

Physiologically Based Pharmacokinetic (PBPK) Modeling to Predict CYP3A-Mediated Drug Interaction between Saxagliptin and Nicardipine: Bridging Rat-to-Human Extrapolation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Saxagliptin Metabolic Stability
2.3. CYP Inhibition Assay by Nicardipine
2.4. Non-Clinical Drug–Drug Interaction Study
2.4.1. Animals
2.4.2. Study Design
2.4.3. Pharmacokinetic Analysis
2.5. LC-MS/MS and HPLC Analysis
2.6. PBPK Model Construction and Simulation
2.7. Sensitivity Analysis
2.8. Evaluation of PBPK Model
3. Results
3.1. Metabolic Stability of Saxagliptin
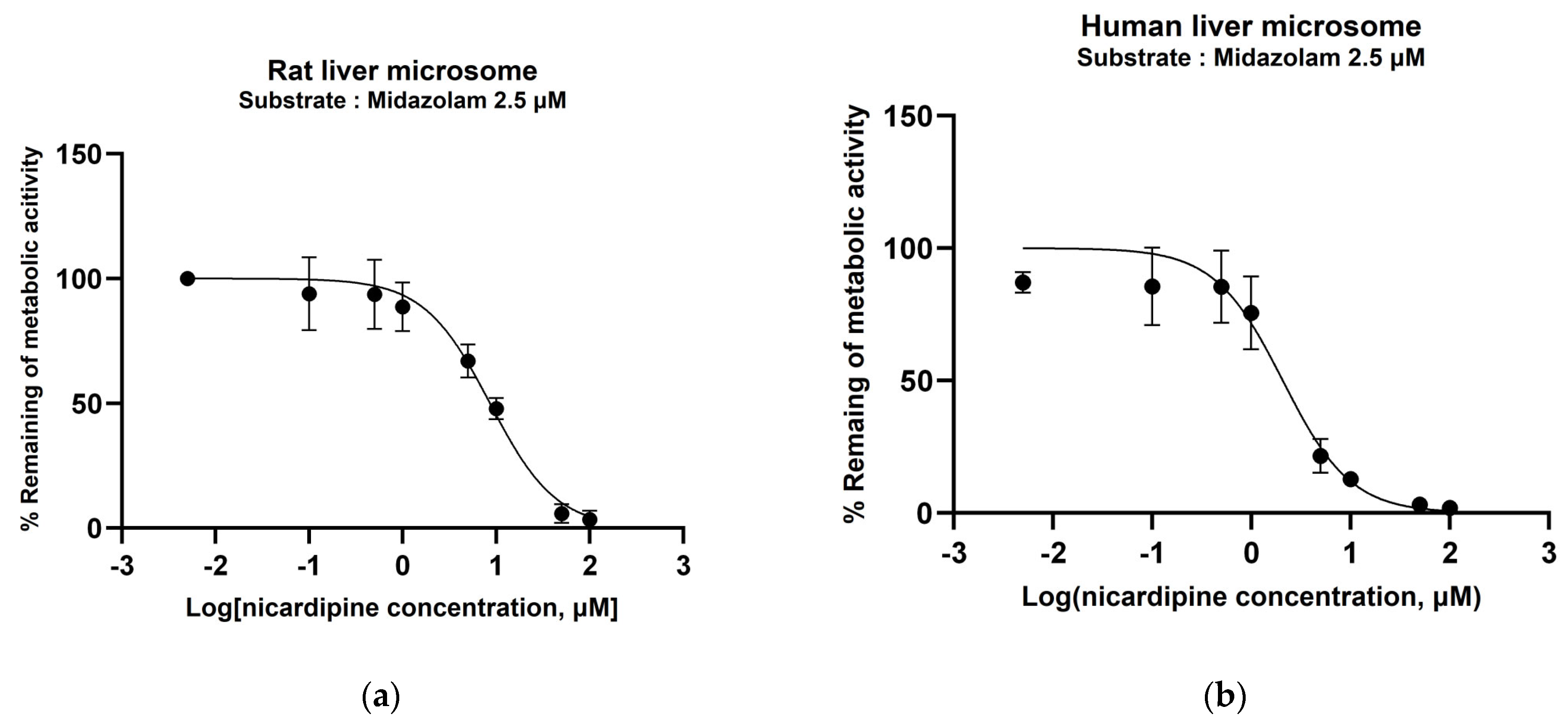
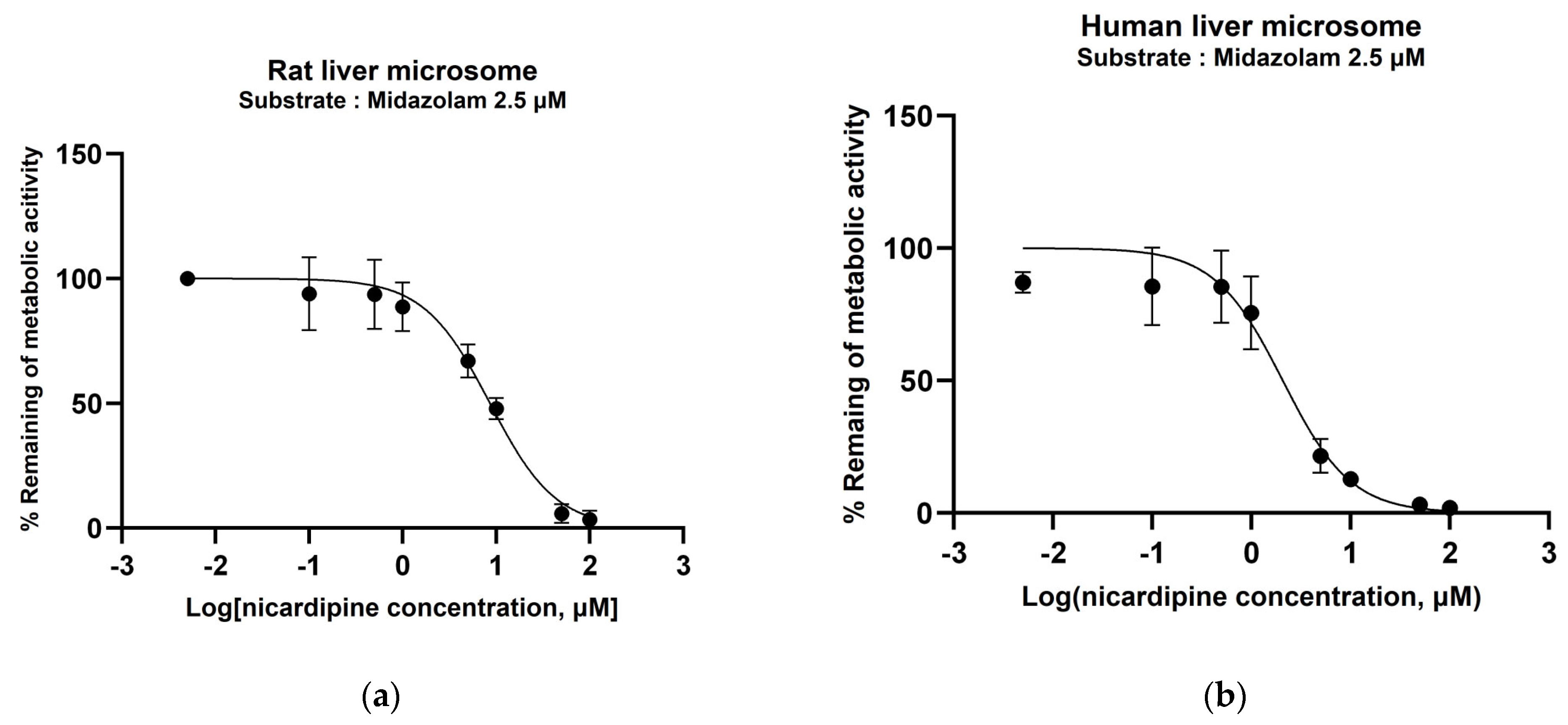
3.2. Determination of CYP450 Inhibition Potential of Nicardipine
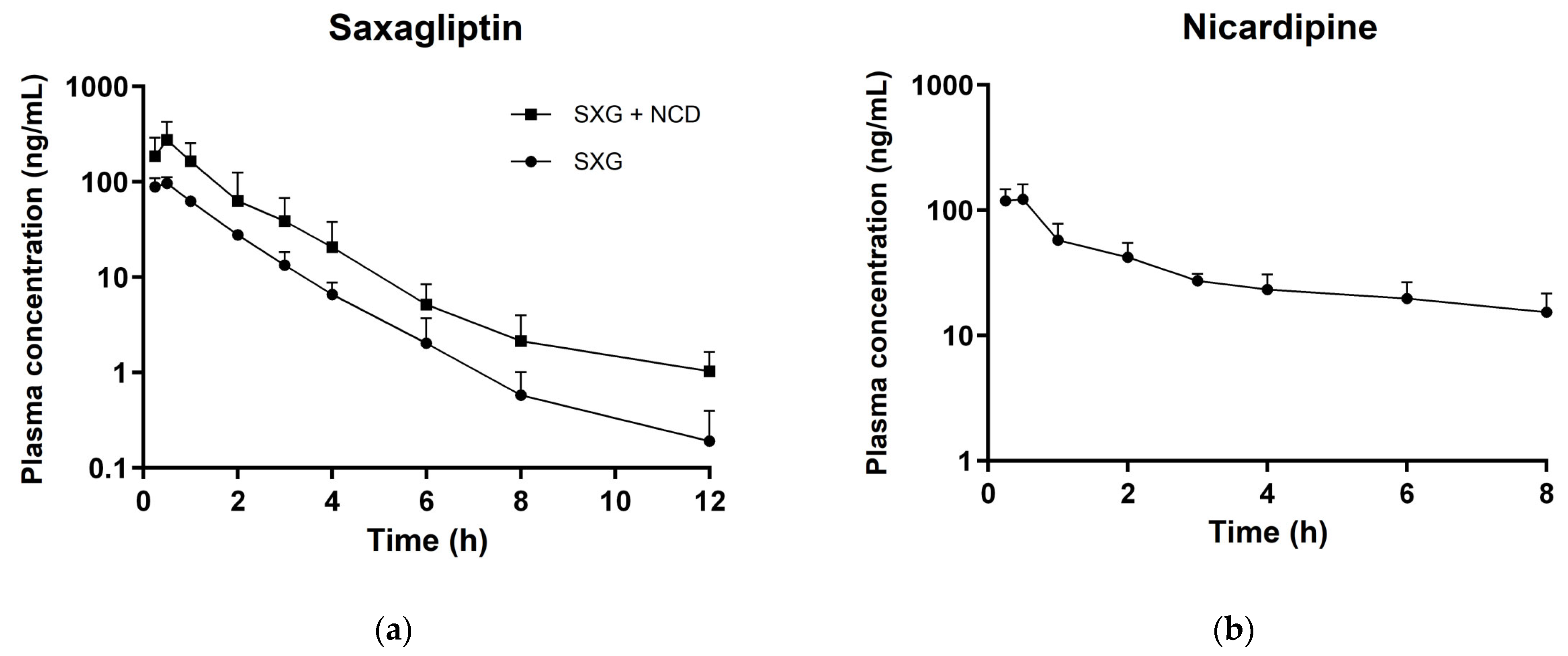
3.3. In Vivo Pharmacokinetic Studies
3.4. PBPK Model Construction
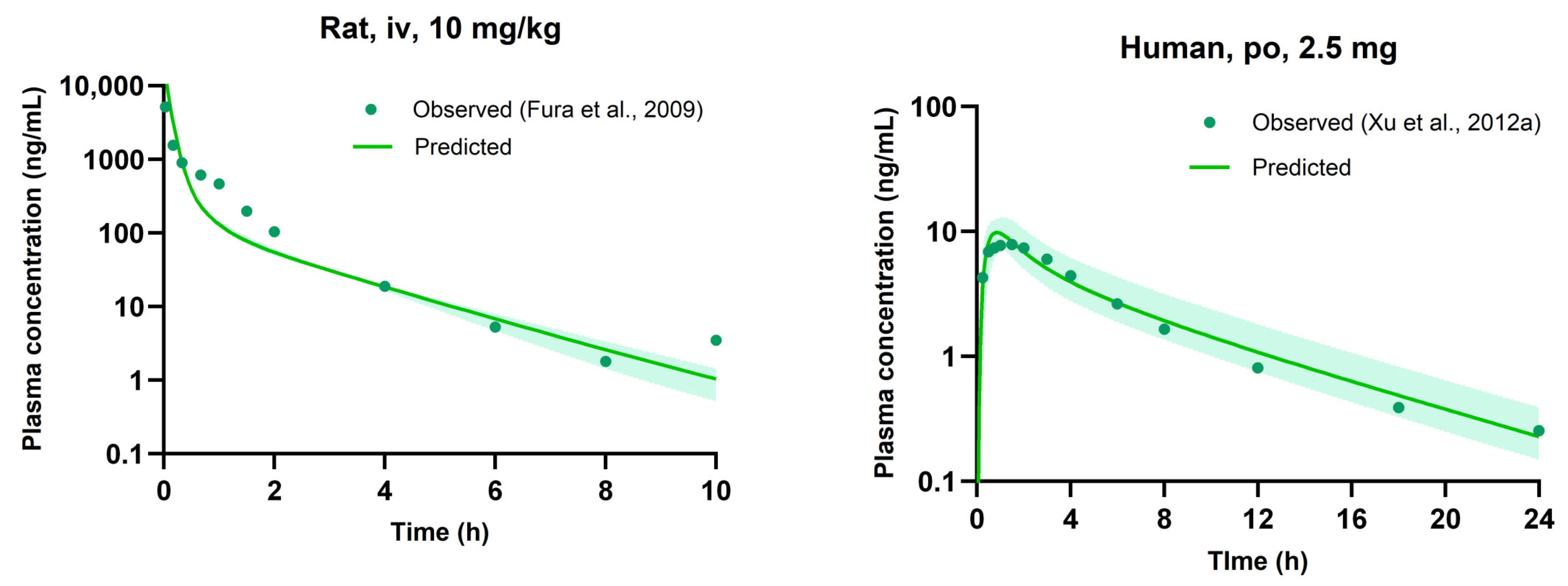
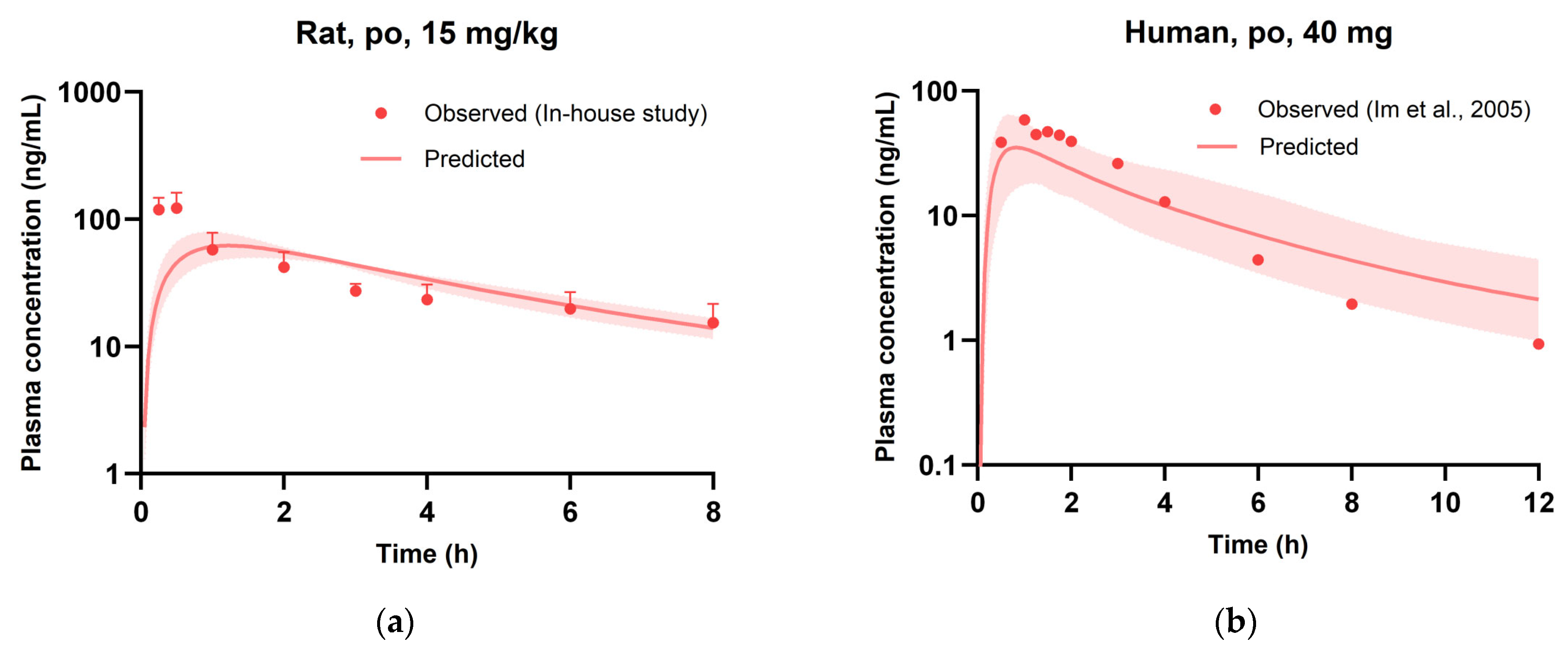
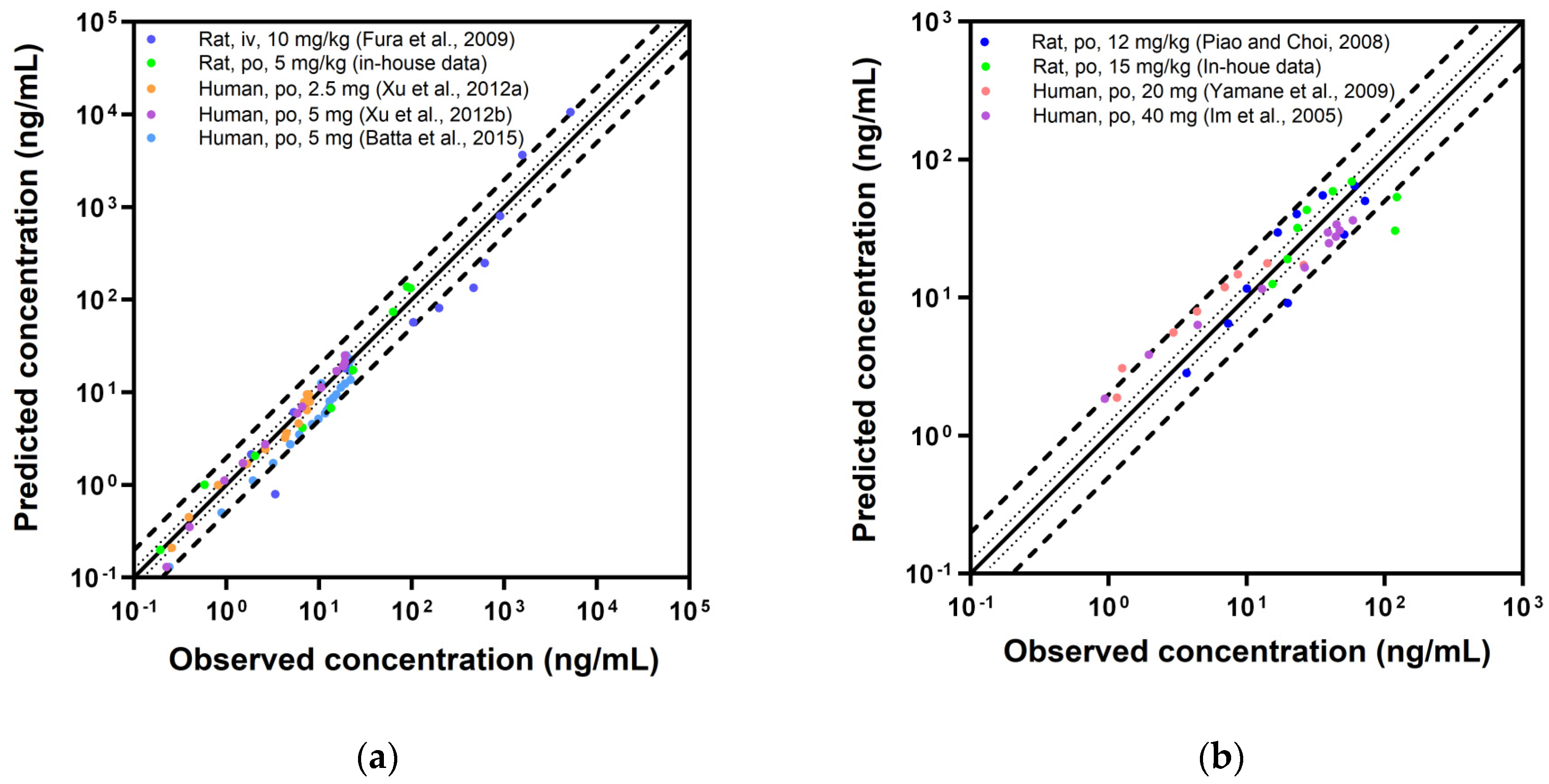
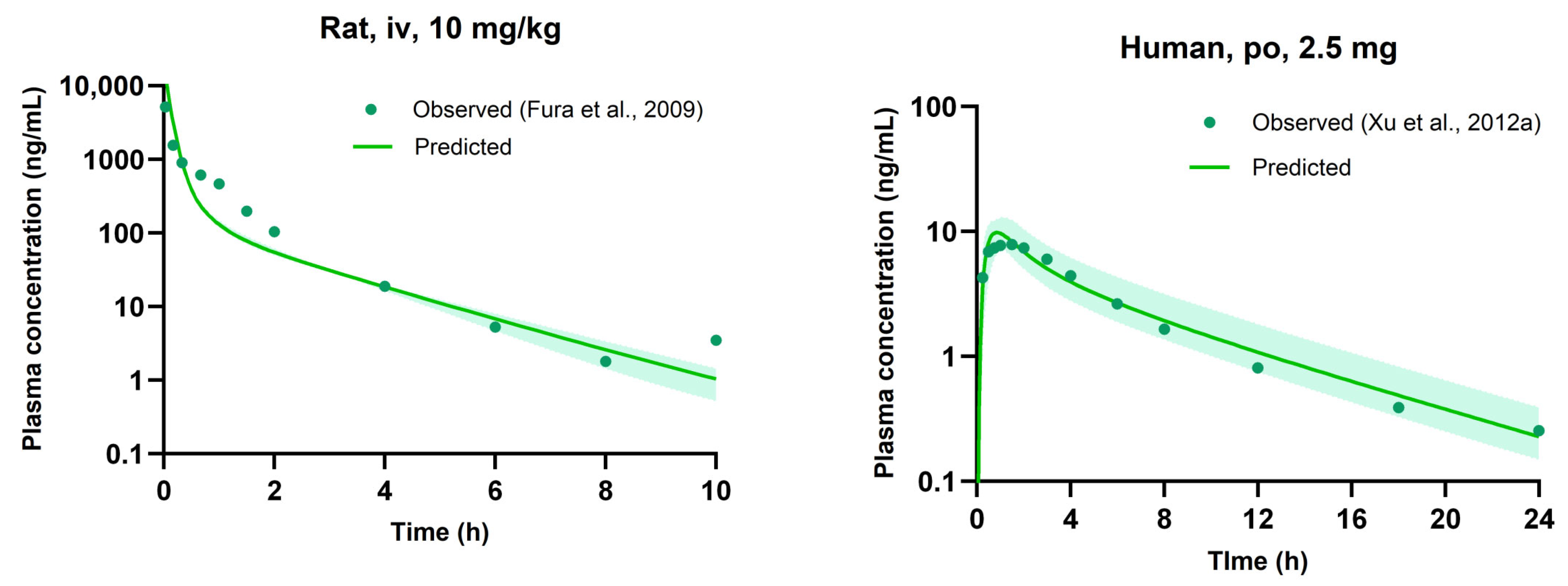
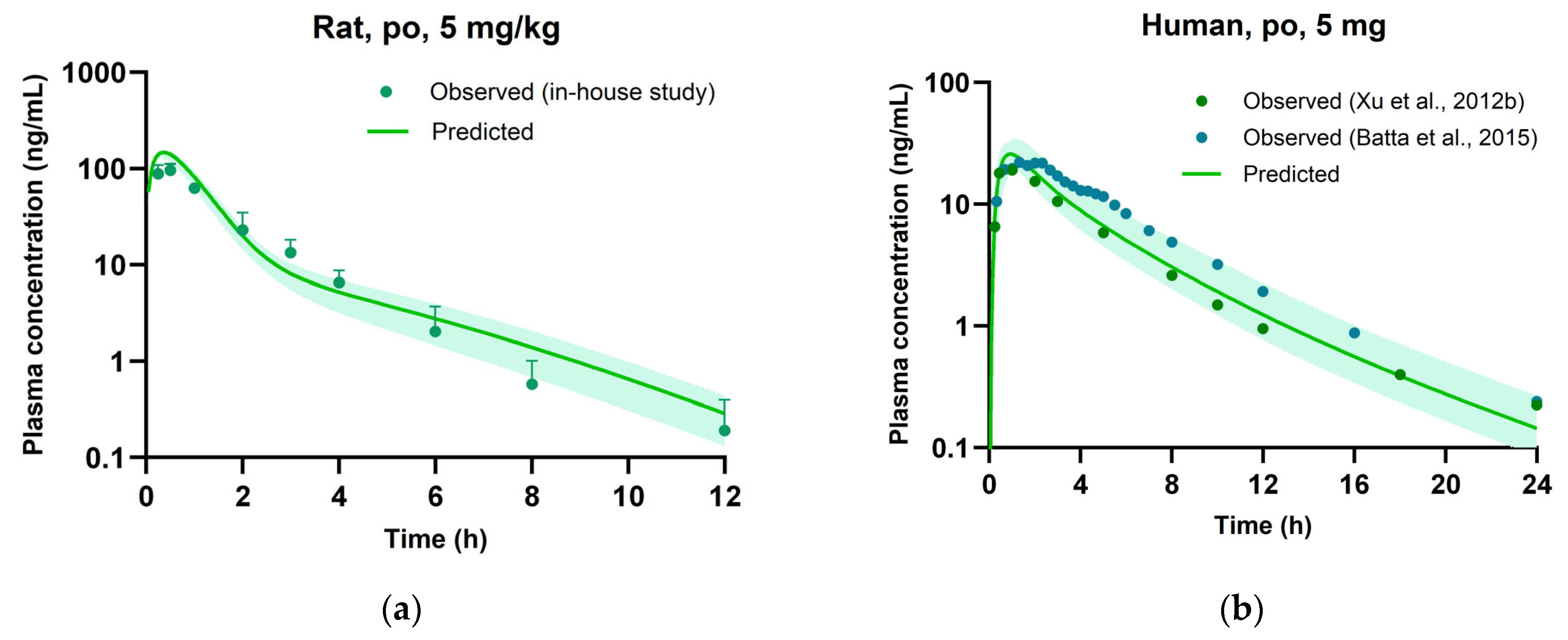
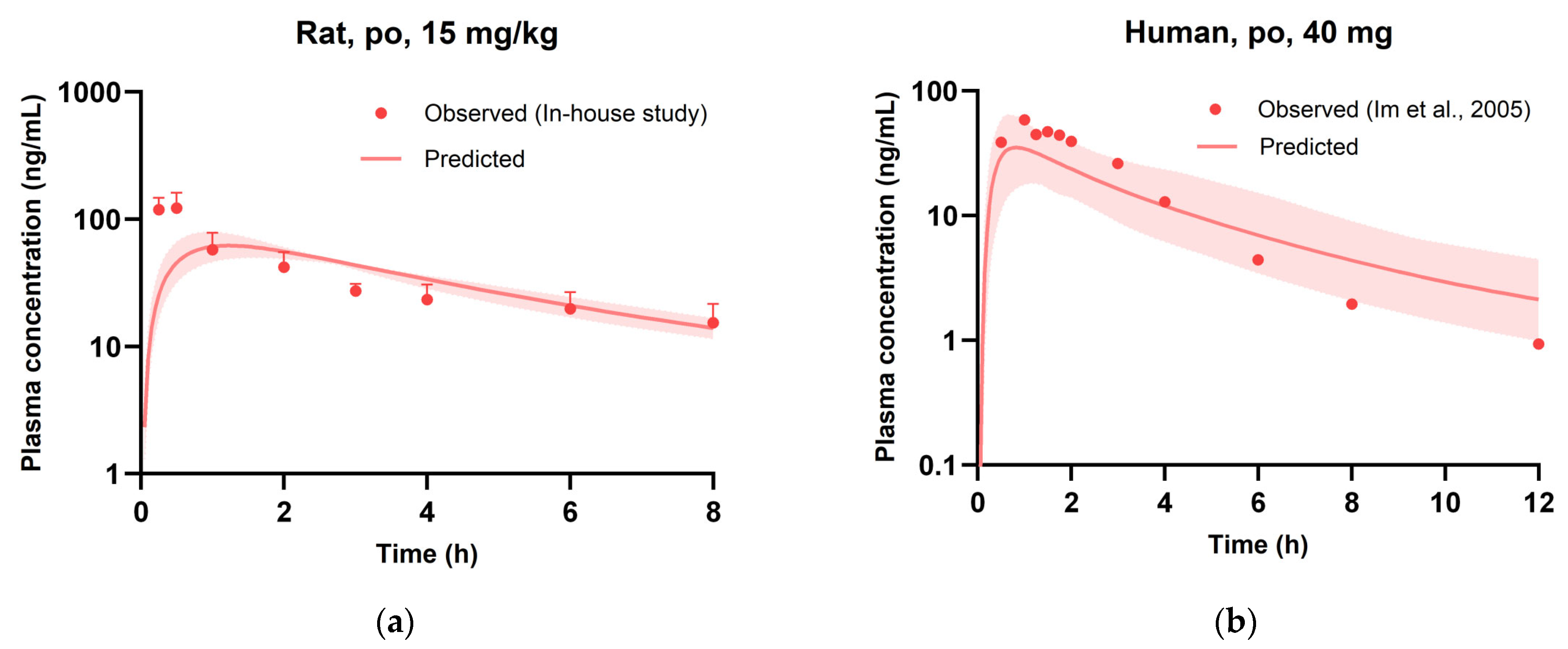
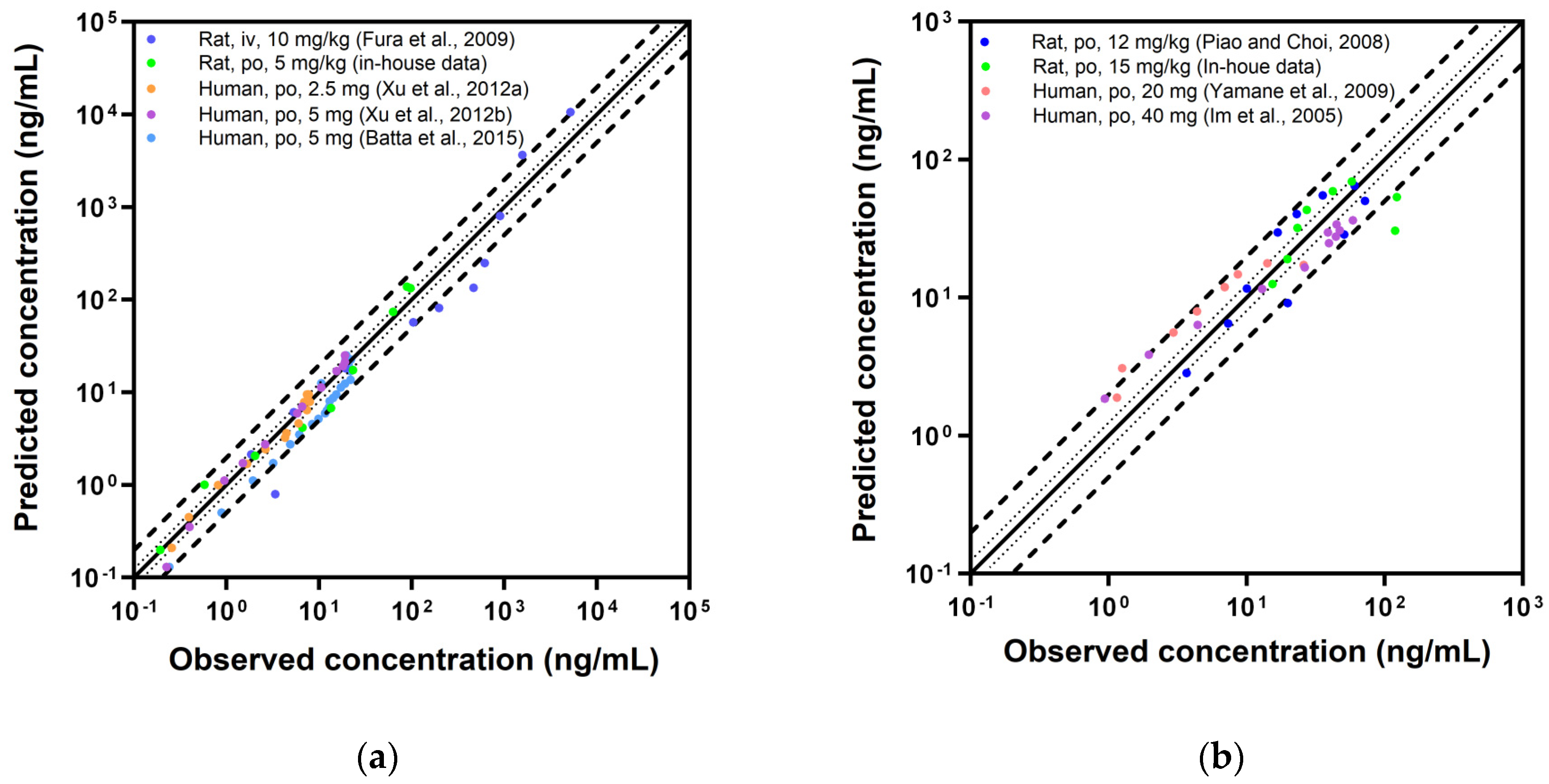
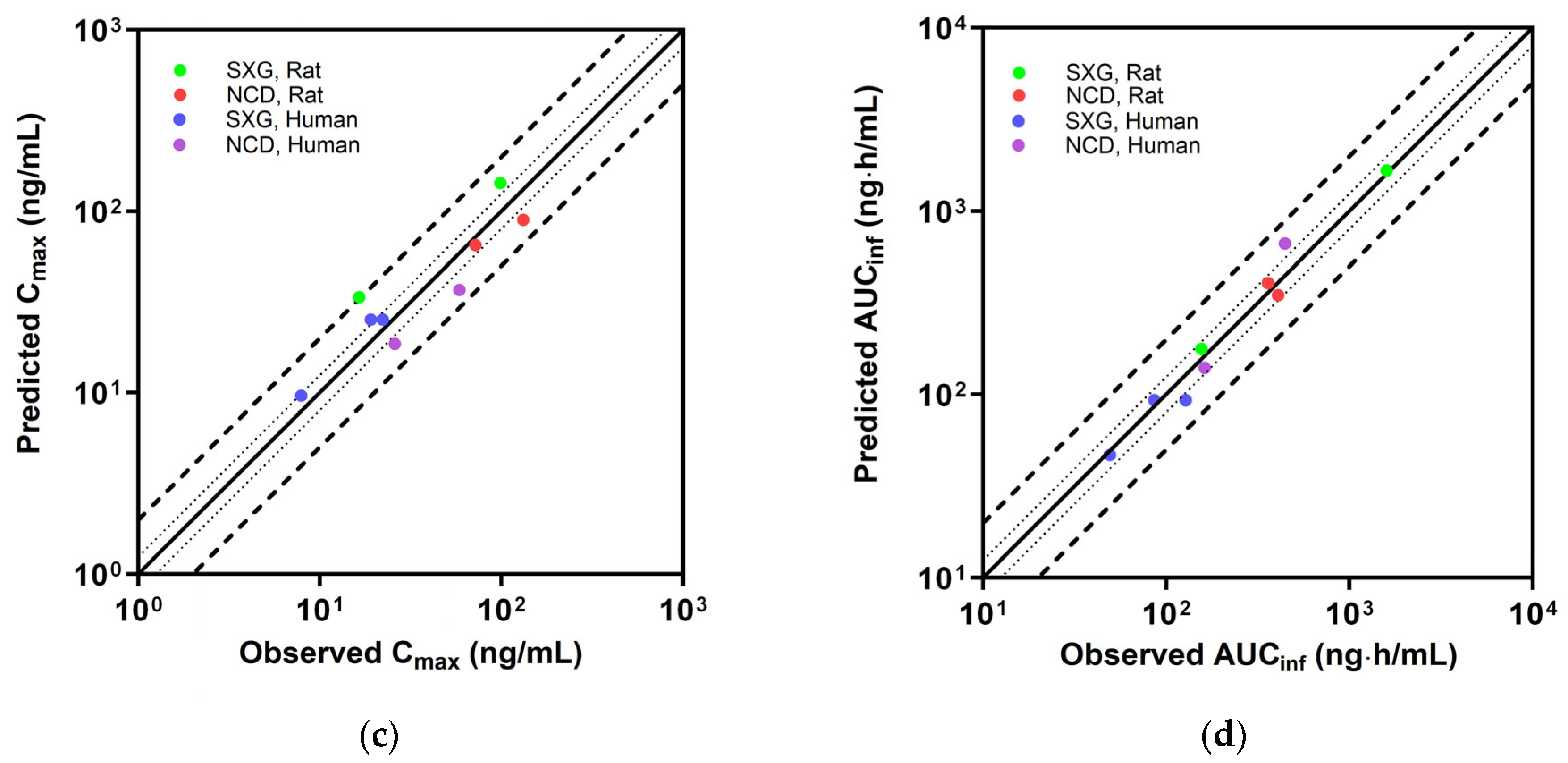
3.5. Evaluation of PBPK Model
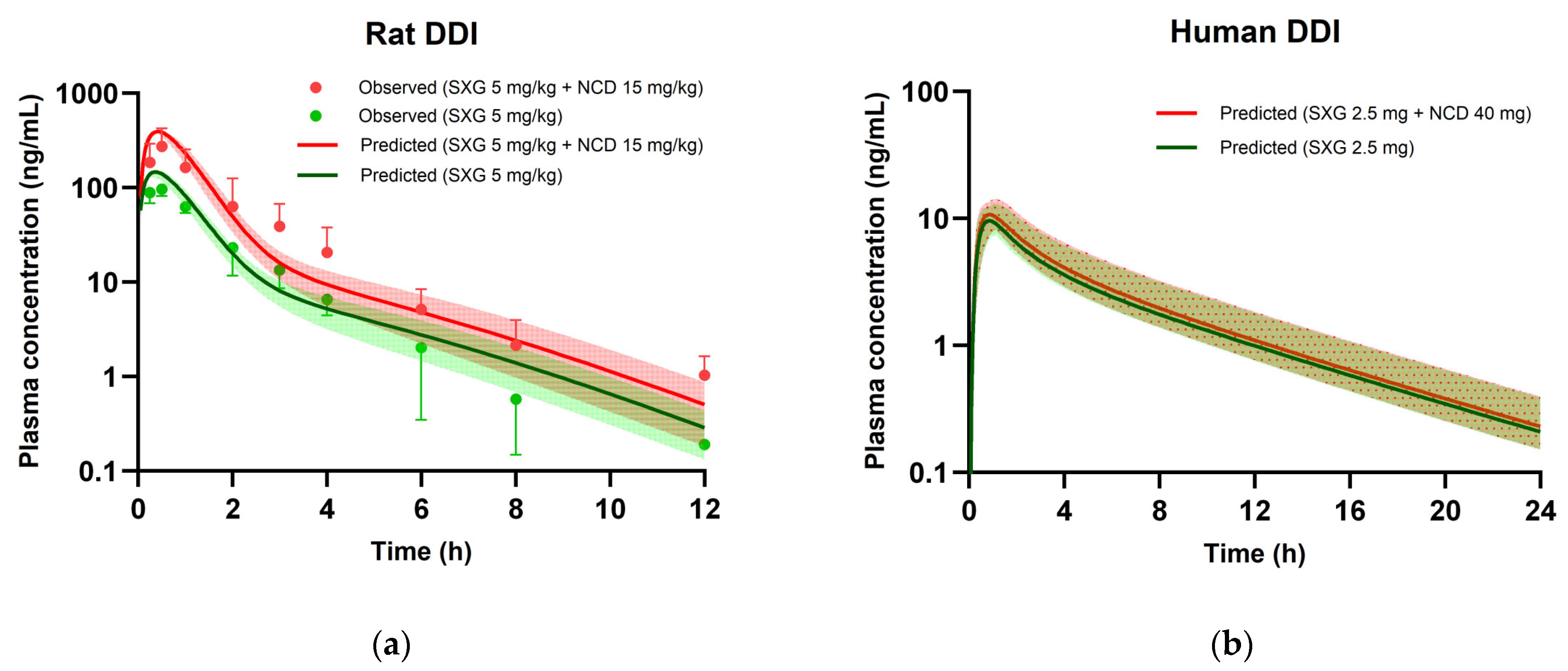
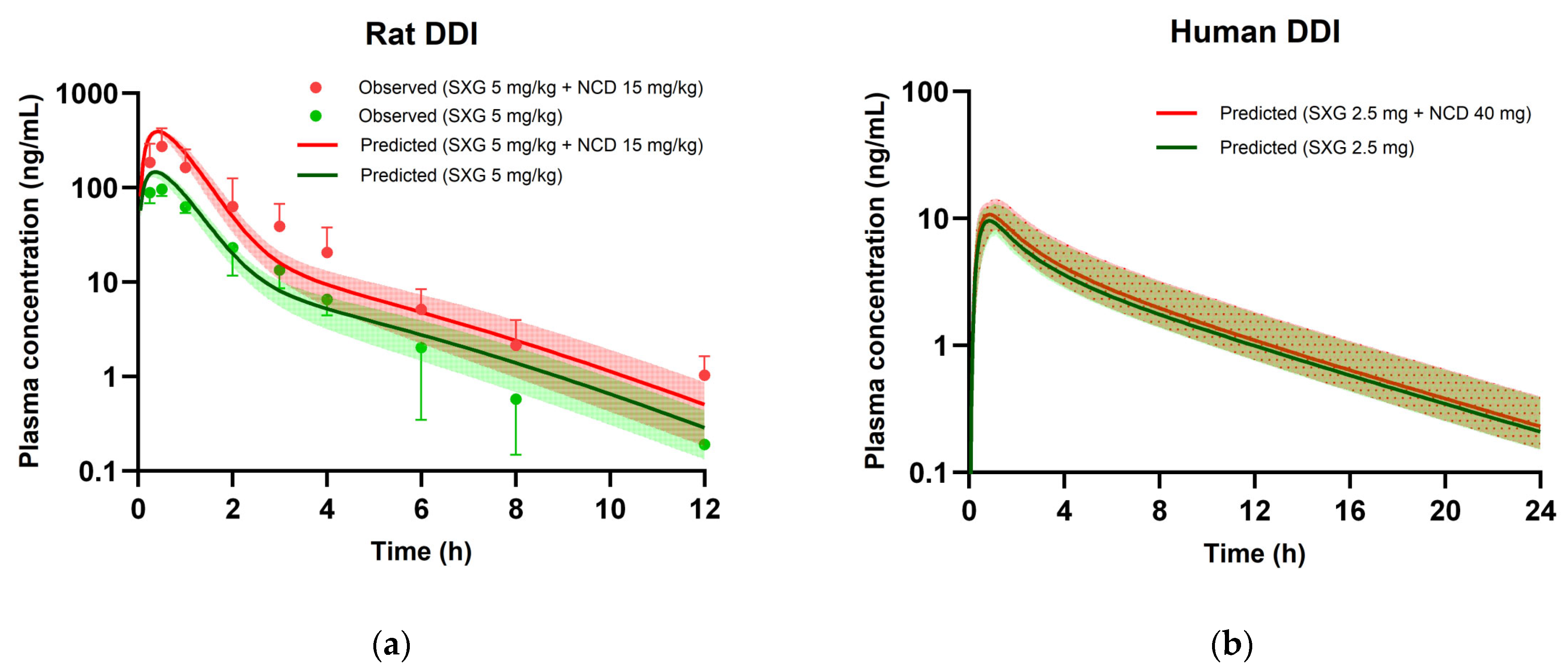
3.6. PBPK Modeling for DDIs
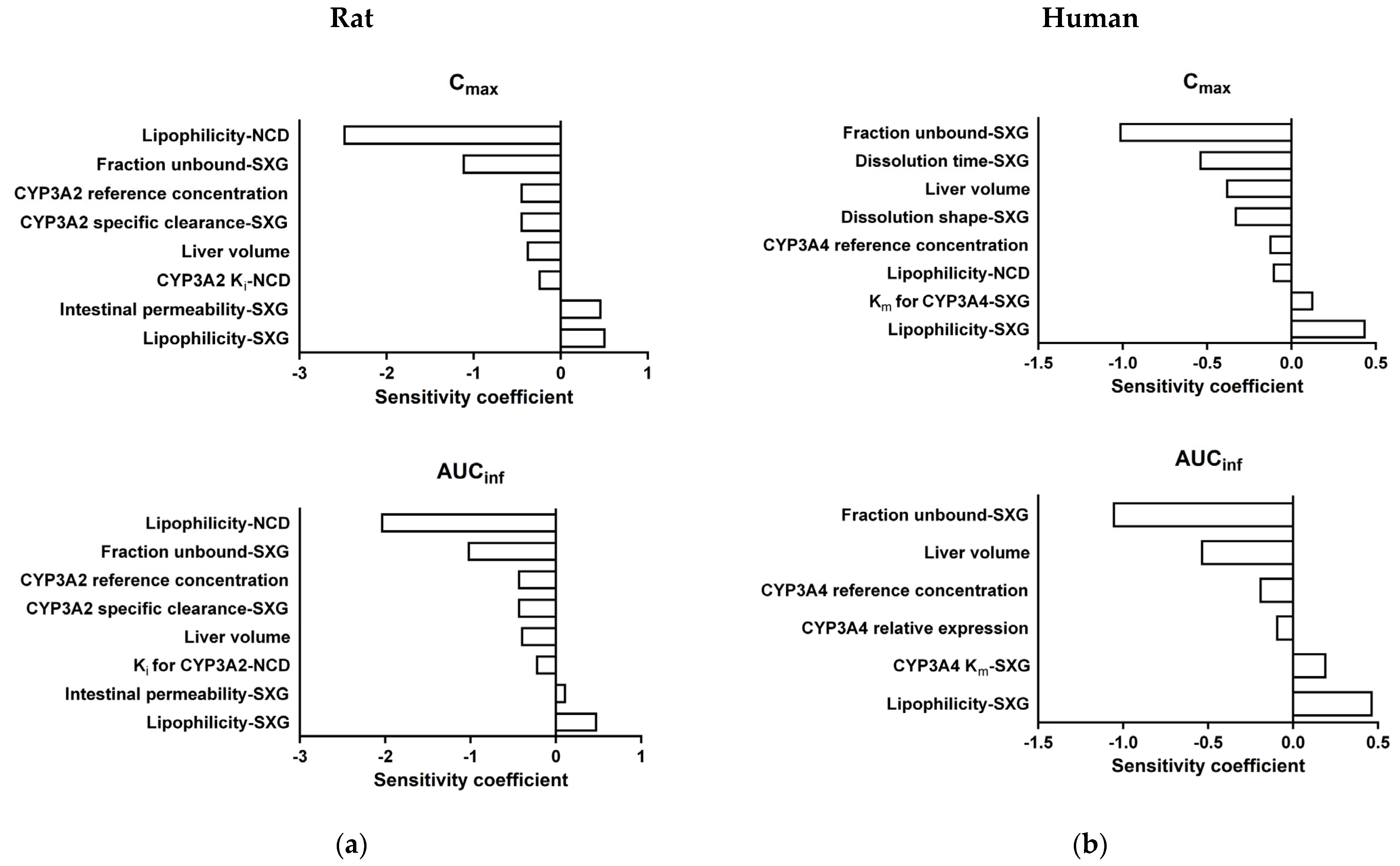
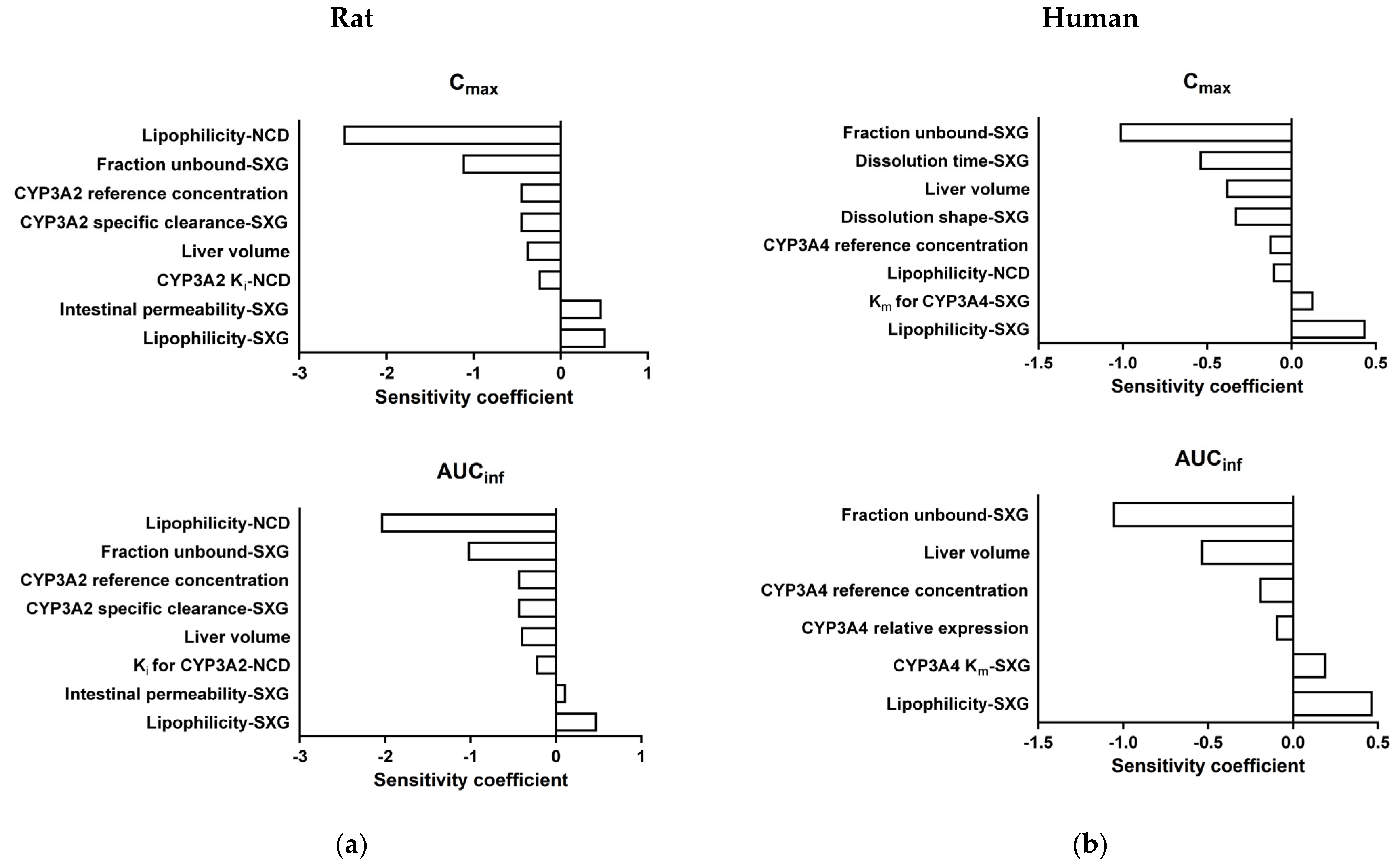
3.7. Sensitivity Analysis of DDI PBPK Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dokken, B.B. The pathophysiology of cardiovascular disease and diabetes: Beyond blood pressure and lipids. Diabetes Spectr. 2008, 21, 160–165. [Google Scholar] [CrossRef]
- Garber, A.J. Attenuating cardiovascular risk factors in patients with type 2 diabetes. Am. Fam. Physician 2000, 62, 2633–2642. [Google Scholar]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Zhao, P.; Huang, S.-M. Predicting drug–drug interactions: An FDA perspective. AAPS J. 2009, 11, 300–306. [Google Scholar] [CrossRef]
- Boulton, D.W. Clinical pharmacokinetics and pharmacodynamics of saxagliptin, a dipeptidyl peptidase-4 inhibitor. Clin. Pharmacokinet. 2017, 56, 11–24. [Google Scholar] [CrossRef]
