
Simultaneously Predicting the Pharmacokinetics of CES1-Metabolized Drugs and Their Metabolites Using Physiologically Based Pharmacokinetic Model in Cirrhosis Subjects


Abstract
:1. Introduction
2. Materials and Methods
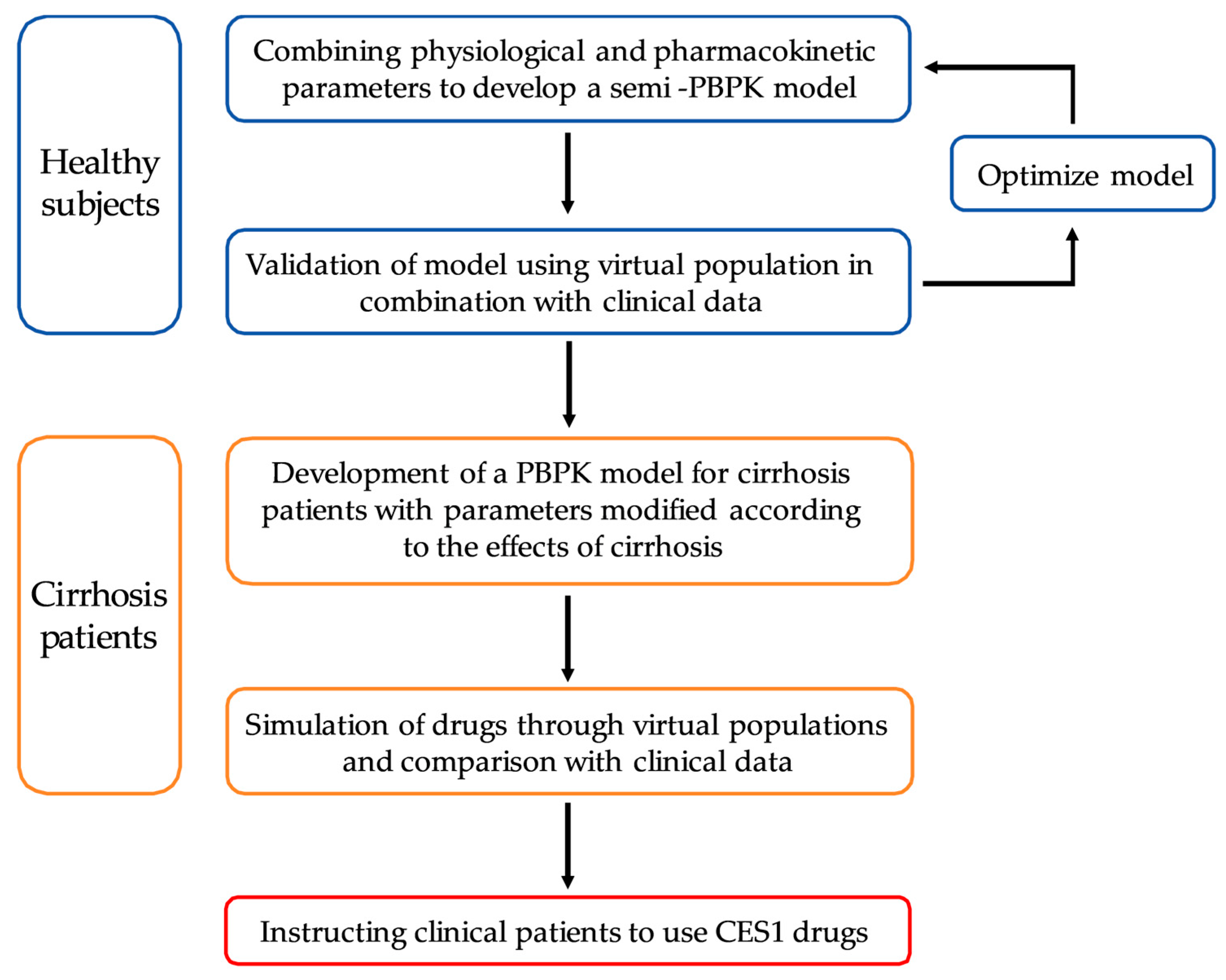
2.1. General Workflow
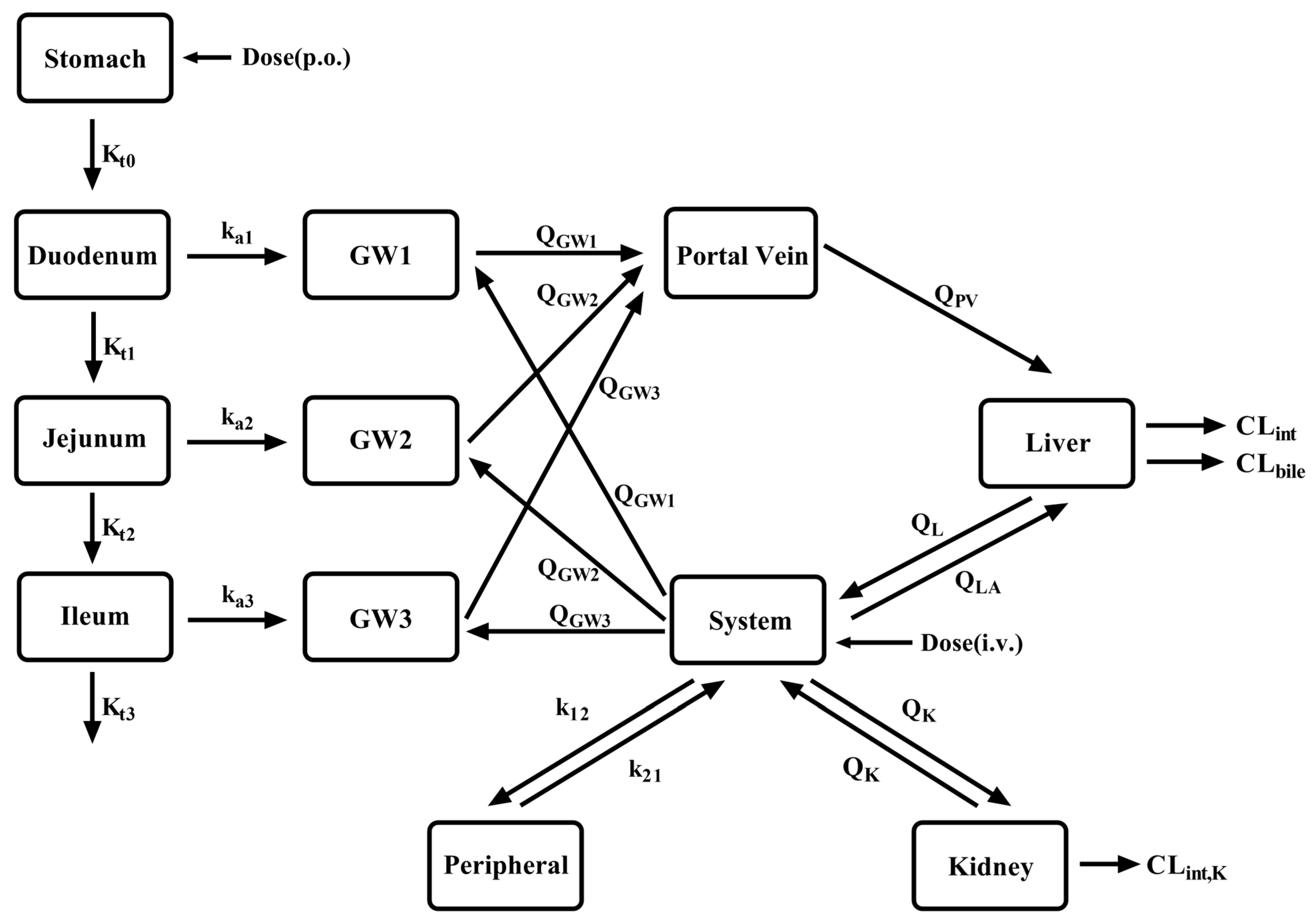
2.2. Model Development
2.3. PBPK Model Development in LC Patients
2.4. Criterion of the Developed PBPK Model
3. Results
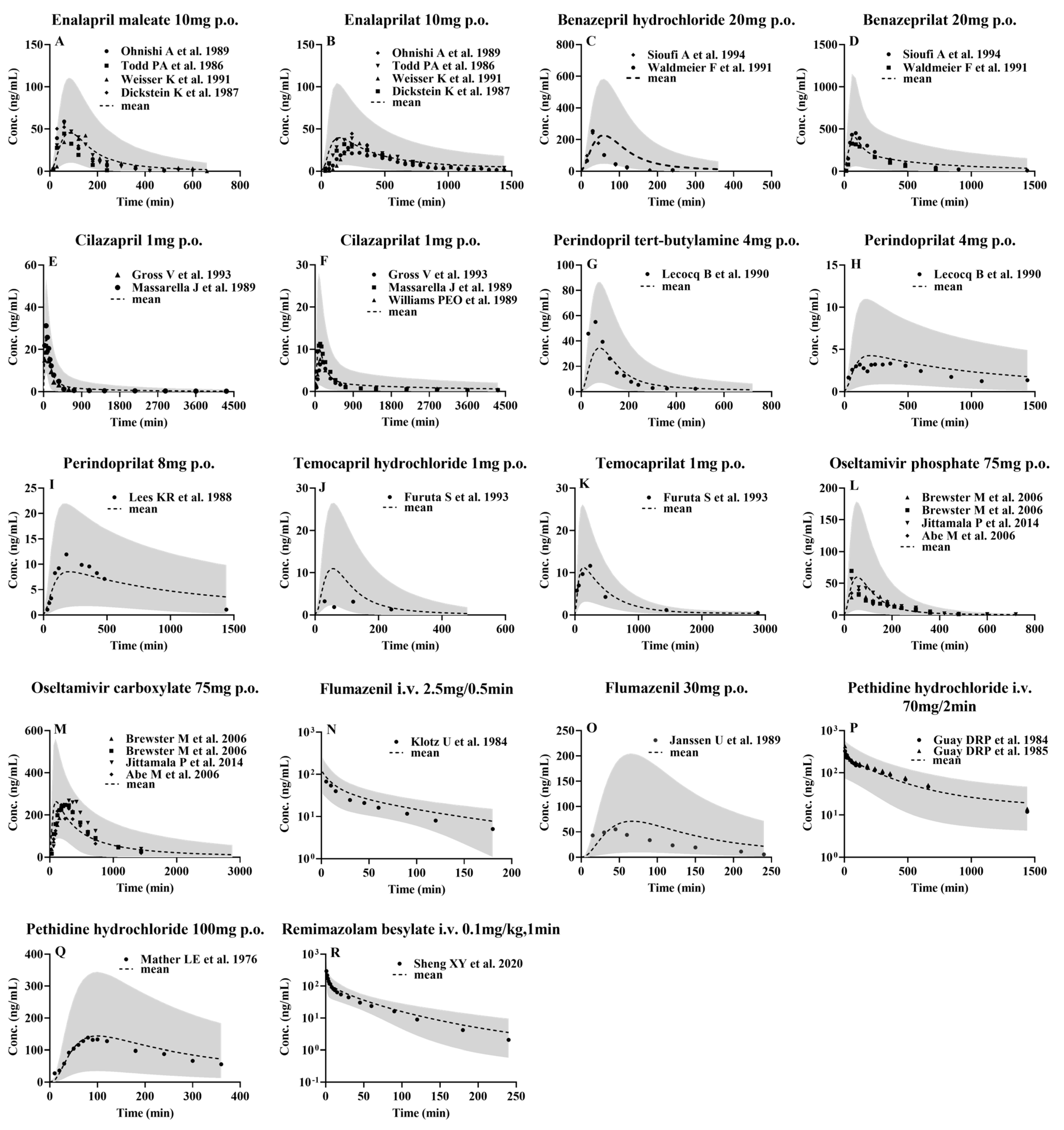
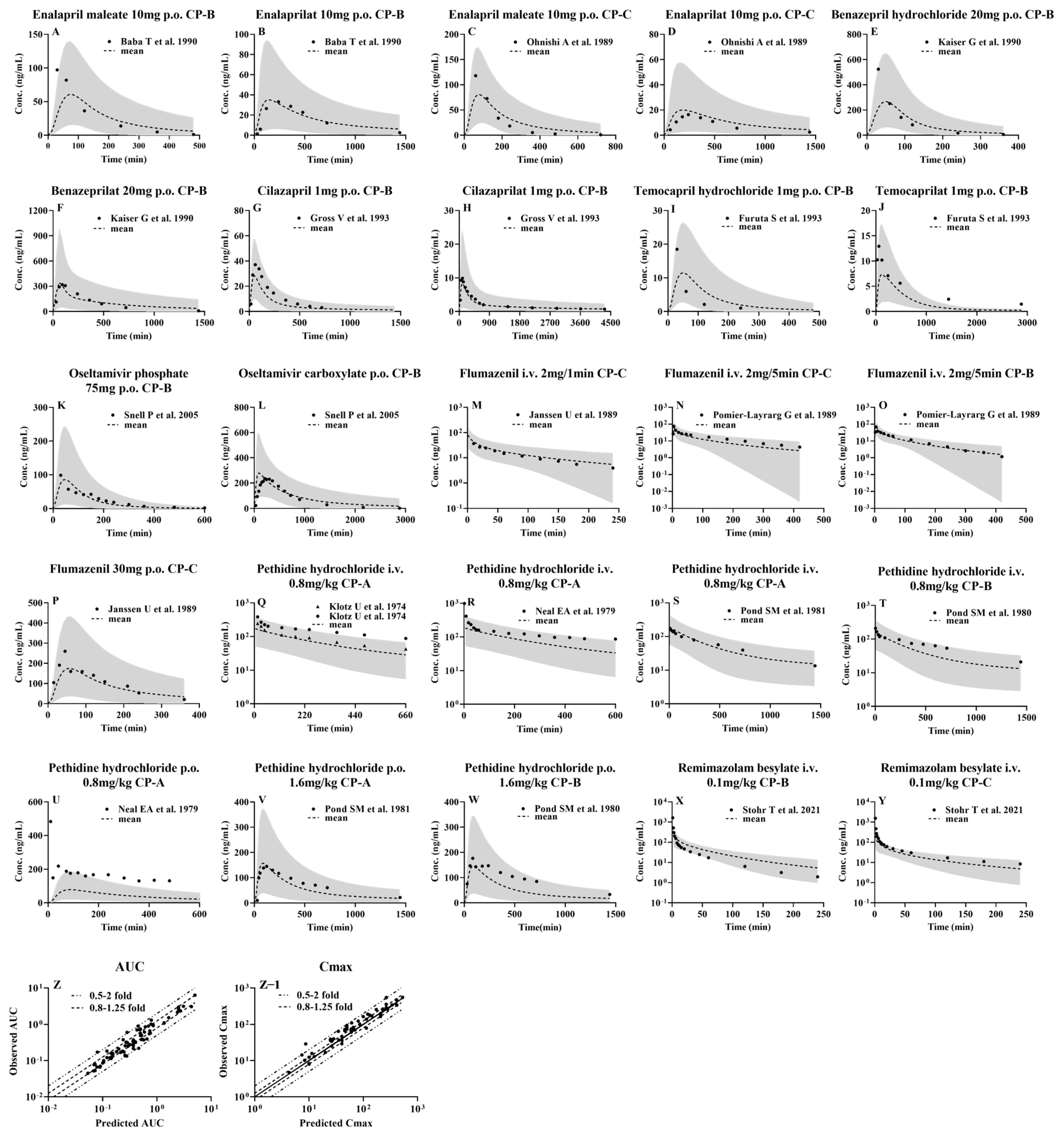
3.1. Drug Data Set
3.1.1. Enalapril and Enalaprilat

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Authors | Drug | Dose (mg) | Analytes | Subjects (n) | Ref |
|---|---|---|---|---|---|---|
| 1 | Ohnishi A et al., 1989 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy (7) | [14] |
| enalapril maleate | 10, p.o | enalapril, enalaprilat | CP-C (7) | |||
| 2 | Todd PA et al., 1986 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy (12) | [104] |
| 3 | Weisser K et al., 1991 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy (8) | [105] |
| 4 | Dickstein K et al., 1987 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy (10) | [106] |
| 5 | Baba T et al., 1990 | enalapril maleate | 10, p.o | enalapril, enalaprilat | CP-B (7) | [107] |
| 6 | Kaiser G et al., 1989 | benazepril HCl | 10, p.o | benazepril, benazeprilat | Healthy (59) | [108] |
| 7 | Schweizer C et al., 1993 | benazepril HCl | 10, p.o | benazepril, benazeprilat | Healthy (11) | [109] |
| 8 | Sioufi A et al., 1994 | benazepril HCl | 20, p.o | benazepril, benazeprilat | Healthy (24) | [110] |
| 9 | Waldmeier F et al., 1991 | benazepril HCl | 20, p.o | benazepril, benazeprilat | Healthy (4) | [111] |
| 10 | Kaiser G et al., 1990 | benazepril HCl | 20, p.o | benazepril, benazeprilat | CP-B (12) | [112] |
| 11 | Macdonald NJ et al., 1993 | benazepril HCl | 10, p.o | benazeprilat | Healthy (18) | [113] |
| 12 | Massarella J et al., 1989 | cilazapril | 1.0, 2.5, 5, p.o | cilazapril, cilazaprilat | Healthy (24) | [51] |
| 13 | Williams PEO et al., 1990 | cilazapril | 2.5, p.o | cilazapril, cilazaprilat | Healthy (13) | [114] |
| 14 | Gross V et al., 1993 | cilazapril | 1, p.o | cilazapril, cilazaprilat | Healthy (10) | [115] |
| cilazapril | 1, p.o | cilazapril, cilazaprilat | CP-B (9) | |||
| 15 | Williams PEO et al., 1989 | cilazapril | 1, p.o | cilazapril, cilazaprilat | Healthy (12) | [116] |
| 16 | Massarella JW et al., 1989 | cilazapril | 5, p.o | cilazapril, cilazaprilat | Healthy (16) | [117] |
| 17 | Francis RJ et al., 1987 | cilazapril | 1.25, 2.5, 5,10, p.o | cilazaprilat | Healthy (12) | [118] |
| 18 | Lecocq B et al., 1990 | perindopril a | 4, p.o | perindopril, perindoprilat | Healthy (12) | [119] |
| 19 | Tsai HH et al., 1989 | perindopril a | 8, p.o | perindopril, perindoprilat | CP-A (8) | [120] |
