


Concomitant Inhibition and Collaring of Dual-Species Biofilms Formed by Candida auris and Staphylococcus aureus by Triazole Based Small Molecule Inhibitors
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
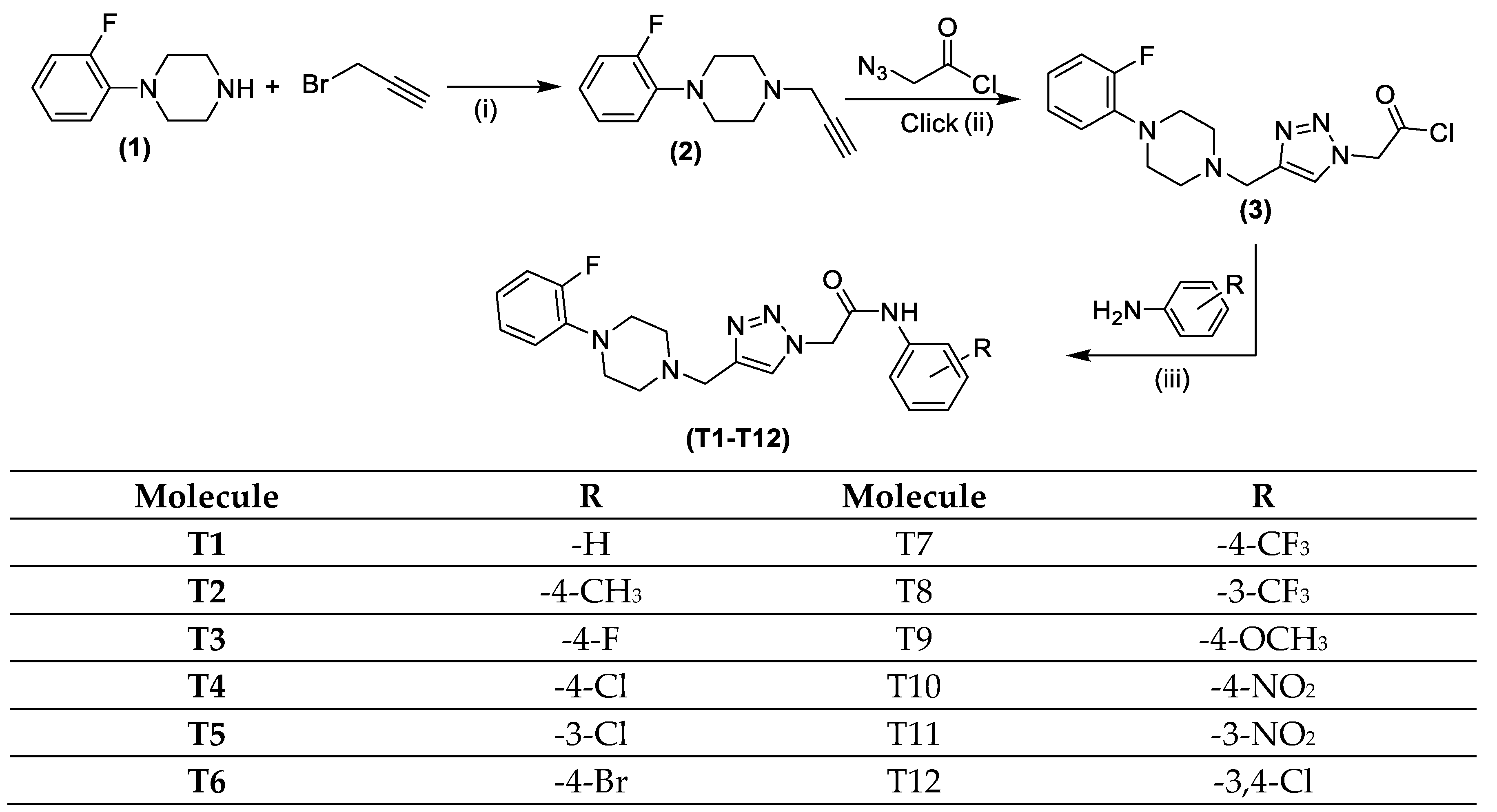
2.1. Chemistry
2.1.1. Click Synthesis of 2-(4-((4-(2-Fluorophenyl)piperazin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)acetyl Chloride (3)
2.1.2. Synthesis of Quinoline Based 1,2,3-Triazole Derivatives (T1–T12)
2.2. Pharmacokinetic Studies/ADMET Profile
2.3. Biological Studies
2.3.1. Fungal and Bacterial Strains Used in Study
2.3.2. Antifungal and Antibacterial Activity Against Individual Pathogen
2.3.3. Antifungal and Antibacterial Activity Against Mixed Pathogen
2.3.4. Anti-Biofilm Activity Against Single—And Mixed Biofilms
2.3.5. Impact on Microbial Viability
2.3.6. Impact on Total Biomass Quantification
2.3.7. T3 Extricates Dual—Species Biofilms Formed by C. auris and S. aureus
2.3.8. Scanning Electron Microscopy (SEM) Analysis
2.3.9. Hemolytic Assay for T3
2.3.10. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
3.2. Physicochemical Properties
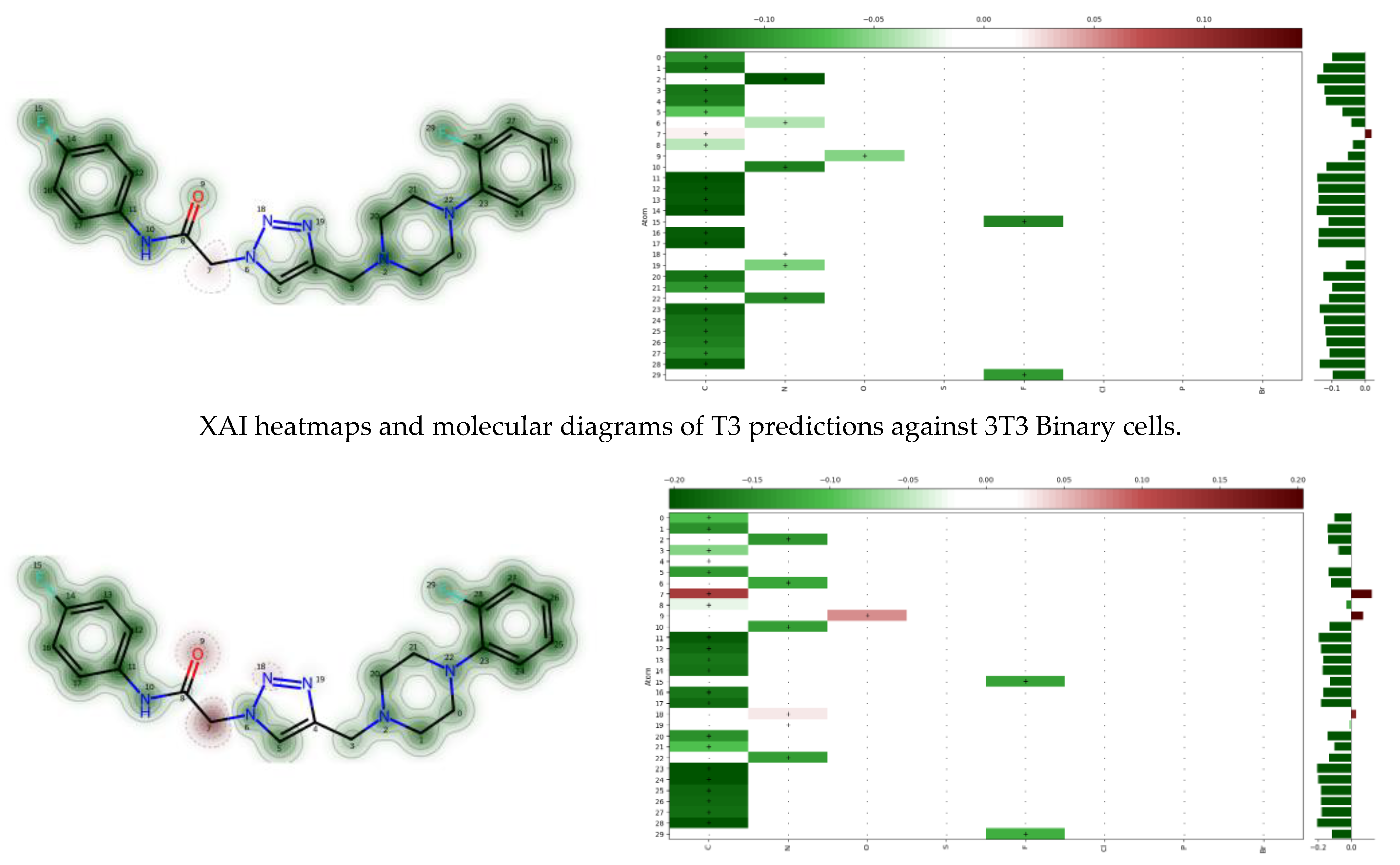
3.2.1. Pharmacokinetic Studies/ADMET Profile
3.2.2. Bioavailability
3.2.3. Toxicity Analysis and Safety Profiling
3.3. Biology
3.3.1. Candidicidal and Bactericidal Activity of the Compounds T1–T12
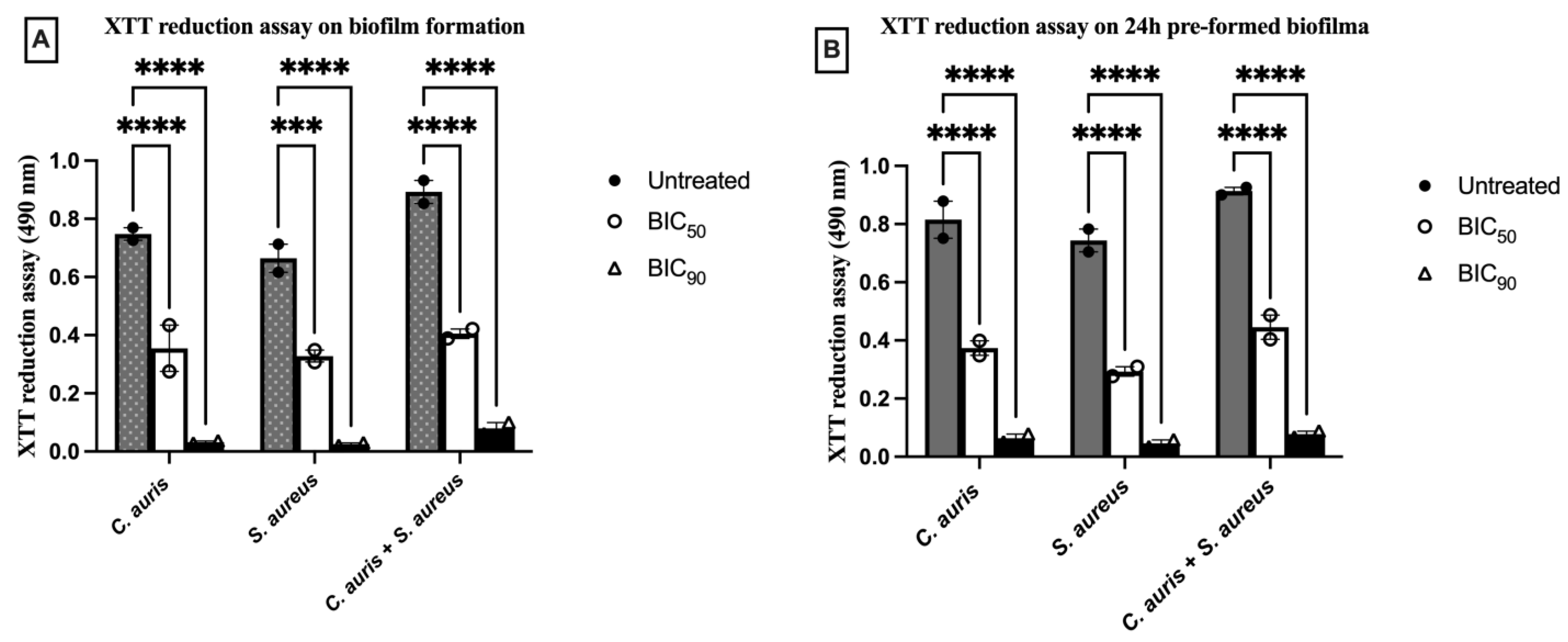
3.3.2. T3 Prohibited Biofilm Formation and Smashed Mature Biofilm Formed by Single and Dual—Species
3.3.3. T3 Impact over Cellular Viability of Pathogens in Single and Dual—Species Biofilm
3.3.4. T3 Drastically Effects Total Biomass in Single and Dual—Species Biofilm
3.3.5. Con-A and FUN-1 Staining Further Confirmed the Anti-Biofilms Property of T3
3.3.6. SEM for Structural Analysis of Dual—Species Biofilms upon Treatment with T3
3.3.7. Cytotoxicity
3.3.8. Cytotoxicity of T3 Against RAW 264.7 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Welsh, R.M.; Bentz, M.L.; Shams, A.; Houston, H.; Lyons, A.; Rose, L.J.; Litvintseva, A.P. Survival, persistence, and isolation of the emerging multidrug-resistant pathogenic yeast Candida auris on a plastic health care surface. J. Clin. Microbiol. 2017, 55, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef] [PubMed]
