
Enhancing Precision in Photodynamic Therapy: Innovations in Light-Driven and Bioorthogonal Activation

 , ,
, , 

Abstract
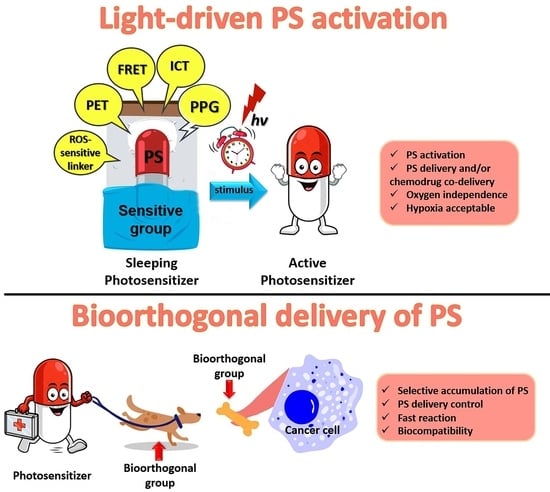
1. Introduction
Photosensitizers
- First-generation PSs
- Second-generation PSs
- Third generation
2. ROS-Activated PS–Drug Conjugates
2.1. Aminoacrylate Linker
2.2. Thioketal Linkage
3. Photoactivated Chemotherapy (Photocleavable Groups)
4. Activated PSs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Requirements | Influence on Fluorescence and ROS Generation | Trigger | Reference | |
|---|---|---|---|---|
| FRET | Overlap of donor emission and acceptor absorption spectra | Increase in acceptor fluorescence, quenching of donor fluorescence | Breaking or changing the bond between donor and acceptor | [96] |
| PET | The presence in the molecule of a donor and an acceptor | Full quenching | Changing pH, addition ions, carbohydrates, phosphates | [97] |
| ICT | The presence in the molecule of a donor and an acceptor, forming a dipole upon excitation | Fluorescence maximum shift, partial quenching | Changing pH or solvent polarity | [98] |
5. Bioorthogonal Delivery
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BODIPY | 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene |
| CA4 | Combretastatin A-4 |
| CPT | Camptothecin |
| cRGD | Cyclic Arg-Gly-Asp peptide |
| CT | Charge transfer |
| DAMPs | Damage-associated molecular patterns |
| DBCO | Dibenzocyclooctyne |
| DLI | Drug–light interval |
| DNBS | 2,4-dinitrobenzenesulfonate |
| EGFR | Epidermal growth factor receptor |
| FRET | Förster resonance energy transfer |
| GSH | Glutathione |
| HAPs | Hypoxia-activatable prodrugs |
| HpD | Hematoporphyrin derivatives |
| ICD | Immunogenic cell death |
| ICT | Intramolecular charge transfer |
| iEDDA | Inverse electron demand Diels–Alder reaction |
| ISC | Intersystem crossing |
| ManNAz | N-azidoacetylmannosamine |
| MLCT | Metal–ligand charge transfer |
| NIR | Near-infrared |
| PACT | Photoactivatable chemotherapy |
| PDT | Photodynamic therapy |
| PET | Photoinduced electron transfer |
| PPG | Photocleavable protective group |
| PRRs | Pattern recognition receptors |
| PS | Photosensitizer |
| PTT | Photothermal therapy |
| ROS | Reactive oxygen species |
| SPAAC | Strain-promoted azide–alkyne cycloaddition |
| STPS | PSs with singlet–triplet (ST) absorption |
| TBET | Through bond energy transfer |
| TCO | Trans-cyclooctenol |
| TTET | Triplet–triplet energy transfer |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Raab, O. On the Effect of Fluorescent Substances on Infusoria. Z. Biol. 1900, 39, 524–526. (In Germany) [Google Scholar]
- Von Tappeiner, H. On the Effect of Photodynamic (Fluorescent) Substances on Protozoa and Enzymes. Arch Klin Med. 1904, 39, 427–487. (In Germany) [Google Scholar]
- Schwartz, S.; Absolo, K.; Vermund, H. Some Relationships of Porphyrins, X-Rays and Tumors. Univ. Minn. Med. Bull. 1955, 27, 7–8. [Google Scholar]
- Lipson, R.L.; Baldes, E.J.; Olsen, A.M. Hematoporphyrin Derivative: A New Aid for Endoscopic Detection of Malignant Disease. J. Thorac. Cardiovasc. Surg. 1961, 42, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Kaufman, J.E.; Goldfarb, A.; Weishaupt, K.R.; Boyle, D.; Mittleman, A. Photoradiation Therapy for the Treatment of Malignant Tumors. Cancer Res. 1978, 38, 2628–2635. [Google Scholar] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA. Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, G.; Gedik, M.E.; Ayan, S. Photodynamic Therapy for the Treatment and Diagnosis of Cancer—A Review of the Current Clinical Status. Front. Chem. 2021, 9, 686303. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic Therapy—Mechanisms, Photosensitizers and Combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Mishchenko, T.; Balalaeva, I.; Gorokhova, A.; Vedunova, M.; Krysko, D.V. Which Cell Death Modality Wins the Contest for Photodynamic Therapy of Cancer? Cell Death Dis. 2022, 13, 455. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting Immunogenic Cancer Cell Death by Photodynamic Therapy: Past, Present and Future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef]
- Dos Santos, A.F.; De Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic Therapy in Cancer Treatment—An Update Review. J. Cancer Metastasis Treat. 2019, 5, 25. [Google Scholar] [CrossRef]
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T.; Yang, Z. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef] [PubMed]
- Abrahamse, H.; Hamblin, M.R. New Photosensitizers for Photodynamic Therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowski, J.M.; Pucelik, B.; Regiel-Futyra, A.; Brindell, M.; Mazuryk, O.; Kyzioł, A.; Stochel, G.; Macyk, W.; Arnaut, L.G. Engineering of Relevant Photodynamic Processes through Structural Modifications of Metallotetrapyrrolic Photosensitizers. Coord. Chem. Rev. 2016, 325, 67–101. [Google Scholar] [CrossRef]
- Donohoe, C.; Senge, M.O.; Arnaut, L.G.; Gomes-da-Silva, L.C. Cell Death in Photodynamic Therapy: From Oxidative Stress to Anti-Tumor Immunity. Biochim. Biophys. Acta-Rev. Cancer 2019, 1872, 188308. [Google Scholar] [CrossRef]