| 20 | Thiollet M et al., 1992 | perindopril a | 8, p.o | perindopril, perindoprilat | CP-B (10) | [121] |
| 21 | Lees KR et al., 1988 | perindopril a | 8, p.o | perindoprilat | Healthy (8) | [122] |
| 22 | Furuta S et al., 1993 | temocapril HCl | 1, p.o | temocapril, temocaprilat | Healthy (6) | [123] |
| temocapril HCl | 1, p.o | temocapril, temocaprilat | CP-C (7) | |||
| 23 | Abe M et al., 2006 | oseltamivir b | 75, p.o | oseltamivir, oseltamivir carboxylate | Healthy (7) | [124] |
| 24 | Brewster M et al., 2006 | oseltamivir b | 75, p.o | oseltamivir, oseltamivir carboxylate | Healthy (18) | [125] |
| 25 | Jittamala P et al., 2014 | oseltamivir b | 75, p.o | oseltamivir, oseltamivir carboxylate | Healthy (12) | [126] |
| oseltamivir b | 150, p.o | oseltamivir, oseltamivir carboxylate | Healthy (12) | |||
| 26 | Snell P et al., 2005 | oseltamivir b | 75, p.o | oseltamivir, oseltamivir carboxylate | CP-B (11) | [15] |
| 27 | Amrei R et al., 1990 | flumazenil | 10 mg, i.v. | flumazenil | Healthy (NA) | [127] |
| 28 | Breimer LTM et al., 1991 | flumazenil | 10/10 min, iv | flumazenil | Healthy (7) | [128] |
| 29 | Pomier-Layrargues G et al., 1989 | flumazenil | 2/5 min, iv | flumazenil | CP-B (8) | [129] |
| flumazenil | 2/5 min, iv | flumazenil | CP-C (8) | |||
| 30 | Klotz U et al., 1984 | flumazenil | 2.5, i.v | flumazenil | Healthy (6) | [81] |
| 31 | Janssen U,et al., 1989 | flumazenil | 30, p.o | flumazenil | Healthy (8) | [130] |
| flumazenil | 2, i.v; 30, p.o | flumazenil | CP-C (8) | |||
| 32 | Verbeeck RK et al., 1981 | pethidine HCl | 25, i.v | pethidine | Healthy (6) | [131] |
| pethidine HCl | 25, p.o | pethidine | Healthy (6) | |||
| 33 | Mather LE et al., 1975 | pethidine HCl | 50, i.v | pethidine | Healthy (4) | [132] |
| 34 | Kuhnert BR et al., 1980 | pethidine HCl | 50, i.v | pethidine | Healthy (7) | [133] |
| 35 | Guay DR et al., 1984 | pethidine HCl | 70, i.v | pethidine | Healthy (8) | [134] |
| 36 | Guay DR et al., 1985 | pethidine HCl | 70, i.v | pethidine | Healthy (8) | [135] |
| 37 | Pond SM et al., 1981 | pethidine HCl | 60, iv; 112, po | pethidine | CP-A (5) | [136] |
| 38 | Pond SM et al., 1980 | pethidine HCl | 54.4, iv; 108.8, po | pethidine | CP-B (4) | [137] |
| 39 | Mather LE et al., 1976 | pethidine HCl | 50, iv; 100, po | pethidine | Healthy (4) | [138] |
| 40 | Klotz U et al., 1974 | pethidine HCl | 63.9, i.v | pethidine | Healthy (8) | [139] |
| pethidine HCl | 53.1, i.v | pethidine | CP-A (10) | |||
| 41 | Neal EA et al., 1979 | pethidine HCl | 56, iv; 56, po | pethidine | Healthy (4) | [140] |
| pethidine HCl | 56, iv; 56, po | pethidine | CP-A (8) | |||
| 42 | Sheng XY et al., 2020 | remimazolam besylate | 1.5425, 3.315, i.v | remimazolam | Healthy (3) | [76] |
| remimazolam besylate | 4.8675, 6.18, i.v | remimazolam | Healthy (7) | |||
