Abstract
According to the latest World Health Organization (WHO) report, an estimated 10.6 million people were diagnosed with tuberculosis (TB) in 2022, and 1.30 million died. A major concern is the emergence of multi-drug-resistant (MDR) and extensively drug-resistant (XDR) strains, fueled by the length of anti-TB treatment and HIV comorbidity. Innovative anti-TB agents acting with new modes of action are the only solution to counteract the spread of resistant infections. To escape starvation and survive inside macrophages, Mtb has evolved to become independent of the host by synthesizing its own amino acids. Therefore, targeting amino acid biosynthesis could subvert the ability of the mycobacterium to evade the host immune system, providing innovative avenues for drug discovery. The aim of this review is to give an overview of the most recent progress in the discovery of amino acid biosynthesis inhibitors. Among the hits discovered over the past five years, tryptophan (Trp) inhibitors stand out as the most advanced and have significantly contributed to demonstrating the feasibility of this approach for future TB drug discovery. Future efforts should be directed at prioritizing the chemical optimization of these hits to enrich the TB drug pipeline with high-quality leads.
1. Introduction
With an estimated 10.6 million people that fell ill and 1.30 million deaths in 2022, tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains the world’s second leading cause of death from a single infectious agent after COVID-19 [1]. Despite significant progress being made over the past ten years, global targets remain off track, and urgent action must be taken to reach the goal adopted by the United Nations (UN) and the World Health Organization (WHO) to end the TB epidemic by 2030. A major concern is the emergence of multi-drug-resistant (MDR) and extensively drug-resistant (XDR) strains; according to the most recent WHO report, in 2022, an estimated 410,000 people developed MDR or rifampicin-resistant (RR) TB. As with other bacterial infections, resistance to standard anti-TB drugs has high economic and social impacts and poses a serious threat to global health. In fact, resistance to commonly used antibiotics, often referred to as a “hidden pandemic”, is a leading cause of death, resulting in increased morbidity, mortality, and healthcare expenses. With the increasing risk of returning to a pre-antibiotic era, the need for novel and effective therapeutic strategies has become more pressing [2]. Over the past decade, intensified research efforts have fueled the TB drug pipeline, offering a positive outlook for the future of TB drug discovery [3]. Despite this progress, drugs that act on novel targets are still unrepresented [4]. Innovative anti-TB agents acting with new modes of action are the only solution to counteract the spread of resistant infections. Encouragingly, a deeper understanding of TB biology has resulted in significant progress in identifying new targets for TB drug discovery. While cell wall biosynthesis has been the major focus for years, metabolic pathways have not received as much attention due to concerns about Mtb’s ability to extract amino acids from the host and potentially reverse their functions [4]. In recent years, several studies have highlighted the central role of amino acids for Mtb survival [5]. Experiments with auxotrophic strains have been useful models to understand metabolic fluxes and the impact of inhibitors, providing growing evidence that auxotrophy for some amino acids makes Mtb less virulent and unable to proliferate in the lungs [6,7,8,9,10,11,12,13]. Indeed, amino acid starvation is a mechanism of innate immunity to limit nutrient availability and eliminate pathogenic microorganisms [6]. To escape starvation and survive inside macrophages, Mtb has evolved to become independent from the host by synthesizing its own amino acids. Such an autarkic metabolic lifestyle is an evolutionary advantage and a virulence mechanism, holding great promise for target discovery. Therefore, targeting amino acid biosynthesis could subvert the ability of mycobacterium to evade the host immune system, leading to rapid killing in vitro and in vivo. An argument further reinforcing the interest in amino acid biosynthesis is the absence of human orthologues, suggesting that they could serve as excellent anti-TB targets.
The aim of this review is to give an overview of the most recent progress in the discovery of amino acid biosynthesis inhibitors. We only report updated information on inhibitors with documented in vitro activity with a focus on those published between 2018 and 2023. Minimal inhibitory concentrations (MICs), 50% inhibitory concentrations (IC50), and 50% cytotoxic concentrations (CC50) were converted to µM to allow for an easier comparison of compounds.
2. Inhibitors of Amino Acid Biosynthesis
2.1. Inhibitors of Aromatic Amino Acid Biosynthesis
Aromatic amino acids are obtained from carbohydrate precursors through seven enzymatic steps, ultimately resulting in chorismate. This is a metabolic node for phenylalanine (Phe), tyrosine (Tyr), and tryptophan (Trp), as well as for important precursors of p-aminobenzoic acid (PABA), p-hydroxybenzoate, and isochorismate, which lead to folates, ubiquinone, and mycobactins, respectively [14] (Figure 1). This pathway, also known as the shikimate pathway, is essential for Mtb survival [15]. It is only conserved among plants and bacteria and is absent in humans, thus garnering interest for its potential in the development of herbicides and antimicrobials.