- Davies, M.J. Singlet Oxygen-Mediated Damage to Proteins and Its Consequences. Biochem. Biophys. Res. Commun. 2003, 305, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowski, J.M. Reactive Oxygen Species in Photodynamic Therapy: Mechanisms of Their Generation and Potentiation. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2017; Volume 70, pp. 343–394. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Invited Review: Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Kou, J.; Dou, D.; Yang, L. Porphyrin Photosensitizers in Photodynamic Therapy and Its Applications. Oncotarget 2017, 8, 81591–81603. [Google Scholar] [CrossRef]
- Allison, R.R. Photodynamic Therapy: Oncologic Horizons. Futur. Oncol. 2014, 10, 123–142. [Google Scholar] [CrossRef]
- Allison, R.R.; Moghissi, K. Photodynamic Therapy (PDT): PDT Mechanisms. Clin. Endosc. 2013, 46, 24–29. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Priefer, R. Non-Porphyrin Dyes Used as Photosensitizers in Photodynamic Therapy. J. Drug Deliv. Sci. Technol. 2020, 60, 101979. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.-G.; Grumezescu, A.M. Photodynamic Therapy—An Up-to-Date Review. Appl. Sci. 2021, 11, 3626. [Google Scholar] [CrossRef]
- Kudinova, N.V.; Berezov, T.T. Photodynamic Therapy of Cancer: Search for Ideal Photosensitizer. Biochem. Suppl. Ser. B Biomed. Chem. 2010, 4, 95–103. [Google Scholar] [CrossRef]
- Sheleg, S.V.; Zhavrid, E.A.; Khodina, T.V.; Kochubeev, G.A.; Istomin, Y.P.; Chalov, V.N.; Zhuravkin, I.N. Photodynamic Therapy with Chlorin e 6 for Skin Metastases of Melanoma. Photodermatol. Photoimmunol. Photomed. 2004, 20, 21–26. [Google Scholar] [CrossRef]
- Lo, P.-C.; Rodríguez-Morgade, M.S.; Pandey, R.K.; Ng, D.K.P.; Torres, T.; Dumoulin, F. The Unique Features and Promises of Phthalocyanines as Advanced Photosensitisers for Photodynamic Therapy of Cancer. Chem. Soc. Rev. 2020, 49, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Otvagin, V.F.; Kuzmina, N.S.; Kudriashova, E.S.; Nyuchev, A.V.; Gavryushin, A.E.; Fedorov, A.Y. Conjugates of Porphyrinoid-Based Photosensitizers with Cytotoxic Drugs: Current Progress and Future Directions toward Selective Photodynamic Therapy. J. Med. Chem. 2022, 65, 1695–1734. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S.K.; Porter, S.L.; Rizk, N.; Sheng, Y.; McKaig, T.; Burnett, K.; White, B.; Nesbitt, H.; Matin, R.N.; McHale, A.P.; et al. Rose Bengal–Amphiphilic Peptide Conjugate for Enhanced Photodynamic Therapy of Malignant Melanoma. J. Med. Chem. 2020, 63, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, N.S.; Otvagin, V.F.; Krylova, L.V.; Nyuchev, A.V.; Romanenko, Y.V.; Koifman, O.I.; Balalaeva, I.V.; Fedorov, A.Y. Synthesis and Antiproliferative Activity of New Chlorin E6 Glycoconjugates. Mendeleev Commun. 2020, 30, 159–161. [Google Scholar] [CrossRef]
- Krylova, L.V.; Peskova, N.N.; Otvagin, V.F.; Kuzmina, N.S.; Nyuchev, A.V.; Fedorov, A.Y.; Balalaeva, I.V. Novel Chlorine E6 Conjugate with Dual Targeting to Cancer Cells. Opera Med. Physiol. 2022, 9, 5–14. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Y.; Song, M.; Zhang, Z.; Xue, J. Silicon Phthalocyanines Axially Disubstituted with Erlotinib toward Small-Molecular-Target-Based Photodynamic Therapy. ChemMedChem 2017, 12, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Sadraeian, M.; Bahou, C.; da Cruz, E.F.; Janini, L.M.R.; Sobhie Diaz, R.; Boyle, R.W.; Chudasama, V.; Eduardo Gontijo Guimarães, F. Photoimmunotherapy Using Cationic and Anionic Photosensitizer-Antibody Conjugates against HIV Env-Expressing Cells. Int. J. Mol. Sci. 2020, 21, 9151. [Google Scholar] [CrossRef] [PubMed]
- Otvagin, V.F.; Nyuchev, A.V.; Kuzmina, N.S.; Grishin, I.D.; Gavryushin, A.E.; Romanenko, Y.V.; Koifman, O.I.; Belykh, D.V.; Peskova, N.N.; Shilyagina, N.Y.; et al. Synthesis and Biological Evaluation of New Water-Soluble Photoactive Chlorin Conjugate for Targeted Delivery. Eur. J. Med. Chem. 2018, 144, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Otvagin, V.F.; Kuzmina, N.S.; Krylova, L.V.; Volovetsky, A.B.; Nyuchev, A.V.; Gavryushin, A.E.; Meshkov, I.N.; Gorbunova, Y.G.; Romanenko, Y.V.; Koifman, O.I.; et al. Water-Soluble Chlorin/Arylaminoquinazoline Conjugate for Photodynamic and Targeted Therapy. J. Med. Chem. 2019, 62, 11182–11193. [Google Scholar] [CrossRef]
- Nyuchev, A.; Otvagin, V.; Gavryushin, A.; Romanenko, Y.; Koifman, O.; Belykh, D.; Schmalz, H.-G.; Fedorov, A. Synthesis of Chlorin–(Arylamino)Quinazoline Hybrids as Models for Multifunctional Drug Development. Synthesis 2015, 47, 3717–3726. [Google Scholar] [CrossRef][Green Version]
- Otvagin, V.F.; Krylova, L.V.; Peskova, N.N.; Kuzmina, N.S.; Fedotova, E.A.; Nyuchev, A.V.; Romanenko, Y.V.; Koifman, O.I.; Vatsadze, S.Z.; Schmalz, H.-G.; et al. A First-in-Class β-Glucuronidase Responsive Conjugate for Selective Dual Targeted and Photodynamic Therapy of Bladder Cancer. Eur. J. Med. Chem. 2024, 269, 116283. [Google Scholar] [CrossRef]
- Kuzmina, N.S.; Otvagin, V.F.; Maleev, A.A.; Urazaeva, M.A.; Nyuchev, A.V.; Ignatov, S.K.; Gavryushin, A.E.; Fedorov, A.Y. Development of Novel Porphyrin/Combretastatin A-4 Conjugates for Bimodal Chemo and Photodynamic Therapy: Synthesis, Photophysical and TDDFT Computational Studies. J. Photochem. Photobiol. A Chem. 2022, 433, 114138. [Google Scholar] [CrossRef]
- Luby, B.M.; Walsh, C.D.; Zheng, G. Advanced Photosensitizer Activation Strategies for Smarter Photodynamic Therapy Beacons. Angew. Chem. Int. Ed. 2019, 58, 2558–2569. [Google Scholar] [CrossRef]
- Liu, M.; Li, C. Recent Advances in Activatable Organic Photosensitizers for Specific Photodynamic Therapy. Chempluschem 2020, 85, 948–957. [Google Scholar] [CrossRef]
