

Attempts to Improve Lipophilic Drugs’ Solubility and Bioavailability: A Focus on Fenretinide



Abstract
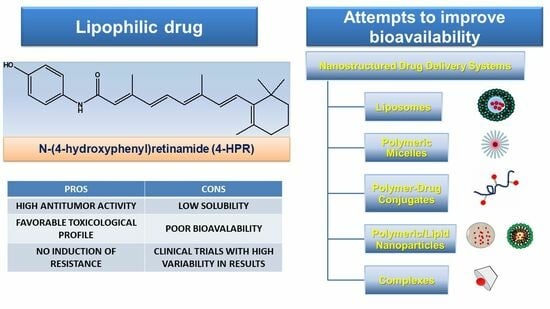
:
1. Introduction
2. Retinoids
2.1. Retinoids Generations
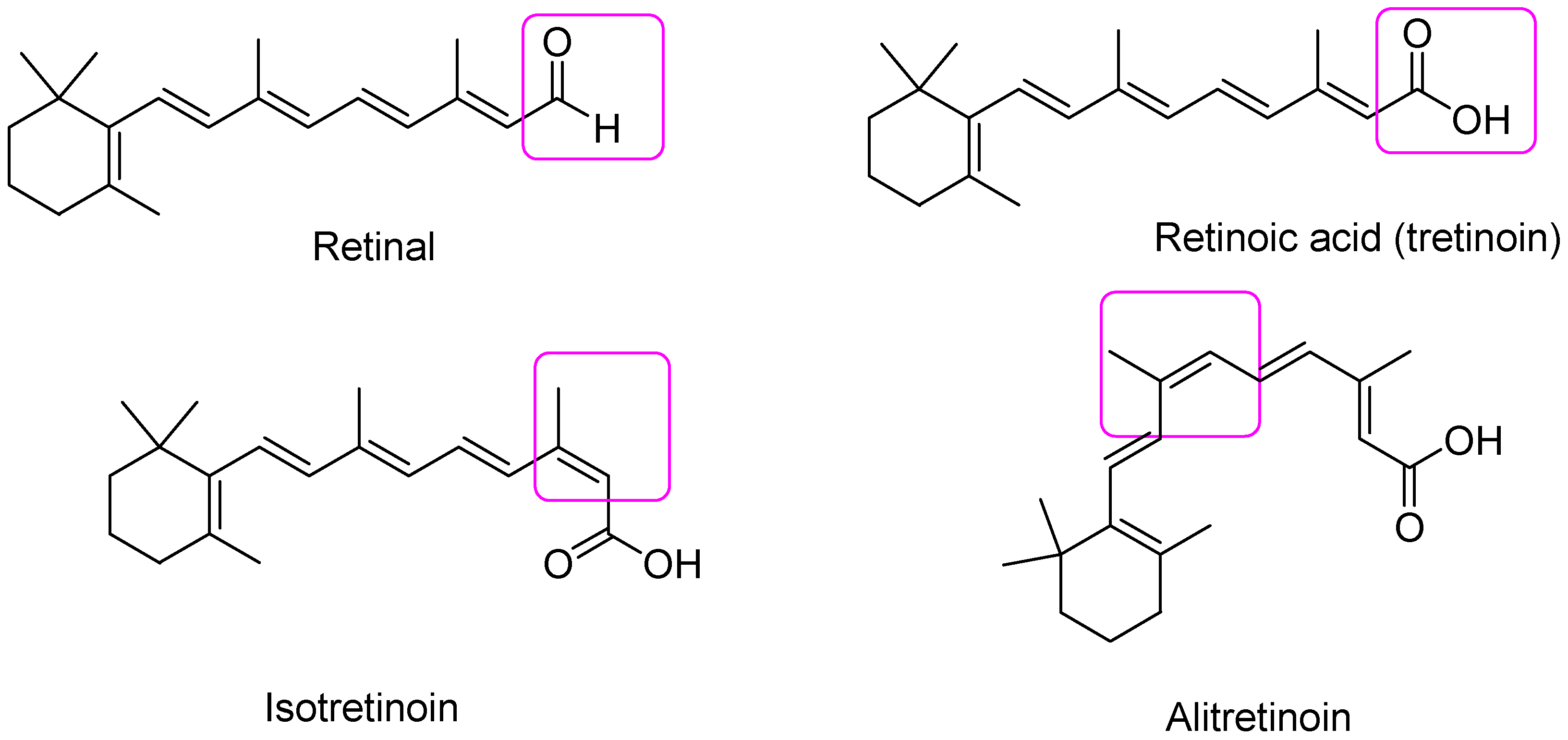
2.1.1. First Generation Retinoids
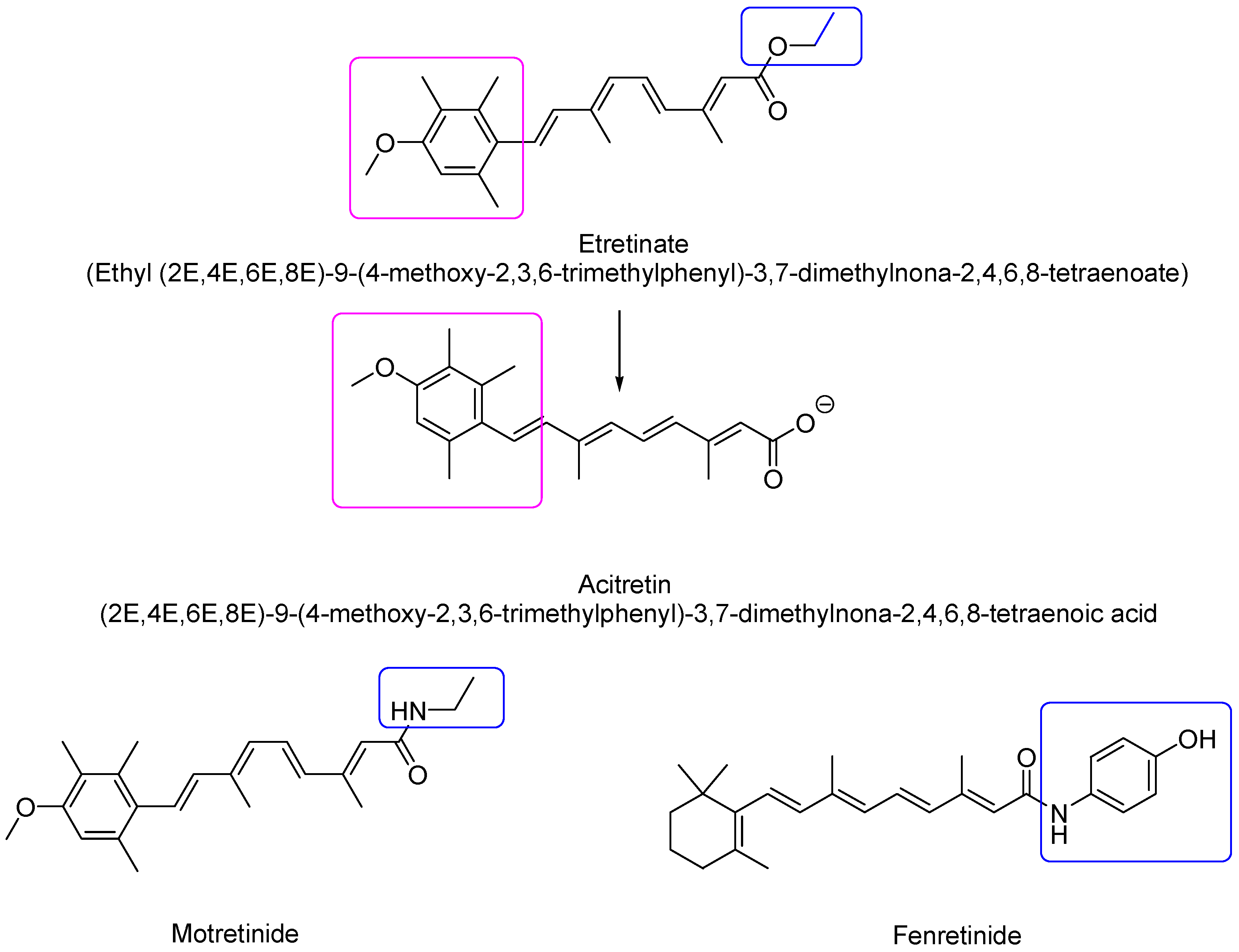
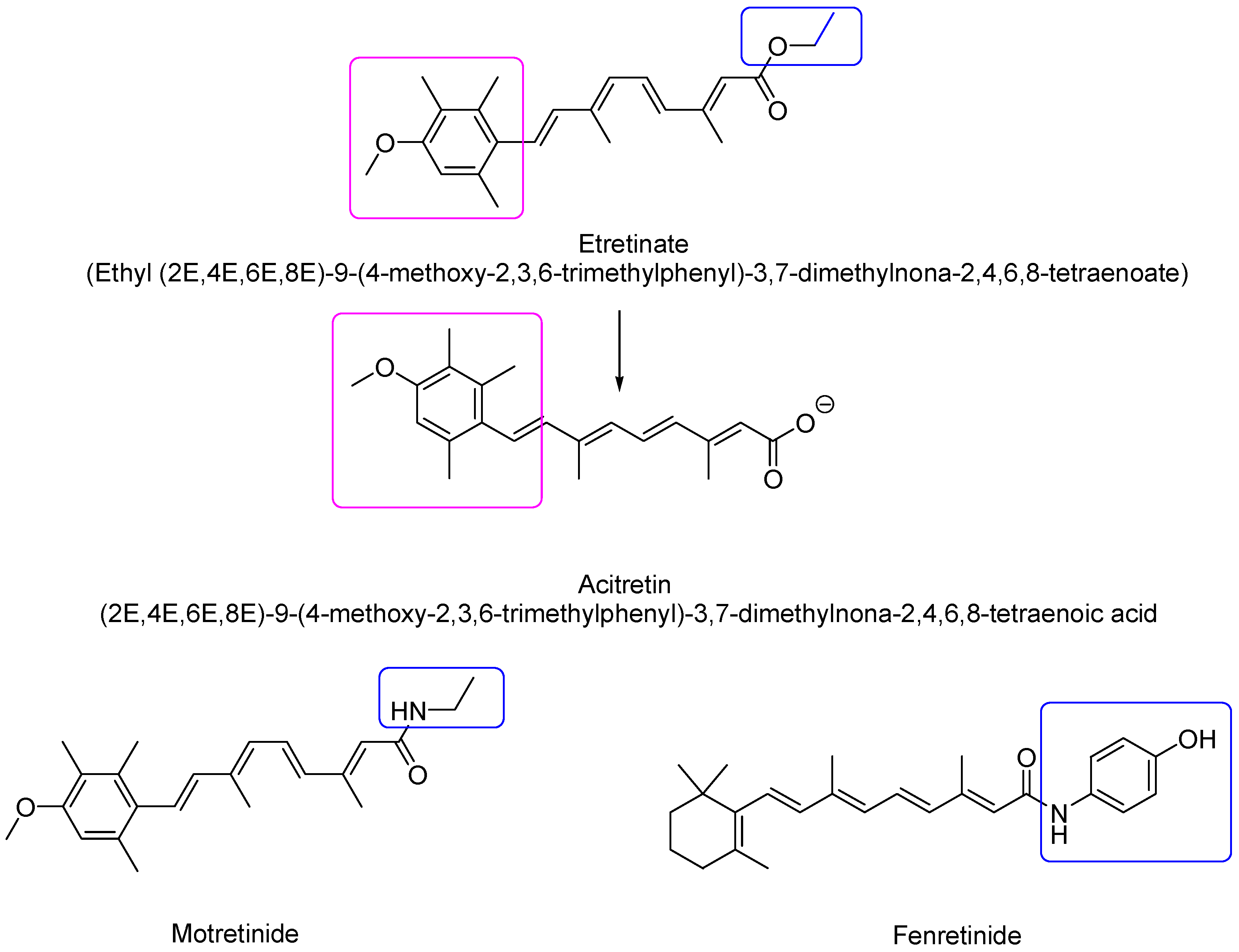
2.1.2. Second-Generation Retinoids
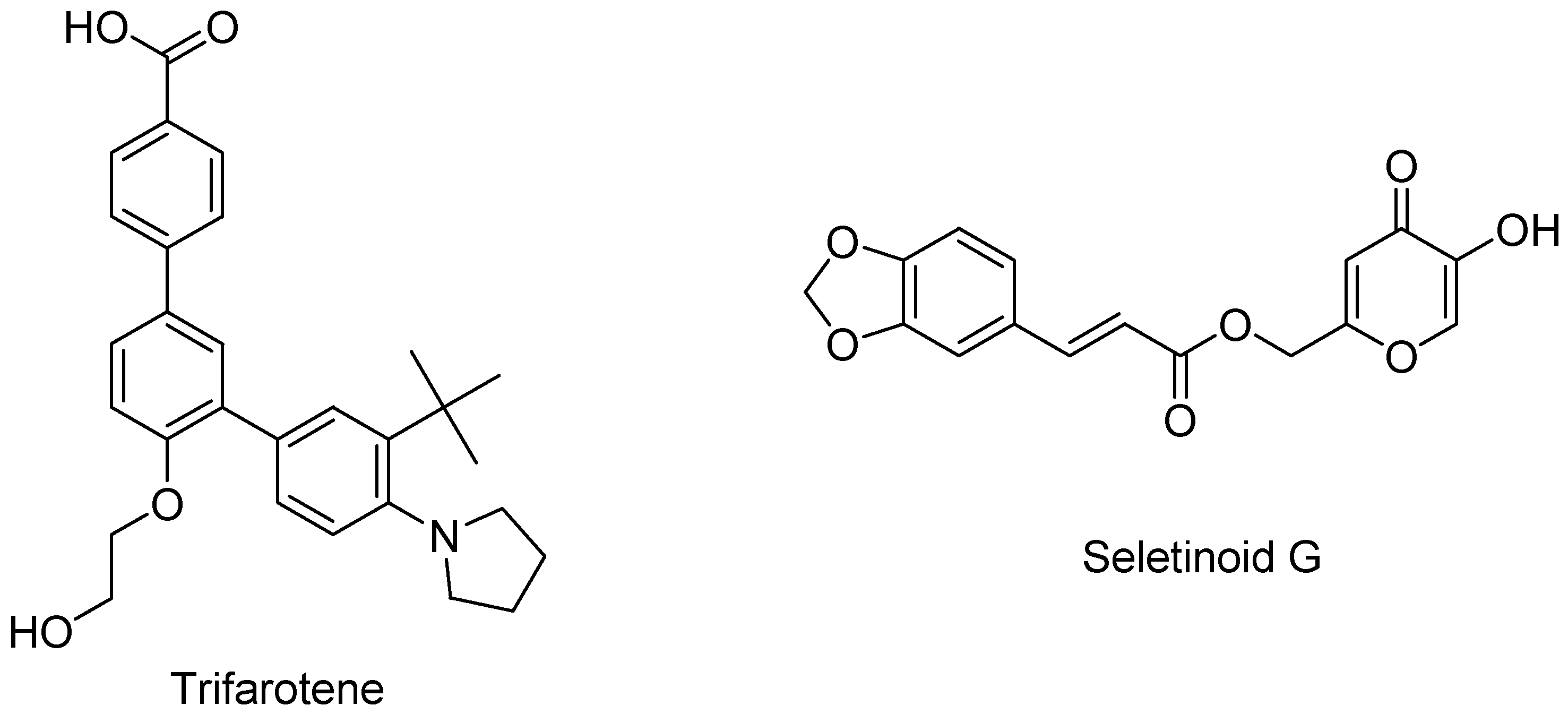
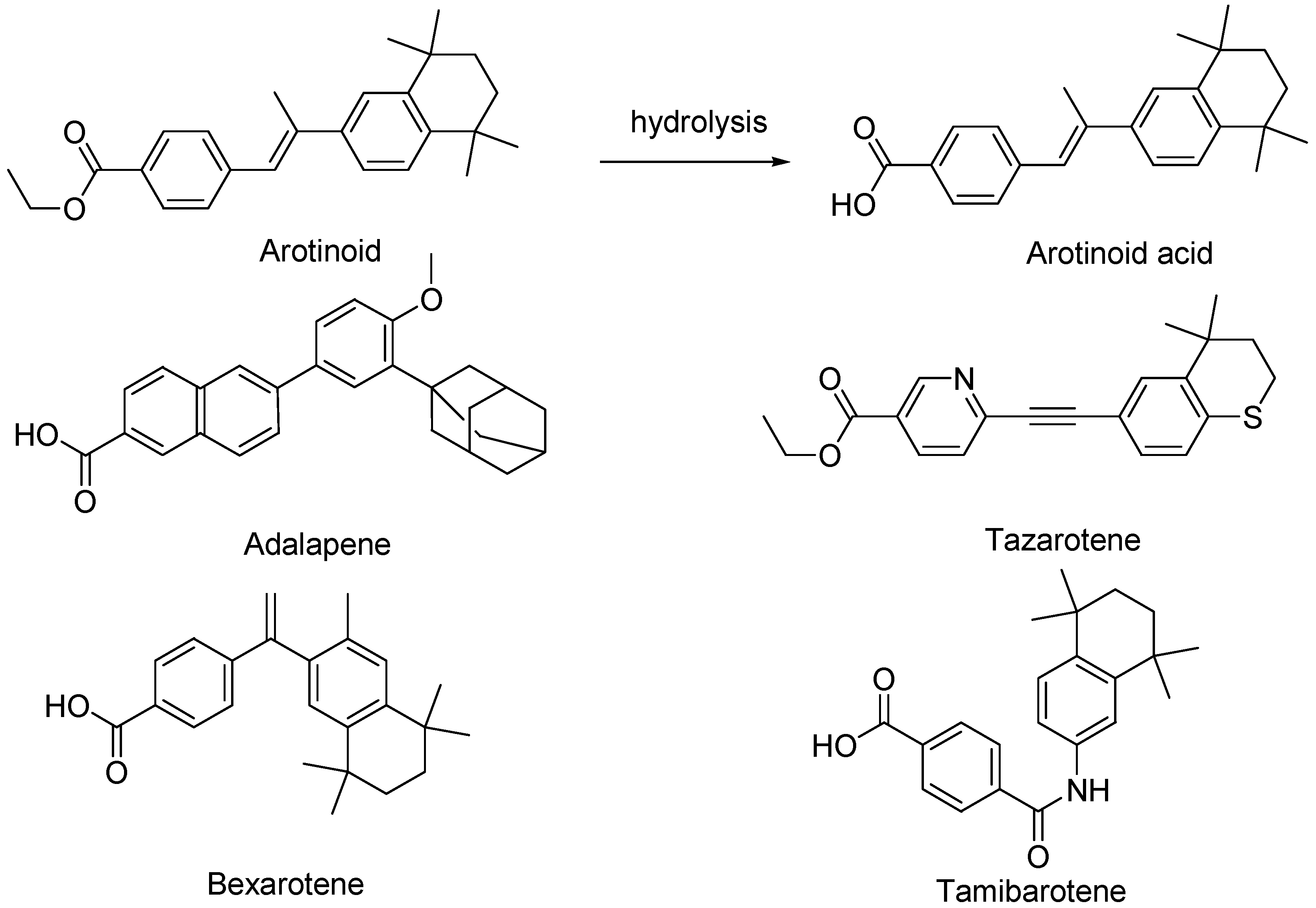
2.1.3. Third-Generation Retinoids
2.1.4. Fourth-Generation Retinoids
3. Fenretinide (4-HPR)
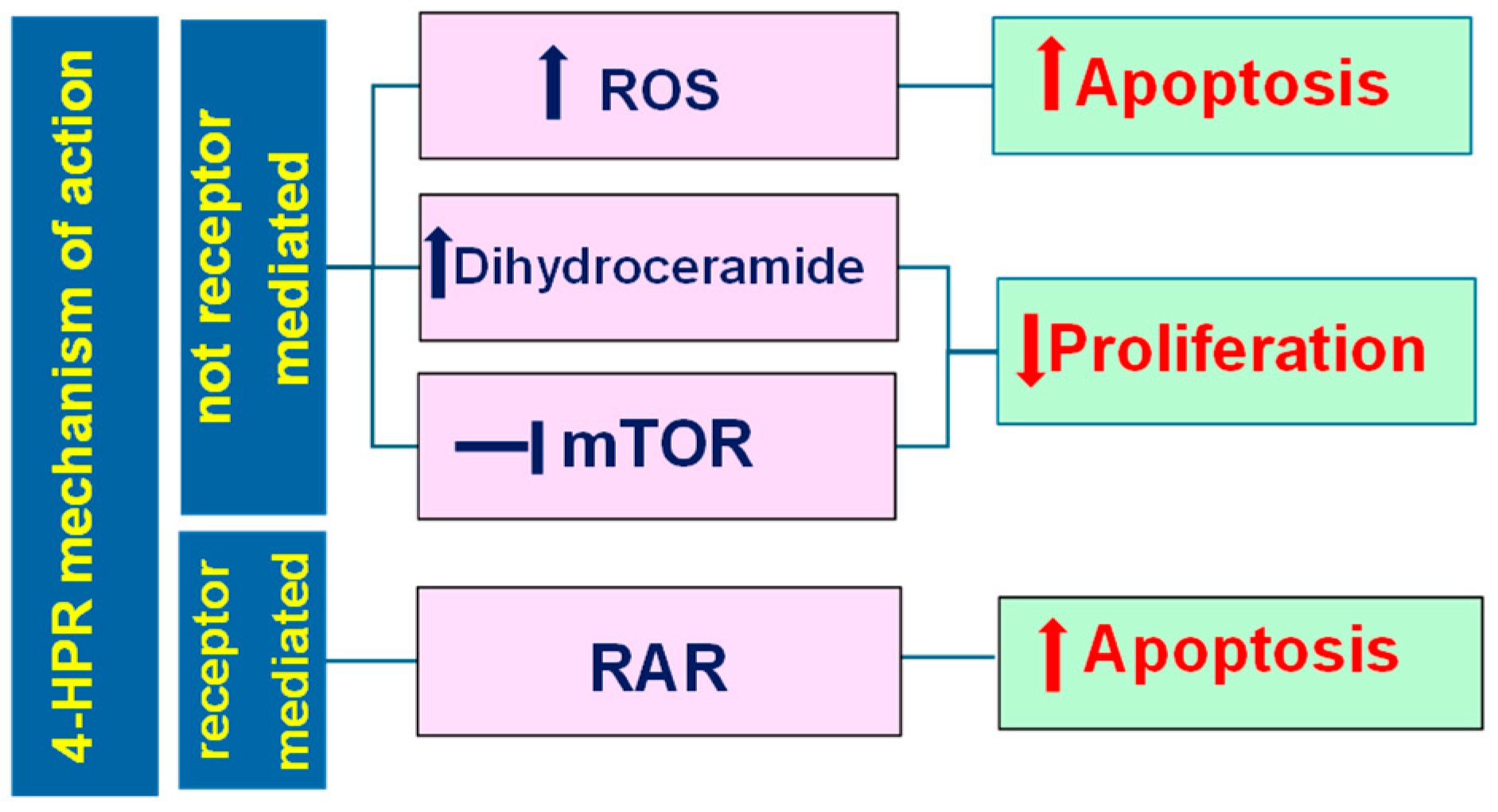
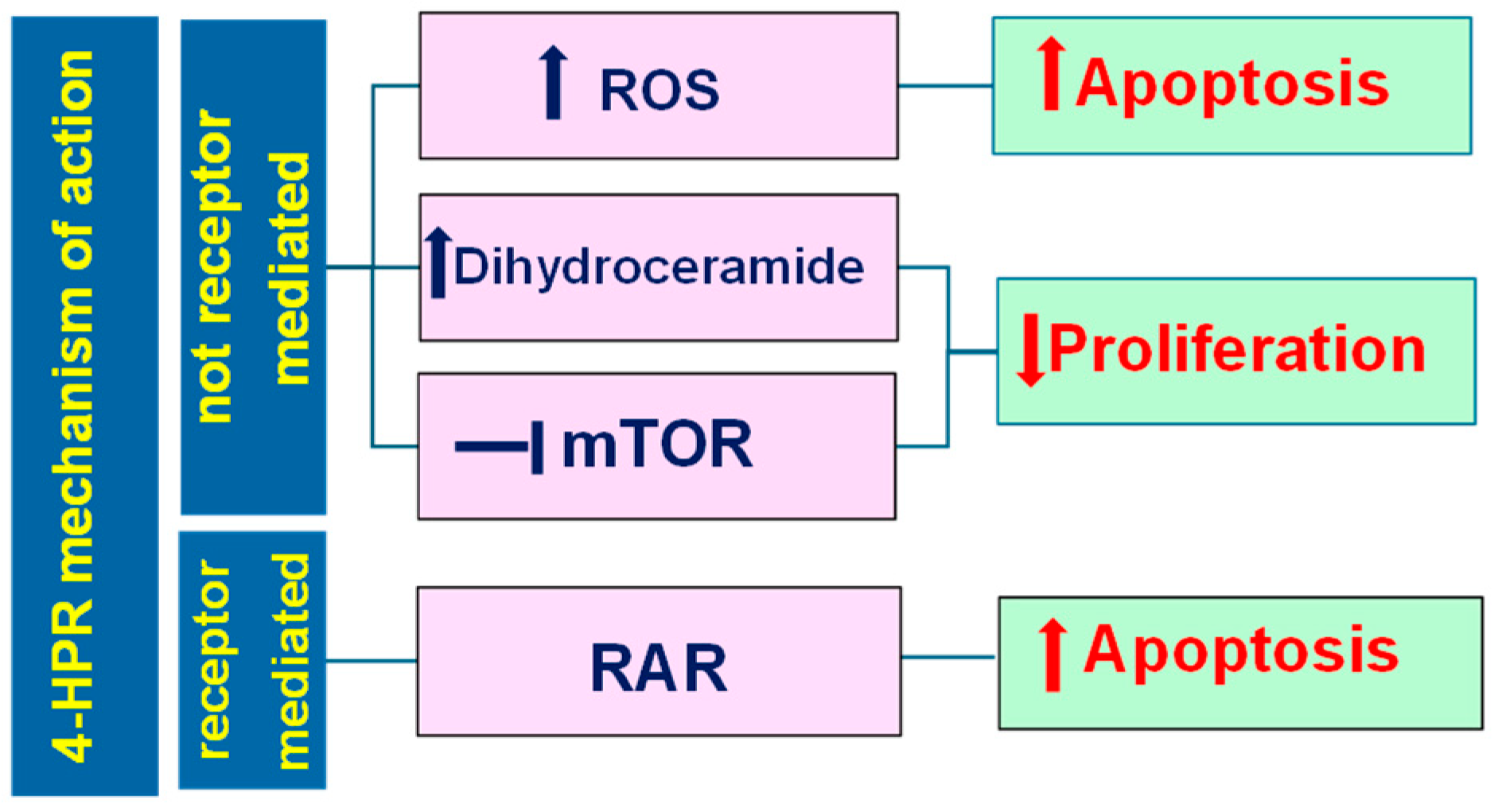
3.1. Pleiotropic Effects Triggered by Fenretinide (4-HPR)
3.2. Results of Capsular 4-HPR Formulation in Early Clinical Trials
3.3. Subsequent Attempts to Raise Drug Bioavailability
3.3.1. Oral Formulations
3.3.2. Parenteral Formulations
3.3.3. Transdermal Formulations
3.3.4. Subcutaneous Formulations
4. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lobo, S. Is There Enough Focus on Lipophilicity in Drug Discovery? Expert Opin. Drug Discov. 2020, 15, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Khalil, S.; Bardawil, T.; Stephan, C.; Darwiche, N.; Abbas, O.; Kibbi, A.G.; Nemer, G.; Kurban, M. Retinoids: A Journey from the Molecular Structures and Mechanisms of Action to Clinical Uses in Dermatology and Adverse Effects. J. Dermatol. Treat. 2017, 28, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.; Reynolds, C.P.; Maurer, B.J. Fenretinide Cytotoxicity for Ewing’s Sarcoma and Primitive Neuroectodermal Tumor Cell Lines Is Decreased by Hypoxia and Synergistically Enhanced by Ceramide Modulators. Cancer Research 2004, 64, 5415–5424. [Google Scholar] [CrossRef] [PubMed]
- Song, M.M.; Makena, M.R.; Hindle, A.; Koneru, B.; Nguyen, T.H.; Verlekar, D.U.; Cho, H.; Maurer, B.J.; Kang, M.H.; Reynolds, C.P. Cytotoxicity and Molecular Activity of Fenretinide and Metabolites in T-Cell Lymphoid Malignancy, Neuroblastoma, and Ovarian Cancer Cell Lines in Physiological Hypoxia. Anticancer Drugs 2019, 30, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zuccari, G.; Russo, E.; Villa, C.; Zorzoli, A.; Marimpietri, D.; Marchitto, L.; Alfei, S. Preparation and Characterization of Amorphous Solid Dispersions for the Solubilization of Fenretinide. Pharmaceuticals 2023, 16, 388. [Google Scholar] [CrossRef] [PubMed]
- Wischke, C.; Zhang, Y.; Mittal, S.; Schwendeman, S.P. Development of PLGA-Based Injectable Delivery Systems For Hydrophobic Fenretinide. Pharm. Res. 2010, 27, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Garaventa, A.; Luksch, R.; Lo Piccolo, M.S.; Cavadini, E.; Montaldo, P.G.; Pizzitola, M.R.; Boni, L.; Ponzoni, M.; Decensi, A.; De Bernardi, B.; et al. Phase I Trial and Pharmacokinetics of Fenretinide in Children with Neuroblastoma. Clin. Cancer Res. 2003, 9, 2032–2039. [Google Scholar] [PubMed]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature of Retinoids. Recommendations 1981. Eur. J. Biochem. 1982, 129, 1–5. [Google Scholar]
- Vivat-Hannah, V.; Zusi, F. Retinoids as Therapeutic Agents: Today and Tomorrow. Mini-Rev. Med. Chem. 2005, 5, 755–760. [Google Scholar] [CrossRef]
- Motamedi, M.; Chehade, A.; Sanghera, R.; Grewal, P. A Clinician’s Guide to Topical Retinoids. J. Cutan. Med. Surg. 2022, 26, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of Retinoid Metabolism and Function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Mcilroy, G.D. The Mechanisms of Fenretinide-Mediated Anti-Cancer Activity and Prevention of Obesity and Type-2 Diabetes. Biochem. Pharmacol. 2014, 91, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Dunlop, N.M.; Newton, D.L.; Henderson, W.R. Relationships between Structure and Activity of Retinoids. Nature 1976, 263, 110–113. [Google Scholar] [CrossRef]
- Willhite, C.C.; Dawson, M.I.; Williams, K.J. Structure-Activity Relationships of Retinoids in Developmental Toxicology. Toxicol. Appl. Pharmacol. 1984, 74, 397–410. [Google Scholar] [CrossRef]