- Mirghani, R.; Saba, T.; Khaliq, H.; Mitchell, J.; Do, L.; Chambi, L.; Diaz, K.; Kennedy, T.; Alkassab, K.; Huynh, T.; et al. Biofilms: Formation, drug resistance and alternatives to conventional approaches. AIMS Microbiol. 2022, 8, 239–277. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Rehman, M.T.; Shahwan, M.; Aatif, M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef] [PubMed]
- Kean, R.; McKloud, E.; Townsend, E.M.; Sherry, L.; Delaney, C.; Jones, B.L.; Williams, C.; Ramage, G. The comparative efficacy of antiseptics against Candida auris biofilms. Int. J. Antimicrob. Agents 2018, 52, 673–677. [Google Scholar] [CrossRef]
- Sharma, S.; Chauhan, A.; Ranjan, A.; Mathkor, D.M.; Haque, S.; Ramniwas, S.; Tuli, H.S.; Yadav, T.J.V. Emerging challenges in antimicrobial resistance: Implications for pathogenic microorganisms, novel antibiotics, and their impact on sustainability. Front. Microbiol. 2024, 15, 1403168. [Google Scholar] [CrossRef]
- Ahmed, S.K.; Hussein, S.; Qurbani, K.; Ibrahim, R.H.; Fareeq, A.; Mahmood, K.A.; Mohamed, M.G. Antimicrobial resistance: Impacts, challenges, and future prospects. J. Med. Surg. Public Health 2024, 2, 100081. [Google Scholar] [CrossRef]
- Vallabhaneni, S.; Kallen, A.; Tsay, S.; Chow, N.; Welsh, R.; Kerins, J.; Kemble, S.K.; Pacilli, M.; Black, S.R.; Landon, E.; et al. Investigation of the first seven reported cases of Candida auris, a globally emerging invasive, multidrug-resistant fungus—United States, May 2013–August 2016. Am. J. Transpl. 2017, 17, 296–299. [Google Scholar] [CrossRef]
- Forsberg, K.; Woodworth, K.; Walters, M.; Berkow, E.L.; Jackson, B.; Chiller, T.; Vallabhaneni, S. Candida auris: The recent emergence of a multidrug-resistant fungal pathogen. Med. Mycol. 2019, 57, 1–12, Erratum in: Med. Mycol. 2019, 57, e7. [Google Scholar] [CrossRef]
- Sherry, L.; Ramage, G.; Kean, R.; Borman, A.; Johnson, E.M.; Richardson, M.D.; Rautemaa-Richardson, R. Biofilm-Forming Capability of Highly Virulent, Multidrug-Resistant Candida auris. Emerg. Infect. Dis. 2017, 23, 328–331. [Google Scholar] [CrossRef]
- Gehrke, A.K.E.; Giai, C.; Gómez, M.I. Staphylococcus aureus Adaptation to the Skin in Health and Persistent/Recurrent Infections. Antibiotics 2023, 12, 1520. [Google Scholar] [CrossRef] [PubMed]
