
Multivariate Approaches in Quantitative Structure–Property Relationships Study for the Photostability Assessment of 1,4-Dihydropyridine Derivatives


 ,
,  , , ,
, , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Instruments
2.2. Software
2.3. Chemicals
2.4. Molecular Descriptors
2.5. Standard Solutions
2.6. Experimental Conditions
3. Results
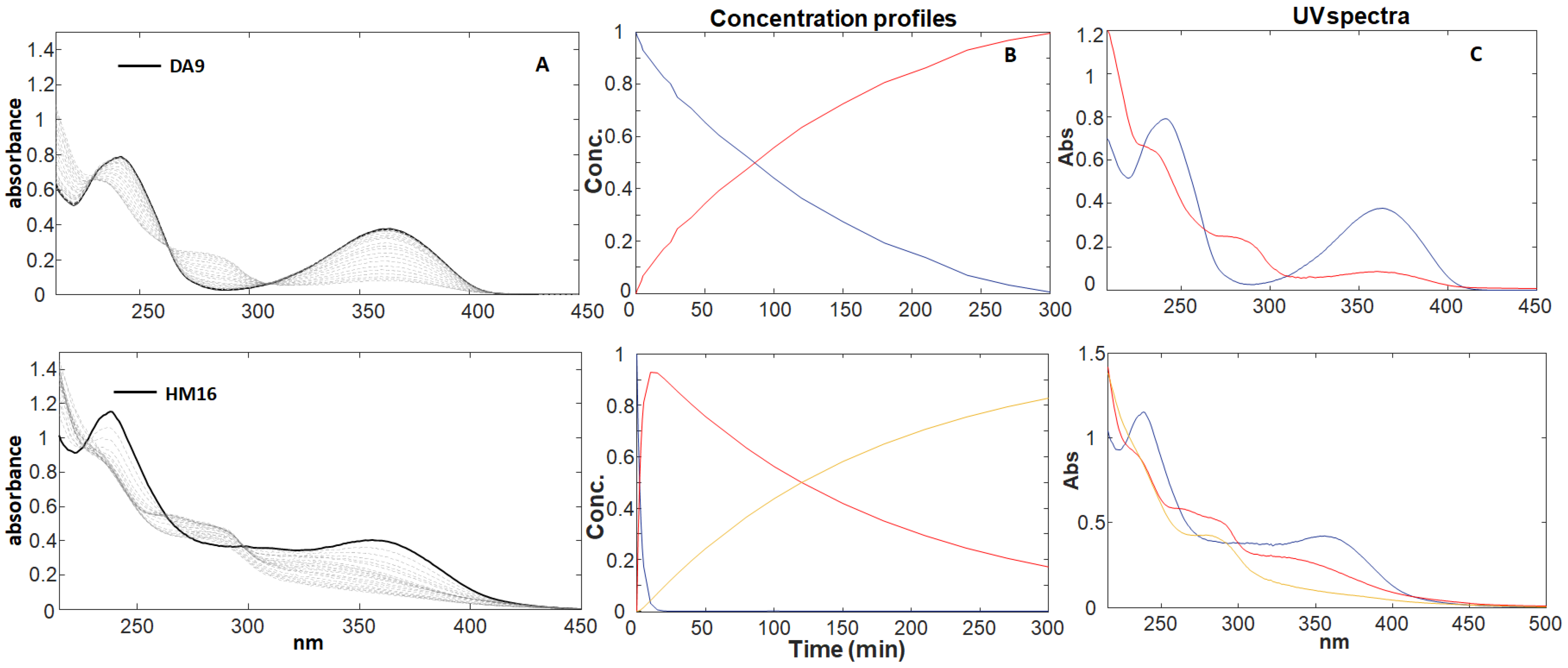
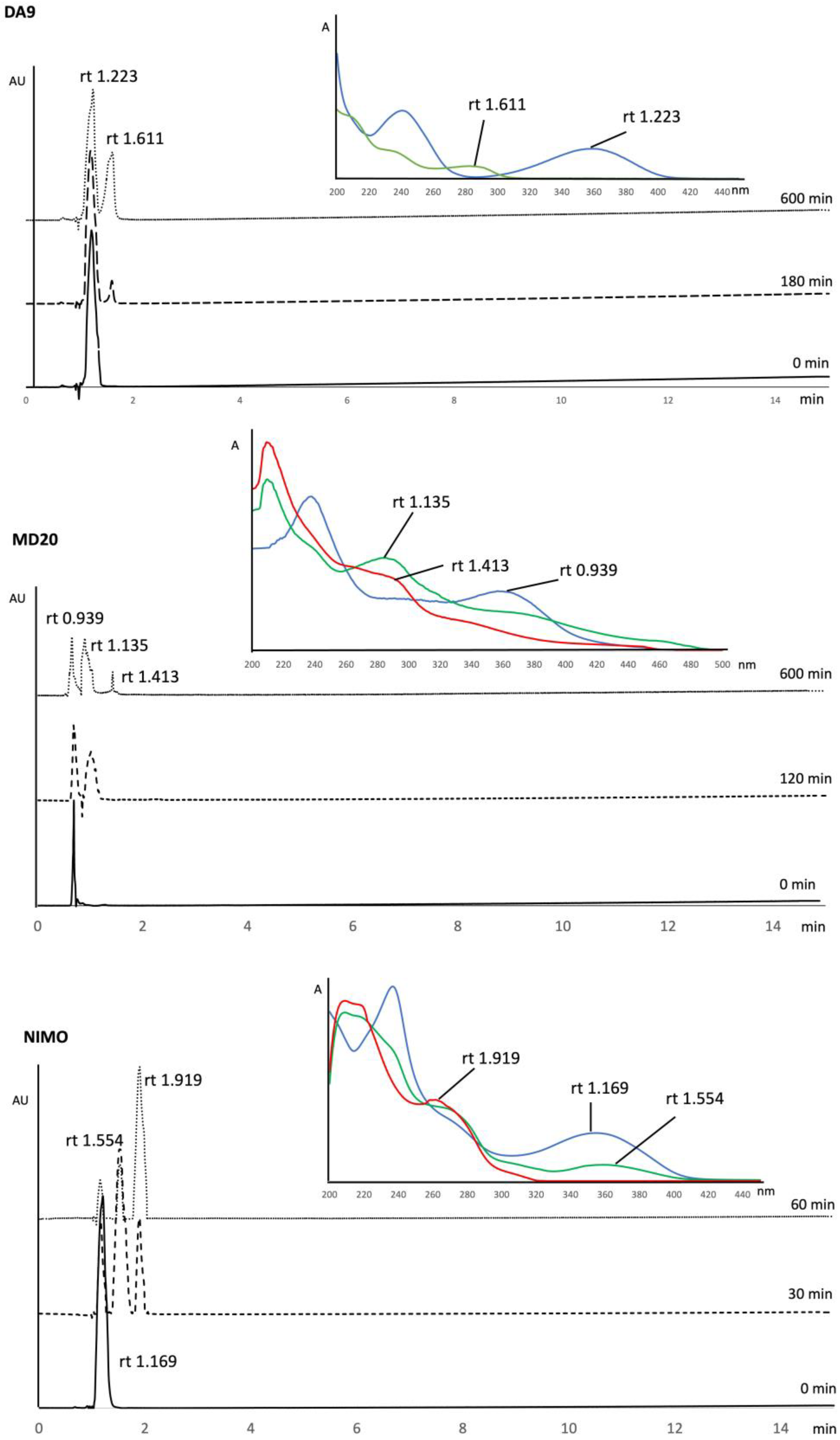
3.1. Photodegradation Studies
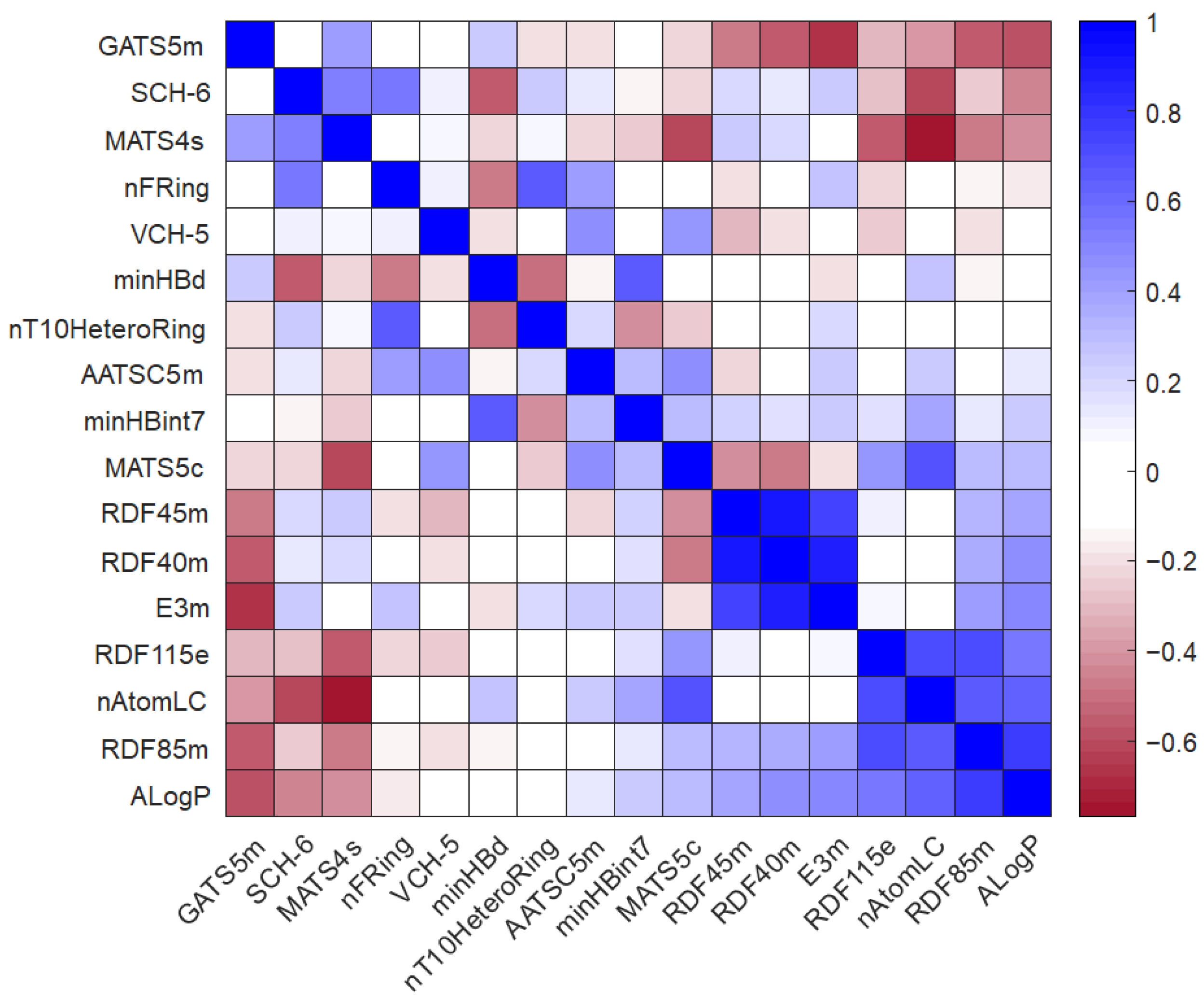
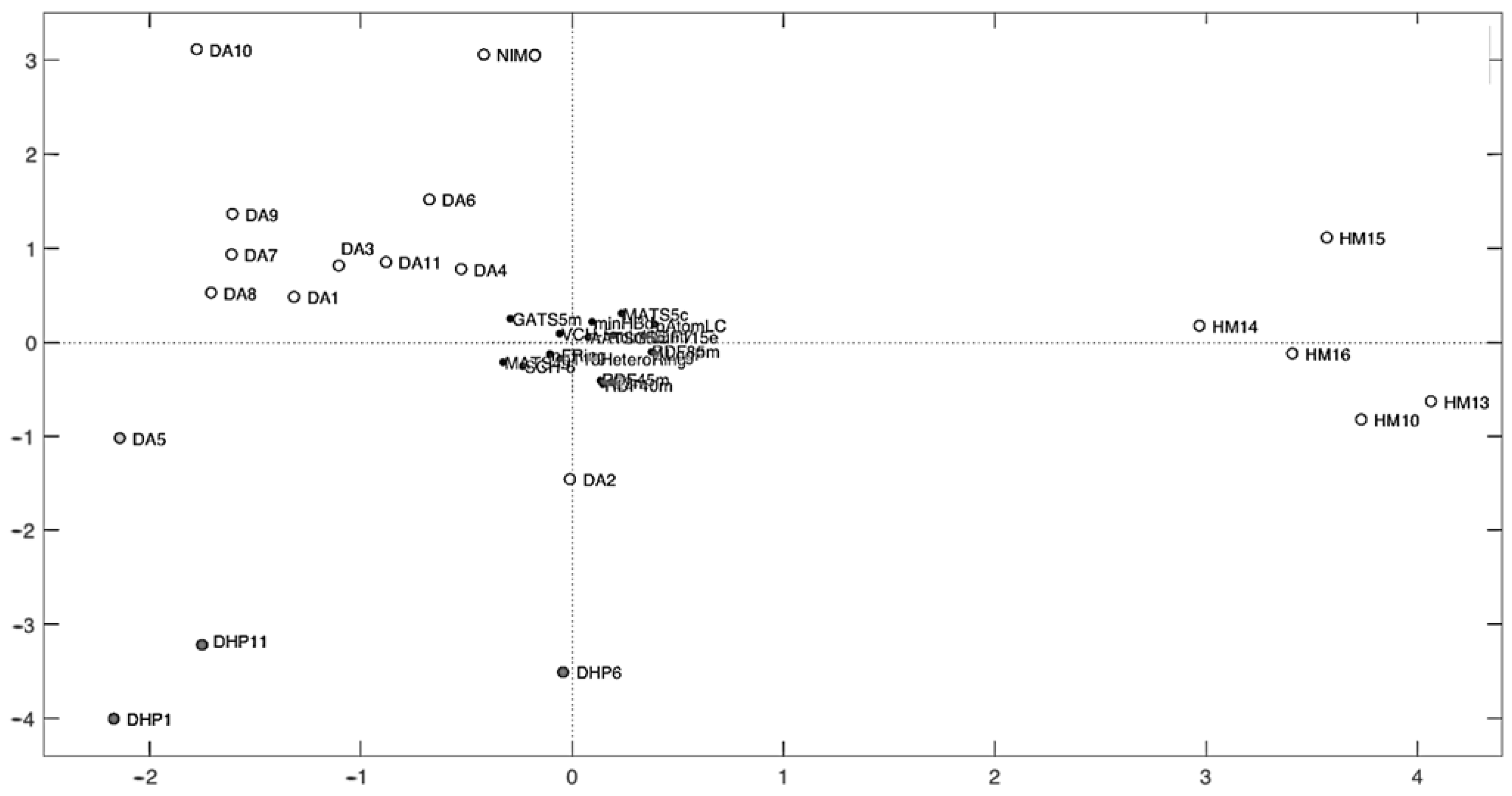
3.2. Selection of Independent Variables for QSPR Method Elaboration
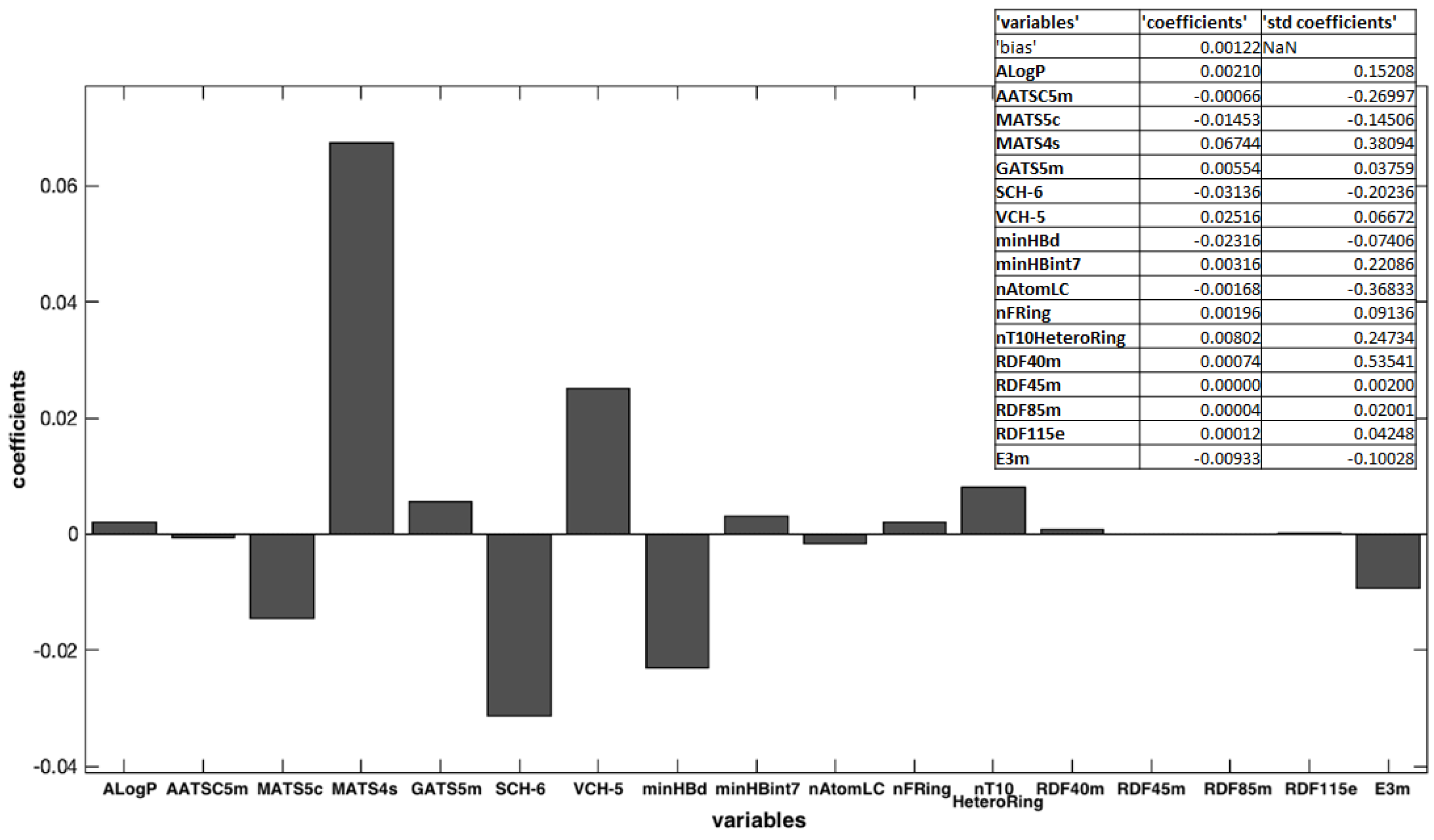
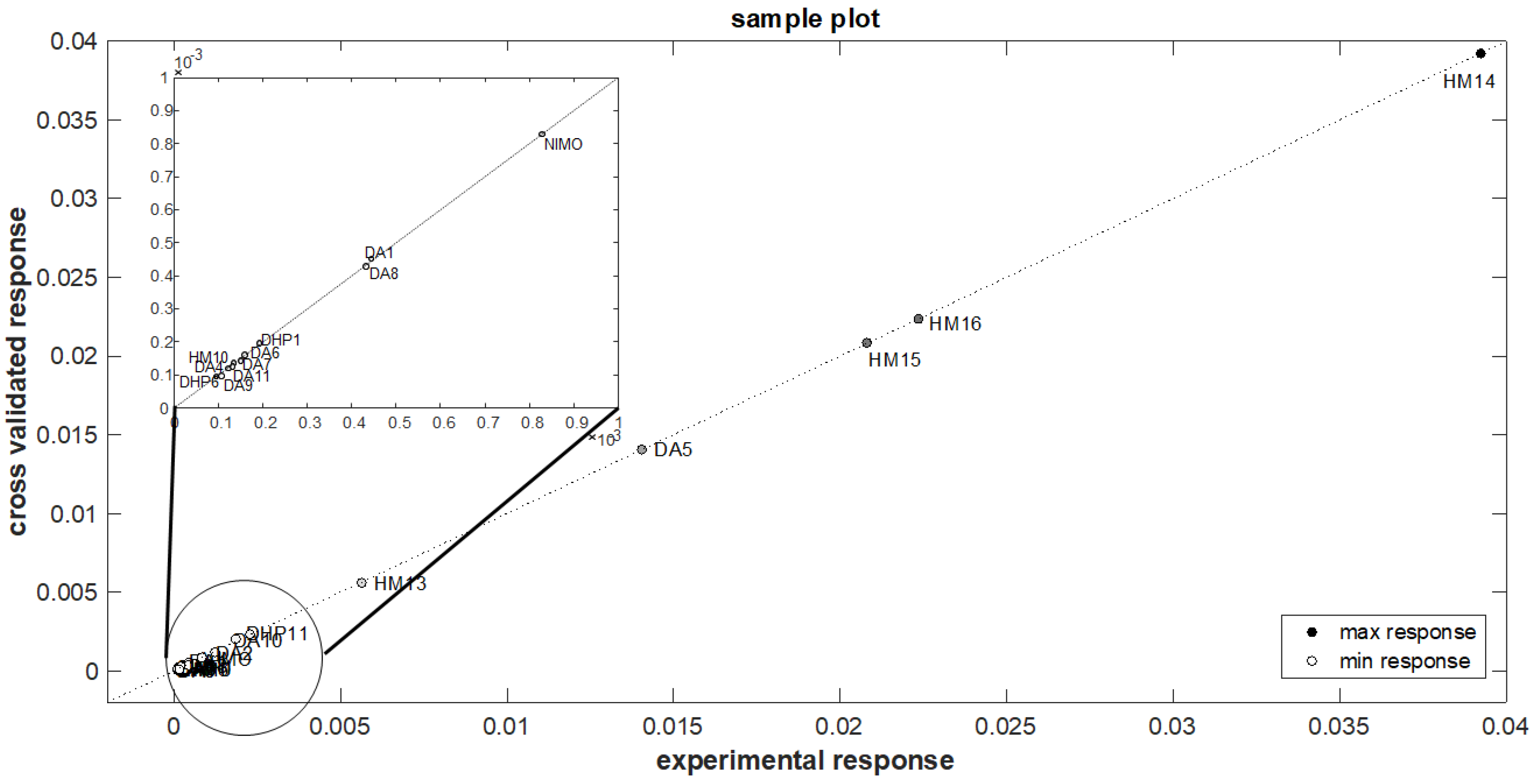
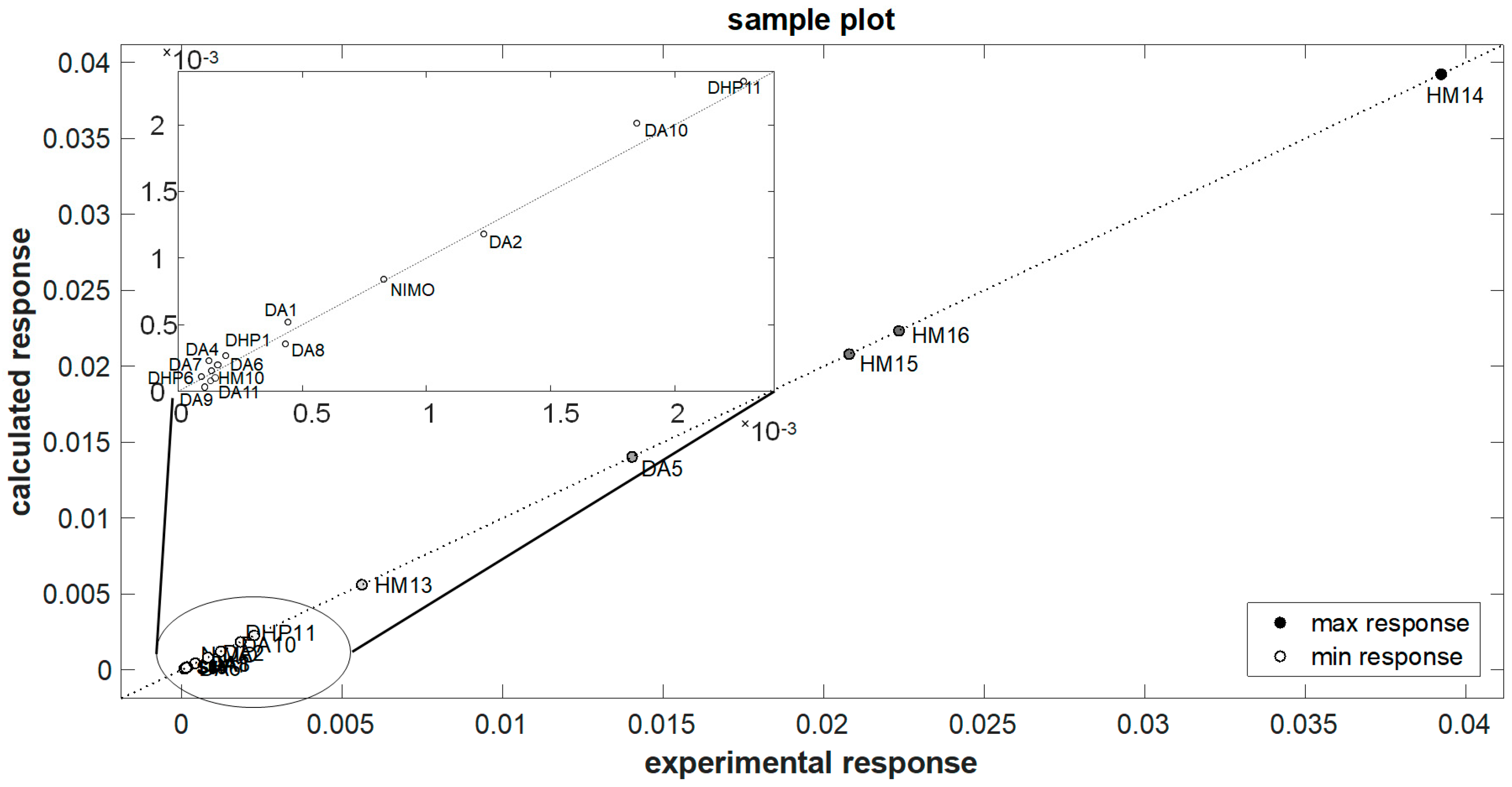
3.3. QSPR Model Elaboration
| K = 1.216 × 103 + 2.100 × 10−3 ALogP − 6.597 × 10−4 AATSC5m − 1.453 × 10−2 MATS5c |
| + 6.744 × 10−2 MATS4s + 5.535 × 10−3 GATS5m − 3.136 × 10−2 SCH-6 + 2.516 × 10−2 VCH-5 |
| − 2.316 × 10−2 minHBd + 3.159 × 10−3 minHBint7 − 1.681 × 10−3 nAtomLC + 1.964 × 10−3 nFRing |
| + 8.021 × 10−3 nT10HeteroRing + 7.442 × 10−4 RDF40m + 3.947 × 10−6 RDF45m |