| remimazolam besylate | 13.26, 24.6, i.v | remimazolam | Healthy (8) | |||
| remimazolam besylate | 18.3, i.v | remimazolam | Healthy (10) | |||
| 43 | Stohr T et al., 2021 | remimazolam besylate | 10.4, i.v | remimazolam | CP-B (8) | [141] |
| remimazolam besylate | 8.2, i.v | remimazolam | CP-C (3) |
3.1.2. Benazepril and Benazeprilat
3.1.3. Cilazapril and Cilazaprilat
3.1.4. Perindopril and Perindoprilat
3.1.5. Temocapril and Temocaprilat
3.1.6. Oseltamivir and Oseltamivir Carboxylate
3.1.7. Flumazenil
3.1.8. Pethidine
3.1.9. Remimazolam
3.2. Development of PBPK Model and Validation Using Pharmacokinetic Parameters from Healthy Subjects following i.v. or Oral Administrations
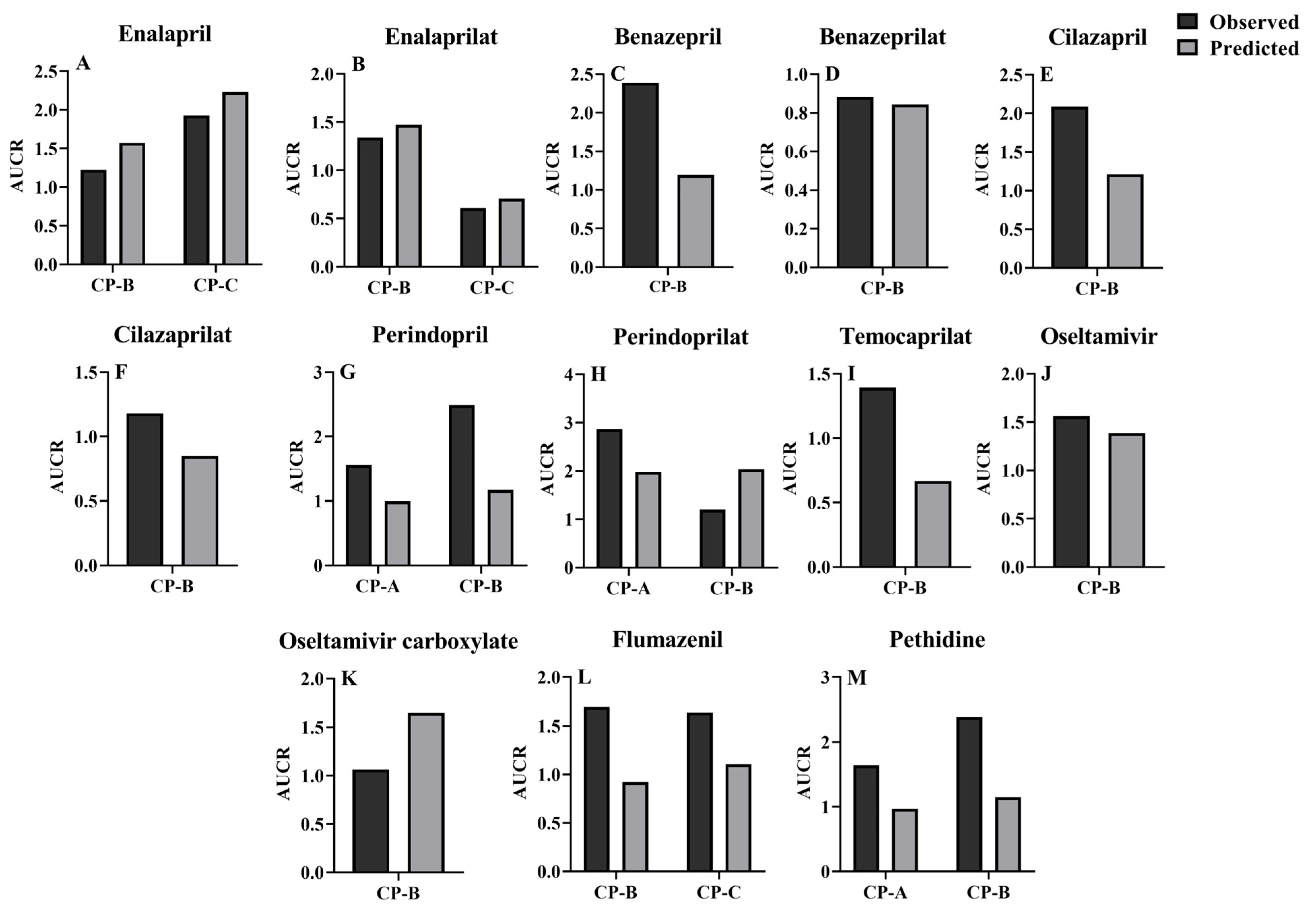
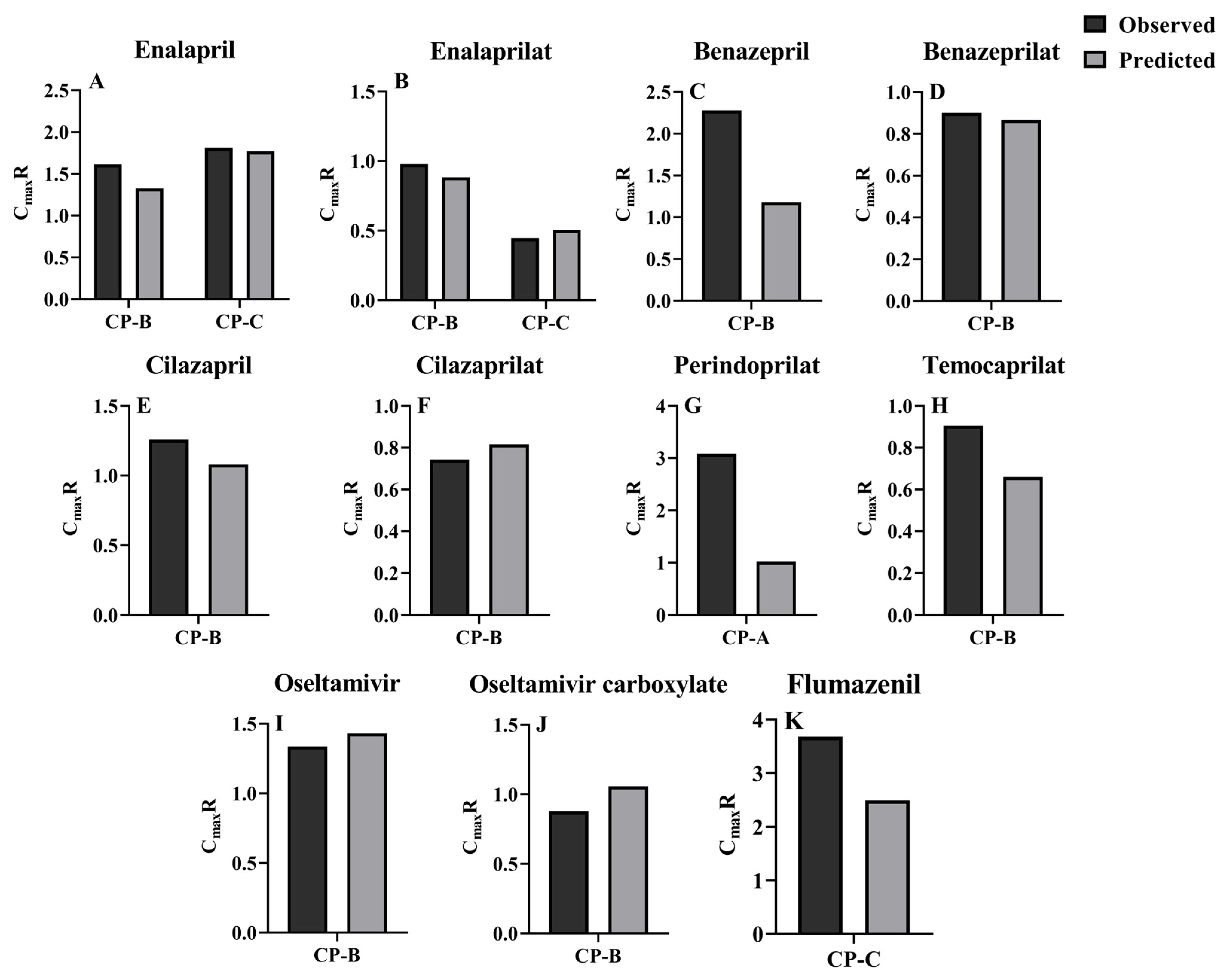
3.3. Prediction of Pharmacokinetic Profiles for CES1 Substrates and Their Active Metabolites following i.v. or Oral Administration to LC Patients Using the Developed PBPK Model
3.4. Sensitivity Analysis of Model Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gines, P.; Krag, A.; Abraldes, J.G.; Sola, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Suk, K.T. The Role of the Gut Microbiome in Liver Cirrhosis Treatment. Int. J. Mol. Sci. 2020, 22, 199. [Google Scholar] [CrossRef] [PubMed]
- El-Khateeb, E.; Darwich, A.S.; Achour, B.; Athwal, V.; Rostami-Hodjegan, A. Review article: Time to revisit Child-Pugh score as the basis for predicting drug clearance in hepatic impairment. Aliment. Pharmacol. Ther. 2021, 54, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Pugh, R.N.; Murray-Lyon, I.M.; Dawson, J.L.; Pietroni, M.C.; Williams, R. Transection of the oesophagus for bleeding oesophageal varices. Br. J. Surg. 1973, 60, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Weersink, R.A.; Burger, D.M.; Hayward, K.L.; Taxis, K.; Drenth, J.P.H.; Borgsteede, S.D. Safe use of medication in patients with cirrhosis: Pharmacokinetic and pharmacodynamic considerations. Expert Opin. Drug Metab. Toxicol. 2020, 16, 45–57. [Google Scholar] [CrossRef]
- Duthaler, U.; Bachmann, F.; Suenderhauf, C.; Grandinetti, T.; Pfefferkorn, F.; Haschke, M.; Hruz, P.; Bouitbir, J.; Krahenbuhl, S. Liver Cirrhosis Affects the Pharmacokinetics of the Six Substrates of the Basel Phenotyping Cocktail Differently. Clin. Pharmacokinet. 2022, 61, 1039–1055. [Google Scholar] [CrossRef]
- Villeneuve, J.P.; Verbeeck, R.K.; Wilkinson, G.R.; Branch, R.A. Furosemide kinetics and dynamics in patients with cirrhosis. Clin. Pharmacol. Ther. 1986, 40, 14–20. [Google Scholar] [CrossRef]
- Food and Drug Administration. Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling. 2003. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pharmacokinetics-patients-impaired-hepatic-function-study-design-data-analysis-and-impact-dosing-and (accessed on 25 January 2024).