- Su, H.; Boulton, D.W.; Barros, A., Jr.; Wang, L.; Cao, K.; Bonacorsi, S.J., Jr.; Iyer, R.A.; Humphreys, W.G.; Christopher, L.J. Characterization of the in vitro and in vivo metabolism and disposition and cytochrome P450 inhibition/induction profile of saxagliptin in human. Drug Metab. Dispos. 2012, 40, 1345–1356. [Google Scholar] [CrossRef]
- Graham, D.J.; Dow, R.J.; Hall, D.J.; Alexander, O.F.; Mroszczak, E.J.; Freedman, D. The metabolism and pharmacokinetics of nicardipine hydrochloride in man. Br. J. Clin. Pharmacol. 1985, 20, 23S–28S. [Google Scholar] [CrossRef] [PubMed]
- Dow, R.J.; Graham, D.J. A review of the human metabolism and pharmacokinetics of nicardipine hydrochloride. Br. J. Clin. Pharmacol. 1986, 22, 195S–202S. [Google Scholar] [CrossRef]
- Higuchi, S.; Shiobara, Y. Metabolic fate of nicardipine hydrochloride, a new vasodilator, by various species in vitro. Xenobiotica 1980, 10, 889–896. [Google Scholar] [CrossRef]
- Katoh, M.; Nakajima, M.; Shimada, N.; Yamazaki, H.; Yokoi, T. Inhibition of human cytochrome P450 enzymes by 1,4-dihydropyridine calcium antagonists: Prediction of in vivo drug–drug interactions. Eur. J. Clin. Pharmacol. 2000, 55, 843–852. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Q.; Zhong, J.; Liu, C.; Zheng, B.; Gong, Q. DPP-4 inhibitors as potential candidates for antihypertensive therapy: Improving vascular inflammation and assisting the action of traditional antihypertensive drugs. Front. Immunol. 2019, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.-F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied concepts in PBPK modeling: How to build a PBPK/PD model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Chen, Y.; Unadkat, J.D.; Zhang, X.; Wu, D.; Heimbach, T. Applications, challenges, and outlook for PBPK modeling and simulation: A regulatory, industrial and academic perspective. Pharm. Res. 2022, 39, 1701–1731. [Google Scholar] [CrossRef]
- Wu, W.; Lin, R.; Ke, M.; Ye, L.; Lin, C. Physiologically based pharmacokinetic modeling characterizes the drug-drug interaction between saxagliptin and rifampicin in patients with renal impairment. J. Clin. Pharmacol. 2023, 63, 848–858. [Google Scholar] [CrossRef]