| + 4.102 × 10−5 RDF85m + 1.163 × 10−5 RDF115e − 9.327 × 10−3 E3m |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, G.Y.; Zou, J.W.; Chen, J.; Hu, G.X.; Jiang, Y.J.; Huang, M. QSPR Study on Hydrophobicity of Pt(II) Complexes with Surface Electrostatic Potential-Based Descriptors. J. Mol. Graph. Model. 2022, 116, 108256. [Google Scholar] [CrossRef]
- Ioele, G.; De Luca, M.; Oliverio, F.; Ragno, G. Prediction of Photosensitivity of 1,4-Dihydropyridine Antihypertensives by Quantitative Structure-Property Relationship. Talanta 2009, 79, 1418–1424. [Google Scholar] [CrossRef]
- Alaoui Mansouri, M.; Kharbach, M.; Bouklouze, A. Current Applications of Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) in Pharmaceutical Analysis: Review. J. Pharm. Sci. 2023; in press. [Google Scholar] [CrossRef]
- Zade, S.V.; Abdollahi, H. The Classification Performance of Multivariate Curve Resolution-Discriminant Analysis: A Comparative Study. Microchem. J. 2023, 191, 108867. [Google Scholar] [CrossRef]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. Dos. Form Des. Parameters 2018, 2, 731–755. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2000; Volume 11, 688p. [Google Scholar]
- Krzemiński, T.F.; Hudziak, D.; Sielańczyk, A.W.; Porc, M.; Kedzia, A. Differential Effects of Furnidipine and Its Active Metabolites in Rat Isolated Working Heart. Vasc. Pharmacol. 2008, 49, 91–96. [Google Scholar] [CrossRef]