- Nguyen, L.; Li, M.; Woo, S.; You, Y. Development of Prodrugs for PDT-Based Combination Therapy Using a Singlet-Oxygen-Sensitive Linker and Quantitative Systems Pharmacology. J. Clin. Med. 2019, 8, 2198. [Google Scholar] [CrossRef]
- Fenical, W.H.; Kearns, D.R.; Radlick, P. Mechanism of the Addition of 1.DELTA.g Excited Oxygen to Olefins. Evidence for a 1,2-Dioxetane Intermediate. J. Am. Chem. Soc. 1969, 91, 3396–3398. [Google Scholar] [CrossRef]
- Hossion, A.M.L.; Bio, M.; Nkepang, G.; Awuah, S.G.; You, Y. Visible Light Controlled Release of Anticancer Drug through Double Activation of Prodrug. ACS Med. Chem. Lett. 2013, 4, 124–127. [Google Scholar] [CrossRef]
- Bio, M.; Rajaputra, P.; Nkepang, G.; Awuah, S.G.; Hossion, A.M.L.; You, Y. Site-Specific and Far-Red-Light-Activatable Prodrug of Combretastatin A-4 Using Photo-Unclick Chemistry. J. Med. Chem. 2013, 56, 3936–3942. [Google Scholar] [CrossRef] [PubMed]
- Bio, M.; Rahman, K.M.M.; Lim, I.; Rajaputra, P.; Hurst, R.E.; You, Y. Singlet Oxygen-Activatable Paclitaxel Prodrugs via Intermolecular Activation for Combined PDT and Chemotherapy. Bioorg. Med. Chem. Lett. 2019, 29, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Bio, M.; Rajaputra, P.; Nkepang, G.; You, Y. Far-Red Light Activatable, Multifunctional Prodrug for Fluorescence Optical Imaging and Combinational Treatment. J. Med. Chem. 2014, 57, 3401–3409. [Google Scholar] [CrossRef] [PubMed]
- Nkepang, G.; Bio, M.; Rajaputra, P.; Awuah, S.G.; You, Y. Folate Receptor-Mediated Enhanced and Specific Delivery of Far-Red Light-Activatable Prodrugs of Combretastatin A-4 to FR-Positive Tumor. Bioconjug. Chem. 2014, 25, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.Y.Y.; Zhou, Y.; Fong, W.-P.; Ng, D.K.P. Multifunctional Molecular Therapeutic Agent for Targeted and Controlled Dual Chemo- and Photodynamic Therapy. J. Med. Chem. 2020, 63, 8512–8523. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, L.; Loredo, A.; Wang, S.; Ada, N.; Xiao, H. Visible Light-Activated Prodrug System with a Novel Heavy-Atom-Free Photosensitizer. Bioorg. Med. Chem. Lett. 2023, 91, 129365. [Google Scholar] [CrossRef]
- Yang, D.-C.; Wang, S.; Weng, X.-L.; Zhang, H.-X.; Liu, J.-Y.; Lin, Z. Singlet Oxygen-Responsive Polymeric Nanomedicine for Light-Controlled Drug Release and Image-Guided Photodynamic–Chemo Combination Therapy. ACS Appl. Mater. Interfaces 2021, 13, 33905–33914. [Google Scholar] [CrossRef]
- He, M.; He, G.; Wang, P.; Jiang, S.; Jiao, Z.; Xi, D.; Miao, P.; Leng, X.; Wei, Z.; Li, Y.; et al. A Sequential Dual-Model Strategy Based on Photoactivatable Metallopolymer for On-Demand Release of Photosensitizers and Anticancer Drugs. Adv. Sci. 2021, 8, 2103334. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Jo, S.D.; Seah, G.L.; Kim, I.; Nam, Y.S. ROS-Induced Biodegradable Polythioketal Nanoparticles for Intracellular Delivery of Anti-Cancer Therapeutics. J. Ind. Eng. Chem. 2015, 21, 1137–1142. [Google Scholar] [CrossRef]
- Liu, B.; Thayumanavan, S. Mechanistic Investigation on Oxidative Degradation of ROS-Responsive Thioacetal/Thioketal Moieties and Their Implications. Cell Rep. Phys. Sci. 2020, 1, 100271. [Google Scholar] [CrossRef]
- Hu, P.; Xu, G.; Yang, D.-C.; Liu, J.-Y.; Chen, Z.; Huang, M. An Advanced Multifunctional Prodrug Combining Photodynamic Therapy with Chemotherapy for Highly Efficient and Precise Tumor Ablation. Dye. Pigment. 2022, 205, 110500. [Google Scholar] [CrossRef]
- Liu, L.; Qiu, W.; Li, B.; Zhang, C.; Sun, L.; Wan, S.; Rong, L.; Zhang, X. A Red Light Activatable Multifunctional Prodrug for Image-Guided Photodynamic Therapy and Cascaded Chemotherapy. Adv. Funct. Mater. 2016, 26, 6257–6269. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, H.-X.; Li, X.-Q.; Yang, D.-C.; Liu, J.-Y. Red Light Triggered Photodynamic-Chemo Combination Therapy Using a Prodrug Caged by Photosensitizer. Eur. J. Med. Chem. 2021, 215, 113251. [Google Scholar] [CrossRef]
- Luo, X.; Chi, X.; Lin, Y.; Yang, Z.; Lin, H.; Gao, J. A Camptothecin Prodrug Induces Mitochondria-Mediated Apoptosis in Cancer Cells with Cascade Activations. Chem. Commun. 2021, 57, 11033–11036. [Google Scholar] [CrossRef]
- Zhu, S.; Li, K.; Qin, S.; Lin, J.; Qiu, L. Cerenkov Radiation Induced Chemo-Photodynamic Therapy Using ROS-Responsive Agent. J. Photochem. Photobiol. A Chem. 2023, 439, 114641. [Google Scholar] [CrossRef]
- Lv, G.; Sun, X.; Qiu, L.; Sun, Y.; Li, K.; Liu, Q.; Zhao, Q.; Qin, S.; Lin, J. PET Imaging of Tumor PD-L1 Expression with a Highly Specific Nonblocking Single-Domain Antibody. J. Nucl. Med. 2020, 61, 117–122. [Google Scholar] [CrossRef]
- Guo, X.; Yang, N.; Ji, W.; Zhang, H.; Dong, X.; Zhou, Z.; Li, L.; Shen, H.; Yao, S.Q.; Huang, W. Mito-Bomb: Targeting Mitochondria for Cancer Therapy. Adv. Mater. 2021, 33, 2007778. [Google Scholar] [CrossRef]
- He, H.; Du, L.; Xue, H.; Wu, J.; Shuai, X. Programmable Therapeutic Nanoscale Covalent Organic Framework for Photodynamic Therapy and Hypoxia-Activated Cascade Chemotherapy. Acta Biomater. 2022, 149, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; del Valle, A.C.; Yeh, H.-P.; He, Y.; Huang, Y.-F. Development of Photo-Activated ROS-Responsive Nanoplatform as a Dual-Functional Drug Carrier in Combinational Chemo-Photodynamic Therapy. Front. Chem. 2019, 6, 647. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Shi, W.; Liu, Y.; Yin, L.; Guo, Z.; Zhou, W.; Fan, Q. Capsaicin-Decorated Semiconducting Polymer Nanoparticles for Light-Controlled Calcium-Overload/Photodynamic Combination Therapy. Small 2022, 18, 2200152. [Google Scholar] [CrossRef] [PubMed]