- Sami, N.; de la Feld, S. Topical Retinoids. In Comprehensive Dermatologic Drug Therapy; Elsevier: Amsterdam, The Netherlands, 2021; pp. 528–540.e4. [Google Scholar]
- Kryczyk-Poprawa, A.; Kwiecień, A.; Opoka, W. Photostability of Topical Agents Applied to the Skin: A Review. Pharmaceutics 2019, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Kircik, L.H. Microsphere Technology: Hype or Help? J. Clin. Aesthet. Dermatol. 2011, 4, 27–31. [Google Scholar]
- Gautam, M.; Tahiliani, H.; Nadkarni, N.; Patil, S.; Godse, K. Acitretin in Pediatric Dermatoses. Indian J. Paediatr. Dermatol. 2016, 17, 87. [Google Scholar] [CrossRef]
- Ward, A.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Etretinate A Review of Its Pharmacological Properties and Therapeutic Efficacy in Psoriasis and Other Skin Disorders. Drugs 1983, 26, 9–43. [Google Scholar] [CrossRef]
- Zimmermann, B.; Stürje, H.; Tsambaos, D. Teratogenicity of Arotinoid Ethyl Ester (RO 13-6298) in Mice. Teratog. Carcinog. Mutagen. 1985, 5, 415–431. [Google Scholar] [CrossRef]
- Tanghetti, E.; Werschler, W.; Lain, T.; Guenin, E.; Martin, G.; Pillai, R. Tazarotene 0.045% Lotion for Once-Daily Treatment of Moderate-to-Severe Acne Vulgaris: Results from Two Phase 3 Trials. J. Drugs Dermatol. 2020, 19, 70–77. [Google Scholar] [CrossRef]
- Gniadecki, R.; Assaf, C.; Bagot, M.; Dummer, R.; Duvic, M.; Knobler, R.; Ranki, A.; Schwandt, P.; Whittaker, S. The Optimal Use of Bexarotene in Cutaneous T-Cell Lymphoma. Br. J. Dermatol. 2007, 157, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A. Retinoid X Receptors. Int. J. Biochem. Cell Biol. 1997, 29, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Xia, Z. The Retinoid X Receptors and Their Ligands. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 21–56. [Google Scholar] [CrossRef] [PubMed]
- De Botton, S.; Cluzeau, T.; Vigil, C.; Cook, R.J.; Rousselot, P.; Rizzieri, D.A.; Liesveld, J.L.; Fenaux, P.; Braun, T.; Banos, A.; et al. Targeting RARA Overexpression with Tamibarotene, a Potent and Selective RARα Agonist, Is a Novel Approach in AML. Blood Adv. 2023, 7, 1858–1870. [Google Scholar] [CrossRef]
- Wagner, N.; Benkali, K.; Alió Sáenz, A.; Poncet, M.; Graeber, M. Clinical Pharmacology and Safety of Trifarotene, a First-in-Class RARγ-Selective Topical Retinoid. J. Clin. Pharmacol. 2020, 60, 660–668. [Google Scholar] [CrossRef]
- Kim, M.-S.; Lee, S.; Rho, H.S.; Kim, D.H.; Chang, I.S.; Chung, J.H. The Effects of a Novel Synthetic Retinoid, Seletinoid G, on the Expression of Extracellular Matrix Proteins in Aged Human Skin in Vivo. Clin. Chim. Acta 2005, 362, 161–169. [Google Scholar] [CrossRef]
- Lee, E.-S.; Ahn, Y.; Bae, I.-H.; Min, D.; Park, N.H.; Jung, W.; Kim, S.-H.; Hong, Y.D.; Park, W.S.; Lee, C.S. Synthetic Retinoid Seletinoid G Improves Skin Barrier Function through Wound Healing and Collagen Realignment in Human Skin Equivalents. Int. J. Mol. Sci. 2020, 21, 3198. [Google Scholar] [CrossRef]
- Ramchatesingh, B.; Martínez Villarreal, A.; Arcuri, D.; Lagacé, F.; Setah, S.A.; Touma, F.; Al-Badarin, F.; Litvinov, I.V. The Use of Retinoids for the Prevention and Treatment of Skin Cancers: An Updated Review. Int. J. Mol. Sci. 2022, 23, 12622. [Google Scholar] [CrossRef]
- Gonçalves, A.; Estevinho, B.N.; Rocha, F. Formulation Approaches for Improved Retinoids Delivery in the Treatment of Several Pathologies. Eur. J. Pharm. Biopharm. 2019, 143, 80–90. [Google Scholar] [CrossRef]
- Moon, R.C.; Thompson, H.J.; Becci, P.J.; Grubbs, C.J.; Gander, R.J.; Newton, D.L.; Smith, J.M.; Phillips, S.L.; Henderson, W.R.; Mullen, L.T.; et al. N-(4-Hydroxyphenyl)Retinamide, a New Retinoid for Prevention of Breast Cancer in the Rat. Cancer Res. 1979, 39, 1339–1346. [Google Scholar]
- Mehta, R.G.; Moon, R.C.; Hawthorne, M.; Formelli, F.; Costa, A. Distribution of Fenretinide in the Mammary Gland of Breast Cancer Patients. Eur. J. Cancer Clin. Oncol. 1991, 27, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Preitner, F.; Mody, N.; Graham, T.E.; Peroni, O.D.; Kahn, B.B. Long-Term Fenretinide Treatment Prevents High-Fat Diet-Induced Obesity, Insulin Resistance, and Hepatic Steatosis. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1420–E1429. [Google Scholar] [CrossRef] [PubMed]