- Samadi, A.; Pour, A.K.; Jamieson, R. Development of Remediation Technologies for Organic Contaminants Informed by QSAR/QSPR Models. Environ. Adv. 2021, 5, 100112. [Google Scholar] [CrossRef]
- Chen, J.; Wu, N.; Qu, R.; Xu, X.; Shad, A.; Pan, X.; Yao, J.; Bin-Jumah, M.; Allam, A.A.; Wang, Z.; et al. Photodegradation of Polychlorinated Diphenyl Sulfides (PCDPSs) under Simulated Solar Light Irradiation: Kinetics, Mechanism, and Density Functional Theory Calculations. J. Hazard. Mater. 2020, 398, 122876. [Google Scholar] [CrossRef]
- Villaverde, J.J.; Sevilla-Morán, B.; Alonso-Prados, J.L.; Sandín-España, P. A Study Using QSAR/QSPR Models Focused on the Possible Occurrence and Risk of Alloxydim Residues from Chlorinated Drinking Water, According to the EU Regulation. Sci. Total Environ. 2022, 839, 156000. [Google Scholar] [CrossRef]
- Buglak, A.A.; Filatov, M.A.; Hussain, M.A.; Sugimoto, M. Singlet Oxygen Generation by Porphyrins and Metalloporphyrins Revisited: A Quantitative Structure-Property Relationship (QSPR) Study. J. Photochem. Photobiol. A Chem. 2020, 403, 112833. [Google Scholar] [CrossRef]