- Li, G.; Yi, B.; Liu, J.; Jiang, X.; Pan, F.; Yang, W.; Liu, H.; Liu, Y.; Wang, G. Effect of CYP3A4 inhibitors and inducers on pharmacokinetics and pharmacodynamics of saxagliptin and active metabolite M2 in humans using physiological-based pharmacokinetic combined DPP-4 occupancy. Front. Pharmacol. 2021, 12, 746594. [Google Scholar] [CrossRef]
- Su, J.; Chang, C.; Xiang, Q.; Zhou, Z.-W.; Luo, R.; Yang, L.; He, Z.-X.; Yang, H.; Li, J.; Bei, Y.; et al. Xyloketal B, a marine compound, acts on a network of molecular proteins and regulates the activity and expression of rat cytochrome P450 3a: A bioinformatic and animal study. Drug Des. Dev. Ther. 2014, 8, 2555–2602. [Google Scholar]
- Mavrevski, R.; Traykov, M.; Trenchev, I.; Trencheva, M. Approaches to modeling of biological experimental data with GraphPad Prism software. WSEAS Trans. Syst. Control 2018, 13, 242–247. [Google Scholar]
- Piao, Y.-J.; Choi, J.-S. Effects of morin on the pharmacokinetics of nicardipine after oral and intravenous administration of nicardipine in rats. J. Pharm. Pharmacol. 2008, 60, 625–629. [Google Scholar] [CrossRef]
- Fura, A.; Khanna, A.; Vyas, V.; Koplowitz, B.; Chang, S.-Y.; Caporuscio, C.; Boulton, D.W.; Christopher, L.J.; Chadwick, K.D.; Hamann, L.G.; et al. Pharmacokinetics of the dipeptidyl peptidase 4 inhibitor saxagliptin in rats, dogs, and monkeys and clinical projections. Drug Metab. Dispos. 2009, 37, 1164–1171. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Willmann, S.; Lippert, J.; Sevestre, M.; Solodenko, J.; Fois, F.; Schmitt, W. PK-Sim®: A physiologically based pharmacokinetic ‘whole-body’ model. Biosilico 2003, 1, 121–124. [Google Scholar] [CrossRef]
- Utsey, K.; Gastonguay, M.S.; Russell, S.; Freling, R.; Riggs, M.M.; Elmokadem, A. Quantification of the impact of partition coefficient prediction methods on physiologically based pharmacokinetic model output using a standardized tissue composition. Drug Metab. Dispos. 2020, 48, 903–916. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab. Pharmacokinet. 2005, 20, 452–477. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab. Pharmacokinet. 