- Mcilroy, G.D.; Delibegovic, M.; Owen, C.; Stoney, P.N.; Shearer, K.D.; McCaffery, P.J.; Mody, N. Fenretinide Treatment Prevents Diet-Induced Obesity in Association With Major Alterations in Retinoid Homeostatic Gene Expression in Adipose, Liver, and Hypothalamus. Diabetes 2013, 62, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Kenel, M.F.; Krayer, J.H.; Merz, E.A.; Fred Pritchard, J. Teratogenicity of N-(4-hydroxyphenyl)-all-Trans.-retinamide in Rats and Rabbits. Teratog. Carcinog. Mutagen. 1988, 8, 1–11. [Google Scholar] [CrossRef]
- Howard, W. Pharmacokinetics, Tissue Distribution, and Placental Permeability of All-Trans- and 13-Cis-N-Ethyl Retinamides in Pregnant Hamsters. Fundam. Appl. Toxicol. 1989, 12, 621–627. [Google Scholar] [CrossRef]
- Tang, X.-H.; Gudas, L.J. Retinoids, Retinoic Acid Receptors, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 345–364. [Google Scholar] [CrossRef]
- Potenza, R.L.; Lodeserto, P.; Orienti, I. Fenretinide in Cancer and Neurological Disease: A Two-Face Janus Molecule. Int. J. Mol. Sci. 2022, 23, 7426. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Shao, Z.-M.; Li, X.-S.; Ordonez, J.V.; Conley, B.A.; Wu, S.; Dawson, M.I.; Han, Q.-X.; Chao, W.; Quick, T.; et al. N-(4-Hydroxyphenyl)Retinamide (4-HPR)-Mediated Biological Actions Involve Retinoid Receptor-Independent Pathways in Human Breast Carcinoma. Carcinogenesis 1995, 16, 2477–2486. [Google Scholar] [CrossRef]
- Hałubiec, P.; Łazarczyk, A.; Szafrański, O.; Bohn, T.; Dulińska-Litewka, J. Synthetic Retinoids as Potential Therapeutics in Prostate Cancer—An Update of the Last Decade of Research: A Review. Int. J. Mol. Sci. 2021, 22, 10537. [Google Scholar] [CrossRef]
- Sani, B.P.; Shealy, Y.F.; Hill, D.L. N-(4-Hydroxyphenyl)Retinamide: Interactions with Retinoid-Binding Proteins/Receptors. Carcinogenesis 1995, 16, 2531–2534. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Kolluri, S.K.; Lin, F.; Liu, W.; Han, Y.-H.; Cao, X.; Dawson, M.I.; Reed, J.C.; Zhang, X. Conversion of Bcl-2 from Protector to Killer by Interaction with Nuclear Orphan Receptor Nur77/TR3. Cell 2004, 116, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, L. Characteristics of Nur77 and Its Ligands as Potential Anticancer Compounds (Review). Mol. Med. Rep. 2018, 18, 4793–4801. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bushue, N.; Bu, P.; Yvonne Wan, Y.-J. Induction and Intracellular Localization of Nur77 Dictate Fenretinide-Induced Apoptosis of Human Liver Cancer Cells. Biochem. Pharmacol. 2010, 79, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Lovat, P.E.; Corazzari, M.; Di Sano, F.; Piacentini, M.; Redfern, C.P.F. The Role of Gangliosides in Fenretinide-Induced Apoptosis of Neuroblastoma. Cancer Lett. 2005, 228, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Lachkar, F.; Ferré, P.; Foufelle, F.; Papaioannou, A. Dihydroceramides: Their Emerging Physiological Roles and Functions in Cancer and Metabolic Diseases. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E122–E130. [Google Scholar] [CrossRef] [PubMed]
- Al-Bustany, H.A.; Muhammad, H.A.; Chawsheen, M.A.; Dash, P.R. Fenretinide Induces Apoptosis and Synergises the Apoptosis Inducing Effect of Gemcitabine through Inhibition of Key Signalling Molecules Involved in A549 Cell Survival in in Silico and in Vitro Analyses. Cell. Signal. 2023, 111, 110885. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Hammerling, U. The Mitochondrial PKCδ/Retinol Signal Complex Exerts Real-Time Control on Energy Homeostasis. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158614. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Bajpai, V.K.; Sahasrabuddhe, A.A.; Kumar, A.; Sinha, R.A.; Behari, S.; Godbole, M.M. Inhibition of N-(4-Hydroxyphenyl)Retinamide-Induced Autophagy at a Lower Dose Enhances Cell Death in Malignant Glioma Cells. Carcinogenesis 2007, 29, 600–609. [Google Scholar] [CrossRef]