- Ioele, G.; Muzzalupo, R.; Gündüz, M.G.; De Luca, M.; Mazzotta, E.; Grande, F.; Occhiuzzi, M.A.; Garofalo, A.; Ragno, G. Use of Pluronic Surfactants in Gel Formulations of Photosensitive 1,4-Dihydropyridine Derivatives: A Potential Approach in the Treatment of Neuropathic Pain. Pharmaceutics 2021, 13, 527. [Google Scholar] [CrossRef]
- Ioele, G.; Gündüz, M.G.; Spatari, C.; De Luca, M.; Grande, F.; Ragno, G. A New Generation of Dihydropyridine Calcium Channel Blockers: Photostabilization of Liquid Formulations Using Nonionic Surfactants. Pharmaceutics 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Ioele, G.; Gündüz, M.G.; De Luca, M.; Şimşek, R.; Şafak, C.; Muzzalupo, R.; Ragno, G. Photodegradation Studies of 1,4-Dihydropyridine Compounds by MCR Analysis on UV Spectral Data. Future Med. Chem. 2016, 8, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zamponi, G.W. T-Type Channel Druggability at a Crossroads. ACS Chem. Neurosci. 2019, 10, 1124–1126. [Google Scholar] [CrossRef]
- ICH Q1A (R2) Stability Testing of New Drug Substances and Drug Products—Scientific Guideline; European Medicines Agency: Amsterdam, The Netherlands, 2003.
- De Juan, A.; Rutan, S.C.; Maeder, M.; Tauler, R. Comprehensive Chemometrics; Elsevier: Amsterdam, The Netherlands, 2009; Volume 2. [Google Scholar]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A Molecular Descriptor Calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Vafaei Molamahmood, H.; Faraji, N.; Long, M. Quantitative Structure-Activity Relationship for the Oxidation of Organic Contaminants by Peracetic Acid Using GA-MLR Method. J. Environ. Manag. 2022, 310, 114747. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley-VCH: Weinheim, Germany, 2009; Volume 2, pp. 1–252. [Google Scholar] [CrossRef]
- Jaumot, J.; de Juan, A.; Tauler, R. MCR-ALS GUI 2.0: New Features and Applications. Chemom. Intell. Lab. Syst. 2015, 140, 1–12. [Google Scholar] [CrossRef]
- Farahani, H.A.; Rahiminezhad, A.; Same, L.; Immannezhad, K. A Comparison of Partial Least Squares (PLS) and Ordinary Least Squares (OLS) Regressions in Predicting of Couples Mental Health Based on Their Communicational Patterns. Procedia Soc. Behav. Sci. 2010, 5, 1459–1463. [Google Scholar] [CrossRef]
- Consonni, V.; Baccolo, G.; Gosetti, F.; Todeschini, R.; Ballabio, D. A MATLAB Toolbox for Multivariate Regression Coupled with Variable Selection. Chemom. Intell. Lab. Syst. 2021, 213, 104313. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- DEDUCT. Database of Endocrine Disrupting Chemicals and Their Toxicity Profiles. Available online: https://cb.imsc.res.in/deduct/ (accessed on 1 March 2023).