- Boxberger, M.; Cenizo, V.; Cassir, N.; Scola, B.L. Challenges in exploring and manipulating the human skin microbiome. Microbiome 2021, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Kean, R.; Rajendran, R.; Haggarty, J.; Townsend, E.M.; Short, B.; Burgess, K.E.; Lang, S.; Millington, O.; Mackay, W.G.; Williams, C.; et al. Candida albicans mycofilms support Staphylococcus aureus colonization and enhances miconazole resistance in dual-species interactions. Front. Microbiol. 2017, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, S.; Du, J.; Wu, M.; Huang, X. Current and prospective therapeutic strategies: Tackling Candida albicans and Streptococcus mutans cross-kingdom biofilm. Front. Cell. Infect. Microbiol. 2023, 13, 1106231. [Google Scholar] [CrossRef]
- Kong, E.F.; Tsui, C.; Kucharikova, S.; Andes, D.; Dijck, P.V.; Jabra-Rizk, M.A. Commensal protection of Staphylococcus aureus against antimicrobials by Candida albicans biofilm matrix. mBio 2016, 7, e01365-16. [Google Scholar] [CrossRef]
- Gülmez, D.; Brown, J.L.; Butcher, M.C.; Delaney, C.; Kean, R.; Ramage, G.; Short, B. Investigating Dual-Species Candida auris and Staphylococcal Biofilm Antiseptic Challenge. Antibiotics 2022, 11, 931. [Google Scholar] [CrossRef]
- Bo, L.; Sun, H.; Li, Y.-D.; Zhu, J.; Wurpel, J.N.D.; Lin, H.; Chen, Z.-S. Combating antimicrobial resistance: The silent war. Front. Pharmacol. 2024, 15, 1347750. [Google Scholar] [CrossRef]
- Zubair, M.; Husain, F.M.; Qais, F.A.; Alam, P.; Ahmad, I.; Albalawi, T.; Ahmad, N.; Alam, M.; Baig, M.H.; Dong, J.-J.; et al. Bio-fabrication of titanium oxide nanoparticles from Ochradenus arabicus to obliterate biofilms of drug-resistant Staphylococcus aureus and Pseudomonas aeruginosa isolated from diabetic foot infections. Appl. Nanosci. 2021, 11, 375–387. [Google Scholar] [CrossRef]
- Abo-Dya, N.E.; Agha, K.A.; Abbas, H.A.; Abu-Kull, M.E.; Alahmdi, M.I.; Osman, N.A. Hybrid N-Acylcysteines as Dual-Acting Matrix Disruptive and Anti-Quorum Sensing Agents Fighting Pseudomonas aeruginosa Biofilms: Design, Synthesis, Molecular Docking Studies, and In Vitro Assays. ACS Omega 2022, 7, 19879–19891. [Google Scholar] [CrossRef]
- Alqahtani, T.T.; Alelyani, A.A.; Yousuf, M.M.; Wejdan, M.K.A.; Husain, F.M.; Zubair, M. Study of Plasmid-Mediated Extended-Spectrum Beta-Lactamase-Producing Clinical Strains of Enterobacteriaceae from Tabuk Region. Cureus 2023, 15, e40183. [Google Scholar] [CrossRef]