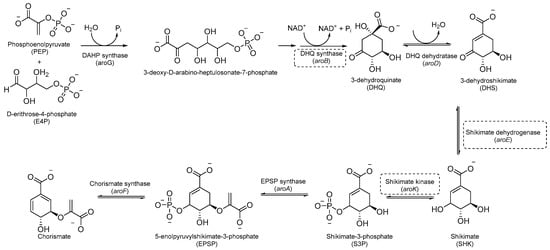
Figure 1.
A schematic representation of the shikimate pathway. The most relevant targets are depicted in dotted boxes.
The first component of the pathway is 3-deoxy-D-arabino-heptulosonate-7-phosphate (DAHP) synthase. This enzyme catalyzes an aldol condensation between D-erithrose-4-phosphate and phospho-enol-pyruvate (PEP) to produce DAHP and inorganic phosphate, and its vulnerability as a drug target has recently been demonstrated [16]. DAHP is then converted into 3-dehydroquinate (DHQ) by dehydroquinate synthase (DHQS). DHQ dehydratase catalyzes DHQ dehydration into 3-dehydroshikimate, which, in turn, is reduced by shikimate dehydrogenase (SD) into the central metabolite shikimate (Figure 1).
The availability of structural information about the first five enzymes of this pathway has raised the possibility of rational structure-based drug design targeting these enzymes. Even though virtual screenings have generated a growing number of potential inhibitors [17,18,19], there are only a few recent reports about their effective inhibition against Mtb [20]. The only two compounds with confirmed activity against Mtb are IMB-T130 and IMB-SD62 [21] (Figure 2), inhibiting DHQS and SD, respectively. IMB-T130 (Figure 2) is the identified hit of a phenotypic screening [22] with very good antimycobacterial activity (0.26 µM), low cytotoxicity, and of the ability to inhibit intracellular growth in a dose-dependent manner. Initially, it was speculated that it exerted anti-Mtb activity via multitarget activity. Further in vitro studies proved that this compound strongly inhibits DHQS (IC50 = 2.86 μM) and has a higher MIC against DHQS-overexpressing strains, suggesting that DHQS could be the target and contribute to its antimycobacterial efficacy. On the other hand, IMB-SD62 (Figure 2) resulted from the lead optimization of a series of 3,6-disubstituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles identified through a target-based screening against SD [22]. This compound improved the MIC (4.8 µM) and cytotoxicity (CC50 = 64.5 µM in Vero cells) compared to its parent compound but showed moderate efficacy in an in vivo model of acute TB infection (1.7 log colony-forming unit (CFU) reduction at 50 mg/kg over a 15-day course of therapy). This could be due to its low solubility and metabolic issues, which limit its absorption and oral bioavailability (14%) and requires further chemical optimization. Moreover, target identification and mechanism of action studies should be carried out to confirm that SD is the direct in vivo target.
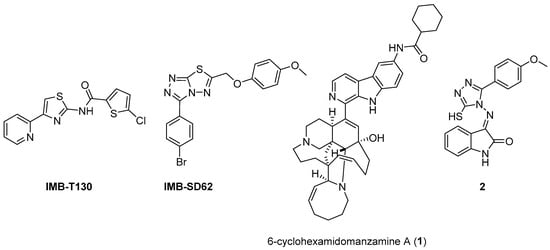
Figure 2.
Chemical structures of shikimate pathway inhibitors.