- Zou, K.H.; Tuncali, K.; Silverman, S.G. Correlation and Simple Linear Regression. Radiology 2003, 227, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Chaple, D.; Masand, V.H.; Zaki, M.E.A.; Al-Hussain, S.A.; Shah, A.; Arora, S.; Jawarkar, R.; Tauqeer, M. In Silico Study to Recognize Novel Angiotensin-Converting-Enzyme-I Inhibitors by 2D-QSAR and Constraint-Based Molecular Simulations. J. Biomol. Struct. Dyn. 2023; epub ahead of print. [Google Scholar] [CrossRef]
- Miranda-Quintana, R.A.; Martínez González, M.; Ayers, P.W. Electronegativity and Redox Reactions. Phys. Chem. Chem. Phys. 2016, 18, 22235–22243. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez del Águila, M.M.; Benítez-Parejo, N. Simple Linear and Multivariate Regression Models. Allergol. Immunopathol. 2011, 39, 159–173. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calibration Set | |||
| Compound | Chemical Structure | IUPAC Name | |
| 1 | DA1 |  | Isobutyl 4-(benzo[d][1,3]dioxol-5-yl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 2 | DA2 |  | Isobutyl 4-(6-bromobenzo[d][1,3]dioxol-5-yl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 3 | DA3 |  | Isobutyl 4-([1,1′-biphenyl]-3-yl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 4 | DA4 |  | Isobutyl 4-([1,1′-biphenyl]-4-yl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 5 | DA5 |  | Isobutyl 2,6,6-trimethyl-5-oxo-4-(4-oxo-4H-chromen-3-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 6 | DA6 |  | Isobutyl 2,6,6-trimethyl-5-oxo-4-(thiophen-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 7 | DA7 |  | Isobutyl 2,6,6-trimethyl-5-oxo-4-(pyridin-3-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 8 | DA8 |  | Isobutyl 2,6,6-trimethyl-4-(naphthalen-2-yl)-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 9 | DA9 |  | Isobutyl 2,6,6-trimethyl-5-oxo-4-phenyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 10 | DA10 |  | Isobutyl 2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 11 | DA11 |  | Isobutyl 4-(4-(1H-imidazol-1-yl)phenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 12 | DHP1 |  | Pyridin-3-ylmethyl 4-(2,3-dichlorophenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 13 | DHP6 |  | Pyridin-3-ylmethyl 4-(2,3-dichlorophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 14 | DHP11 |  | Pyridin-3-ylmethyl 4-(2,3-dichlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 15 | HM10 |  | 2-(Methacryloyloxy)ethyl 4-(3,5-dichloro-2-hydroxyphenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 16 | HM13 |  | 2-(Methacryloyloxy)ethyl 4-(3-bromo-5-chloro-2-hydroxyphenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 17 | HM14 |  | 2-(Methacryloyloxy)ethyl 4-(5-bromo-2-hydroxy-3-nitrophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 18 | HM15 |  | 2-(Methacryloyloxy)ethyl 4-(3-bromo-2-hydroxy-5-nitrophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 19 | HM16 |  | 2-(Methacryloyloxy)ethyl 4-(2-hydroxy-3,5-dinitrophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 20 | NIMO |  | 3-isopropyl 5-(2-methoxyethyl) 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate |
| Prediction Set | |||
| Compound | Chemical Structure | IUPAC Name | |
| 1 | DA12 |  | Isobutyl 4-(4-(1H-1,2,4-triazol-1-yl)phenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 2 | DHP5 |  | Pyridin-3-ylmethyl 4-(2-chloro-3-(trifluoromethyl)phenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 3 | DHP7 |  | Pyridin-3-ylmethyl 4-(3-chloro-2-fluorophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 4 | DHP9 |  | Pyridin-3-ylmethyl 4-(2-fluoro-3-(trifluoromethyl)phenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 5 | DHP10 |  | Pyridin-3-ylmethyl 4-(2-chloro-3-(trifluoromethyl)phenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 6 | DHP12 |  | Pyridin-3-ylmethyl 4-(3-chloro-2-fluorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 7 | HM8 |  | Benzyl 4-(2-hydroxy-3,5-dinitrophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 8 | M3 |  | Benzyl 4-(2-hydroxy-5-nitrophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 9 | MD20 |  | Isobutyl 4-(2-hydroxy-3,5-dinitrophenyl)-2,6,6-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate |
| 10 | NICA |  | 3-(2-(benzyl(methyl)amino)ethyl) 5-methyl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate |
| Drugs | k1 (×10−4) | SD (±) k1 (×10−4) | k2 (×10−4) | SD (±) k2 (×10−4) | R2 | t0.1 (min) | % lof |