- Phua, S.Z.F.; Xue, C.; Lim, W.Q.; Yang, G.; Chen, H.; Zhang, Y.; Wijaya, C.F.; Luo, Z.; Zhao, Y. Light-Responsive Prodrug-Based Supramolecular Nanosystems for Site-Specific Combination Therapy of Cancer. Chem. Mater. 2019, 31, 3349–3358. [Google Scholar] [CrossRef]
- Reeßing, F.; Szymanski, W. Beyond Photodynamic Therapy: Light-Activated Cancer Chemotherapy. Curr. Med. Chem. 2018, 24, 4905–4950. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.; Denning, C.A.; Heidary, D.K.; Wachter, E.; Nease, L.A.; Ruiz, J.; Glazer, E.C. Ruthenium-Containing P450 Inhibitors for Dual Enzyme Inhibition and DNA Damage. Dalt. Trans. 2017, 46, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Wang, F.; Lu, S.; Chen, X. Recent Progress in Studies of Photocages. Smart Mol. 2023, 1, e20220003. [Google Scholar] [CrossRef]
- Lameijer, L.N.; Ernst, D.; Hopkins, S.L.; Meijer, M.S.; Askes, S.H.C.; Le Dévédec, S.E.; Bonnet, S. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed. 2017, 56, 11549–11553. [Google Scholar] [CrossRef]
- Toupin, N.P.; Arora, K.; Shrestha, P.; Peterson, J.A.; Fischer, L.J.; Rajagurubandara, E.; Podgorski, I.; Winter, A.H.; Kodanko, J.J. BODIPY-Caged Photoactivated Inhibitors of Cathepsin B Flip the Light Switch on Cancer Cell Apoptosis. ACS Chem. Biol. 2019, 14, 2833–2840. [Google Scholar] [CrossRef]
- Janeková, H.; Russo, M.; Ziegler, U.; Štacko, P. Photouncaging of Carboxylic Acids from Cyanine Dyes with Near-Infrared Light. Angew. Chem. Int. Ed. 2022, 61, e202204391. [Google Scholar] [CrossRef] [PubMed]
- Egyed, A.; Németh, K.; Molnár, T.Á.; Kállay, M.; Kele, P.; Bojtár, M. Turning Red without Feeling Embarrassed─Xanthenium-Based Photocages for Red-Light-Activated Phototherapeutics. J. Am. Chem. Soc. 2023, 145, 4026–4034. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.C. The Ligand Field Photosubstitution Reactions of D6 Hexacoordinate Metal Complexes. Coord. Chem. Rev. 1982, 44, 61–82. [Google Scholar] [CrossRef]
- Bonnet, S. Ruthenium-Based Photoactivated Chemotherapy. J. Am. Chem. Soc. 2023, 145, 23397–23415. [Google Scholar] [CrossRef] [PubMed]
- Bretin, L.; Husiev, Y.; Ramu, V.; Zhang, L.; Hakkennes, M.; Abyar, S.; Johns, A.C.; Le Dévédec, S.E.; Betancourt, T.; Kornienko, A.; et al. Red-Light Activation of a Microtubule Polymerization Inhibitor via Amide Functionalization of the Ruthenium Photocage. Angew. Chem. Int. Ed. 2024, 63, e202316425. [Google Scholar] [CrossRef] [PubMed]
- He, G.; He, M.; Wang, R.; Li, X.; Hu, H.; Wang, D.; Wang, Z.; Lu, Y.; Xu, N.; Du, J.; et al. A Near-Infrared Light-Activated Photocage Based on a Ruthenium Complex for Cancer Phototherapy. Angew. Chem. Int. Ed. 2023, 62, e202218768. [Google Scholar] [CrossRef] [PubMed]
- Toupin, N.P.; Steinke, S.J.; Herroon, M.K.; Podgorski, I.; Turro, C.; Kodanko, J.J. Unlocking the Potential of Ru(II) Dual-action Compounds with the Power of the Heavy-atom Effect. Photochem. Photobiol. 2022, 98, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Sitkowska, K.; Feringa, B.L.; Szymański, W. Green-Light-Sensitive BODIPY Photoprotecting Groups for Amines. J. Org. Chem. 2018, 83, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Kand, D.; Weinstain, R.; Winter, A.H. Meso -Methyl BODIPY Photocages: Mechanisms, Photochemical Properties, and Applications. J. Am. Chem. Soc. 2023, 145, 17497–17514. [Google Scholar] [CrossRef]
- Peterson, J.A.; Wijesooriya, C.; Gehrmann, E.J.; Mahoney, K.M.; Goswami, P.P.; Albright, T.R.; Syed, A.; Dutton, A.S.; Smith, E.A.; Winter, A.H. Family of BODIPY Photocages Cleaved by Single Photons of Visible/Near-Infrared Light. J. Am. Chem. Soc. 2018, 140, 7343–7346. [Google Scholar] [CrossRef]
- Slanina, T.; Shrestha, P.; Palao, E.; Kand, D.; Peterson, J.A.; Dutton, A.S.; Rubinstein, N.; Weinstain, R.; Winter, A.H.; Klán, P. In Search of the Perfect Photocage: Structure–Reactivity Relationships in Meso -Methyl BODIPY Photoremovable Protecting Groups. J. Am. Chem. Soc. 2017, 139, 15168–15175. [Google Scholar] [CrossRef] [PubMed]
- Zlatić, K.; Popović, M.; Uzelac, L.; Kralj, M.; Basarić, N. Antiproliferative Activity of Meso-Substituted BODIPY Photocages: Effect of Electrophiles vs Singlet Oxygen. Eur. J. Med. Chem. 2023, 259, 115705. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Kohn, A.W.; Van Voorhis, T. Toward Prediction of Nonradiative Decay Pathways in Organic Compounds II: Two Internal Conversion Channels in BODIPYs. J. Phys. Chem. C 2020, 124, 3925–3938. [Google Scholar] [CrossRef]
- Briggs, E.A.; Besley, N.A.; Robinson, D. QM/MM Excited State Molecular Dynamics and Fluorescence Spectroscopy of BODIPY. J. Phys. Chem. A 2013, 117, 2644–2650. [Google Scholar] [CrossRef]
- Shrestha, P.; Dissanayake, K.C.; Gehrmann, E.J.; Wijesooriya, C.S.; Mukhopadhyay, A.; Smith, E.A.; Winter, A.H. Efficient Far-Red/Near-IR Absorbing BODIPY Photocages by Blocking Unproductive Conical Intersections. J. Am. Chem. Soc. 2020, 142, 15505–15512. [Google Scholar] [CrossRef]
- Lv, W.; Long, K.; Yang, Y.; Chen, S.; Zhan, C.; Wang, W. A Red Light-Triggered Drug Release System Based on One-Photon Upconversion-Like Photolysis. Adv. Healthc. Mater. 2020, 9, 2001118. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Lv, W.; Wang, Z.; Zhang, Y.; Chen, K.; Fan, N.; Li, F.; Zhang, Y.; Wang, W. Near-Infrared Light-Triggered Prodrug Photolysis by One-Step Energy Transfer. Nat. Commun. 2023, 14, 8112. [Google Scholar] [CrossRef]