- Ribatti, D.; Alessandri, G.; Baronio, M.; Raffaghello, L.; Cosimo, E.; Marimpietri, D.; Montaldo, P.G.; De Falco, G.; Caruso, A.; Vacca, A.; et al. Inhibition of Neuroblastoma-Induced Angiogenesis by Fenretinide. Int. J. Cancer 2001, 94, 314–321. [Google Scholar] [CrossRef]
- Breitman, T.; Collins, S.; Keene, B. Terminal Differentiation of Human Promyelocytic Leukemic Cells in Primary Culture in Response to Retinoic Acid. Blood 1981, 57, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of Acute Promyelocytic Leukemia: Updated Recommendations from an Expert Panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Veal, G.J.; Tweddle, D.A.; Visser, J.; Errington, J.; Buck, H.; Marange, J.; Moss, J.; Joseph, S.; Mulla, H. Pharmacokinetics and Safety of a Novel Oral Liquid Formulation of 13-Cis Retinoic Acid in Children with Neuroblastoma: A Randomized Crossover Clinical Trial. Cancers 2021, 13, 1868. [Google Scholar] [CrossRef] [PubMed]
- Kreissman, S.G.; Seeger, R.C.; Matthay, K.K.; London, W.B.; Sposto, R.; Grupp, S.A.; Haas-Kogan, D.A.; LaQuaglia, M.P.; Yu, A.L.; Diller, L.; et al. Purged versus Non-Purged Peripheral Blood Stem-Cell Transplantation for High-Risk Neuroblastoma (COG A3973): A Randomised Phase 3 Trial. Lancet Oncol. 2013, 14, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.J.; Kang, M.H.; Villablanca, J.G.; Janeba, J.; Groshen, S.; Matthay, K.K.; Sondel, P.M.; Maris, J.M.; Jackson, H.A.; Goodarzian, F.; et al. Phase I Trial of Fenretinide Delivered Orally in a Novel Organized Lipid Complex in Patients with Relapsed/Refractory Neuroblastoma: A Report from the New Approaches to Neuroblastoma Therapy (NANT) Consortium. Pediatr. Blood Cancer 2013, 60, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Nitani, C.; Hara, J.; Kawamoto, H.; Taguchi, T.; Kimura, T.; Yoshimura, K.; Hamada, A.; Kitano, S.; Hattori, N.; Ushijima, T.; et al. Phase I Study of Tamibarotene Monotherapy in Pediatric and Young Adult Patients with Recurrent/Refractory Solid Tumors. Cancer Chemother. Pharmacol. 2021, 88, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkins, E.S.; Haas-Kogan, D.; Gerbing, R.B.; London, W.B.; Villablanca, J.G. Long-Term Results for Children with High-Risk Neuroblastoma Treated on a Randomized Trial of Myeloablative Therapy Followed by 13-Cis-Retinoic Acid: A Children’s Oncology Group Study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef]
- Modiano, M.R.; Dalton, W.S.; Lippman, S.M.; Joffe, L.; Booth, A.R.; Meyskens, F.L. Phase II Study of Fenretinide (N-[4-Hydroxyphenyl]Retinamide) in Advanced Breast Cancer and Melanoma. Investig. New Drugs 1990, 8, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U.; Heilbrun, L.K.; Parchment, R.E.; Jain, V.; Zwiebel, J.; Boinpally, R.R.; LoRusso, P.; Hussain, M. Phase II Trial of Fenretinide in Advanced Renal Carcinoma. Investig. New Drugs 2005, 23, 179–185. [Google Scholar] [CrossRef]
- Villablanca, J.G.; Krailo, M.D.; Ames, M.M.; Reid, J.M.; Reaman, G.H.; Reynolds, C.P. Phase I Trial of Oral Fenretinide in Children With High-Risk Solid Tumors: A Report From the Children’s Oncology Group (CCG 09709). J. Clin. Oncol. 2006, 24, 3423–3430. [Google Scholar] [CrossRef]
- Zuccari, G.; Alfei, S.; Marimpietri, D.; Iurilli, V.; Barabino, P.; Marchitto, L. Mini-Tablets: A Valid Strategy to Combine Efficacy and Safety in Pediatrics. Pharmaceuticals 2022, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, J.G.; London, W.B.; Naranjo, A.; McGrady, P.; Ames, M.M.; Reid, J.M.; McGovern, R.M.; Buhrow, S.A.; Jackson, H.; Stranzinger, E.; et al. Phase II Study of Oral Capsular 4-Hydroxyphenylretinamide (4-HPR/Fenretinide) in Pediatric Patients with Refractory or Recurrent Neuroblastoma: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2011, 17, 6858–6866. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, N.; Boddy, A.; Daly, A.; Veal, G. Characterization of the Metabolism of Fenretinide by Human Liver Microsomes, Cytochrome P450 Enzymes and UDP-glucuronosyltransferases. Br. J. Pharmacol. 2011, 162, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, U.; Mariani, L.; Decensi, A.; Formelli, F.; Camerini, T.; Miceli, R.; Di Mauro, M.G.; Costa, A.; Marubini, E.; Sporn, M.B.; et al. Fifteen-Year Results of a Randomized Phase III Trial of Fenretinide to Prevent Second Breast Cancer. Ann. Oncol. 2006, 17, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.M.; Stockler, M.; Lim, R.; Mok, T.; Millward, M.; Boyer, M. Phase II Study of High-Dose Fenretinide for Advanced or Metastatic Hormone-Refractory Prostate Cancer [Abstract]. In Proceedings of the ASCO Genitourinary Cancers Symposium, Orlando, FL, USA, 26–28 February 2009; Volume 189. [Google Scholar]