- Chen, Y.; Ke, M.; Xu, J.; Lin, C. Simulation of the Pharmacokinetics of Oseltamivir and Its Active Metabolite in Normal Populations and Patients with Hepatic Cirrhosis Using Physiologically Based Pharmacokinetic Modeling. AAPS PharmSciTech 2020, 21, 98. [Google Scholar] [CrossRef]
- Her, L.; Zhu, H.J. Carboxylesterase 1 and Precision Pharmacotherapy: Pharmacogenetics and Nongenetic Regulators. Drug Metab. Dispos. 2020, 48, 230–244. [Google Scholar] [CrossRef]
- Hosokawa, M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules 2008, 13, 412–431. [Google Scholar] [CrossRef]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.K.; Streit, T.M.; Herring, K.L. Carboxylesterases: Dual roles in lipid and pesticide metabolism. J. Pestic. Sci. 2010, 35, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, A.; Tsuboi, Y.; Ishizaki, T.; Kubota, K.; Ohno, T.; Yoshida, H.; Kanezaki, A.; Tanaka, T. Kinetics and dynamics of enalapril in patients with liver cirrhosis. Clin. Pharmacol. Ther. 1989, 45, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Snell, P.; Dave, N.; Wilson, K.; Rowell, L.; Weil, A.; Galitz, L.; Robson, R. Lack of effect of moderate hepatic impairment on the pharmacokinetics of oral oseltamivir and its metabolite oseltamivir carboxylate. Br. J. Clin. Pharmacol. 2005, 59, 598–601. [Google Scholar] [CrossRef]
- Gertz, M.; Harrison, A.; Houston, J.B.; Galetin, A. Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab. Dispos. 2010, 38, 1147–1158. [Google Scholar] [CrossRef]
- Edginton, A.N.; Willmann, S. Physiology-based simulations of a pathological condition: Prediction of pharmacokinetics in patients with liver cirrhosis. Clin. Pharmacokinet. 2008, 47, 743–752. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Li, R.; Barton, H.A.; Maurer, T.S. A Mechanistic Pharmacokinetic Model for Liver Transporter Substrates Under Liver Cirrhosis Conditions. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 338–349. [Google Scholar] [CrossRef]
- Perdaems, N.; Blasco, H.; Vinson, C.; Chenel, M.; Whalley, S.; Cazade, F.; Bouzom, F. Predictions of metabolic drug-drug interactions using physiologically based modelling: Two cytochrome P450 3A4 substrates coadministered with ketoconazole or verapamil. Clin. Pharmacokinet. 2010, 49, 239–258. [Google Scholar] [CrossRef]
- Johnson, T.N.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.T.; Rostami-Hodjegan, A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 2010, 49, 189–206. [Google Scholar] [CrossRef]
- Gertz, M.; Houston, J.B.; Galetin, A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab. Dispos. 2011, 39, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Badhan, R.; Penny, J.; Galetin, A.; Houston, J.B. Methodology for development of a physiological model incorporating CYP3A and P-glycoprotein for the prediction of intestinal drug absorption. J. Pharm. Sci. 2009, 98, 2180–2197. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, S.; Fynne, L.; Gronbaek, H.; Krogh, K. Small intestinal transit in patients with liver cirrhosis and portal hypertension: A descriptive study. BMC Gastroenterol. 2012, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Rodriquez, A.; Martin, A.; Oterino, J.A.; Blanco, I.; Jimenez, M.; Perez, A.; Novoa, J.M. Renal function in compensated hepatic cirrhosis: Effects of an amino acid infusion and relationship with nitric acid. Dig. Dis. 1999, 17, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, M.J.; Menzies, I.S.; Ho, H.; Gregory, G.G.; Casner, N.A.; Crane, R.S.; Hernandez, J.A. Assessment of intestinal permeability and absorption in cirrhotic patients with ascites using combined sugar probes. Dig. Dis. Sci. 2004, 49, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.C.; Nielsen, S.; Brandl, M.; Bauer-Brandl, A. Drug Permeability Profiling Using the Novel Permeapad(R) 96-Well Plate. Pharm. Res. 2020, 37, 93. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.S.; Yoon, C.H.; Balthasar, J.P.; Choi, B.Y.; Hong, S.H.; Kim, H.J.; Lee, J.B.; Hwang, S.W.; Yoo, S.D. Prediction of drug bioavailability in humans using immobilized artificial membrane phosphatidylcholine column chromatography and in vitro hepatic metabolic clearance. Biomed. Chromatogr. 2009, 23, 764–769. [Google Scholar] [CrossRef]
- Holford, N.H.G. Basic Principles. In Basic & Clinical Pharmacology, 12th ed.; Katzung, B.G., Masters, S.B., Trevor, A.J., Eds.; McGraw·Hill: New York City, NY, USA, 2012; p. 39. [Google Scholar]
- Dahlgren, D.; Roos, C.; Sjogren, E.; Lennernas, H. Direct In Vivo Human Intestinal Permeability (Peff) Determined with Different Clinical Perfusion and Intubation Methods. J. Pharm. Sci. 2015, 104, 2702–2726. [Google Scholar] [CrossRef]
- Tarkiainen, E.K.; Tornio, A.; Holmberg, M.T.; Launiainen, T.; Neuvonen, P.J.; Backman, J.T.; Niemi, M. Effect of carboxylesterase 1 c.428G > A single nucleotide variation on the pharmacokinetics of quinapril and enalapril. Br. J. Clin. Pharmacol. 2015, 80, 1131–1138. [Google Scholar] [CrossRef]
- Gangnus, T.; Burckhardt, B.B.; Consortium, C. Low-volume LC-MS/MS method for the pharmacokinetic investigation of carvedilol, enalapril and their metabolites in whole blood and plasma: Application to a paediatric clinical trial. Drug Test. Anal. 2021, 13, 694–708. [Google Scholar] [CrossRef]
- Claassen, K.; Willmann, S.; Eissing, T.; Preusser, T.; Block, M. A detailed physiologically based model to simulate the pharmacokinetics and hormonal pharmacodynamics of enalapril on the circulating endocrine Renin-Angiotensin-aldosterone system. Front. Physiol. 2013, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Faisal, M.; Cawello, W.; Burckhardt, B.B.; de Hoon, J.; Laer, S.; Consortium, L. Simultaneous Semi-Mechanistic Population Pharmacokinetic Modeling Analysis of Enalapril and Enalaprilat Serum and Urine Concentrations From Child Appropriate Orodispersible Minitablets. Front. Pediatr. 2019, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Hockings, N.; Ajayi, A.A.; Reid, J.L. Age and the pharmacokinetics of angiotensin converting enzyme inhibitors enalapril and enalaprilat. Br. J. Clin. Pharmacol. 1986, 21, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Jogiraju, V.K.; Avvari, S.; Gollen, R.; Taft, D.R. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm. Drug Dispos. 2017, 38, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Jhee, S.S.; Yen, M.; Ereshefsky, L.; Leibowitz, M.; Schulte, M.; Kaeser, B.; Boak, L.; Patel, A.; Hoffmann, G.; Prinssen, E.P.; et al. Low penetration of oseltamivir and its carboxylate into cerebrospinal fluid in healthy Japanese and Caucasian volunteers. Antimicrob. Agents Chemother. 2008, 52, 3687–3693. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Massarella, J.; Ward, P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin. Pharmacokinet. 1999, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Snell, P.; Oo, C.; Dorr, A.; Barrett, J. Lack of pharmacokinetic interaction between the oral anti-influenza neuraminidase inhibitor prodrug oseltamivir and antacids. Br. J. Clin. Pharmacol. 2002, 54, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, S.; Lee, H.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S.; Lim, K.S. The novel carboxylesterase 1 variant c.662A>G may decrease the bioactivation of oseltamivir in humans. PLoS ONE 2017, 12, e0176320. [Google Scholar] [CrossRef]
- Hsueh, C.H.; Hsu, V.; Zhao, P.; Zhang, L.; Giacomini, K.M.; Huang, S.M. PBPK Modeling of the Effect of Reduced Kidney Function on the Pharmacokinetics of Drugs Excreted Renally by Organic Anion Transporters. Clin. Pharmacol. Ther. 2018, 103, 485–492. [Google Scholar] [CrossRef]
- Remko, M. Acidity, lipophilicity, solubility, absorption, and polar surface area of some ACE inhibitors. Chem. Pap. 2007, 61, 133–141. [Google Scholar] [CrossRef]
- Nishimuta, H.; Houston, J.B.; Galetin, A. Hepatic, intestinal, renal, and plasma hydrolysis of prodrugs in human, cynomolgus monkey, dog, and rat: Implications for in vitro-in vivo extrapolation of clearance of prodrugs. Drug Metab. Dispos. 2014, 42, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; Cipriano, A.; Chan, K.; John, V.A. Pharmacokinetic interaction study between benazepril and amlodipine in healthy subjects. Eur. J. Clin. Pharmacol. 1994, 47, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Chan, E.; Chen, X.; Liao, X.X.; Tang, C.; Zhou, Z.W.; Huang, M.; Zhou, S.F. Simultaneous and rapid quantitation of benazepril and benazeprilat in human plasma by high performance liquid chromatography with ultraviolet detection. J. Pharm. Biomed. Anal. 2007, 44, 224–230. [Google Scholar] [CrossRef]
- Gatarić, B.B. Primena Tehnika za Naprednu Analizu Podataka u Biofarmaceutskoj Karakterizaciji Lekova: Identifikacija, Klasifikacija i Predviđanje Faktora Koji Utiču na Intestinalnu Apsorpciju Lekovitih Supstanci. Ph.D. Thesis, University of Belgrade, Belgrade, Serbia, 2021. [Google Scholar]
- Gengo, F.M.; Brady, E. The pharmacokinetics of benazepril relative to other ACE inhibitors. Clin. Cardiol. 1991, 14, IV44–IV55. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Buch, A.; Glazer, R.D.; John, V.A.; Barr, W.H. Site-differential gastrointestinal absorption of benazepril hydrochloride in healthy volunteers. Pharm. Res. 1994, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Navia, M.; Chaturvedi, P. Design principles for orally bioavailable drugs. Drug Discov. Today 1996, 1, 179–189. [Google Scholar] [CrossRef]
- Wu, L.P.; Cui, Y.; Xiong, M.J.; Wang, S.R.; Chen, C.; Ye, L.M. Mixed micellar liquid chromatography methods: Modelling quantitative retention-activity relationships of angiotensin converting enzyme inhibitors. Biomed. Chromatogr. 2008, 22, 1243–1251. [Google Scholar] [CrossRef]
- Massarella, J.; DeFeo, T.; Lin, A.; Limjuco, R.; Brown, A. The pharmacokinetics and dose proportionality of cilazapril. Br. J. Clin. Pharmacol. 1989, 27 (Suppl. 2), 199S–204S. [Google Scholar] [CrossRef]