- Amemori, S.; Sasaki, Y.; Yanai, N.; Kimizuka, N. Near-Infrared-to-Visible Photon Upconversion Sensitized by a Metal Complex with Spin-Forbidden yet Strong S0–T1 Absorption. J. Am. Chem. Soc. 2016, 138, 8702–8705. [Google Scholar] [CrossRef]
- Li, X.; Kwon, N.; Guo, T.; Liu, Z.; Yoon, J. Innovative Strategies for Hypoxic-Tumor Photodynamic Therapy. Angew. Chem. Int. Ed. 2018, 57, 11522–11531. [Google Scholar] [CrossRef]
- Rapozzi, V.; Beverina, L.; Salice, P.; Pagani, G.A.; Camerin, M.; Xodo, L.E. Photooxidation and Phototoxicity of π-Extended Squaraines. J. Med. Chem. 2010, 53, 2188–2196. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, C.; Zheng, Q.; Jia, Q.; Wang, Z.; Shi, P.; Guo, Z. Harnessing Hypoxia-Dependent Cyanine Photocages for In Vivo Precision Drug Release. Angew. Chem. Int. Ed. 2021, 60, 9553–9561. [Google Scholar] [CrossRef] [PubMed]
- Scherer, K.M.; Bisby, R.H.; Botchway, S.W.; Hadfield, J.A.; Parker, A.W. Anticancer Phototherapy Using Activation of E -Combretastatins by Two-Photon–Induced Isomerization. J. Biomed. Opt. 2014, 20, 051004. [Google Scholar] [CrossRef][Green Version]
- Sekhar, A.R.; Chitose, Y.; Janoš, J.; Dangoor, S.I.; Ramundo, A.; Satchi-Fainaro, R.; Slavíček, P.; Klán, P.; Weinstain, R. Porphyrin as a Versatile Visible-Light-Activatable Organic/Metal Hybrid Photoremovable Protecting Group. Nat. Commun. 2022, 13, 3614. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Cheng, H.; Xie, B.-R.; Qiu, W.-X.; Song, L.-L.; Zhuo, R.-X.; Zhang, X.-Z. A Ratiometric Theranostic Probe for Tumor Targeting Therapy and Self-Therapeutic Monitoring. Biomaterials 2016, 104, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, C.; Emery, B.P.; Sedgwick, A.C.; Bull, S.D.; He, X.-P.; Tian, H.; Yoon, J.; Sessler, J.L.; James, T.D. Förster Resonance Energy Transfer (FRET)-Based Small-Molecule Sensors and Imaging Agents. Chem. Soc. Rev. 2020, 49, 5110–5139. [Google Scholar] [CrossRef] [PubMed]
- Daly, B.; Ling, J.; de Silva, A.P. Current Developments in Fluorescent PET (Photoinduced Electron Transfer) Sensors and Switches. Chem. Soc. Rev. 2015, 44, 4203–4211. [Google Scholar] [CrossRef]
- Sasaki, S.; Drummen, G.P.C.; Konishi, G. Recent Advances in Twisted Intramolecular Charge Transfer (TICT) Fluorescence and Related Phenomena in Materials Chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef]
- Bai, Z.; Liu, Y.; Zhang, P.; Guo, J.; Ma, Y.; Yun, X.; Zhao, X.; Zhong, R.; Zhang, F. Fluorescence Resonance Energy Transfer between Bovine Serum Albumin and Fluoresceinamine. Luminescence 2016, 31, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.F.; Chen, J.; Jarvi, M.T.; Cao, W.-G.; Allen, A.D.; Liu, Y.; Tidwell, T.T.; Wilson, B.C.; Zheng, G. FRET Quenching of Photosensitizer Singlet Oxygen Generation. J. Phys. Chem. B 2009, 113, 3203–3211. [Google Scholar] [CrossRef]
- Li, X.; Gao, X.; Shi, W.; Ma, H. Design Strategies for Water-Soluble Small Molecular Chromogenic and Fluorogenic Probes. Chem. Rev. 2014, 114, 590–659. [Google Scholar] [CrossRef]
- Xue, Y.; Tian, J.; Liu, Z.; Chen, J.; Wu, M.; Shen, Y.; Zhang, W. A Redox Stimulation-Activated Amphiphile for Enhanced Photodynamic Therapy. Biomacromolecules 2019, 20, 2796–2808. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, L.; Cui, X.; Li, S.; Wu, H. Maximizing the Thiol-Activated Photodynamic and Fluorescence Imaging Functionalities of Theranostic Reagents by Modularization of Bodipy-Based Dyad Triplet Photosensitizers. J. Mater. Chem. B 2015, 3, 9194–9211. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Lo, P.; Ng, D.K.P. A Glutathione-Activated Phthalocyanine-Based Photosensitizer for Photodynamic Therapy. Chem.—A Eur. J. 2014, 20, 6241–6245. [Google Scholar] [CrossRef]
- Cao, J.-J.; Zhang, M.-S.; Li, X.-Q.; Yang, D.-C.; Xu, G.; Liu, J.-Y. A Glutathione-Responsive Photosensitizer with Fluorescence Resonance Energy Transfer Characteristics for Imaging-Guided Targeting Photodynamic Therapy. Eur. J. Med. Chem. 2020, 193, 112203. [Google Scholar] [CrossRef] [PubMed]
- Vangara, A.; Pramanik, A.; Gao, Y.; Gates, K.; Begum, S.; Chandra Ray, P. Fluorescence Resonance Energy Transfer Based Highly Efficient Theranostic Nanoplatform for Two-Photon Bioimaging and Two-Photon Excited Photodynamic Therapy of Multiple Drug Resistance Bacteria. ACS Appl. Bio Mater. 2018, 1, 298–309. [Google Scholar] [CrossRef]
- Cao, H.; Wang, L.; Yang, Y.; Li, J.; Qi, Y.; Li, Y.; Li, Y.; Wang, H.; Li, J. An Assembled Nanocomplex for Improving Both Therapeutic Efficiency and Treatment Depth in Photodynamic Therapy. Angew. Chem. 2018, 130, 7885–7889. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Yu, Z.; Zhu, X.; Yu, J.; Wu, Z.; Wang, S.; Zhou, H. Molecular Tailoring Based on Forster Resonance Energy Transfer for Initiating Two-Photon Theranostics with Amplified Reactive Oxygen Species. Anal. Chem. 2022, 94, 14029–14037. [Google Scholar] [CrossRef]
- Lioret, V.; Bellaye, P.-S.; Arnould, C.; Collin, B.; Decréau, R.A. Dual Cherenkov Radiation-Induced Near-Infrared Luminescence Imaging and Photodynamic Therapy toward Tumor Resection. J. Med. Chem. 2020, 63, 9446–9456. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, M.; Fan, J.; Peng, X. Activity-Based Sensing and Theranostic Probes Based on Photoinduced Electron Transfer. Acc. Chem. Res. 2019, 52, 2818–2831. [Google Scholar] [CrossRef]