- Cheung, E.; Pinski, J.; Dorff, T.; Groshen, S.; Quinn, D.I.; Reynolds, C.P.; Maurer, B.J.; Lara, P.N.; Tsao-Wei, D.D.; Twardowski, P.; et al. Oral Fenretinide in Biochemically Recurrent Prostate Cancer: A California Cancer Consortium Phase II Trial. Clin. Genitourin. Cancer 2009, 7, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Puduvalli, V.K.; Yung, W.K.A.; Hess, K.R.; Kuhn, J.G.; Groves, M.D.; Levin, V.A.; Zwiebel, J.; Chang, S.M.; Cloughesy, T.F.; Junck, L.; et al. Phase II Study of Fenretinide (NSC 374551) in Adults With Recurrent Malignant Gliomas: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2004, 22, 4282–4289. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Frgala, T.; Tsao-Wei, D.D.; Groshen, S.; Morgan, R.; McNamara, M.; Scudder, S.; Zwiebel, J.A.; Lenz, H.J.; Garcia, A.A. High Plasma Levels of Fenretinide (4-HPR) Were Associated with Improved Outcome in a Phase II Study of Recurrent Ovarian Cancer: A Study by the California Cancer Consortium. J. Clin. Oncol. 2007, 25, 5555. [Google Scholar] [CrossRef]
- Jasti, B.R.; LoRousso, P.; Parchment, R.E.; Wozniak, A.J.; Flaherty, L.E.; Shields, L.F.; Zalupski, M.; Zweibel, J. Phase I Clinical Trial of Fenretinide (NSC374551) in Advanced Solid Tumors. Proc. Am. Soc. Clin. Oncol. 2001, 20, 485. [Google Scholar]
- Schneider, B.J.; Worden, F.; Gadgeel, S.; Hodges, C.; Parchment, R.; Zwiebel, J.; Kraut, M.; Kalemkerian, G. Phase II Study of Fenretinide in Patients with Small Cell Lung Cancer (SCLC) with Progression after First- or Second-Line Chemotherapy. J. Clin. Oncol. 2004, 22, 7299. [Google Scholar] [CrossRef]
- Cooper, J.P.; Hwang, K.; Singh, H.; Wang, D.; Reynolds, C.P.; Curley Jr, R.W.; Williams, S.C.; Maurer, B.J.; Kang, M.H. Fenretinide Metabolism in Humans and Mice: Utilizing Pharmacological Modulation of Its Metabolic Pathway to Increase Systemic Exposure. Br. J. Pharmacol. 2011, 163, 1263–1275. [Google Scholar] [CrossRef]
- Mohrbacher, A.M.; Yang, A.S.; Groshen, S.; Kummar, S.; Gutierrez, M.E.; Kang, M.H.; Tsao-Wei, D.; Reynolds, C.P.; Newman, E.M.; Maurer, B.J. Phase I Study of Fenretinide Delivered Intravenously in Patients with Relapsed or Refractory Hematologic Malignancies: A California Cancer Consortium Trial. Clin. Cancer Res. 2017, 23, 4550–4555. [Google Scholar] [CrossRef]
- Bensa, V.; Calarco, E.; Giusto, E.; Perri, P.; Corrias, M.V.; Ponzoni, M.; Brignole, C.; Pastorino, F. Retinoids Delivery Systems in Cancer: Liposomal Fenretinide for Neuroectodermal-Derived Tumors. Pharmaceuticals 2021, 14, 854. [Google Scholar] [CrossRef]
- Apraiz, A.; Boyano, M.D.; Asumendi, A. Cell-Centric View of Apoptosis and Apoptotic Cell Death-Inducing Antitumoral Strategies. Cancers 2011, 3, 1042–1080. [Google Scholar] [CrossRef]
- Maurer, B.J.; Kalous, O.; Yesair, D.W.; Wu, X.; Janeba, J.; Maldonado, V.; Khankaldyyan, V.; Frgala, T.; Sun, B.-C.; McKee, R.T.; et al. Improved Oral Delivery of N-(4-Hydroxyphenyl)Retinamide with a Novel LYM-X-SORB Organized Lipid Complex. Clin. Cancer Res. 2007, 13, 3079–3086. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.E.; Maurer, B.J.; Reynolds, C.P.; Kang, M.; Singh, H.; Crandon, S.; Murgo, A.J.; Doroshow, J.H. Phase I Trial of Fenretinide Lym-x-Sorb Oral Powder in Adults with Solid Tumors and Lymphomas. Anticancer Res. 2011, 31, 961–966. [Google Scholar] [PubMed]
- Orienti, I.; Meco, D.; Di Francesco, A.M.; Cusano, G.; Popoli, P.; Potenza, R.L.; Armida, M.; Falconi, M.; Teti, G.; Gotti, R.; et al. Nanoencapsulation of Fenretinide in Glucosamine Butyrate—Gelatin Matrices as a Mean to Improve Its Oral Bioavailability. J. Nanomed. Nanotechnol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Betancourt, A.O.; Lemieux, M.; Thibert, R. Solid Oral Formulation of Fenretinide. U.S. Patent No. 10,512,619, 24 December 2016. [Google Scholar]