- Fillastre, J.P.; Moulin, B.; Godin, M.; Williams, P.E.; Brown, A.N.; Francis, R.J.; Pinta, P.; Manfredi, R. Pharmacokinetics of cilazapril in patients with renal failure. Br. J. Clin. Pharmacol. 1989, 27 (Suppl. 20), 275S–282S. [Google Scholar] [CrossRef]
- Kleinbloesem, C.H.; van Brummelen, P.; Francis, R.J.; Wiegand, U.W. Clinical pharmacology of cilazapril. Drugs 1991, 41 (Suppl. 1), 3–10. [Google Scholar] [CrossRef]
- Williams, P.E.; Brown, A.N.; Rajaguru, S.; Francis, R.J.; Walters, G.E.; McEwen, J.; Durnin, C. The pharmacokinetics and bioavailability of cilazapril in normal man. Br. J. Clin. Pharmacol. 1989, 27 (Suppl. 2), 181S–188S. [Google Scholar] [CrossRef]
- Sugihara, M.; Takeuchi, S.; Sugita, M.; Higaki, K.; Kataoka, M.; Yamashita, S. Analysis of Intra- and Intersubject Variability in Oral Drug Absorption in Human Bioequivalence Studies of 113 Generic Products. Mol. Pharm. 2015, 12, 4405–4413. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Takeda, J.; Sato, S. pH-dependent inhibitory effects of angiotensin-converting enzyme inhibitors on cefroxadine uptake by rabbit small intestinal brush-border membrane vesicles and their relationship with hydrophobicity and the ratio of zwitterionic species. Biol. Pharm. Bull. 1999, 22, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Ohura, K. Evaluation of the Oral Absorption of Ester-type Prodrugs. Yakugaku Zasshi 2020, 140, 369–376. [Google Scholar] [CrossRef]
- Maeda, K.; Ieiri, I.; Yasuda, K.; Fujino, A.; Fujiwara, H.; Otsubo, K.; Hirano, M.; Watanabe, T.; Kitamura, Y.; Kusuhara, H.; et al. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin. Pharmacol. Ther. 2006, 79, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Puchler, K.; Eckl, K.M.; Fritsche, L.; Renneisen, K.; Neumayer, H.H.; Sierakowski, B.; Lavrijssen, A.T.; Thomsen, T.; Roots, I. Pharmacokinetics of temocapril and temocaprilat after 14 once daily oral doses of temocapril in hypertensive patients with varying degrees of renal impairment. Br. J. Clin. Pharmacol. 1997, 44, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Ohura, K.; Nozawa, T.; Murakami, K.; Imai, T. Evaluation of transport mechanism of prodrugs and parent drugs formed by intracellular metabolism in Caco-2 cells with modified carboxylesterase activity: Temocapril as a model case. J. Pharm. Sci. 2011, 100, 3985–3994. [Google Scholar] [CrossRef]
- Vistoli, G.; Pedretti, A.; Testa, B. Chemodiversity and molecular plasticity: Recognition processes as explored by property spaces. Future Med. Chem. 2011, 3, 995–1010. [Google Scholar] [CrossRef]
- Oguchi, H.; Miyasaka, M.; Koiwai, T.; Tokunaga, S.; Hora, K.; Sato, K.; Yoshie, T.; Shioya, H.; Furuta, S. Pharmacokinetics of temocapril and enalapril in patients with various degrees of renal insufficiency. Clin. Pharmacokinet. 1993, 24, 421–427. [Google Scholar] [CrossRef]
- Song, J.C.; White, C.M. Clinical pharmacokinetics and selective pharmacodynamics of new angiotensin converting enzyme inhibitors: An update. Clin. Pharmacokinet. 2002, 41, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kawaratani, T.; Shioya, H.; Uji, Y.; Saruta, T. Study on pharmacokinetics of a new biliary excreted oral angiotensin converting enzyme inhibitor, temocapril (CS-622) in humans. Biopharm. Drug Dispos. 1993, 14, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Helal, F.; Lane, M.E. Transdermal delivery of Angiotensin Converting Enzyme inhibitors. Eur. J. Pharm. Biopharm. 2014, 88, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Tomono, T.; Ogihara, T.; Terada, K.; Sugano, K. Investigation of biopharmaceutical drug properties suitable for orally disintegrating tablets. ADMET DMPK 2016, 4, 335–360. [Google Scholar] [CrossRef]
- Sun, H. Capture hydrolysis signals in the microsomal stability assay: Molecular mechanisms of the alkyl ester drug and prodrug metabolism. Bioorg. Med. Chem. Lett. 2012, 22, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Hurst, M.; Jarvis, B. Perindopril: An updated review of its use in hypertension. Drugs 2001, 61, 867–896. [Google Scholar] [CrossRef] [PubMed]
- Devissaguet, J.P.; Ammoury, N.; Devissaguet, M.; Perret, L. Pharmacokinetics of perindopril and its metabolites in healthy volunteers. Fundam. Clin. Pharmacol. 1990, 4, 175–189. [Google Scholar] [CrossRef]
- Vrhovac, B.; Sarapa, N.; Bakran, I.; Huic, M.; Macolic-Sarinic, V.; Francetic, I.; Wolf-Coporda, A.; Plavsic, F. Pharmacokinetic changes in patients with oedema. Clin. Pharmacokinet. 1995, 28, 405–418. [Google Scholar] [CrossRef]
- Ghiadoni, L. Perindopril for the treatment of hypertension. Expert Opin. Pharmacother. 2011, 12, 1633–1642. [Google Scholar] [CrossRef]
- Li, Q.; Hao, Z.; Yu, Y.; Tang, Y. Bioequivalence study of two perindopril tert-butylamine tablet formulations in healthy Chinese subjects under fasting and fed conditions: A randomized, open-label, single-dose, crossover trial. Biomed. Pharmacother. 2021, 135, 111221. [Google Scholar] [CrossRef]
- Ogawa, R.; Stachnik, J.M.; Echizen, H. Clinical pharmacokinetics of drugs in patients with heart failure: An update (part 2, drugs administered orally). Clin. Pharmacokinet. 2014, 53, 1083–1114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Curd, L.; Lohmer, L.L.; Ossig, J.; Schippers, F.; Stoehr, T.; Schmith, V. Population Pharmacokinetics of Remimazolam in Procedural Sedation with Nonhomogeneously Mixed Arterial and Venous Concentrations. Clin. Transl. Sci. 2021, 14, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.Y.; Liang, Y.; Yang, X.Y.; Li, L.E.; Ye, X.; Zhao, X.; Cui, Y.M. Safety, pharmacokinetic and pharmacodynamic properties of single ascending dose and continuous infusion of remimazolam besylate in healthy Chinese volunteers. Eur. J. Clin. Pharmacol. 2020, 76, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M. Remimazolam: Pharmacological characteristics and clinical applications in anesthesiology. Anesth. Pain Med. 2022, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Jiang, L.; Chen, T.M.; Hwang, K.K. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur. J. Med. Chem. 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Gottipati, G. Prediction of Human Systemic, Biologically Relevant Pharmacokinetic (PK) Properties Using Quantitative Structure Pharmacokinetic Relationships (QSPKR) and Interspecies Pharmacokinetic Allometric Scaling (PK-AS) Approaches for Four Different Pharmacological Classes of Compounds. Ph.D. Thesis, Virginia Commonwealth University, Richmond, VA, USA, 2014. [Google Scholar]
- Ellison, C.A. Structural and functional pharmacokinetic analogs for physiologically based pharmacokinetic (PBPK) model evaluation. Regul. Toxicol. Pharmacol. 2018, 99, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Klotz, U.; Ziegler, G.; Reimann, I.W. Pharmacokinetics of the selective benzodiazepine antagonist Ro 15-1788 in man. Eur. J. Clin. Pharmacol. 1984, 27, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.D.; Kumar, S.P.; Patel, C.N.; Shankar, S.S.; Pandya, H.A.; Solanki, H.A. Parallel screening of drug-like natural compounds using Caco-2 cell permeability QSAR model with applicability domain, lipophilic ligand efficiency index and shape property: A case study of HIV-1 reverse transcriptase inhibitors. J. Mol. Struct. 2017, 1146, 80–95. [Google Scholar] [CrossRef]
- Karavokiros, K.A.; Tsipis, G.B. Flumazenil: A benzodiazepine antagonist. DICP 1990, 24, 976–981. [Google Scholar] [CrossRef]
- Paixao, P.; Gouveia, L.F.; Morais, J.A. Prediction of the in vitro intrinsic clearance determined in suspensions of human hepatocytes by using artificial neural networks. Eur. J. Pharm. Sci. 2010, 39, 310–321. [Google Scholar] [CrossRef]
- Klotz, U.; Kanto, J. Pharmacokinetics and clinical use of flumazenil (Ro 15-1788). Clin. Pharmacokinet. 1988, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ghafourian, T.; Barzegar-Jalali, M.; Hakimiha, N.; Cronin, M.T. Quantitative structure-pharmacokinetic relationship modelling: Apparent volume of distribution. J. Pharm. Pharmacol. 2004, 56, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, W.E.; Castle, M.C. Species differences in the hydrolysis of meperidine and its inhibition by organophosphate compounds. Fundam. Appl. Toxicol. 1988, 11, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Alsmadi, M.M.; Idkaidek, N. The Analysis of Pethidine Pharmacokinetics in Newborn Saliva, Plasma, and Brain Extracellular Fluid After Prenatal Intrauterine Exposure from Pregnant Mothers Receiving Intramuscular Dose Using PBPK Modeling. Eur. J. Drug Metab. Pharmacokinet. 2023, 48, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.M.; Kretschzmar, K.M. Effect of phenytoin on meperidine clearance and normeperidine formation. Clin. Pharmacol. Ther. 1981, 30, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Tse, J.; Jennings, F.; Orme, M.L. Pharmacokinetics of low-dose intravenous pethidine in patients with renal dysfunction. J. Clin. Pharmacol. 1987, 27, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Paixao, P.; Gouveia, L.F.; Morais, J.A. Prediction of the human oral bioavailability by using in vitro and in silico drug related parameters in a physiologically based absorption model. Int. J. Pharm. 2012, 429, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, S.C.; Kress, D.R.; Bertz, R.J.; Pau, A.; Davey, R. The effect of ritonavir on the pharmacokinetics of meperidine and normeperidine. Pharmacotherapy 2000, 20, 549–553. [Google Scholar] [CrossRef]
- Toutain, P.L.; Lefebvre, H.P.; King, J.N. Benazeprilat disposition and effect in dogs revisited with a pharmacokinetic/pharmacodynamic modeling approach. J. Pharmacol. Exp. Ther. 2000, 280, 1087–1093. [Google Scholar]
- Pan, D.Q.; Jiang, M.; Liu, T.T.; Wang, Q.; Shi, J.H. Combined spectroscopies and molecular docking approach to characterizing the binding interaction of enalapril with bovine serum albumin. Luminescence 2017, 32, 481–490. [Google Scholar] [CrossRef]