2006, 21, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Bastian, F.; Parmentier, G.; Roux, J.; Moretti, S.; Laudet, V.; Robinson-Rechavi, M. Bgee: Integrating and comparing heterogeneous transcriptome data among species. In Proceedings of the Data Integration in the Life Sciences: 5th International Workshop, DILS 2008, Evry, France, 25–27 June 2008; Proceedings 5. Springer: Berlin/Heidelberg, Germany, 2008; pp. 124–131. [Google Scholar]
- Naritomi, Y.; Terashita, S.; Kimura, S.; Suzuki, A.; Kagayama, A.; Sugiyama, Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab. Dispos. 2001, 29, 1316–1324. [Google Scholar] [PubMed]
- Shimada, M.; Murayama, N.; Nagata, K.; Hashimoto, H.; Ishikawa, H.; Yamazoe, Y. A specific loss of growth hormone abolished sex-dependent expression of hepatic cytochrome P450 in dwarf rats: Reversal of the profiles by growth hormone-treatment. Arch. Biochem. Biophys. 1997, 337, 34–42. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Bode, J.W. Reactor ChemAxon Ltd., Maramaros Koz 2/a, Budapest, 1037 Hungary. www.chemaxon.com. Contact ChemAxon for pricing information. J. Am. Chem. Soc. 2004, 126, 15317. [Google Scholar] [CrossRef]
- EMA. Onglyza. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/onglyza (accessed on 31 July 2023).
- Saxagliptin Hydrate. Available online: https://www.pharmacodia.com/yaodu/html/v1/chemicals/17ed8abedc255908be746d245e50263a.html (accessed on 31 July 2023).
- Barr, J.T.; Lade, J.M.; Tran, T.B.; Dahal, U.P. Fraction unbound for liver microsome and hepatocyte incubations for all major species can be approximated using a single-species surrogate. Drug Metab. Dispos. 2019, 47, 419–423. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Krishnaiah, Y.S.; Satyanarayana, V.; Karthikeyan, R.S. Effect of the solvent system on the in vitro permeability of nicardipine hydrochloride through excised rat epidermis. J. Pharm. Pharm. Sci. 2002, 5, 123–130. [Google Scholar]
- Obach, R.S.; Lombardo, F.; Waters, N.J. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dipos. 2008, 36, 1385–1405. [Google Scholar] [CrossRef]
- Li, C.; Liu, T.; Cui, X.; Uss, A.S.; Cheng, K.-C. Development of in vitro pharmacokinetic screens using Caco-2, human hepatocyte, and Caco-2/human hepatocyte hybrid systems for the prediction of oral bioavailability in humans. J. Biomol. Screen. 2007, 12, 1084–1091. [Google Scholar] [CrossRef]