- Orienti, I.; Francescangeli, F.; De Angelis, M.L.; Fecchi, K.; Bongiorno-Borbone, L.; Signore, M.; Peschiaroli, A.; Boe, A.; Bruselles, A.; Costantino, A.; et al. A New Bioavailable Fenretinide Formulation with Antiproliferative, Antimetabolic, and Cytotoxic Effects on Solid Tumors. Cell Death Dis. 2019, 10, 529. [Google Scholar] [CrossRef] [PubMed]
- Orienti, I.; Salvati, V.; Sette, G.; Zucchetti, M.; Bongiorno-Borbone, L.; Peschiaroli, A.; Zolla, L.; Francescangeli, F.; Ferrari, M.; Matteo, C.; et al. A Novel Oral Micellar Fenretinide Formulation with Enhanced Bioavailability and Antitumour Activity against Multiple Tumours from Cancer Stem Cells. J. Exp. Clin. Cancer Res. 2019, 38, 373. [Google Scholar] [CrossRef] [PubMed]
- Orienti, I.; Zuccari, G.; Bergamante, V.; Carosio, R.; Gotti, R.; Cilli, M.; Montaldo. Fenretinide-Polyvinylalcohol Conjugates: New Systems Allowing Fenretinide Intravenous Administration. Biomacromolecules 2007, 8, 3258–3262. [Google Scholar] [CrossRef]
- Okuda, T.; Kawakami, S.; Higuchi, Y.; Satoh, T.; Oka, Y.; Yokoyama, M.; Yamashita, F.; Hashida, M. Enhanced In Vivo Antitumor Efficacy of Fenretinide Encapsulated in Polymeric Micelles. Int. J. Pharm. 2009, 373, 100–106. [Google Scholar] [CrossRef]
- Di Paolo, D.; Pastorino, F.; Zuccari, G.; Caffa, I.; Loi, M.; Marimpietri, D.; Brignole, C.; Perri, P.; Cilli, M.; Nico, B.; et al. Enhanced Anti-Tumor and Anti-Angiogenic Efficacy of a Novel Liposomal Fenretinide on Human Neuroblastoma. J. Control. Release 2013, 170, 445–451. [Google Scholar] [CrossRef]
- Pignatta, S.; Orienti, I.; Falconi, M.; Teti, G.; Arienti, C.; Medri, L.; Zanoni, M.; Carloni, S.; Zoli, W.; Amadori, D.; et al. Albumin Nanocapsules Containing Fenretinide: Pre-Clinical Evaluation of Cytotoxic Activity in Experimental Models of Human Non-Small Cell Lung Cancer. Nanomedicine 2015, 11, 263–273. [Google Scholar] [CrossRef]
- Durante, S.; Orienti, I.; Teti, G.; Salvatore, V.; Focaroli, S.; Tesei, A.; Pignatta, S.; Falconi, M. Anti-Tumor Activity of Fenretinide Complexed with Human Serum Albumin in Lung Cancer Xenograft Mouse Model. Oncotarget 2014, 5, 4811–4820. [Google Scholar] [CrossRef]
- Zhang, Y.; Wischke, C.; Mittal, S.; Mitra, A.; Schwendeman, S.P. Design of Controlled Release PLGA Microspheres for Hydrophobic Fenretinide. Mol. Pharm. 2016, 13, 2622–2630. [Google Scholar] [CrossRef]
- Thomas, J.S.; El-Khoueiry, A.B.; Maurer, B.J.; Groshen, S.; Pinski, J.K.; Cobos, E.; Gandara, D.R.; Lenz, H.J.; Kang, M.H.; Reynolds, C.P.; et al. A Phase I Study of Intravenous Fenretinide (4-HPR) for Patients with Malignant Solid Tumors. Cancer Chemother. Pharmacol. 2021, 87, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Orienti, I.; Cripe, T.P.; Currier, M.A.; Cavallari, C.; Teti, G.; Falconi, M. A Cationic Nanomicellar Complex of the Quaternary Amphiphilic Amine RC16+ with Fenretinide as a New Multitasking System for Antitumor Therapy. Curr. Drug Deliv. 2019, 16, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Desai, K.-G.H.; Mallery, S.R.; Holpuch, A.S.; Phelps, M.P.; Schwendeman, S.P. Mucoadhesive Fenretinide Patches for Site-Specific Chemoprevention of Oral Cancer: Enhancement of Oral Mucosal Permeation of Fenretinide by Coincorporation of Propylene Glycol and Menthol. Mol. Pharm. 2012, 9, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Nieto, K.; Mallery, S.R.; Schwendeman, S.P. Microencapsulation of Amorphous Solid Dispersions of Fenretinide Enhances Drug Solubility and Release from PLGA in Vitro and in Vivo. Int. J. Pharm. 2020, 586, 119475. [Google Scholar] [CrossRef]
- Salata, G.C.; Malagó, I.D.; Carvalho Dartora, V.F.M.; Marçal Pessoa, A.F.; Fantini, M.C.d.A.; Costa, S.K.P.; Machado-Neto, J.A.; Lopes, L.B. Microemulsion for Prolonged Release of Fenretinide in the Mammary Tissue and Prevention of Breast Cancer Development. Mol. Pharm. 2021, 18, 3401–3417. [Google Scholar] [CrossRef]