- Lee, A.; Shirley, M. Remimazolam: A Review in Procedural Sedation. Drugs 2021, 81, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Blei, A.T. Albumin dialysis for the treatment of hepatic encephalopathy. J. Gastroenterol. Hepatol. 2004, 19, S224–S228. [Google Scholar] [CrossRef]
- Nafisi, S.; Vishkaee, T.S. Study on the interaction of tamiflu and oseltamivir carboxylate with human serum albumin. J. Photochem. Photobiol. B 2011, 105, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, D.; Radan, M.; Dikic, T.; Nikolic, M.P.; Oljacic, S.; Nikolic, K. The evaluation of drug-plasma protein binding interaction on immobilized human serum albumin stationary phase, aided by different computational approaches. J. Pharm. Biomed. Anal. 2022, 211, 114593. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.J.; Critchley, J.A.; Tomlinson, B.; Resplandy, G. Comparison of the pharmacokinetics and pharmacodynamics of oral doses of perindopril in normotensive Chinese and Caucasian volunteers. Br. J. Clin. Pharmacol. 1995, 39, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Pijls, K.E.; Jonkers, D.M.; Elamin, E.E.; Masclee, A.A.; Koek, G.H. Intestinal epithelial barrier function in liver cirrhosis: An extensive review of the literature. Liver Int. 2013, 33, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Ladumor, M.K.; Storelli, F.; Liang, X.; Lai, Y.; Enogieru, O.J.; Chothe, P.P.; Evers, R.; Unadkat, J.D. Predicting changes in the pharmacokinetics of CYP3A-metabolized drugs in hepatic impairment and insights into factors driving these changes. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 261–273. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, B.; Parker, R.B.; Laizure, S.C. Clinical implications of genetic variation in carboxylesterase drug metabolism. Expert Opin. Drug Metab. Toxicol. 2018, 14, 131–142. [Google Scholar] [CrossRef]
- Gomez, H.J.; Cirillo, V.J.; Irvin, J.D. Enalapril: A review of human pharmacology. Drugs 1985, 30 (Suppl. 1), 13–24. [Google Scholar] [CrossRef]
- Todd, P.A.; Heel, R.C. Enalapril. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension and congestive heart failure. Drugs 1986, 31, 198–248. [Google Scholar] [CrossRef]
- Weisser, K.; Schloos, J.; Lehmann, K.; Dusing, R.; Vetter, H.; Mutschler, E. Pharmacokinetics and converting enzyme inhibition after morning and evening administration of oral enalapril to healthy subjects. Eur. J. Clin. Pharmacol. 1991, 40, 95–99. [Google Scholar] [CrossRef]
- Dickstein, K.; Till, A.E.; Aarsland, T.; Tjelta, K.; Abrahamsen, A.M.; Kristianson, K.; Gomez, H.J.; Gregg, H.; Hichens, M. The pharmacokinetics of enalapril in hospitalized patients with congestive heart failure. Br. J. Clin. Pharmacol. 1987, 23, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Murabayashi, S.; Tomiyama, T.; Takebe, K. The pharmacokinetics of enalapril in patients with compensated liver cirrhosis. Br. J. Clin. Pharmacol. 1990, 29, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, G.; Ackermann, R.; Brechbuhler, S.; Dieterle, W. Pharmacokinetics of the angiotensin converting enzyme inhibitor benazepril.HCl (CGS 14 824 A) in healthy volunteers after single and repeated administration. Biopharm. Drug Dispos. 1989, 10, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, C.; Kaiser, G.; Dieterle, W.; Mann, J. Pharmacokinetics and pharmacodynamics of benazepril hydrochloride in patients with major proteinuria. Eur. J. Clin. Pharmacol. 1993, 44, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Sioufi, A.; Pommier, F.; Gauducheau, N.; Godbillon, J.; Choi, L.; John, V. The absence of a pharmacokinetic interaction between aspirin and the angiotensin-converting enzyme inhibitor benazepril in healthy volunteers. Biopharm. Drug Dispos. 1994, 15, 451–461. [Google Scholar] [CrossRef]
- Waldmeier, F.; Kaiser, G.; Ackermann, R.; Faigle, J.W.; Wagner, J.; Barner, A.; Lasseter, K.C. The disposition of [14C]-labelled benazepril HCl in normal adult volunteers after single and repeated oral dose. Xenobiotica 1991, 21, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, G.; Ackermann, R.; Gschwind, H.P.; James, I.M.; Sprengers, D.; McIntyre, N.; Defalco, A.; Holmes, I.B. The influence of hepatic cirrhosis on the pharmacokinetics of benazepril hydrochloride. Biopharm. Drug Dispos. 1990, 11, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, N.J.; Sioufi, A.; Howie, C.A.; Wade, J.R.; Elliott, H.L. The effects of age on the pharmacokinetics and pharmacodynamics of single oral doses of benazepril and enalapril. Br. J. Clin. Pharmacol. 1993, 36, 205–209. [Google Scholar] [CrossRef]
- Williams, P.E.; Brown, A.N.; Rajaguru, S.; Francis, R.J.; Bell, A.J.; Dewland, P.M. Pharmacokinetics of cilazapril during repeated oral dosing in healthy young volunteers. Eur. J. Drug Metab. Pharmacokinet. 1990, 15, 63–67. [Google Scholar] [CrossRef]
- Gross, V.; Treher, E.; Haag, K.; Neis, W.; Wiegand, U.; Scholmerich, J. Angiotensin-converting enzyme (ACE)-inhibition in cirrhosis. Pharmacokinetics and dynamics of the ACE-inhibitor cilazapril (Ro 31-2848). J. Hepatol. 1993, 17, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.E.; Brown, A.N.; Rajaguru, S.; Walters, G.E.; McEwen, J.; Durnin, C. A pharmacokinetic study of cilazapril in elderly and young volunteers. Br. J. Clin. Pharmacol. 1989, 27 (Suppl. 2), 211S–215S. [Google Scholar] [CrossRef] [PubMed]
- Massarella, J.W.; DeFeo, T.M.; Brown, A.N.; Lin, A.; Wills, R.J. The influence of food on the pharmacokinetics and ACE inhibition of cilazapril. Br. J. Clin. Pharmacol. 1989, 27 (Suppl. 2), 205S–209S. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.J.; Brown, A.N.; Kler, L.; Fasanella d’Amore, T.; Nussberger, J.; Waeber, B.; Brunner, H.R. Pharmacokinetics of the converting enzyme inhibitor cilazapril in normal volunteers and the relationship to enzyme inhibition: Development of a mathematical model. J. Cardiovasc. Pharmacol. 1987, 9, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lecocq, B.; Funck-Brentano, C.; Lecocq, V.; Ferry, A.; Gardin, M.E.; Devissaguet, M.; Jaillon, P. Influence of food on the pharmacokinetics of perindopril and the time course of angiotensin-converting enzyme inhibition in serum. Clin. Pharmacol. Ther. 1990, 47, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Lees, K.R.; Howden, C.W.; Reid, J.L. The pharmacokinetics and pharmacodynamics of perindopril in patients with hepatic cirrhosis. Br. J. Clin. Pharmacol. 1989, 28, 53–59. [Google Scholar] [CrossRef]
- Thiollet, M.; Funck-Brentano, C.; Grange, J.D.; Midavaine, M.; Resplandy, G.; Jaillon, P. The pharmacokinetics of perindopril in patients with liver cirrhosis. Br. J. Clin. Pharmacol. 1992, 33, 326–328. [Google Scholar] [CrossRef]
- Lees, K.R.; Green, S.T.; Reid, J.L. Influence of age on the pharmacokinetics and pharmacodynamics of perindopril. Clin. Pharmacol. Ther. 1988, 44, 418–425. [Google Scholar] [CrossRef]
- Furuta, S.; Kiyosawa, K.; Higuchi, M.; Kasahara, H.; Saito, H.; Shioya, H.; Oguchi, H. Pharmacokinetics of temocapril, an ACE inhibitor with preferential biliary excretion, in patients with impaired liver function. Eur. J. Clin. Pharmacol. 1993, 44, 383–385. [Google Scholar] [CrossRef]
- Abe, M.; Smith, J.; Urae, A.; Barrett, J.; Kinoshita, H.; Rayner, C.R. Pharmacokinetics of oseltamivir in young and very elderly subjects. Ann. Pharmacother. 2006, 40, 1724–1730. [Google Scholar] [CrossRef]
- Brewster, M.; Smith, J.R.; Dutkowski, R.; Robson, R. Active metabolite from Tamiflu solution is bioequivalent to that from capsule delivery in healthy volunteers: A cross-over, randomised, open-label study. Vaccine 2006, 24, 6660–6663. [Google Scholar] [CrossRef] [PubMed]
- Jittamala, P.; Pukrittayakamee, S.; Tarning, J.; Lindegardh, N.; Hanpithakpong, W.; Taylor, W.R.; Lawpoolsri, S.; Charunwattana, P.; Panapipat, S.; White, N.J.; et al. Pharmacokinetics of orally administered oseltamivir in healthy obese and nonobese Thai subjects. Antimicrob. Agents Chemother. 2014, 58, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Amrein, R.; Hetzel, W. Pharmacology of Dormicum (Midazolam) and Anexate (Flumazenil). Acta Anaesth. Scand. 1990, 34, 6–15. [Google Scholar] [CrossRef]
- Breimer, L.T.; Hennis, P.J.; Burm, A.G.; Danhof, M.; Bovill, J.G.; Spierdijk, J.; Vletter, A.A. Pharmacokinetics and EEG effects of flumazenil in volunteers. Clin. Pharmacokinet. 1991, 20, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Pomier-Layrargues, G.; Giguere, J.F.; Lavoie, J.; Willems, B.; Butterworth, R.F. Pharmacokinetics of benzodiazepine antagonist Ro 15-1788 in cirrhotic patients with moderate or severe liver dysfunction. Hepatology 1989, 10, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Janssen, U.; Walker, S.; Maier, K.; von Gaisberg, U.; Klotz, U. Flumazenil disposition and elimination in cirrhosis. Clin. Pharmacol. Ther. 1989, 46, 317–323. [Google Scholar] [CrossRef]
- Verbeeck, R.K.; Branch, R.A.; Wilkinson, G.R. Meperidine disposition in man: Influence of urinary pH and route of administration. Clin. Pharmacol. Ther. 1981, 30, 619–628. [Google Scholar] [CrossRef]
- Mather, L.E.; Tucker, G.T.; Pflug, A.E.; Lindop, M.J.; Wilkerson, C. Meperidine kinetics in man. Intravenous injection in surgical patients and volunteers. Clin. Pharmacol. Ther. 1975, 17, 21–30. [Google Scholar] [CrossRef]
- Kuhnert, B.R.; Kuhnert, P.M.; Prochaska, A.L.; Sokol, R.J. Meperidine disposition in mother, neonate, and nonpregnant females. Clin. Pharmacol. Ther. 1980, 27, 486–491. [Google Scholar] [CrossRef]
- Guay, D.R.; Meatherall, R.C.; Chalmers, J.L.; Grahame, G.R. Cimetidine alters pethidine disposition in man. Br. J. Clin. Pharmacol. 1984, 18, 907–914. [Google Scholar] [CrossRef]