- Raychaudhuri, S. Introduction to Monte Carlo simulation. In Proceedings of the 2008 Winter Simulation Conference, Miami, FL, USA, 7–10 December 2008; pp. 91–100. [Google Scholar]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 inducer carbamazepine and its drug-drug interaction potential: A physiologically based pharmacokinetic modeling approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef]
- Xu, X.S.; Demers, R.; Gu, H.; Christopher, L.J.; Su, H.; Cojocaru, L.; Boulton, D.W.; Kirby, M.; Stouffer, B.; Humphreys, W.G.; et al. Liquid chromatography and tandem mass spectrometry method for the quantitative determination of saxagliptin and its major pharmacologically active 5-monohydroxy metabolite in human plasma: Method validation and overcoming specific and non-specific binding at low concentrations. J. Chromatogr. B Biomed. Appl. 2012, 889, 77–86. [Google Scholar]
- Xu, X.S.; Dueker, S.R.; Christopher, L.J.; Lohstroh, P.N.; Keung, C.F.; Cao, K.; Bonacorsi, S.J.; Cojocaru, L.; Shen, J.X.; Humphreys, W.G.; et al. Overcoming bioanalytical challenges in an Onglyza® intravenous [14C] microdose absolute bioavailability study with accelerator MS. Bioanalysis 2012, 4, 1855–1870. [Google Scholar] [CrossRef] [PubMed]
- Batta, N.; Pilli, N.R.; Derangula, V.R.; Vurimindi, H.B.; Damaramadugu, R.; Yejella, R.P. A rapid and sensitive LC-MS/MS Assay for the determination of saxagliptin and its active metabolite 5-hydroxy saxagliptin in human plasma and its application to a pharmacokinetic study. Drug Res. 2015, 65, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Yamane, N.; Takami, T.; Tozuka, Z.; Sugiyama, Y.; Yamazaki, A.; Kumagai, Y. Microdose clinical trial: Quantitative determination of nicardipine and prediction of metabolites in human plasma. Drug Metab. Pharmacokinet. 2009, 24, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Im, H.-T.; Cho, S.-H.; Lee, H.-W.; Park, W.-S.; Rew, J.-H.; Choi, Y.-W.; Yong, C.-S.; Lee, K.-T. Validation of a simple HPLC method for determination of nicardipine in human plasma and its application to single-dose pharmacokinetics. J. Pharm. Investig. 2005, 35, 461–465. [Google Scholar]