- Tang, Y.; Xue, L.; Yu, Q.; Chen, D.; Cheng, Z.; Wang, W.; Shao, J.; Dong, X. Smart Aza-BODIPY Photosensitizer for Tumor Microenvironment-Enhanced Cancer Phototherapy. ACS Appl. Bio Mater. 2019, 2, 5888–5897. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, R.; Liu, J.; Kong, H.; Zhang, K.; Zhang, Y.-N.; Kong, X.; Zhang, Q.; Zhao, Y. Hierarchical Nano-to-Molecular Disassembly of Boron Dipyrromethene Nanoparticles for Enhanced Tumor Penetration and Activatable Photodynamic Therapy. Biomaterials 2021, 275, 120945. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhou, J.; Shen, Z.; Ding, L.; Yu, J.-S.; Ju, H. A PH-Activatable and Aniline-Substituted Photosensitizer for near-Infrared Cancer Theranostics. Chem. Sci. 2015, 6, 5969–5977. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.T.F.; Lo, P.-C.; Jiang, X.-J.; Wang, Q.; Ng, D.K.P. A Dual Activatable Photosensitizer toward Targeted Photodynamic Therapy. J. Med. Chem. 2014, 57, 4088–4097. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.K.B.; Yu, L.; Wong, R.C.H.; Fong, W.-P.; Ng, D.K.P.; Lo, P.-C. Dual Cathepsin B and Glutathione-Activated Dimeric and Trimeric Phthalocyanine-Based Photodynamic Molecular Beacons for Targeted Photodynamic Therapy. J. Med. Chem. 2021, 64, 17455–17467. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, F.; Shi, W.; Gurzadyan, G.; Yin, H.; Song, B.; Liang, R.; Peng, X. Nitroreductase-Activatable Theranostic Molecules with High PDT Efficiency under Mild Hypoxia Based on a TADF Fluorescein Derivative. ACS Appl. Mater. Interfaces 2019, 11, 15426–15435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-F.; Yang, X. Photosensitizer That Selectively Generates Singlet Oxygen in Nonpolar Environments: Photophysical Mechanism and Efficiency for a Covalent BODIPY Dimer. J. Phys. Chem. B 2013, 117, 9050–9055. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, W.; Zhao, J. Switching of the Triplet Excited State of Styryl 2,6-Diiodo-Bodipy and Its Application in Acid-Activatable Singlet Oxygen Photosensitizing. J. Org. Chem. 2014, 79, 10240–10255. [Google Scholar] [CrossRef]
- Kamkaew, A.; Lim, S.H.; Lee, H.B.; Kiew, L.V.; Chung, L.Y.; Burgess, K. BODIPY Dyes in Photodynamic Therapy. Chem. Soc. Rev. 2013, 42, 77–88. [Google Scholar] [CrossRef]
- Lim, S.H.; Thivierge, C.; Nowak-Sliwinska, P.; Han, J.; van den Bergh, H.; Wagnières, G.; Burgess, K.; Lee, H.B. In Vitro and In Vivo Photocytotoxicity of Boron Dipyrromethene Derivatives for Photodynamic Therapy. J. Med. Chem. 2010, 53, 2865–2874. [Google Scholar] [CrossRef]
- Epelde-Elezcano, N.; Palao, E.; Manzano, H.; Prieto-Castañeda, A.; Agarrabeitia, A.R.; Tabero, A.; Villanueva, A.; de la Moya, S.; López-Arbeloa, Í.; Martínez-Martínez, V.; et al. Rational Design of Advanced Photosensitizers Based on Orthogonal BODIPY Dimers to Finely Modulate Singlet Oxygen Generation. Chem.-A Eur. J. 2017, 23, 4837–4848. [Google Scholar] [CrossRef]
- Lu, S.; Lei, X.; Ren, H.; Zheng, S.; Qiang, J.; Zhang, Z.; Chen, Y.; Wei, T.; Wang, F.; Chen, X. PEGylated Dimeric BODIPY Photosensitizers as Nanocarriers for Combined Chemotherapy and Cathepsin B-Activated Photodynamic Therapy in 3D Tumor Spheroids. ACS Appl. Bio Mater. 2020, 3, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Y.; Chen, L.; Hu, X.; Xie, Z. A Glutathione-Activatable Photodynamic and Fluorescent Imaging Monochromatic Photosensitizer. J. Mater. Chem. B 2017, 5, 4239–4245. [Google Scholar] [CrossRef] [PubMed]
- Radunz, S.; Wedepohl, S.; Röhr, M.; Calderón, M.; Tschiche, H.R.; Resch-Genger, U. PH-Activatable Singlet Oxygen-Generating Boron-Dipyrromethenes (BODIPYs) for Photodynamic Therapy and Bioimaging. J. Med. Chem. 2020, 63, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Simões, J.C.S.; Sarpaki, S.; Papadimitroulas, P.; Therrien, B.; Loudos, G. Conjugated Photosensitizers for Imaging and PDT in Cancer Research. J. Med. Chem. 2020, 63, 14119–14150. [Google Scholar] [CrossRef] [PubMed]
- Solban, N.; Rizvi, I.; Hasan, T. Targeted Photodynamic Therapy. Lasers Surg. Med. 2006, 38, 522–531. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Agarwal, P.; van der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A Pictet-Spengler Ligation for Protein Chemical Modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Scinto, S.L.; Bilodeau, D.A.; Hincapie, R.; Lee, W.; Nguyen, S.S.; Xu, M.; am Ende, C.W.; Finn, M.G.; Lang, K.; Lin, Q.; et al. Bioorthogonal Chemistry. Nat. Rev. Methods Prim. 2021, 1, 30. [Google Scholar] [CrossRef]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef]
- Tam, L.K.B.; Ng, D.K.P. “Click” for Precise Photodynamic Therapy. Mater. Chem. Front. 2023, 7, 3184–3193. [Google Scholar] [CrossRef]
- Kozma, E.; Bojtár, M.; Kele, P. Bioorthogonally Assisted Phototherapy: Recent Advances and Prospects. Angew. Chem. Int. Ed. 2023, 62, e202303198. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zou, S.; Liao, X.; Chen, Y.; Luo, D.; Ji, L.; Chao, H. Ruthenium(II) Complexes as Bioorthogonal Two-Photon Photosensitizers for Tumour-Specific Photodynamic Therapy against Triple-Negative Breast Cancer Cells. Chem. Commun. 2021, 57, 4408–4411. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, S.T.; Bertozzi, C.R. Metabolic Labeling of Glycans with Azido Sugars and Subsequent Glycan-Profiling and Visualization via Staudinger Ligation. Nat. Protoc. 2007, 2, 2930–2944. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Dong, Y.; Tu, Y.; Luo, S.; Pang, X.; Zhang, W.; Yao, W.; Tang, W.; Yang, H.; Wei, X.; et al. Bioorthogonal Pretargeting Strategy for Anchoring Activatable Photosensitizers on Plasma Membranes for Effective Photodynamic Therapy. ACS Appl. Mater. Interfaces 2021, 13, 14004–14014. [Google Scholar] [CrossRef] [PubMed]