- Zubair, M. Antimicrobial and Anti-Biofilm Activities of Citrus sinensis and Moringa oleifera Against the Pathogenic Pseudomonas aeruginosa and Staphylococcus aureus. Cureus 2020, 12, e12337. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Zahra, A.; Kamthan, M.; Husain, F.M.; Albalawi, T.; Zubair, M.; Alatawy, R.; Abid, M.; Noorani, M.S. Microbial Biofilms: Applications, Clinical Consequences, and Alternative Therapies. Microorganisms 2023, 11, 1934. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef] [PubMed]
- Reena, F.J.; Phelan, J.P.; Gallagher, L.; Wood, D.F.; Shanahan, R.M.; Cano, R.; Muimhneachain, E.Ó.; McGlacken, G.P.; O’Gara, F. Exploiting Interkingdom Interactions for Development of Small-Molecule Inhibitors of Candida albicans Biofilm Formation. Antimicrob. Agents Chemother. 2016, 60, 5894–5905. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Sheng, Y.; Zhang, J.; Xu, Y.; Yu, J.; Wang, B.; Zhao, H.; Wang, X.; Guo, Y.; Wu, X.; et al. Small-Molecule Compound SYG-180-2-2 to Effectively Prevent the Biofilm Formation of Methicillin-Resistant Staphylococcus aureus. Front. Microbiol. 2022, 12, 770657. [Google Scholar] [CrossRef] [PubMed]
- Qvortrup, K.; Hultqvist, L.D.; Nilsson, M.; Jakobsen, T.H.; Cu, C.U.J.; Uhd, J.; Andersen, J.B.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T. Small Molecule Anti-biofilm Agents Developed on the Basis of Mechanistic Understanding of Biofilm Formation. Front. Chem. 2019, 7, 742. [Google Scholar] [CrossRef]
- Matin, M.M.; Matin, P.; Rahman, M.R.; Hadda, T.B.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 864286. [Google Scholar] [CrossRef]
- Peyton, L.R.; Gallagher, S.; Hashemzadeh, M. Triazole antifungals: A review. Drugs Today 2015, 51, 705–718. [Google Scholar] [CrossRef]
- Lengerli, D.; Ibis, K.; Nural, Y.; Banoglu, E. The 1,2,3-triazole ‘all-in-one’ ring system in drug discovery: A good bioisostere, a good pharmacophore, a good linker, and a versatile synthetic tool. Expert Opin. Drug Dis. 2022, 17, 1209–1236. [Google Scholar] [CrossRef]
- Faizan, M.; Kumar, R.; Mazumder, A.; Salahuddin; Kukreti, N.; Kumar, A.; Chaitanya, M.V.N.L. The medicinal chemistry of piperazines: A review. Chem. Biol. Drug Des. 2024, 103, e14537. [Google Scholar] [CrossRef]