|---|---|---|---|---|---|---|---|
| DA1 | 4.435 | 0.107 | 0.389 | 0.013 | 99.89 | 4.134 | 3.281 |
| DA2 | 12.31 | 0.494 | 1.771 | 0.060 | 99.73 | 1.491 | 5.153 |
| DA3 | 1.217 | 0.022 | - | - | 99.93 | 15.064 | 2.693 |
| DA4 | 1.226 | 0.020 | - | - | 99.77 | 14.954 | 4.758 |
| DA5 | 140.4 | 6.183 | - | - | 99.17 | 0.131 | 6.085 |
| DA6 | 1.597 | 0.071 | - | - | 99.57 | 11.483 | 6.553 |
| DA7 | 1.345 | 0.012 | - | - | 99.96 | 13.633 | 1.963 |
| DA8 | 4.322 | 0.091 | - | - | 99.96 | 4.242 | 2.105 |
| DA9 | 1.061 | 0.005 | - | - | 99.99 | 17.273 | 0.919 |
| DA10 | 18.48 | 0.775 | - | - | 99.36 | 0.992 | 6.981 |
| DA11 | 1.315 | 0.016 | - | - | 99.94 | 13.939 | 2.406 |
| DA12 | 1.389 | 0.009 | - | - | 99.99 | 13.204 | 0.999 |
| DHP1 | 392.4 | 7.800 | - | - | 99.95 | 1.171 | 1.910 |
| DHP5 | 376.3 | 4.900 | - | - | 99.89 | 1.220 | 1.730 |
| DHP6 | 208.0 | 5.000 | - | - | 99.87 | 2.200 | 2.550 |
| DHP7 | 91.00 | 1.900 | - | - | 99.67 | 5.030 | 1.751 |
| DHP9 | 143.3 | 4.300 | - | - | 99.83 | 3.190 | 2.392 |
| DHP10 | 222.3 | 8.100 | - | - | 99.58 | 2.060 | 2.251 |
| DHP11 | 223.5 | 8.900 | - | - | 99.64 | 2.051 | 1.751 |
| DHP12 | 81.30 | 1.800 | - | - | 99.78 | 5.631 | 1.232 |
| HM8 | 101.0 | 4.798 | 2.354 | 0.070 | 99.71 | 0.182 | 5.341 |
| HM10 | 1.918 | 0.060 | 0.752 | 0.032 | 99.94 | 9.562 | 2.456 |
| HM13 | 0.950 | 0.300 | - | - | 99.94 | 19.329 | 2.512 |
| HM14 | 22.77 | 0.501 | - | - | 99.97 | 0.805 | 3.643 |
| HM15 | 1.494 | 0.039 | - | - | 99.32 | 12.274 | 6.263 |
| HM16 | 56.28 | 1.262 | 0.781 | 0.013 | 99.95 | 0.326 | 2.199 |
| MD20 | 78.12 | 2.442 | 2.335 | 0.050 | 99.84 | 0.235 | 4.035 |
| M3 | 2.642 | 0.030 | 1.271 | 0.017 | 99.97 | 6.940 | 1.836 |
| NICA | 6.044 | 0.211 | - | - | 99.84 | 3.030 | 3.941 |
| NIMO | 8.290 | 0.494 | 8.290 | 0.762 | 99.98 | 2.211 | 1.501 |
| Compound | ALogP | AATSC5m | MATS5c | MATS4s | GATS5m | SCH-6 | VCH-5 | minHBd | minHBint7 | nAtomLC | nFRing | nT10HeteroRing | RDF40m | RDF45m | RDF85m | RDF115e | E3m |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Calibration set | |||||||||||||||||
| DA1 | 0.895 | 0.5349 | −0.0763 | −0.091 | 0.960 | 0.328 | 0.029 | 0.277 | 1.541 | 7 | 2 | 1 | 12.672 | 13.082 | 6.3882 | 5.3440 | 0.219 |
| DA2 | 1.784 | 12.455 | −0.1343 | −0.092 | 0.928 | 0.316 | 0.029 | 0.266 | 1.500 | 7 | 2 | 1 | 23.441 | 14.137 | 6.3882 | 5.3440 | 0.468 |
| DA3 | 1.198 | 0.6475 | −0.1391 | −0.070 | 0.995 | 0.279 | 0 | 0.252 | 0 | 7 | 1 | 1 | 12.991 | 12.828 | 8.4192 | 11.175 | 0.108 |
| DA4 | 1.198 | 0.3788 | −0.1377 | −0.071 | 1.005 | 0.279 | 0 | 0.255 | 0 | 7 | 1 | 1 | 12.987 | 11.390 | 10.488 | 15.520 | 0.172 |
| DA5 | 0.531 | 1.0708 | −0.3468 | −0.070 | 0.966 | 0.232 | 0 | 0.287 | 0 | 7 | 2 | 2 | 18.319 | 12.670 | 5.6093 | 7.2459 | 0.244 |
| DA6 | 1.721 | 7.3378 | −0.0287 | −0.071 | 0.914 | 0.177 | 0.118 | 0.262 | 0 | 7 | 1 | 1 | 14.711 | 8.9512 | 5.1640 | 5.3099 | 0.201 |
| DA7 | 1.198 | 1.0262 | −0.2355 | −0.079 | 0.991 | 0.196 | 0 | 0.279 | 0 | 7 | 1 | 1 | 11.716 | 11.609 | 4.6429 | 5.3100 | 0.179 |
| DA8 | 1.198 | 0.7416 | −0.1399 | −0.074 | 0.996 | 0.245 | 0 | 0.257 | 0 | 7 | 2 | 1 | 11.954 | 11.300 | 6.8782 | 5.4597 | 0.190 |
| DA9 | 1.198 | 1.1070 | −0.1436 | −0.070 | 1.001 | 0.196 | 0 | 0.269 | 0 | 7 | 1 | 1 | 11.656 | 11.066 | 4.8526 | 5.3092 | 0.129 |
| DA10 | 1.588 | −2.2961 | −0.2223 | −0.119 | 1.087 | 0.104 | 0 | 0.307 | 0 | 7 | 1 | 1 | 8.6260 | 4.6878 | 3.0658 | 5.3097 | 0.051 |
| DA11 | 1.211 | −0.0733 | −0.0964 | −0.074 | 0.997 | 0.260 | 0.039 | 0.264 | 0 | 7 | 1 | 1 | 12.545 | 14.596 | 10.913 | 9.0331 | 0.154 |
| DHP1 | 2.151 | −6.9717 | −0.4033 | 0.094 | 1.013 | 0.264 | 0 | 0.248 | 0.183 | 4 | 1 | 1 | 31.851 | 23.834 | 9.9394 | 4.3322 | 0.366 |
| DHP6 | 2.113 | 1.0891 | −0.3044 | −0.084 | 0.869 | 0.264 | 0 | 0.261 | 0.191 | 4 | 1 | 1 | 33.429 | 25.382 | 8.1966 | 7.1145 | 0.364 |
| DHP11 | 0.884 | −3.0051 | −0.3548 | −0.033 | 0.939 | 0.284 | 0 | 0.288 | 0.215 | 4 | 1 | 1 | 31.568 | 23.373 | 7.8238 | 4.0094 | 0.386 |
| HM10 | 2.728 | −0.5443 | −0.0922 | −0.184 | 0.850 | 0.149 | 0 | 0.268 | 0.121 | 11 | 1 | 1 | 26.156 | 19.072 | 14.842 | 13.939 | 0.379 |
| HM13 | 2.872 | −3.5382 | −0.0939 | −0.181 | 0.791 | 0.149 | 0 | 0.266 | 0.163 | 11 | 1 | 1 | 22.243 | 15.955 | 22.571 | 11.192 | 0.322 |
| HM14 | 2.788 | 7.1796 | −0.0891 | −0.092 | 0.941 | 0.149 | 0 | 0.308 | 1.532 | 11 | 1 | 1 | 21.555 | 19.234 | 13.523 | 11.140 | 0.306 |