- Sudsakorn, S.; Bahadduri, P.; Fretland, J.; Lu, C. 2020 FDA Drug-drug interaction guidance: A comparison analysis and action plan by pharmaceutical industrial scientists. Curr. Drug Metab. 2020, 21, 403–426. [Google Scholar] [CrossRef]
- Ito, K.; Iwatsubo, T.; Kanamitsu, S.; Ueda, K.; Suzuki, H.; Sugiyama, Y. Prediction of pharmacokinetic alterations caused by drug-drug interactions: Metabolic interaction in the liver. Pharmacol. Rev. 1998, 50, 387–412. [Google Scholar] [PubMed]
- US Food Drug Administration. Guidance for Industry, In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 25 January 2024).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Rat | Human | Reference/ Comment | |||
|---|---|---|---|---|---|---|---|
| Reference | Input | Reference | Input | ||||
| Molecular weight | g/mol | 315.41 | |||||
| Lipophilicity | logD7.4 | −0.35 | −0.7 | −0.35 | −0.7 | [28]/Optimization | |
| pKa (Compound type) | 7.9 (base) | [29] | |||||
| Solubility (ref-pH) | mg/mL | 17.6 (pH 7) | [30] | ||||
| Fraction unbound | 0.82 | 0.82 | 1 | 1 | [19] | ||
| Specific intestinal permeability | 10−6 cm/s | 2.73 | 1.7 | 2.73 | 1.7 | [31]/Optimization | |
| Partition coefficient | Table S2 | PK-Sim standard | |||||
| Cellular permeability | 10−6 cm/s | 13 | 13 | PK-Sim standard | |||
| rCYP3A2 hCYP3A4 hCYP3A5 | μL/min/pmol CYP | 1.99 | 1.99 | In-house data | |||
| CLint | 0.38 | 0.38 | [6] | ||||
| 0.09 | 0.09 | [6] | |||||
| Renal clearance | mL/min/kg | 38 | 38 | 1.9 | 1.9 | [19] | |
| Dissolution shape (tablet 2.5 mg) | 0.68 | Optimization | |||||
| Dissolution time (tablet 2.5 mg) | min | 120.5 | Optimization | ||||
| Dissolution shape (tablet 5 mg) | 0.8 | Optimization | |||||
| Dissolution time (tablet 5 mg) | min | 71.0 | Optimization | ||||
| Parameter | Unit | Rat | Human | Reference/ Comment | |||
|---|---|---|---|---|---|---|---|
| Reference Value | Input Value | Reference Value | Input Value | ||||
| Molecular weight | g/mol | 479.59 | |||||
| Lipophilicity | logD7.4 | 4.6 | [32] | ||||
| pKa (Compound type) | 8.1 (base) | [33] | |||||
| Solubility (ref-pH) | mg/mL | 7.9 (pH 7) | [34] | ||||