- Wu, Y.-J. Heterocycles and Medicine: A Survey of the Heterocyclic Drugs Approved by the U.S. FDA from 2000 to Present. In Progress in Heterocyclic Chemistry; Gordon, W., Gribble, J., Joule, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 24, pp. 1–53. ISBN 9780080968070. [Google Scholar] [CrossRef]
- Wani, M.Y.; Alghamidi, M.S.S.; Srivastava, V.; Ahmad, A.; Aqlan, F.M.; Al Bogami, A.S. Click synthesis of pyrrolidine-based 1,2,3-triazole derivatives as antifungal agents causing cell cycle arrest and apoptosis in Candida auris. Bioorg. Chem. 2023, 136, 106562. [Google Scholar] [CrossRef]
- Parveen, H.; Wani, M.Y.; Mukhtar, S.; Ahmad, A. Synthesis, in vitro and in silico screening of novel ferrocenyl substituted cyclohexenone and indazole derivatives as effective antimycobacterial agents. J. Mol. Struct. 2024, 1311, 138472. [Google Scholar] [CrossRef]
- Wani, M.Y.; Srivastava, V.; El-Said, W.A.; Al-Bogami, A.S.; Ahmad, A. Inhibition of apoptosis and biofilm formation in Candida auris by click-synthesized triazole-bridged quinoline derivatives. RSC Adv. 2024, 14, 21190–21202. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, N.; Yao, Z.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.; Cao, D. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef]
- Pa, C. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeast, 4th ed.; CLSI Standard M27; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Patel, J.B.; Cockerill, F.R.; Bradford, P.A. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fourth Informational Supplement, M100-S24; Institute CLS: Wayne, PA, USA, 2014. [Google Scholar]
- Srivastava, V.; Ahmad, A. Abrogation of pathogenic attributes in drug resistant Candida auris strains by farnesol. PLoS ONE 2020, 15, e0233102. [Google Scholar] [CrossRef]
- Li, H.; Zhang, C.; Liu, P.; Liu, W.; Gao, Y.; Sun, S. In vitro interactions between fluconazole and minocycline against mixed cultures of Candida albicans and Staphylococcus aureus. J. Microbiol. Immunol. Infect. 2015, 48, 655–661. [Google Scholar] [CrossRef]
- Fernandes, L.; Fortes, B.N.; Lincopan, N.; Ishida, K. Caspofungin and polymyxin B reduce the cell viability and total biomass of mixed biofilms of carbapenem-resistant Pseudomonas aeruginosa and Candida spp. Front. Microbiol. 2020, 11, 573263. [Google Scholar] [CrossRef]
