang, W.; Zhao, Z. Licorice: Resources, applications in ancient and modern times. J. Ethnopharmacol. 2022, 298, 115594. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, R.; Yuan, B.; Liu, Y.; Liu, C. The antiviral and antimicrobial activities of licorice, a widely-used Chinese herb. Acta Pharm. Sin. B 2015, 5, 310–315. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Lv, J. Effects of glycyrrhizin on the pharmacokinetics of paeoniflorin in rats and its potential mechanism. Pharm. Biol. 2019, 57, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sheng, Z.; Xiao, J.; Li, Y.; Huang, J.; Jia, J.; Zeng, X.; Li, L. Advances in the roles of glycyrrhizic acid in cancer therapy. Front. Pharmacol. 2023, 14, 1265172. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, J.; Mu, Y.; Foda, M.F.; Han, H. Activation of TRPV1 by capsaicin-loaded CaCO3 nanoparticle for tumor-specific therapy. Biomaterials 2022, 284, 121520. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Mann, S. Emergent Nanostructures: Water-Induced Mesoscale Transformation of Surfactant-Stabilized Amorphous Calcium Carbonate Nanoparticles in Reverse Microemulsions. Adv. Funct. Mater. 2002, 12, 773–779. [Google Scholar] [CrossRef]
- Tong, H.; Ma, W.; Wang, L.; Wan, P.; Hu, J.; Cao, L. Control over the crystal phase, shape, size and aggregation of calcium carbonate via a l-aspartic acid inducing process. Biomaterials 2004, 25, 3923–3929. [Google Scholar] [CrossRef]
- Zhao, Y.; Luo, Z.; Li, M.; Qu, Q.; Ma, X.; Yu, S.; Zhao, Y. A Preloaded Amorphous Calcium Carbonate/Doxorubicin@Silica Nanoreactor for pH-Responsive Delivery of an Anticancer Drug. Angew. Chem. Int. Ed. 2014, 54, 919–922. [Google Scholar] [CrossRef]
- Wang, C.; Han, M.; Liu, X.; Chen, S.; Hu, F.; Sun, J.; Yuan, H. Mitoxantrone-preloaded water-responsive phospholipid-amorphous calcium carbonate hybrid nanoparticles for targeted and effective cancer therapy. Int. J. Nanomed. 2019, 14, 1503–1517. [Google Scholar] [CrossRef]
- Moulin, P.; Roques, H. Zeta potential measurement of calcium carbonate. J. Colloid Interface Sci. 2003, 261, 115–126. [Google Scholar] [CrossRef]
- Zhou, F.; Li, H.; Liu, Y.; Deng, H.; Rong, J.; Zhao, J. Hyaluronan derivative decorated calcium carbonate nanoparticle as a potential platform for breast cancer synergistic therapy via blood coagulation and drug delivery. J. Drug Deliv. Sci. Technol. 2023, 83, 104406. [Google Scholar] [CrossRef]
- Sadeghi, M.; Ordway, B.; Rafiei, I.; Borad, P.; Fang, B.; Koomen, J.L.; Zhang, C.; Yoder, S.; Johnson, J.; Damaghi, M. Integrative Analysis of Breast Cancer Cells Reveals an Epithelial-Mesenchymal Transition Role in Adaptation to Acidic Microenvironment. Front. Oncol. 2020, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, Y.; Wu, Y.; Chen, J.; Seto, S.-W.; Leung, G.P.-H.; Cai, Y.; Li, J.; Zhang, J. Glycyrrhizic Acid-Lipid Framework Nanovehicle Loading Triptolide for Combined Immunochemotherapy. ACS Appl. Mater. Interfaces 2023, 15, 41337–41350. [Google Scholar] [CrossRef]
- Selyutina, O.Y.; Apanasenko, I.; Kim, A.; Shelepova, E.A.; Khalikov, S.S.; Polyakov, N.E. Spectroscopic and molecular dynamics characterization of glycyrrhizin membrane-modifying activity. Colloids Surf. B Biointerfaces 2016, 147, 459–466. [Google Scholar] [CrossRef]
- Su, X.; Wu, L.; Hu, M.; Dong, W.; Xu, M.; Zhang, P. Glycyrrhizic acid: A promising carrier material for anticancer therapy. Biomed. Pharmacother. 2017, 95, 670–678. [Google Scholar] [CrossRef]
- Selyutina, O.Y.; Polyakov, N.E.; Korneev, D.V.; Zaitsev, B.N. Influence of glycyrrhizin on permeability and elasticity of cell membrane: Perspectives for drugs delivery. Drug Deliv. 2014, 23, 848–855. [Google Scholar] [CrossRef]
- Ye, S.; Luo, W.; Khan, Z.A.; Wu, G.; Xuan, L.; Shan, P.; Lin, K.; Chen, T.; Wang, J.; Hu, X.; et al. Celastrol Attenuates Angiotensin II–Induced Cardiac Remodeling by Targeting STAT3. Circ. Res. 2020, 126, 1007–1023. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y.; Luo, W.; Chen, S.; Lin, F.; Zhang, X.; Fan, S.; Shen, X.; Wang, Y.; Liang, G. Celastrol induces ROS-mediated apoptosis via directly targeting peroxiredoxin-2 in gastric cancer cells. Theranostics 2020, 10, 10290–10308. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Zhang, Q.; Shen, S.; An, Y.; Yuan, L.; Wong, Y.-K.; Huang, S.; Huang, S.; Huang, J.; Cheng, G.; et al. Mechanistic engineering of celastrol liposomes induces ferroptosis and apoptosis by directly targeting VDAC2 in hepatocellular carcinoma. Asian J. Pharm. Sci. 2023, 18, 100874. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Li, X.; Xu, H.; Yang, J.; Wang, C.; Zhang, C.; Deng, Y.; Lu, A.; Zheng, C.; et al. Carrier-free self-assembled nanomedicine based on celastrol and galactose for targeting therapy of hepatocellular carcinoma via inducing ferroptosis. Eur. J. Med. Chem. 2024, 267, 116183. [Google Scholar] [CrossRef]