- Kali, G.; Haddadzadegan, S.; Bernkop-Schnürch, A. Cyclodextrins and Derivatives in Drug Delivery: New Developments, Relevant Clinical Trials, and Advanced Products. Carbohydr. Polym. 2024, 324, 121500. [Google Scholar] [CrossRef]
- Muccio, D.D.; Atigadda, V.R.; Brouillette, W.J.; Bland, K.I.; Krontiras, H.; Grubbs, C.J. Translation of a Tissue-Selective Rexinoid, UAB30, to the Clinic for Breast Cancer Prevention. Curr. Top. Med. Chem. 2017, 17, 676–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, X.; Zhou, J.; Su, C. Extracellular Vesicles for Drug Delivery in Cancer Treatment. Biol. Proced. Online 2023, 25, 28. [Google Scholar] [CrossRef] [PubMed]

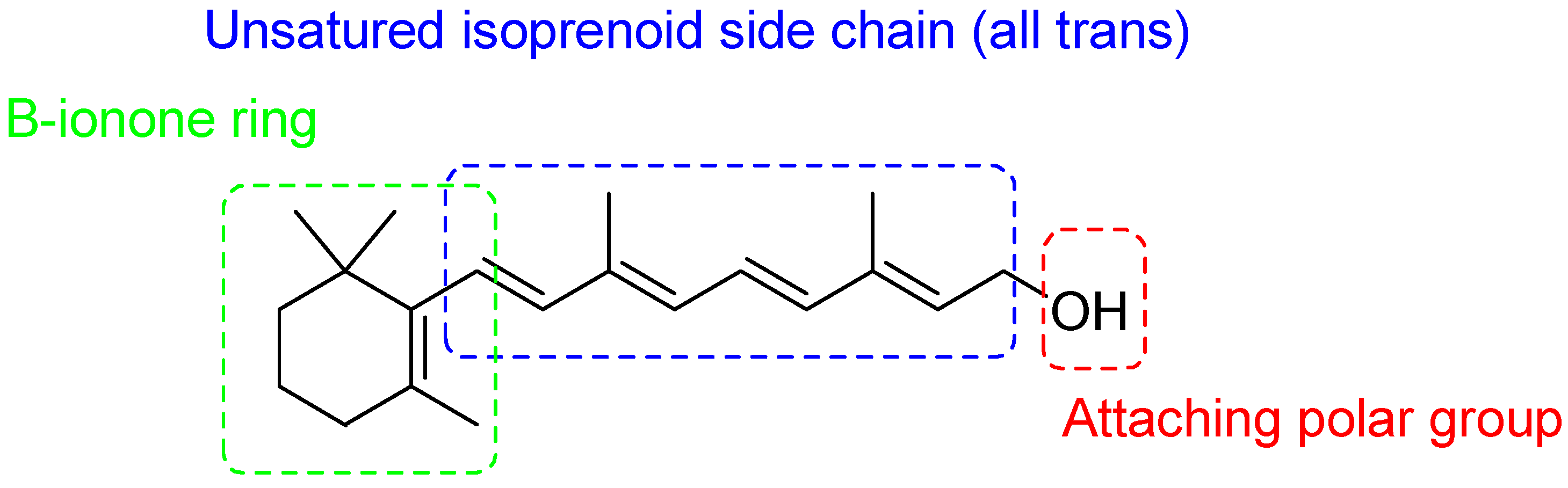


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generation | Description | Compounds |
|---|---|---|
| First | Naturally occurring compounds and their isomers | Retinol, retinal, tretinoin *, isotretinoin, Alitretinoin |
| Second | Mono-aromatic synthetic analogs formulated exclusively for oral dosing | Etretinate, acitretin **, motretinide, Fenretinide |
| Third | Retinoidal polyaromatic compounds | Arotinoids, adapalene, bexarotene, Tazarotene, tamibarotene |
| Fourth | Pyranone derivatives for topical use with selectivity towards the RARs located in the epidermis | Trifarotene, seletinoid G |
| Compound | Agency | Year | Indication | Actual Use in Cancer | Ref. |
|---|---|---|---|---|---|
| ATRA | FDA | 1995 | APL | As approval | [53] |
| EMA | |||||
| 13-cis retinoic acid | FDA | 1982 | Severe acne | Neuroblastoma | [54] |
| EMA | 2003 | ||||
| Bexarotene | FDA | 1999 | Refractory cases of cutaneous T-cell lymphoma | As approval | [55] |
| EMA | 2001 | ||||
| 4-HPR | In clinical trials | Neuroblastoma | [56] | ||
| Tamibarotene | Neuroblastoma | [57] | |||
| Indication | Sponsor | Phase | Formulation | Daily Dose | Enrolment | PL a (μmol/L) | Response | Ref. |
|---|---|---|---|---|---|---|---|---|
| Prostate cancer | CTRG | II | Capsule | 1800 mg | 27 | b | ≥50% reduction in PSA in 1/27 | [66] |
| Prostate cancer | CCC | II | Capsule | 1800 mg | 23 | b | No objective response | [67] |
| Brain tumors | NABTC | II | Capsule | 1600–1800 mg | 45 | 2 ± 0.9 | No activity | [68] |
| Ovarian cancer | CCC | II | Capsule | 1800 mg | 31 | 12.5 | OS at 18 mo 66% in >9 µmol/L | [69] |
| Solid tumors | BAKCI | I | Capsule | 500–4800 mg/m2 | 31 | Unknown | No activity | [70] |
| Renal cell carcinoma | BAKCI | II | Capsule | 1800 mg/m2 | 19 | b | No activity | [60] |
| Small cell lung cancer | UMCC | II | Capsule | 1800 mg/m2 | 19 | 7 ± 4 | No objective responses | [71] |
| Pediatric neuroblastoma | IG & INT | I | Capsule | 100–4000 mg/m2 | 54 | 13 ± 6 c | No CR/PR, 41 SD | [8] |
| Pediatric solid tumors | COG | I | Capsule | 350–3300 mg/m2 | 54 | 10 ± 3 | 1 CR, 13 SD | [61] |
| Pediatric neuroblastoma | COG | II | Capsule | 2475/1800 mg/m2 fixed | 58 | 8 ± 3 | 1 PR, 7 SD | [63] |
| Pediatric neuroblastoma | NANT | I | 4-HPR/LXS | 352–2210 mg/m2 | 32 | 16 ± 3 | 4 CR, 6 SD | [56] |
| Pediatric neuroblastoma | NANT | I | 4-HPR/LXS + ketoconazole | 1500 mg/m2 fixed | N.R. | 18 ± 4 | On-going | [72] |
| Hematologic malignancies | NCI | I | 4-HPR-ILE | 80–1810 mg/m2 | 19 | > 50 µmol/L | 36% CR + PR | [73] |
| Formulation | Ingredients | Study Type | Treatment Plan | Results | Other | Ref. |
|---|---|---|---|---|---|---|
| Soft gel capsules | Corn oil TWEEN 80 | Phase II NB | 2475 mg/m2/d or 1800 mg/m2/d 7 days every 21 days Max 30 cycles | Cmax = 6–13 µM | ⬆ Tolerability ⬇ Compliance Interpatient variability | [63] |
| 4-HPR LYM-X-SORB Lipid matrix | LPCH MG FFA | Preclinical | PK study 120–360 mg/kg/d twice a day 9 doses | Cmax > 3-fold vs capsular 4-HPR in mice | ⬇ Toxicity Interpatient variability ⬇ Tolerability in adults GIT irritation | [56,76,77] |
| Phase I NB | 352 mg to 2210 mg 4-HPR/m2/day BID, 7 days, every 3 weeks | Cmax = 21 µM | ||||
| Phase I solid tumors | 1000 mg/m2/day TID, 7 days, every 3 weeks | MTD = 800 mg/m2/day | ||||