- Lone, S.A.; Ahmad, A. Inhibitory effect of novel Eugenol Tosylate Congeners on pathogenicity of Candida albicans. BMC Complement. Med. Ther. 2020, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Shameer, M.K.; Shubham, B.; Ce, Y.; Dongning, L.; Zach, F.; Binghe, W. Click chemistry and drug delivery: A bird’s-eye view. Acta Pharmaceut. Sin. B 2022, 13, 1990–2016. [Google Scholar] [CrossRef]
- Agalave, S.G.; Suleman, R.M.; Pore, V.S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Jafri, H.; Banerjee, G.; Khan, M.S.A.; Ahmad, I.; Abulreesh, H.H.; Althubiani, A.S. Synergistic interaction of eugenol and antimicrobial drugs in eradication of single and mixed biofilms of Candida albicans and Streptococcus mutans. AMB Express 2020, 10, 185. [Google Scholar] [CrossRef]
- Tsui, C.; Kong, E.F.; Jabra-Rizk, M.A. Pathogenesis of Candida albicans biofilm. Pathog. Dis. 2016, 74, ftw018. [Google Scholar] [CrossRef]
- Young, T.; Alshanta, O.; Kean, R.; Bradshaw, D.; Pratten, J.; Williams, C.; Woodall, C.; Ramage, G.; Brown, J.L. Candida albicans as an essential “keystone” component within polymicrobial oral biofilm models? Microorganisms 2020, 9, 59. [Google Scholar] [CrossRef]
- Carolus, H.; Dyck, K.V.; Dijck, P.V. Candida albicans and Staphylococcus species: A threatening twosome. Front. Microbiol. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Luo, Y.; McAuley, D.F.; Fulton, C.R.; Pessoa, J.S.; McMullan, R.; Lundy, F.T. Targeting Candida albicans in dual-species biofilms with antifungal treatment reduces Staphylococcus aureus and MRSA in vitro. PLoS ONE 2021, 16, e0249547. [Google Scholar] [CrossRef]
- Lara, H.H.; Lopez-Ribot, J.L. Inhibition of mixed biofilms of Candida albicans and methicillin-resistant Staphylococcus aureus by positively charged silver nanoparticles and functionalized silicone elastomers. Pathogens 2020, 9, 784. [Google Scholar] [CrossRef] [PubMed]
- Sabaeifard, P.; Abdi-Ali, A.; Soudi, M.R.; Dinarvand, R. Optimization of tetrazolium salt assay for Pseudomonas aeruginosa biofilm using microtiter plate method. J. Microbiol. Methods 2014, 105, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Zhou, G.; Munyon, R.; Ghannoum, M.A. Candida biofilm: A well-designed protected environment. Med. Mycol. 2005, 43, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Locock, K.; Verma-Gaur, J.; Hay, I.D.; Meagher, L.; Traven, A. Searching for new strategies against polymicrobial biofilm infections: Guanylated polymethacrylates kill mixed fungal/bacterial biofilms. J. Antimicrob. Chemother. 