| Fraction unbound | 0.084 | 0.084 | 0.01 | 0.01 | [9,35] | ||
| Specific intestinal permeability | 10−6 cm/s | 1.15 | [36] | ||||
| Partition coefficient | Table S2 | PK-Sim standard | |||||
| Cellular permeability | cm/s | 0.09 | 0.09 | PK-Sim standard | |||
| Total hepatic clearance | t1/2 | min | 0.62 | 0.62 | 4.5 | 4.5 | [26] |
| Ki | CYP3A2 | μM | 0.39 | 0.39 | In-house data | ||
| CYP3A4 | 0.06 | 0.06 | [10] | ||||
| Dissolution shape (tablet) | 1.18 | Optimization | |||||
| Dissolution time (tablet) | min | 5.59 | Optimization | ||||
| Group | SXG 5 mg/kg (n = 6) | SXG 5 mg/kg + NCD 15 mg/kg (n = 7) | ||
|---|---|---|---|---|
| Parameter | Unit | Value | DDI Ratio | |
| Cmax | ng/mL | 98.5 ± 15.1 | 281 ± 147 * | 2.85 |
| Tmax | h | 0.417 ± 0.129 | 0.464 ± 0.094 | |
| t1/2 | h | 1.82 ± 0.737 | 2.97 ± 1.23 | 1.63 |
| AUCinf | ng·h/mL | 157 ± 23.1 | 408 ± 213 * | 2.60 |
| CL/F | L/h/kg | 32.5 ± 5.15 | 14.3 ± 4.86 ** | 0.44 |
| Parameter | Unit | Value |
|---|---|---|
| Cmax | ng/mL | 132 ± 35.3 |
| Tmax | h | 0.357 ± 0.134 |
| t1/2 | h | 5.76 ± 1.45 |
| AUCinf | ng·h/mL | 408 ± 121 |
| CL/F | L/h/kg | 39.7 ± 11.9 |
| Parameter | Rat po 5 mg/kg | Human po 2.5 mg | |||||
|---|---|---|---|---|---|---|---|
| Observed | Predicted | Predicted/ Observed | Observed [39] | Predicted | Predicted/ Observed | ||
| Cmax | ng/mL | 98.5 ± 15.1 | 144 | 1.46 | 7.89 | 9.64 | 1.22 |
| t1/2 | h | 1.82 ± 0.737 | 1.63 | 0.90 | 7.21 | 5.33 | 0.74 |
| AUCinf | ng·h/mL | 157 ± 23.1 | 177 | 1.13 | 49.1 | 46.9 | 0.96 |
| Parameter | Rat po 15 mg/kg | Human po 40 mg | |||||
|---|---|---|---|---|---|---|---|
| Observed | Predicted | Predicted/ Observed | Observed [43] | Predicted | Predicted/ Observed | ||
| Cmax | ng/mL | 132 ± 35.3 | 69.3 | 0.53 | 58.8 | 36.4 | 0.63 |
| t1/2 | h | 5.76 ± 1.45 | 2.96 | 0.51 | 2.80 | 3.44 | 1.23 |
| AUCinf | ng·h/mL | 408 ± 121 | 333 | 0.82 | 163 | 138 | 0.85 |
| Species | Dose (Route) | MRD | Pred/Obs Ratio | Reference | |
|---|---|---|---|---|---|
| Cmax | AUCinf | ||||
| Saxagliptin | |||||
| Rat | 10 mg/kg (iv) | 1.92 | 2.05 | 1.04 | [19] |
| 5 mg/kg (po) | 1.14 | 1.46 | 1.13 | In-house data | |
| GMFE | 1.73 | 1.08 | |||
| Human | 2.5 mg (po) | 1.32 | 1.22 | 0.96 | [39] |