- Guay, D.R.; Meatherall, R.C.; Chalmers, J.L.; Grahame, G.R.; Hudson, R.J. Ranitidine does not alter pethidine disposition in man. Br. J. Clin. Pharmacol. 1985, 20, 55–59. [Google Scholar] [CrossRef]
- Pond, S.M.; Tong, T.; Benowitz, N.L.; Jacob, P.; Rigod, J. Presystemic metabolism of meperidine to normeperidine in normal and cirrhotic subjects. Clin. Pharmacol. Ther. 1981, 30, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.M.; Tong, T.; Benowitz, N.L.; Jacob, P. Enhanced bioavailability of pethidine and pentazocine in patients with cirrhosis of the liver. Aust. N. Z. J. Med. 1980, 10, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Mather, L.E.; Tucker, G.T. Systemic availability of orally administered meperidine. Clin. Pharmacol. Ther. 1976, 20, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Klotz, U.; McHorse, T.S.; Wilkinson, G.R.; Schenker, S. The effect of cirrhosis on the disposition and elimination of meperidine in man. Clin. Pharmacol. Ther. 1974, 16, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.A.; Meffin, P.J.; Gregory, P.B.; Blaschke, T.F. Enhanced Bioavailability and Decreased Clearance of Analgesics in Patients with Cirrhosis. Gastroenterology 1979, 77, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Stohr, T.; Colin, P.J.; Ossig, J.; Pesic, M.; Borkett, K.; Winkle, P.; Struys, M.; Schippers, F. Pharmacokinetic properties of remimazolam in subjects with hepatic or renal impairment. Br. J. Anaesth. 2021, 127, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Grislain, L.; Mocquard, M.T.; Dabe, J.F.; Bertrand, M.; Luijten, W.; Marchand, B.; Resplandy, G.; Devissaguet, M. Interspecies comparison of the metabolic pathways of perindopril, a new angiotensin-converting enzyme (ACE) inhibitor. Xenobiotica 1990, 20, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Duthaler, U.; Bachmann, F.; Ozbey, A.C.; Umehara, K.; Parrott, N.; Fowler, S.; Krahenbuhl, S. The Activity of Members of the UDP-Glucuronosyltransferase Subfamilies UGT1A and UGT2B is Impaired in Patients with Liver Cirrhosis. Clin. Pharmacokinet. 2023, 62, 1141–1155. [Google Scholar] [CrossRef]
- Ishizuka, H.; Konno, K.; Naganuma, H.; Sasahara, K.; Kawahara, Y.; Niinuma, K.; Suzuki, H.; Sugiyama, Y. Temocaprilat, a novel angiotensin-converting enzyme inhibitor, is excreted in bile via an ATP-dependent active transporter (cMOAT) that is deficient in Eisai hyperbilirubinemic mutant rats (EHBR). J. Pharmacol. Exp. Ther. 1997, 280, 1304–1311. [Google Scholar]
- Gao, G.; Law, F.; Wong, R.N.S.; Mak, N.K.; Yang, M.S.M. A physiologically-based pharmacokinetic model of oseltamivir phosphate and its carboxylate metabolite for rats and humans. ADMET DMPK 2019, 7, 22–43. [Google Scholar] [CrossRef]
- Kleingeist, B.; Bocker, R.; Geisslinger, G.; Brugger, R. Isolation and pharmacological characterization of microsomal human liver flumazenil carboxylesterase. J. Pharm. Pharm. Sci. A Publ. Can. Soc. Pharm. Sci. Soc. Can. Des. Sci. Pharm. 1998, 1, 38–46. [Google Scholar]
- Tegeder, I.; Lötsch, J.; Geisslinger, G. Pharmacokinetics of Opioids in Liver Disease. Clin. Pharmacokinet. 1999, 37, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Ikeda, M.; Kishimoto, M.; Tsujino, T.; Kaneto, H.; Matsuhisa, M.; Kajimoto, Y.; Watarai, T.; Yamasaki, Y.; Hori, M. Evaluation of gut motility in type II diabetes by the radiopaque marker method. J. Gastroenterol. Hepatol. 2000, 15, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Brightman, F.; Gill, H.; Lee, S.; Pufong, B. Simulation modelling of human intestinal absorption using Caco-2 permeability and kinetic solubility data for early drug discovery. J. Pharm. Sci. 2008, 97, 4557–4574. [Google Scholar] [CrossRef]
- Chaturvedi, P.R.; Decker, C.J.; Odinecs, A. Prediction of pharmacokinetic properties using experimental approaches during early drug discovery. Curr. Opin. Chem. Biol. 2001, 5, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Okino, M.S.; Mavrovouniotis, M.L. Simplification of Mathematical Models of Chemical Reaction Systems. Chem. Rev. 1998, 98, 391–408. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2014, 79, 48–55. [Google Scholar] [CrossRef]
- Riccardi, K.; Cawley, S.; Yates, P.D.; Chang, C.; Funk, C.; Niosi, M.; Lin, J.; Di, L. Plasma Protein Binding of Challenging Compounds. J. Pharm. Sci. 2015, 104, 2627–2636. [Google Scholar] [CrossRef]
- Turpeinen, M.; Zanger, U.M. Cytochrome P450 2B6: Function, genetics, and clinical relevance. Drug Metab. Drug Interact. 2012, 27, 185–197. [Google Scholar] [CrossRef]
- Danziger, L.H.; Martin, S.J.; Blum, R.A. Central Nervous System Toxicity Associated with Meperidine Use in Hepatic Disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1994, 14, 235–238. [Google Scholar] [CrossRef]
- Soleimanpour, H.; Safari, S.; Shahsavari Nia, K.; Sanaie, S.; Alavian, S.M. Opioid Drugs in Patients with Liver Disease: A Systematic Review. Hepat. Mon. 2016, 16, e32636. [Google Scholar] [CrossRef]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.; Mathias, A.; et al. Abundance of Phase 1 and 2 Drug-Metabolizing Enzymes in Alcoholic and Hepatitis C Cirrhotic Livers: A Quantitative Targeted Proteomics Study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef]
- Kapczinski, F.; Sherman, D.; Williams, R.; Lader, M.; Curran, V. Differential effects of flumazenil in alcoholic and nonalcoholic cirrhotic patients. Psychopharmacology 1995, 120, 220–226. [Google Scholar] [CrossRef]
- Shi, J.; Wang, X.; Nguyen, J.H.; Bleske, B.E.; Liang, Y.; Liu, L.; Zhu, H.J. Dabigatran etexilate activation is affected by the CES1 genetic polymorphism G143E (rs71647871) and gender. Biochem. Pharmacol. 2016, 119, 76–84. [Google Scholar] [CrossRef]
- Gines, P.; Schrier, R.W. Renal failure in cirrhosis. N. Engl. J. Med. 2009, 361, 1279–1290. [Google Scholar] [CrossRef] [PubMed]

| Normal | Child–Pugh Class | Units | |||
|---|---|---|---|---|---|
| A | B | C | |||
| Blood flow rates | |||||
| Liver a | 1450 [17,18] | 1436.5 | 1176.9 | 1656.3 | mL/min |
| Hepatic arterial | 300 [18] | 390 [17,18] | 486.9 [9] | 1020 [17] | mL/min |
| Portal vein | 1150 [18] | 1046.5 [9] | 690 [19] | 636.3 [9] | mL/min |
| Kidney | 1240 [18] | 1091.2 [17] | 806 [17] | 595.2 [17] | mL/min |
| Duodenum b | 45 [20] | 45 | 45 | 45 | mL/min |
| Jejunum b | 173 [20] | 173 | 173 | 173 | mL/min |
| Ileum b | 102 [20] | 102 | 102 | 102 | mL/min |
| Volume | |||||
| Liver | 1690 [18] | 1368.9 [21] | 1098.5 [21] | 895.7 [21] | mL |
| Portal vein b | 70 [18] | 70 | 70 | 70 | mL |
| Kidney b | 280 [18] | 280 | 280 | 280 | mL |
| Duodenum b | 21 [22] | 21 | 21 | 21 | mL |
| Jejunum b | 63 [22] | 63 | 63 | 63 | mL |
| Ileum b | 42 [22] | 42 | 42 | 42 | mL |
| Transit rates c | |||||
| Stomach | 0.04 [23] | 0.0504 [24] | 0.0504 [24] | 0.0504 [24] | min−1 |
| Duodenum | 0.07 [23] | 0.0889 [24] | 0.0889 [24] | 0.0889 [24] | min−1 |
| Jejunum | 0.03 [23] | 0.0381 [24] | 0.0381 [24] | 0.0381 [24] | min−1 |
| Ileum | 0.04 [23] | 0.0508 [24] | 0.0508 [24] | 0.0508 [24] | min−1 |
| Gut radius | |||||
| r1 b | 2 [23] | 2 | 2 | 2 | cm |
| r2 b | 1.63 [23] | 1.63 | 1.63 | 1.63 | cm |
| r3 b | 1.45 [23] | 1.45 | 1.45 | 1.45 | cm |
| Glomerular filtration rate | 105 [25] | 82 [25] | 82 [25] | 82 [25] | mL/min |
| Albumin | 44.7 [9] | 36.2 [17] | 30.4 [17] | 26.3 [9] | g/L |
| α1-acid glycoprotein | 0.8 [21] | 0.57 [21] | 0.52 [21] | 0.46 [21] | g/L |
| CES1 | 2.45 [9] | 2.45 [9] | 1.715 [9] | 0.735 [9] | mg/g Liver |
| CYP2B6 | 17 [21] | 17 [21] | 15.3 [21] | 13.6 [21] | pmol/mg |
| Lactulose/Rhamnose ratio | 0.037 [26] | 0.046 [26] | 0.052 [26] | 0.057 [26] | / |
| MRP2 ratio | 1 | 0.54 [19] | 0.54 [19] | 0.54 [19] | / |
| Drug | logP | pka | CLint | Vmax | Km | KL;P d | KG;P d | KK;P d | CLb | Vsys | K12 | K21 | Peff,A–B | CLint,K | Rb | fu,b | F | ka |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mL/min | nmoL/min/ mg protein | μmol/L | mL/min | L | min−1 | min−1 | 10−4 cm/s | mL/min | ||||||||||
| Enalapril | 0.59 [27] | 5.20 [27] | 784 [28] | / | / | 1.66 | 2.29 | 1.79 | / | 40 [29] | / | / | 1.60 [30] | 624.6 [31] | 0.74 [32] | 0.74 [33] | / | |
| Enalaprilat | −0.74 [33] | 2.03 [33] | / | / | / | 1.12 | 1.04 | 1.25 | / | 46.1 [34] | 0.001 [34] | 0.0009 [34] | / | 186.4 [35] | 0.73 [32] | 0.68 [33] | / | |
| Oseltamivir | 0.36 [36] | 7.7 [36] | 20,255.4 [36] | / | / | 1.19 | 1.12 | 1.29 | / | 61.289 [37] f | / | / | / | 1357.95 [38] | 1 e | 0.58 [36] | / | 0.061 [39] g |
| Oseltamivir carboxylate | −1.3 a | 4.19 a | / | / | / | 1.71 | 1.89 | 1.91 | / | 160.729 [40] f | / | / | / | 438.5 [41] | 1 e | 0.97 [36] | / | |
| Benazepril | 1.11 [42] | 4.74 [42] | 6696 [43] | / | / | 0.087 | 0.122 | 0.088 | 385.8 [44] g | 4.8 [45] g | 0.0215 [45] g | 0.0238 [45] g | 1.21 [46] | 8391.6 c | 1 e | 0.03 [47] | 0.35 [29] | |
| Benazeprilat | 0.56 [42] | 1.97 [42] | / | / | / | 0.093 | 0.088 | 0.101 | / | 1.204 [48] f | 0.0438 [48] f | 0.00837 [48] f | / | 447.9 [47] | 1 e | 0.05 [47] | / | |
| Cilazapril | 0.55 [49] | 3.3 [50] | 199.7 c | / | / | 1.32 | 1.31 | 1.43 | 205 a | 18.23 [51] f | 0.00325 [51] f | 0.00155 [51] f | / | 118.095 [52] | 1 e | 0.7 [49] | / | 0.099 [53] g |
| Cilazaprilat | −0.48 a | 3.17 a | / | / | / | 1.28 | 1.22 | 1.42 | / | 10.3517 [51] f | 0.00084 [51] f | 0.008 [51] f | / | 75.48 [52] | 1 e | 0.76 [54] | / | / |
| Temocapril | 2.102 [55] | 2.8 [56] | 5359.7 [57] | / | / | 2.82 | 3.17 | 2.47 | / | 15.398 [58] g | / | / | / | 110.2 [59] | 1 e | 0.3 [60] | 0.65 [61] | 0.065 [58] g |
| Temocaprilat | 2.215 [62] | 2.09 [60] | / | / | / | 0.289 | 0.322 | 0.251 | / | 58.535 [63] f | 0.00184 [63] f | 0.000078 [63] f | / | 949.84 [64] 1899.68 [65] b | 1 e | 0.025 [60] | / | / |
| Perindopril | −1.31 [66] | 3.2 [67] | 1011.15 [68] 156.47 [69] c,j | / | / | 0.665 | 0.633 | 0.742 | / | 13.119 [70] g | 0.0028 [70] g | 0.0024 [70] g | 1.34 [43] | 130.2 [71] | 1 e | 0.4 [72] | 0.66 [64] | / |
| Perindoprilat | −0.08 a | 3.08 a | / | / | / | 1.45 | 1.38 | 1.61 | / | 53.44 [73] f | 0.271 [73] f | 0.0996 [73] f | / | 231.78 [74] | 1 e | 0.85 [72] | / | / |