- Wu, L.; Lin, Y.; Song, J.; Li, L.; Rao, X.; Wan, W.; Wei, G.; Hua, F.; Ying, J. TMEM175: A lysosomal ion channel associated with neurological diseases. Neurobiol. Dis. 2023, 185, 106244. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, S.; Xu, H. Not just protons: Chloride also activates lysosomal acidic hydrolases. J. Cell Biol. 2023, 222, e202305007. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, G.K.; Heisner, J.S.; Camara, A.K.; Kwok, W.-M. Electrophysiological characterization of mitochondrial currents at the contact site of the inner and outer membrane. Biophys. J. 2022, 121, 315a–316a. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 2020, 36, 101678. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Czabotar, P.E. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family Effector Proteins. Cold Spring Harb. Perspect. Biol. 2019, 12, a036319. [Google Scholar] [CrossRef]
- Murad, F.; Garcia-Saez, A.J. Bcl-xL inhibits tBid and Bax via distinct mechanisms. Faraday Discuss. 2020, 232, 86–102. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Manfioletti, G.; Fedele, M. Epithelial–Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2023, 24, 11386. [Google Scholar] [CrossRef]
- Shin, A.E.; Giancotti, F.G.; Rustgi, A.K. Metastatic colorectal cancer: Mechanisms and emerging therapeutics. Trends Pharmacol. Sci. 2023, 44, 222–236. [Google Scholar] [CrossRef]
- Xue, W.; Hao, J.; Zhang, Q.; Jin, R.; Luo, Z.; Yang, X.; Liu, Y.; Lu, Q.; Ouyang, Y.; Guo, H. Chlorogenic Acid Inhibits Epithelial-Mesenchymal Transition and Invasion of Breast Cancer by Down-Regulating LRP6. J. Pharmacol. Exp. Ther. 2022, 384, 254–264. [Google Scholar] [CrossRef]
- Chen, T.; Jia, W.; Zhang, B.; Xie, H.; Wu, Q. EMT transcription factors activated circuits: A novel tool to study EMT dynamics and its therapeutic implications. Synth. Syst. Biotechnol. 2023, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Pedrosa, L.; Vila, I.; Chen, Y.; Győrffy, B.; Sánchez-Moral, L.; Siles, L.; Lozano, J.J.; Esteve-Codina, A.; Darling, D.S.; et al. The EMT factor ZEB1 paradoxically inhibits EMT in BRAF-mutant carcinomas. J. Clin. Investig. 2023, 8, e164629. [Google Scholar] [CrossRef]
- Arab-Bafrani, Z.; Zabihi, E.; Jafari, S.M.; Khoshbin-Khoshnazar, A.; Mousavi, E.; Khalili, M.; Babaei, A. Enhanced radiotherapy efficacy of breast cancer multi cellular tumor spheroids through in-situ fabricated chitosan-zinc oxide bio-nanocomposites as radio-sensitizing agents. Int. J. Pharm. 2021, 605, 120828. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Song, J.; Shi, F.; Zhang, Z.; Zhu, F.; Xue, J.; Tan, X.; Zhang, L.; Jia, X. Formulation and Evaluation of Celastrol-Loaded Liposomes. Molecules 2011, 16, 7880–7892. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, X.; Hu, J.; Qiu, Z.; Yuan, M.; Zheng, G. Celastrol-Loaded Galactosylated Liposomes Effectively Inhibit AKT/c-Met-Triggered Rapid Hepatocarcinogenesis in Mice. Mol. Pharm. 2020, 17, 738–747. [Google Scholar] [CrossRef]
- Xiao, L.; Zhou, B.; Luo, S.; Deng, L.; Xue, Y.; Zhang, L.; Du, Z.; Li, P.; Wang, L.; Tian, B.; et al. Liposomal co-delivery system encapsulating celastrol and paclitaxel displays highly enhanced efficiency and low toxicity against pancreatic cancer. J. Drug Deliv. Sci. Technol. 2022, 78, 103947. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Li, J.; Yue, L.; Ji, C. Targeted Delivery of Celastrol by GA-Modified Liposomal Calcium Carbonate Nanoparticles to Enhance Antitumor Efficacy Against Breast Cancer. Pharmaceutics 2024, 16, 1382. https://doi.org/10.3390/pharmaceutics16111382
Zhang W, Li J, Yue L, Ji C. Targeted Delivery of Celastrol by GA-Modified Liposomal Calcium Carbonate Nanoparticles to Enhance Antitumor Efficacy Against Breast Cancer. Pharmaceutics. 2024; 16(11):1382. https://doi.org/10.3390/pharmaceutics16111382
Chicago/Turabian StyleZhang, Wei, Jiping Li, Liling Yue, and Chenfeng Ji. 2024. "Targeted Delivery of Celastrol by GA-Modified Liposomal Calcium Carbonate Nanoparticles to Enhance Antitumor Efficacy Against Breast Cancer" Pharmaceutics 16, no. 11: 1382. https://doi.org/10.3390/pharmaceutics16111382
APA StyleZhang, W., Li, J., Yue, L., & Ji, C. (2024). Targeted Delivery of Celastrol by GA-Modified Liposomal Calcium Carbonate Nanoparticles to Enhance Antitumor Efficacy Against Breast Cancer. Pharmaceutics, 16(11), 1382. https://doi.org/10.3390/pharmaceutics16111382