| HM15 | 2.788 | 2.2566 | −0.1016 | −0.183 | 0.988 | 0.149 | 0 | 0.291 | 1.532 | 11 | 1 | 1 | 20.069 | 13.500 | 19.511 | 14.199 | 0.256 |
| HM16 | 2.559 | 1.2276 | −0.1192 | −0.119 | 0.880 | 0.149 | 0 | 0.336 | 1.648 | 11 | 1 | 1 | 23.994 | 21.070 | 11.162 | 14.199 | 0.323 |
| NIMO | 0.727 | −4.1293 | −0.1769 | −0.095 | 1.057 | 0.129 | 0 | 0.390 | 1.906 | 7 | 0 | 0 | 16.456 | 15.744 | 4.2296 | 6.2738 | 0.111 |
| Prediction set | |||||||||||||||||
| DA12 | 1.211 | −0.4667 | −0.1001 | −0.078 | 0.997 | 0.260 | 0.029 | 0.269 | 0 | 7 | 1 | 1 | 12.708 | 12.200 | 10.202 | 14.360 | 0.175 |
| DHP5 | 2.505 | −5.7584 | −0.2736 | 0.013 | 1.016 | 0.264 | 0 | 0.285 | 0 | 4 | 1 | 1 | 35.806 | 22.011 | 12.323 | 5.1042 | 0.271 |
| DHP7 | 1.849 | 0.5783 | −0.3282 | −0.106 | 0.882 | 0.264 | 0 | 0.292 | 0.092 | 4 | 1 | 1 | 22.168 | 18.126 | 8.0499 | 7.1145 | 0.342 |
| DHP9 | 2.203 | 1.4613 | −0.2254 | 0.062 | 0.873 | 0.264 | 0 | 0.329 | 0 | 4 | 1 | 1 | 17.679 | 16.303 | 10.610 | 7.8853 | 0.253 |
| DHP10 | 2.467 | 1.9612 | −0.2013 | −0.082 | 0.861 | 0.264 | 0 | 0.298 | 0 | 4 | 1 | 1 | 37.457 | 23.559 | 10.756 | 7.8853 | 0.258 |
| DHP12 | 0.619 | −3.2561 | −0.3772 | −0.087 | 0.954 | 0.284 | 0 | 0.319 | 0.105 | 4 | 1 | 1 | 21.045 | 16.118 | 7.6773 | 4.0094 | 0.359 |
| HM8 | 1.485 | 4.3519 | −0.2163 | −0.054 | 0.832 | 0.251 | 0 | 0.321 | 0 | 4 | 1 | 1 | 24.438 | 19.468 | 13.238 | 10.310 | 0.343 |
| M3 | 0.825 | 3.5196 | −0.2307 | −0.114 | 0.874 | 0.264 | 0 | 0.302 | 0 | 4 | 1 | 1 | 17.510 | 12.141 | 7.5004 | 6.4756 | 0.302 |
| MD20 | 2.058 | 3.0868 | −0.2009 | −0.043 | 0.838 | 0.149 | 0 | 0.316 | 0 | 7 | 1 | 1 | 22.172 | 15.548 | 8.1647 | 5.3099 | 0.418 |
| NICA | 0.906 | −3.4479 | −0.2082 | −0.037 | 1.091 | 0.231 | 0 | 0.364 | 0.741 | 8 | 0 | 0 | 19.970 | 15.665 | 9.4702 | 10.748 | 0.115 |
| Compound | k (×10−3) | k (×10−3) | Error % |
|---|---|---|---|
| Experimental Value | Predicted Value | ||
| Calibration set | |||
| DA1 | 0.443 | 0.452 | 1.878 |
| DA2 | 1.231 | 1.226 | −0.400 |
| DA3 | 0.122 | 0.120 | −1.215 |
| DA4 | 0.123 | 0.129 | 4.884 |
| DA5 | 14.042 | 14.043 | 0.007 |
| DA6 | 0.160 | 0.161 | 0.632 |
| DA7 | 0.134 | 0.136 | 0.788 |
| DA8 | 0.432 | 0.429 | −0.788 |
| DA9 | 0.106 | 0.097 | −9.039 |
| DA10 | 1.848 | 1.852 | 0.221 |
| DA11 | 0.132 | 0.126 | −4.333 |
| DHP1 | 39.240 | 39.239 | −0.002 |
| DHP6 | 20.800 | 20.801 | 0.006 |
| DHP11 | 22.350 | 22.351 | 0.005 |
| HM10 | 0.192 | 0.197 | 2.814 |
| HM13 | 0.095 | 0.095 | 0.299 |
| HM14 | 2.277 | 2.292 | 0.623 |
| HM15 | 0.149 | 0.143 | −4.165 |
| HM16 | 5.628 | 5.616 | −0.221 |
| NIMO | 0.829 | 0.830 | 0.118 |
| Prediction set | |||
| DA12 | 0.139 | 0.135 | −2.704 |
| DHP5 | 37.630 | 35.454 | −5.783 |
| DHP7 | 9.100 | 9.683 | 6.403 |
| DHP9 | 14.330 | 15.072 | 5.179 |
| DHP10 | 22.230 | 22.264 | 0.154 |
| DHP12 | 8.130 | 8.770 | 7.876 |
| HM8 | 10.099 | 10.365 | 2.635 |
| M3 | 0.264 | 0.228 | −13.662 |
| MD20 | 7.812 | 7.978 | 2.123 |
| NICA | 0.604 | 0.640 | 5.813 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chieffallo, M.; De Luca, M.; Grande, F.; Occhiuzzi, M.A.; Gündüz, M.G.; Garofalo, A.; Ioele, G. Multivariate Approaches in Quantitative Structure–Property Relationships Study for the Photostability Assessment of 1,4-Dihydropyridine Derivatives. Pharmaceutics 2024, 16, 206. https://doi.org/10.3390/pharmaceutics16020206
Chieffallo M, De Luca M, Grande F, Occhiuzzi MA, Gündüz MG, Garofalo A, Ioele G. Multivariate Approaches in Quantitative Structure–Property Relationships Study for the Photostability Assessment of 1,4-Dihydropyridine Derivatives. Pharmaceutics. 2024; 16(2):206. https://doi.org/10.3390/pharmaceutics16020206
Chicago/Turabian StyleChieffallo, Martina, Michele De Luca, Fedora Grande, Maria Antonietta Occhiuzzi, Miyase Gözde Gündüz, Antonio Garofalo, and Giuseppina Ioele. 2024. "Multivariate Approaches in Quantitative Structure–Property Relationships Study for the Photostability Assessment of 1,4-Dihydropyridine Derivatives" Pharmaceutics 16, no. 2: 206. https://doi.org/10.3390/pharmaceutics16020206
APA StyleChieffallo, M., De Luca, M., Grande, F., Occhiuzzi, M. A., Gündüz, M. G., Garofalo, A., & Ioele, G. (2024). Multivariate Approaches in Quantitative Structure–Property Relationships Study for the Photostability Assessment of 1,4-Dihydropyridine Derivatives. Pharmaceutics, 16(2), 206. https://doi.org/10.3390/pharmaceutics16020206