| Remimazolam | 3.68 a | 5.99 a | 79,212.96 c | / | / | 36.34 | 63.19 | 31.2 | 1180 [75] | 15.0768 [76] f | 0.01638 [76] f 0.3117 [76] f (K13) | 0.000476 [76] f 0.5057 [76] f (K31) | / | / | 1 e | 0.08 [77] | / | |
| Flumazenil | 1.64 [78] | 0.86 [79] | 8169.9 c | / | / | 2.57 | 2.71 | 2.41 | 1120 [80] | 24.054 [81] g | 0.0376 [81] g | 0.0427 [81] g | 3.78 [82] | 1.67 [83] | 1 [84] | 0.6 [85] | / | |
| Pethidine | 2.35 [86] | 8.7 [86] | / | 1.56 [87] h 5.382 [88] i | 261 [87] h 356 [88] i | 14.82 | 4.18 | 12.02 | / | 328.676 [89] f | 0.002224 [89] f | 0.0003697 [89] f | / | 58.78 [90] | 0.87 [91] | 0.48 [88] | / | 0.117 [92] g |
| Drug | Dose | Subjects | AUC0–t (μg × h/mL) | Cmax (ng/mL) | ||||
|---|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | |||
| Enalapril | 10 mg [14] | HT | 0.1229 | 0.1467 | 0.84 | 66.9 | 45.6 | 1.47 |
| 10 mg [104] | HT | NR | 0.1151 | / | NR | 45.6 | / | |
| 10 mg [105] | HT | 0.1600 | 0.1526 | 1.05 | 72.1 | 45.6 | 1.58 | |
| 10 mg [105] | HT | 0.1480 | 0.1547 | 0.96 | 65.4 | 45.6 | 1.43 | |
| 10 mg [106] | HT | NR | 0.1467 | / | NR | 45.6 | / | |
| 10 mg [107] | CP-B | 0.1761 | 0.2253 | 0.78 | 110.1 | 60.6 | 1.82 | |
| 10 mg [14] | CP-C | 0.2769 | 0.3195 | 0.87 | 123.4 | 80.7 | 1.53 | |
| Enalaprilat | 10 mg [14] | HT | 0.3754 | 0.3683 | 1.02 | 46.1 | 39.7 | 1.16 |
| 10 mg [104] | HT | NR | 0.3683 | / | NR | 39.7 | / | |
| 10 mg [105] | HT | 0.2170 | 0.3683 | 0.59 | 29.3 | 39.7 | 0.74 | |
| 10 mg [105] | HT | 0.2600 | 0.3683 | 0.71 | 37.3 | 39.7 | 0.94 | |
| 10 mg [106] | HT | NR | 0.2776 | / | NR | 39.7 | / | |
| 10 mg [107] | CP-B | 0.3812 | 0.5154 | 0.74 | 36.8 | 35.1 | 1.05 | |
| 10 mg [14] | CP-C | 0.1733 | 0.2476 | 0.70 | 16.8 | 20.1 | 0.84 | |
| Drug | Dose | Subjects | AUC0–t (μg × h/mL) | Cmax (ng/mL) | ||||
|---|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | |||
| Benazepril | 10 mg [108] | HT | 0.1390 | 0.2571 | 0.54 | 139.139 | 113.545 | 1.23 |
| 10 mg [109] | HT | 0.1380 | 0.2665 | 0.52 | 78.957 | 113.545 | 0.70 | |
| 20 mg [110] | HT | 0.2195 | 0.4611 | 0.48 | 265.313 | 227.089 | 1.17 | |
| 20 mg [111] | HT | NR | 0.4611 | / | 252.98 | 227.089 | 1.11 | |
| 20 mg [112] | CP-B | 0.6159 | 0.5883 | 1.05 | 543.472 | 268.130 | 2.03 | |
| Benazeprilat | 10 mg [113] | HT | 1.5330 | 1.6492 | 0.93 | 188.704 | 198.97 | 0.95 |
| 10 mg [108] | HT | 1.0787 | 1.3554 | 0.80 | 200.410 | 198.97 | 1.01 | |
| 10 mg [109] | HT | 1.1039 | 1.3554 | 0.81 | 164.520 | 198.97 | 0.83 | |
| 20 mg [110] | HT | 2.3800 | 2.7107 | 0.88 | 463.830 | 397.95 | 1.17 | |
| 20 mg [111] | HT | NR | 2.7107 | / | 342.164 | 397.95 | 0.86 | |
| 20 mg [112] | CP-B | 2.1650 | 2.3870 | 0.91 | 345.010 | 344.85 | 1.00 | |
| Drug | Dose | Subjects | AUC0–t (μg × h/mL) | Cmax (ng/mL) | ||||
|---|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | |||
| Cilazapril | 1 mg [51] | HT | 0.0998 | 0.1044 | 0.96 | 33.9 | 26.2 | 1.29 |
| 2.5 mg [51] | HT | 0.2560 | 0.2610 | 0.98 | 82.7 | 65.4 | 1.26 | |
| 5 mg [51] | HT | 0.4960 | 0.5221 | 0.95 | 182.0 | 130.8 | 1.39 | |
| 2.5 mg [114] | HT | 0.1830 | 0.2341 | 0.78 | 75.7 | 65.4 | 1.16 | |
| 1 mg [115] | HT | 0.0657 | 0.0890 | 0.74 | 25.2 | 26.2 | 0.96 | |
| 1 mg [115] | CP-B | 0.1840 | 0.1201 | 1.53 | 40.0 | 28.3 | 1.41 | |
| Cilazaprilat | 1 mg [51] | HT | 0.0791 | 0.0725 | 1.09 | 12.4 | 10.2 | 1.22 |
| 1 mg [116] | HT | NR | 0.1158 | / | 8.3 | 10.2 | 0.81 | |
| 2.5 mg [51] | HT | 0.175 | 0.1811 | 0.97 | 37.7 | 25.4 | 1.48 | |
| 5 mg [51] | HT | 0.342 | 0.3623 | 0.94 | 94.2 | 50.8 | 1.85 | |
| 2.5 mg [114] | HT | 0.178 | 0.1811 | 0.98 | 39.3 | 25.4 | 1.55 | |
| 5 mg [117] | HT | 0.398 | 0.6580 | 0.60 | 83.4 | 50.8 | 1.64 | |
| 1.25 mg [118] | HT | 0.070 | 0.0906 | 0.77 | 13.0 | 12.7 | 1.02 | |
| 2.5 mg [118] | HT | 0.170 | 0.1811 | 0.94 | 36.0 | 25.4 | 1.42 | |
| 5 mg [118] | HT | 0.280 | 0.3623 | 0.77 | 74.0 | 50.8 | 1.46 | |
| 10 mg [118] | HT | 0.550 | 0.7246 | 0.76 | 165.0 | 101.5 | 1.63 | |
| 1 mg [115] | HT | 0.053 | 0.0725 | 0.73 | 7.96 | 10.2 | 0.78 | |
| 1 mg [115] | CP-B | 0.0775 | 0.0695 | 1.12 | 10.2 | 8.3 | 1.23 | |
| Drug | Dose | Subjects | AUC0–t (μg × h/mL) | Cmax (ng/mL) | ||||
|---|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | |||
| Perindopril | 4 mg [119] | HT | 0.121 | 0.120 | 1.01 | 64.2 | 34.6 | 1.86 |
| 8 mg [120] | CP-A | 0.377 | 0.239 | 1.58 | NR | 70.4 | / | |
| 8 mg [121] | CP-B | 0.602 | 0.281 | 2.14 | NR | 77.0 | / | |
| Perindoprilat | 4 mg [119] | HT | 0.0520 | 0.0681 | 0.76 | 4.7 | 4.3 | 1.09 |
| 8 mg [122] | HT | 0.1197 | 0.1362 | 0.88 | NR | 8.5 | / | |
| 8 mg [120] | CP-A | 0.3210 | 0.2695 | 1.19 | 29 | 8.8 | 3.33 | |
| 8 mg [121] | CP-B | 0.1340 | 0.2777 | 0.48 | NR | 8.6 | / | |
| Drug | Dose | Subjects | AUC0–t (μg × h/mL) | Cmax (ng/mL) | ||||
|---|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Ore | Obs | Pre | Obs/Ore | |||
| Temocapril | 1 mg [123] | HT | NR | 0.0257 | / | NR | 11.0 | / |
| 1 mg [123] | CP-B | NR | 0.0271 | / | NR | 11.6 | / | |
| Temocaprilat | 1 mg [123] | HT | 0.1230 | 0.1199 | 1.03 | 15.8 | 11.2 | 1.41 |
| 1 mg [123] | CP-B | 0.1714 | 0.0800 | 2.14 | 14.3 | 7.4 | 1.93 | |
| Drug | Dose | Subjects | AUC0–t (μg × h/mL) | Cmax (ng/mL) | ||||
|---|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | |||
| Oseltamivir | 75 mg [126] | HT | 0.1590 | 0.1430 | 1.11 | 74.4 | 59.8 | 1.24 |
| 75 mg [125] | HT | 0.1240 | 0.1430 | 0.87 | 75.1 | 59.8 | 1.26 | |
| 75 mg [125] | HT | 0.1140 | 0.1430 | 0.80 | 67.6 | 59.8 | 1.13 | |
| 75 mg [124] | HT | 0.1188 | 0.1442 | 0.82 | 61.0 | 59.8 | 1.02 | |
| 150 mg [126] | HT | 0.3130 | 0.2860 | 1.09 | 192.0 | 119.5 | 1.61 | |
| 75 mg [15] | CP-B | 0.2100 | 0.1985 | 1.06 | 100.0 | 85.6 | 1.17 | |
| Oseltamivir carboxylate | 75 mg [126] | HT | 3.0200 | 2.5068 | 1.20 | 291.00 | 264.93 | 1.10 |
| 75 mg [125] | HT | 2.6500 | 2.5068 | 1.06 | 276.00 | 264.93 | 1.04 | |
| 75 mg [125] | HT | 2.5600 | 2.5068 | 1.02 | 278.00 | 264.93 | 1.05 | |
| 75 mg [124] | HT | 3.1763 | 3.0861 | 1.03 | 360.31 | 264.93 | 1.36 | |
| 150 mg [126] | HT | 6.3100 | 5.0135 | 1.26 | 550.00 | 529.86 | 1.04 | |
| 75 mg [15] | CP-B | 3.1000 | 4.3235 | 0.72 | 260.00 | 279.86 | 0.93 | |
| Dose | Subjects | AUC0–t or CL | Cmax | ||||
|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | ||
| 10 mg, 1 min, i.v. [127] | HT | 0.9000 a | 0.4486 a | 2.01 | |||
| 10 mg, 10 min, i.v. [128] | HT | 0.8967 a | 0.4549 a | 1.97 | |||
| 2.5 mg, 0.5 min, i.v. [81] | HT | 0.7160 a | 0.4766 a | 1.50 | |||
| 2 mg, 5 min, i.v. [129] | CP-B | 0.4932 a | 0.4988 a | 0.99 | |||
| 2 mg, 5 min, i.v. [129] | CP-C | 0.3165 a | 0.4030 a | 0.79 | |||
| 2 mg, 1 min, i.v. [130] | CP-C | 0.7050 a | 0.4295 a | 1.64 | |||
| 30 mg, p.o. [130] | HT | NR | 0.1741 b | / | 70.1 | 71.0 | 0.99 |
| 30 mg, p.o. [130] | CP-C | NR | 0.5139 b | / | 258.0 | 174.7 | 1.48 |
| Dose | Subjects | AUC0–t or CL | Cmax | ||||
|---|---|---|---|---|---|---|---|
| Obs | Pre | Obs/Pre | Obs | Pre | Obs/Pre | ||
| 25 mg, 1 min, i.v. [131] | HT | 0.5624 a | 0.4956 a | 1.13 | |||
| 50 mg, 1 min, i.v. [132] | HT | 1.0200 a | 0.7784 a | 1.31 | |||
| 50 mg, 1 min, i.v. [133] | HT | 0.9640 a | 0.8532 a | 1.13 | |||
| 70 mg, 2 min, i.v. [134] | HT | 0.7505 a | 0.4952 a | 1.52 | |||
| 70 mg, 2 min, i.v. [135] | HT | 0.7226 a | 0.4952 a | 1.46 | |||
| 0.8 mg/kg, 1 min, i.v. [139] | HT | 1.3160 a | 0.7887 a | 1.67 | |||
| 0.8 mg/kg, 5 min, i.v. [140] | HT | 0.9000 a | 0.6972 a | 1.29 | |||
| 0.8 mg/kg, 1 min, i.v. [136] | CP-A | 0.3920 a | 0.5349 a | 0.73 | |||
| 0.8 mg/kg, 1 min, i.v. [139] | CP-A | 0.6640 a | 0.7405 a | 0.90 | |||
| 0.8 mg/kg, 5 min, i.v. [140] | CP-A | 0.5730 a | 0.7560 a | 0.76 | |||
| 0.8 mg/kg, 1 min, i.v. [137] | CP-B | 0.3730 a | 0.5724 a | 0.65 | |||
| 25 mg, p.o. [131] | HT | 0.9270 a | 0.8563 a | 1.08 | NR | 36.0 | / |
| 100 mg, p.o. [138] | HT | 0.8600 b | 0.6097 b | 1.41 | 170.0 | 143.9 | 1.18 |
| 0.8 mg/kg, p.o. [140] | HT | NR | 0.4649 b | / | NR | 80.6 | / |
| 1.6 mg/kg, p.o. [136] | CP-A | NR | 1.2681 b | / | NR | 157.0 | / |
| 0.8 mg/kg, p.o. [140] | CP-A | NR | 0.4439 b | / | NR | 78.5 | / |
| 1.6 mg/kg, p.o. [137] | CP-B | NR | 1.1983 b | / | NR | 146.3 | / |
| Dose | Subjects | AUC0–t (μg × h/mL) | ||
|---|---|---|---|---|
| Obs | Pre | Obs/Pre | ||
| 0.05 mg/kg [76] | HT | 0.0447 | 0.0536 | 0.83 |
| 0.075 mg/kg | HT | 0.0665 | 0.0787 | 0.84 |
| 0.1 mg/kg | HT | 0.0860 | 0.1000 | 0.86 |
| 0.2 mg/kg | HT | 0.1683 | 0.2145 | 0.78 |
| 0.3 mg/kg | HT | 0.2517 | 0.2960 | 0.85 |
| 0.4 mg/kg | HT | 0.3317 | 0.3979 | 0.83 |
| 10.4 mg [141] | CP-B | NR | 0.1277 | / |
| 8.2 mg | CP-C | NR | 0.0805 | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, X.; Zhang, Z.; Mu, R.; Hu, G.; Liu, L.; Liu, X. Simultaneously Predicting the Pharmacokinetics of CES1-Metabolized Drugs and Their Metabolites Using Physiologically Based Pharmacokinetic Model in Cirrhosis Subjects. Pharmaceutics 2024, 16, 234. https://doi.org/10.3390/pharmaceutics16020234
Luo X, Zhang Z, Mu R, Hu G, Liu L, Liu X. Simultaneously Predicting the Pharmacokinetics of CES1-Metabolized Drugs and Their Metabolites Using Physiologically Based Pharmacokinetic Model in Cirrhosis Subjects. Pharmaceutics. 2024; 16(2):234. https://doi.org/10.3390/pharmaceutics16020234
Chicago/Turabian StyleLuo, Xin, Zexin Zhang, Ruijing Mu, Guangyu Hu, Li Liu, and Xiaodong Liu. 2024. "Simultaneously Predicting the Pharmacokinetics of CES1-Metabolized Drugs and Their Metabolites Using Physiologically Based Pharmacokinetic Model in Cirrhosis Subjects" Pharmaceutics 16, no. 2: 234. https://doi.org/10.3390/pharmaceutics16020234
APA StyleLuo, X., Zhang, Z., Mu, R., Hu, G., Liu, L., & Liu, X. (2024). Simultaneously Predicting the Pharmacokinetics of CES1-Metabolized Drugs and Their Metabolites Using Physiologically Based Pharmacokinetic Model in Cirrhosis Subjects. Pharmaceutics, 16(2), 234. https://doi.org/10.3390/pharmaceutics16020234