| 4-HPR Nanocapsules | GB GEL | Preclinical NB SC model | Efficacy study: 200 mg/kg, 3 days /week for 3 weeks Adsorption study: 100 mg/kg, 5 days | Cmax = 6 µM ITC = 12 µM ⬇ Tumor growth in fasting conditions | Mean size = 213 mm EE% = 87% DL% = 12% | [78] |
| PVP 4-HPR Spray-dried Amorphous solid dispersion | PVP | Preclinical | PK study SD (20 mg/kg) | AUC0–48 = 5911 ng × h/mL Cmax = 505 ng/mL | ⬆ Bioavailability Storage stability at 5 °C and 60% RH for up to 6 months | [79] |
| NanoFEN Inclusion complex | 2-HP-βCD | Preclinical | PK study SD (5 mg/kg) | Cmax = 730 ng/mL (2-fold ⬆ vs. capsular 4-HPR) AUC0–last = 9378 h × mg/mL (3-fold ⬆ vs. capsular 4-HPR) | ⬆ Solubility by 1409-fold ⬆ Anticancer activity in vitro | [80] |
| Bio-nFeR Micelles | PCH GTB | Preclinical Lung Melanoma Colon SC model | PK study SD =1,050,100 or 200 mg/kg CA = 150 mg/kg, 5 days on and 2 days off for 3 weeks | SD (200 mg/kg): Cmax = 9.2 µM AUC0–last = 112,957 ng × h/mL CA: Cmax = 12 µM AUCinf = 85,378 ng × h/mL ITC = 5 μM | Mean size = 300 nm EE% = 92% DL% = 9% 300 mg/mL Bio-nFeR provide 25 mg/mL 4-HPR in solution | [81] |
| Formulation | Ingredients | Study Type | Treatment Plan | Results | Other | Ref. |
|---|---|---|---|---|---|---|
| 4-HPR-PVA Polymeric micelles | PVA | Preclinical Metastatic NB | Efficacy study 13.5 mg/mL, 5 times, every 3 days (i.v.) | ⬆ Solubility by 200-fold ⬆ MST | Mean size = 350 nm Constant in vitro release | [82] |
| 4-HPR Bz-PEG-PAS Polymeric micelles | PEG-cPA-Bz | Preclinical Melanoma SC model | PK study SD (75 mg/kg, i.v.) Efficacy study 75 mg/kg, 3 times every 2 days (i.v.) | ⬆ Residence time ITC = 56.6 μg/g after 6 h | EE% = 70% Mean size = 173 nm | [83] |
| Liposomal 4-HPR | HSPC, CHE DSPE-PEG2000 NGR * | Preclinical Orthotopic NB | Efficacy study 1 mg/kg twice a week, 6 weeks (i.v.) | Vascular targeting ⬆ Efficacy | Mean size = 142 nm EE% = 69% ⬇ Drug leakage | [84] |
| 4-HPR HSA Nanocapsules | HSA | Preclinical Lung adenocarcinoma SC model | Efficacy study 1 mg/kg, 12 times every 3 days (i.v.) | ⬇ 63% tumor volume vs. CTR ITC = 5.7 µM ⬆ Bioavailability | Size = 80–100 nm DL% = 14% | [85,86] |
| 4-HPR PLGA Microspheres | PLGA Brij 98 MgCO3 | Preclinical | PK study SD (4.2 mg, i.m.) | Cmax = 0.24 µM on 10th day 101% Bioavailability Tmax = 240 h | Size = 5–10 µm Sustained release | [87] |
| 20% 4-HPR-soy O/W | Soy oil Egg phospholipids | Phase I Hematologic malignancies | 80–1810 mg/m2/d, on days 1 to 5, 21-day cycles (c.i.v.) | MTD = 1280 mg/m2/d for 5 days ⬆ Cmax 5-7-fold vs capsular 4-HPR | Dose-limiting toxicity ⬆ Time drug infusion ⬇ Potency For T-cell lymphomas | [73] |
| Phase I Solid tumors | 1280 mg/m2/d for 5 consecutive days in a 21-day cycle (c.i.v.) | Dose reduction needed Cmax = 9.9–10.8 µg/mL | ⬆ Adverse events ⬇ Activity | [88] | ||
| NanoFEN Inclusion complex | 2-HP-βCD | Preclinical Lung Colorectal SC model | PK study Single dose (5 mg/kg, i.v.) Efficacy study 10 mg/kg three times/week (i.p.) | Cmax = 6932 ng/mL AUCinf = 13,657 ng × h/mL Relapse prevention | ⬆ Efficacy in vitro ⬇ Tumor mass | [80] |
| Fen-RC16+ Micelles | C16-ceramide+ | Preclinical NB SC model | Efficacy study 1.02 mg/kg 8 times every other day (i.v.) | ⬇ Tumor mass Relapses prevention | Size = 20–40 nm Solubility = 1.5 mg/mL ⬆ Intracellular uptake | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfei, S.; Zuccari, G. Attempts to Improve Lipophilic Drugs’ Solubility and Bioavailability: A Focus on Fenretinide. Pharmaceutics 2024, 16, 579. https://doi.org/10.3390/pharmaceutics16050579
Alfei S, Zuccari G. Attempts to Improve Lipophilic Drugs’ Solubility and Bioavailability: A Focus on Fenretinide. Pharmaceutics. 2024; 16(5):579. https://doi.org/10.3390/pharmaceutics16050579
Chicago/Turabian StyleAlfei, Silvana, and Guendalina Zuccari. 2024. "Attempts to Improve Lipophilic Drugs’ Solubility and Bioavailability: A Focus on Fenretinide" Pharmaceutics 16, no. 5: 579. https://doi.org/10.3390/pharmaceutics16050579
APA StyleAlfei, S., & Zuccari, G. (2024). Attempts to Improve Lipophilic Drugs’ Solubility and Bioavailability: A Focus on Fenretinide. Pharmaceutics, 16(5), 579. https://doi.org/10.3390/pharmaceutics16050579