2016, 71, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Liu, Y.; Benhamou, R.I.; Sanchez, H.; Simon-Soro, A.; Li, Y.; Hwang, G.; Fridman, M.; Andes, D.R.; Koo, H. Bacterial-derived exopolysaccharides enhance antifungal drug tolerance in a cross-kingdom oral biofilm. ISME J. 2018, 12, 1427–1442. [Google Scholar] [CrossRef] [PubMed]
- Scheunemann, G.; Fortes, B.N.; Lincopan, N.; Ishida, K. Caspofungin Inhibits Mixed Biofilms of Candida albicans and Methicillin-Resistant Staphylococcus aureus and Displays Effectiveness in Coinfected Galleria mellonella Larvae. Microbiol. Spectr. 2021, 9, e00744-21. [Google Scholar] [CrossRef]
- Ponde, N.O.; Lortal, L.; Ramage, G.; Naglik, J.R.; Richardson, J.P. Candida albicans biofilms and polymicrobial interactions. Crit. Rev. Microbiol. 2021, 47, 91–111. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | M.Wt | Clog P * | Log D # | HBA | HBD | RBs | tPSA | Ro5 (Y/N) |
|---|---|---|---|---|---|---|---|---|
| T1 | 394.45 | 2.378 | 2.38 | 5 | 1 | 7 | 66.29 | Y |
| T2 | 408.47 | 2.510 | 2.74 | 5 | 1 | 7 | 66.29 | Y |
| T3 | 412.44 | 2.517 | 2.29 | 6 | 1 | 7 | 66.29 | Y |
| T4 | 428.89 | 3.489 | 3.34 | 5 | 1 | 7 | 66.29 | Y |
| T5 | 428.89 | 3.031 | 2.82 | 5 | 1 | 7 | 66.29 | Y |
| T6 | 473.34 | 3.140 | 2.95 | 5 | 1 | 7 | 66.29 | Y |
| T7 | 462.18 | 3.396 | 3.19 | 7 | 1 | 8 | 66.29 | Y |
| T8 | 462.44 | 3.396 | 3.17 | 8 | 1 | 8 | 66.29 | Y |
| T9 | 424.47 | 2.386 | 2.48 | 6 | 1 | 8 | 75.52 | Y |
| T10 | 439.44 | 2.286 | 2.46 | 7 | 1 | 8 | 112.11 | Y |
| T11 | 439.44 | 2.286 | 2.33 | 7 | 1 | 8 | 112.11 | Y |
| T12 | 463.34 | 3.684 | 3.19 | 5 | 1 | 7 | 66.29 | Y |
| † Fluconazole | 306.27 | 0.50 | 0.56 | 7 | 1 | 5 | 81.93 | Y |
| Test | C. auris MRL6057 | S. aureus ATCC29213 | C. auris MRL6057 + S. aureus ATCC29213 | |||
|---|---|---|---|---|---|---|
| Median MIC (µg/mL) | Median MFC (µg/mL) | Median MIC (µg/mL) | Median MBC (µg/mL) | Median MIC (µg/mL) | Median MFC/MBC (µg/mL) | |
| T1 | 156.25 | 625.0 | 78.12 | 156.25 | 1250 | 2500 |
| T2 | 312.5 | 625.0 | 156.25 | 312.5 | 1250 | 2500 |
| T3 | 9.76 | 19.53 | 2.44 | 2.44 | 78.12 | 78.12 |
| T4 | 78.12 | 78.12 | 78.12 | 78.12 | 625.0 | 625.0 |
| T5 | 19.53 | 39.06 | 19.53 | 19.53 | 156.25 | 312.5 |