| 5 mg (po) | 1.17 | 1.32 | 1.08 | [40] | |
| 5 mg (po) | 1.60 | 1.15 | 0.73 | [41] | |
| GMFE | 1.23 | 1.16 | |||
| Nicardipine | |||||
| Rat | 12 mg/kg (po) | 1.69 | 0.91 | 1.33 | [18] |
| 15 mg/kg (po) | 2.08 | 0.68 | 0.85 | In-house data | |
| GMFE | 1.27 | 1.15 | |||
| Human | 20 mg (po) | 1.92 | 0.72 | 1.50 | [42] |
| 40 mg (po) | 1.66 | 0.63 | 0.86 | [43] | |
| GMFE | 1.48 | 1.33 | |||
| Parameter | Rat | Human | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Observed | Predicted by PBPK Model | Predicted by PBPK Model | |||||||
| SXG | SXG + NCD | DDI Ratio | SXG | SXG + NCD | DDI Ratio | SXG | SXG + NCD | DDI Ratio | |
| Cmax (ng/mL) | 98.5 ± 15.1 | 281 ± 147 | 2.85 | 147 (130–154) | 395 (377–407) | 2.69 | 9.81 (7.36–12.9) | 10.8 (8.15–14.2) | 1.10 |
| AUCinf (ng·h/mL) | 157 ± 23.1 | 408 ± 213 | 2.60 | 193 (152–222) | 490 (381–577) | 2.54 | 50.5 (35.2–78.2) | 52.9 (36.8–82.3) | 1.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-M.; Yoon, J.-H.; Maeng, H.-J.; Kim, Y.C. Physiologically Based Pharmacokinetic (PBPK) Modeling to Predict CYP3A-Mediated Drug Interaction between Saxagliptin and Nicardipine: Bridging Rat-to-Human Extrapolation. Pharmaceutics 2024, 16, 280. https://doi.org/10.3390/pharmaceutics16020280
Lee J-M, Yoon J-H, Maeng H-J, Kim YC. Physiologically Based Pharmacokinetic (PBPK) Modeling to Predict CYP3A-Mediated Drug Interaction between Saxagliptin and Nicardipine: Bridging Rat-to-Human Extrapolation. Pharmaceutics. 2024; 16(2):280. https://doi.org/10.3390/pharmaceutics16020280
Chicago/Turabian StyleLee, Jeong-Min, Jin-Ha Yoon, Han-Joo Maeng, and Yu Chul Kim. 2024. "Physiologically Based Pharmacokinetic (PBPK) Modeling to Predict CYP3A-Mediated Drug Interaction between Saxagliptin and Nicardipine: Bridging Rat-to-Human Extrapolation" Pharmaceutics 16, no. 2: 280. https://doi.org/10.3390/pharmaceutics16020280
APA StyleLee, J.-M., Yoon, J.-H., Maeng, H.-J., & Kim, Y. C. (2024). Physiologically Based Pharmacokinetic (PBPK) Modeling to Predict CYP3A-Mediated Drug Interaction between Saxagliptin and Nicardipine: Bridging Rat-to-Human Extrapolation. Pharmaceutics, 16(2), 280. https://doi.org/10.3390/pharmaceutics16020280