- Harty, J.I.; Amin, M.; Wieman, T.J.; Tseng, M.T.; Ackerman, D.; Broghamer, W. Complications of Whole Bladder Dihematoporphyrin Ether Photodynamic Therapy. J. Urol. 1989, 141, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Bio, M.; Rajaputra, P.; You, Y. Photodynamic Therapy via FRET Following Bioorthogonal Click Reaction in Cancer Cells. Bioorg. Med. Chem. Lett. 2016, 26, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, S.; Shi, L.; Teh, C.; Qi, G.; Liu, B. Cancer-Cell-Activated in Situ Synthesis of Mitochondria-Targeting AIE Photosensitizer for Precise Photodynamic Therapy. Angew. Chem. Int. Ed. 2021, 60, 14945–14953. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.L.; Carter, S.L.; Wileyto, E.P.; Miller, J.; Yuan, M.; Yu, G.; Durham, A.C.; Busch, T.M. Tumor Vascular Microenvironment Determines Responsiveness to Photodynamic Therapy. Cancer Res. 2012, 72, 2079–2088. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic Therapy and Anti-Tumour Immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef]
- Zhang, R.; Feng, L.; Dong, Z.; Wang, L.; Liang, C.; Chen, J.; Ma, Q.; Zhang, R.; Chen, Q.; Wang, Y.; et al. Glucose & Oxygen Exhausting Liposomes for Combined Cancer Starvation and Hypoxia-Activated Therapy. Biomaterials 2018, 162, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Liu, Y.; Dong, Y.; Wang, K.; Yuan, Y. Bioorthogonal Chemistry and Illumination Controlled Programmed Size-Changeable Nanomedicine for Synergistic Photodynamic and Hypoxia-Activated Therapy. Biomaterials 2022, 284, 121480. [Google Scholar] [CrossRef] [PubMed]
- Yaghini, E.; Dondi, R.; Tewari, K.M.; Loizidou, M.; Eggleston, I.M.; MacRobert, A.J. Endolysosomal Targeting of a Clinical Chlorin Photosensitiser for Light-Triggered Delivery of Nano-Sized Medicines. Sci. Rep. 2017, 7, 6059. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse Electron Demand Diels–Alder Reactions in Chemical Biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.C.T.; Meimetis, L.G.; Hilderbrand, S.A.; Weissleder, R. BODIPY–Tetrazine Derivatives as Superbright Bioorthogonal Turn-on Probes. Angew. Chem. Int. Ed. 2013, 52, 6917–6920. [Google Scholar] [CrossRef] [PubMed]
- Linden, G.; Zhang, L.; Pieck, F.; Linne, U.; Kosenkov, D.; Tonner, R.; Vázquez, O. Conditional Singlet Oxygen Generation through a Bioorthogonal DNA-targeted Tetrazine Reaction. Angew. Chem. Int. Ed. 2019, 58, 12868–12873. [Google Scholar] [CrossRef] [PubMed]
- Linden, G.; Vázquez, O. Bioorthogonal Turn-On BODIPY-Peptide Photosensitizers for Tailored Photodynamic Therapy. Chem.—A Eur. J. 2020, 26, 10014–10023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wong, R.C.H.; Dai, G.; Ng, D.K.P. A Bioorthogonally Activatable Photosensitiser for Site-Specific Photodynamic Therapy. Chem. Commun. 2020, 56, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wong, R.C.H.; Zhou, Y.; Ng, D.K.P.; Lo, P.-C. A Novel Distyryl Boron Dipyrromethene with Two Functional Tags for Site-Specific Bioorthogonal Photosensitisation towards Targeted Photodynamic Therapy. Chem. Commun. 2019, 55, 13518–13521. [Google Scholar] [CrossRef]
- Devaraj, N.K.; Hilderbrand, S.; Upadhyay, R.; Mazitschek, R.; Weissleder, R. Bioorthogonal Turn-On Probes for Imaging Small Molecules inside Living Cells. Angew. Chem. Int. Ed. 2010, 49, 2869–2872. [Google Scholar] [CrossRef]
- Xiong, J.; Xue, E.Y.; Wu, Q.; Lo, P.-C.; Ng, D.K.P. A Tetrazine-Responsive Isonitrile-Caged Photosensitiser for Site-Specific Photodynamic Therapy. J. Control. Release 2023, 353, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Zeng, W.; Zhang, J.; Liu, Y.; Miao, Y.; Liu, S.; Yang, Y.; Xu, J.; Ye, D. Cascade In Situ Self-Assembly and Bioorthogonal Reaction Enable the Enrichment of Photosensitizers and Carbonic Anhydrase Inhibitors for Pretargeted Cancer Theranostics. Angew. Chem. Int. Ed. 2024, 63, e202314039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, S.; Gao, Z.; Zhou, J.; Xia, Y.; Tian, J.; Shi, C.; Wang, Z. Liposome Trade-off Strategy in Mitochondria-Targeted NIR-Cyanine: Balancing Blood Circulation and Cell Retention for Enhanced Anti-Tumor Phototherapy in Vivo. Nano Res. 2021, 14, 2432–2440. [Google Scholar] [CrossRef]
- Svatunek, D.; Wilkovitsch, M.; Hartmann, L.; Houk, K.N.; Mikula, H. Uncovering the Key Role of Distortion in Bioorthogonal Tetrazine Tools That Defy the Reactivity/Stability Trade-Off. J. Am. Chem. Soc. 2022, 144, 8171–8177. [Google Scholar] [CrossRef]
- Tu, J.; Svatunek, D.; Parvez, S.; Liu, A.C.; Levandowski, B.J.; Eckvahl, H.J.; Peterson, R.T.; Houk, K.N.; Franzini, R.M. Stable, Reactive, and Orthogonal Tetrazines: Dispersion Forces Promote the Cycloaddition with Isonitriles. Angew. Chem. Int. Ed. 2019, 58, 9043–9048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Trout, W.S.; Liu, S.; Andrade, G.A.; Hudson, D.A.; Scinto, S.L.; Dicker, K.T.; Li, Y.; Lazouski, N.; Rosenthal, J.; et al. Rapid Bioorthogonal Chemistry Turn-on through Enzymatic or Long Wavelength Photocatalytic Activation of Tetrazine Ligation. J. Am. Chem. Soc. 2016, 138, 5978–5983. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, H.; Zhang, T.; Zou, X.; Wang, H.; Rosenberger, J.E.; Vannam, R.; Trout, W.S.; Grimm, J.B.; Lavis, L.D.; et al. Enabling In Vivo Photocatalytic Activation of Rapid Bioorthogonal Chemistry by Repurposing Silicon-Rhodamine Fluorophores as Cytocompatible Far-Red Photocatalysts. J. Am. Chem. Soc. 2021, 143, 10793–10803. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, D.; Johnson, M.; Devaraj, N.K. Light-Activated Tetrazines Enable Precision Live-Cell Bioorthogonal Chemistry. Nat. Chem. 2022, 14, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Knittel, C.; Chadwick, S.; Kuehling, C.; Devaraj, N. Enzymatic Activation of Caged Tetrazines for Cell-Specific Bioconjugation. ChemRxiv 2024. [Google Scholar] [CrossRef]