| T6 | 39.06 | 78.12 | 39.06 | 78.12 | 312.5 | 312.5 |
| T7 | 156.25 | 312.5 | 156.25 | 312.5 | 312.5 | 625.0 |
| T8 | 78.12 | 312.5 | 39.06 | 312.5 | 625.0 | 625.0 |
| T9 | 312.5 | 625.0 | 156.25 | 312.5 | 625.0 | 1200 |
| T10 | 78.12 | 312.5 | 19.53 | 39.06 | 156.25 | 625.0 |
| T11 | 78.12 | 156.25 | 156.25 | 312.5 | 312.5 | 625.0 |
| T12 | 312.5 | 1250 | 312.5 | 625 | 1250 | 2500 |
| AmB | 4.0 | 8.0 | NA | NA | NT | NT |
| AZI | NT | NT | 0.5 | 0.5 | NT | NT |
| T3 | Biofilm Formation | 24 h Preformed Biofilm | ||||
|---|---|---|---|---|---|---|
| C. auris | S. aureus | C. auris + S. aureus | C. auris | S. aureus | C. auris + S. aureus | |
| BIC50 | 9.76 μg/mL (0.25× MIC) | 4.88 μg/mL (0.25× MIC) | 9.76 μg/mL (0.25× MIC) | 39.06 μg/mL (MIC) | 9.76 μg/mL (0.5× MIC) | 78.12 μg/mL (2× MIC) |
| BIC90 | 19.53 μg/mL (0.5× MIC) | 9.76 μg/mL (0.5× MIC) | 19.53 μg/mL (0.5× MIC) | 78.12 μg/mL (2× MIC) | 39.06 μg/mL (2× MIC) | 156.25 μg/mL (4× MIC) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parveen, H.; Mukhtar, S.; Albalawi, M.O.; Khasim, S.; Ahmad, A.; Wani, M.Y. Concomitant Inhibition and Collaring of Dual-Species Biofilms Formed by Candida auris and Staphylococcus aureus by Triazole Based Small Molecule Inhibitors. Pharmaceutics 2024, 16, 1570. https://doi.org/10.3390/pharmaceutics16121570
Parveen H, Mukhtar S, Albalawi MO, Khasim S, Ahmad A, Wani MY. Concomitant Inhibition and Collaring of Dual-Species Biofilms Formed by Candida auris and Staphylococcus aureus by Triazole Based Small Molecule Inhibitors. Pharmaceutics. 2024; 16(12):1570. https://doi.org/10.3390/pharmaceutics16121570
Chicago/Turabian StyleParveen, Humaira, Sayeed Mukhtar, Mona O. Albalawi, Syed Khasim, Aijaz Ahmad, and Mohmmad Younus Wani. 2024. "Concomitant Inhibition and Collaring of Dual-Species Biofilms Formed by Candida auris and Staphylococcus aureus by Triazole Based Small Molecule Inhibitors" Pharmaceutics 16, no. 12: 1570. https://doi.org/10.3390/pharmaceutics16121570
APA StyleParveen, H., Mukhtar, S., Albalawi, M. O., Khasim, S., Ahmad, A., & Wani, M. Y. (2024). Concomitant Inhibition and Collaring of Dual-Species Biofilms Formed by Candida auris and Staphylococcus aureus by Triazole Based Small Molecule Inhibitors. Pharmaceutics, 16(12), 1570. https://doi.org/10.3390/pharmaceutics16121570