- Hu, L.; Li, B.; Liao, Y.; Wang, S.; Hou, P.; Cheng, Y.; Zhang, S. Nitroreductase-Induced Bioorthogonal Ligation for Prodrug Activation: A Traceless Strategy for Cancer-Specific Imaging and Therapy. Bioorg. Chem. 2022, 129, 106167. [Google Scholar] [CrossRef]
- Li, J.; Chen, P.R. Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat. Chem. Biol. 2016, 12, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, N.K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.C.T.; Mikula, H.; Weissleder, R. Unraveling Tetrazine-Triggered Bioorthogonal Elimination Enables Chemical Tools for Ultrafast Release and Universal Cleavage. J. Am. Chem. Soc. 2018, 140, 3603–3612. [Google Scholar] [CrossRef]
- Wu, H.; Alexander, S.C.; Jin, S.; Devaraj, N.K. A Bioorthogonal Near-Infrared Fluorogenic Probe for MRNA Detection. J. Am. Chem. Soc. 2016, 138, 11429–11432. [Google Scholar] [CrossRef] [PubMed]











































| PSs | Commercial Name | λmax. (nm) | λmax.·10−3 (M−1·cm−1) | Application | Refs. |
|---|---|---|---|---|---|
| First generation | |||||
HpD (ether form) | Photofrin | 630 | 3 | Lung, bladder, esophagus, cervical, brain, gastrointestinal cancer | [12] |
| Second generation | |||||
Verteporfin | Visudyne | 690 | 34 | Skin, pancreas AMD, basal cell carcinoma | [6,8,12] |
ALA-induced protoporphyrin IX | Ameluz | 630 | - | Actinic keratosis and basal cell carcinoma | [12] |
| AlaCare | 630 | - | Actinic keratosis | [12] | |
| Levulan | 635 | 5 | Actinic keratosis | [12] | |
2-(1-hexyloxyethyl)-2-devinyl pyropheophorbide-a, HPPH | Photochlor | 665 | 47.5 | Lung, esophagus, head and neck cancer | [12] |
m-tetra(hydroxyphenyl)chlorin | Foscan | 652 | 30 | Prostate, bronchus, pancreas, head and neck cancer | [12,13] |
Chlorin-e6 derivatives | Photoditazine | 668 | 48 | Skin, breast, lung, prostate | [25] |
| Photolon | 665 | 50 | Skin, breast, nasopharyngeal sarcoma | [12,13] | |
| Radachlorin | 662 | 34.2 | Skin, lung, brain | [6,12] | |
Talaporfin | Laserphyrin | 664 | 40 | Lung, esophagus, brain, liver, colon | [6,12,24] |
Redaporfin | LUZ11 | 749 | 140 | Biliary tract, head and neck cancer | [12,13,14] |
Tin ethyl etiopurpurin | Purlytin | 660 | 28 | Breast, skin, prostate, Kaposi’s sarcoma | [8,14] |
Lutetium texaphyrin | LUTRIN | 732 | 42 | Cervical, prostate, brain, AMD | [14,25] |
Padeliporfin | TOOKAD, WST11 | 762 | 110 | Prostate | [13,14] |
Zinc phthalocyanine | CGP 55847 | 670 | 200 | Squamous cell carcinoma of upper aerodigestive tract | [13,14] |
Tetrasulfonated chloroaluminum phthalocyanine | Photosens | 675 | 200 | Skin, breast, lung, cervical, larynx, head and neck cancer, liver and gastrointestinal cancer | [14,25] |
Silicon phthalocyanine | Pc4 | 675 | 200 | Cutaneous neoplasms | [14,15] |
| Photoprotective Group (LG-leaving Group) | Wavelength Range, nm | Release Quantum Yield (Фr), % | Applicability to PDT | Reference |
|---|---|---|---|---|
Ru-complex | 350–760 | 1·10−2–22 | Yes | [70] |
BODIPY | 514–709 | 1·10−3–3.8 | Yes | [71] |
Cyanine | 640–817 | Up to 14 | No | [72] |
Xantene | 540–640 | Up to 18 | No | [73] |
| Click Reaction | iEDDA | |
|---|---|---|
| Rate constant, k (M−1s−1) | 10–102 | 1–106 |
| Bioorthogonal handles |  |  |
| Catalyst | yes—Cu (I), except for the SPAAC | no |
| In vivo application | yes | yes |
| Limitations | requirement for toxic copper (I) ions or limitation by the structure of a hindered cyclic alkyne | need for precise tetrazine design due to effects on reaction rates, limited stability of tetrazines in water |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzmina, N.S.; Fedotova, E.A.; Jankovic, P.; Gribova, G.P.; Nyuchev, A.V.; Fedorov, A.Y.; Otvagin, V.F. Enhancing Precision in Photodynamic Therapy: Innovations in Light-Driven and Bioorthogonal Activation. Pharmaceutics 2024, 16, 479. https://doi.org/10.3390/pharmaceutics16040479
Kuzmina NS, Fedotova EA, Jankovic P, Gribova GP, Nyuchev AV, Fedorov AY, Otvagin VF. Enhancing Precision in Photodynamic Therapy: Innovations in Light-Driven and Bioorthogonal Activation. Pharmaceutics. 2024; 16(4):479. https://doi.org/10.3390/pharmaceutics16040479
Chicago/Turabian StyleKuzmina, Natalia S., Ekaterina A. Fedotova, Petar Jankovic, Galina P. Gribova, Alexander V. Nyuchev, Alexey Yu. Fedorov, and Vasilii F. Otvagin. 2024. "Enhancing Precision in Photodynamic Therapy: Innovations in Light-Driven and Bioorthogonal Activation" Pharmaceutics 16, no. 4: 479. https://doi.org/10.3390/pharmaceutics16040479
APA StyleKuzmina, N. S., Fedotova, E. A., Jankovic, P., Gribova, G. P., Nyuchev, A. V., Fedorov, A. Y., & Otvagin, V. F. (2024). Enhancing Precision in Photodynamic Therapy: Innovations in Light-Driven and Bioorthogonal Activation. Pharmaceutics, 16(4), 479. https://doi.org/10.3390/pharmaceutics16040479