The most extensively studied enzyme of this pathway is the shikimate synthase (SK), which catalyzes the fifth step. This protein enables the conversion of shikimate to shikimate-3-phosphate and belongs to the nucleoside monophosphate (NMP) kinase family, which is known for large conformational changes during catalysis. It has an α/β architecture and presents four distinct binding domains: (i) a first NMP binding domain; (ii) a nucleotide binding site; (iii) an LID domain, which is responsible for the binding of Adenosine Triphosphate (ATP); and (iv) a reduced core domain [14]. This was the first enzyme that was proven to be essential for Mtb viability [15] and has drawn significant attention over the past 15 years [20]. To date, no SK inhibitors have been tested for their in vivo activity. Indeed, the most relevant studies published in the past five years aimed to identify new chemical entities rather than focusing on the chemical optimization of existing hits. However, different chemical scaffolds, which could be starting points for future optimization studies, have been identified. 6-Cyclohexamidomanzamine A (compound 1, Figure 2) emerged as the most potent and less toxic inhibitor of 26 marine-derived alkaloids against SK. The marine environment is an important source of biomolecules with a high chemical diversity and a wide range of biological activities [23]. This compound was a mixed non-competitive inhibitor of all forms of SK (free enzyme, enzyme–substrate binary, and tertiary complex). Kinetic profiling predicted a very slow binding time-dependent inhibition mechanism, where compound 1 induces the slow isomerization of the enzyme and the generation of a greater affinity complex. Such a time-dependent component could translate into an enhanced duration of the in vivo effect, which could be a great therapeutic advantage. Therefore, despite the complex structure, this class of compounds is worthy of future investigations, and these alkaloids could serve as novel scaffolds for future chemical optimization [24]. The availability of the crystal structure of SK has facilitated the design of structure-based inhibitors [25,26]. In two independent studies, Dadlani and De Freitas [19,27] adopted a mixed approach by integrating structural data, in vitro testing, and docking simulations. This approach provided a promising compound (MIC = 2.3 µM, SI = 341.63) characterized by a triazole-isatin scaffold (compound 2, Figure 2). Preliminary structure–activity relationship (SAR) investigations highlighted the importance of the nature, size, and lipophilicity of the substituent on the phenyl ring, paving the way for future research focused on improving the antimycobacterial activities and drug-like properties of this class of molecules.
The last two steps of the shikimate pathway are catalyzed by 5-enolpyruvylshikimate-3-phosphate synthase (EPSP) and chorismate mutase. Even though genetic studies have assessed their vulnerability as drug targets and crystal structures could aid in the design of structure-based inhibitors [28,29], to date, the development of inhibitors is still in its infancy [30,31].
Inhibitors of Trp Biosynthesis
Chorismate is a key metabolic intermediate for Trp biosynthesis. Its conversion to anthranilate is catalyzed by the enzyme anthranilate synthase (AS). AS is a heterodimeric enzyme composed of TrpE (AS-I), which catalyzes the production of anthranilate from chorismate and ammonia, and TrpG (AS-II), a glutamine (Gln) aminotransferase (GAT), which provides the required ammonia by converting Gln to glutamate (Glu) (Figure 3). The anthranilate is then transformed into phosphoribosyl anthranilate (PRA) by the enzyme anthranilate phosphoribosyltransferase (TrpD), which transfers a 5’-phosphoribose unit from phosphoribosyl pyrophosphate (PRPP) onto the amino group of the anthranilate. The ribose ring of PRA is then opened by the isomerase TrpF to give the isomer 1-carboxyphenylamino-1′-deoxyribulose-5′-phosphate (CdRP, Figure 3). This then undergoes a ring closure reaction catalyzed by TrpC, resulting in the indole heterocyclic ring system. The last step is catalyzed by Trp synthase (TrpAB), a bienzymatic complex that exists in tetrameric (αββα) form [32] and is thought to be essential for the survival of Mtb in vivo [3]. First, TrpA splits indole-3-glycerol-phosphate (IGP) to form indole and glyceraldehyde-3-phosphate (G3P); then, TrpB condenses indole with L-serine (L-Ser), forming Trp (Figure 3).
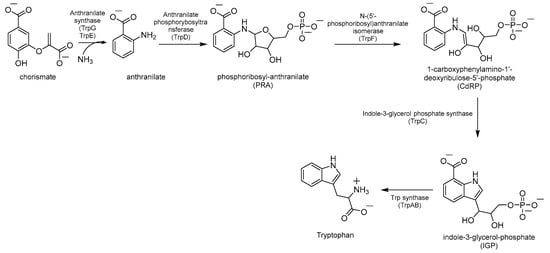
Figure 3.
Schematic representation of Trp biosynthesis.
In macrophages, Mtb has restricted access to Trp. For this reason, Trp biosynthesis is one of the most investigated and attractive amino acid metabolic pathways for TB drug development [33]. Indeed, Trp auxotrophic strains were less virulent and failed to cause disease in immunocompetent and immunocompromised mice [6]. Trp starvation driven by CD4 T cells is one of the host immune responses after Mtb infection [6]. When infected, the macrophage actualizes Trp starvation by expressing indoleamine 2,3-dioxygenase (IDO), an enzyme that catabolizes Trp to kynurenine and other metabolites. IDO catalyzes the first and rate-limiting step of the Trp degradative process, also known as the kynurenine pathway [34,35]. It is one of the most induced genes in both human and mice macrophages infected with Mtb. Indeed, Trp and kynurenine levels are extremely different from other metabolites in patients with TB and are highly altered after treatment; in a patient’s plasma, the Trp levels were lower than in those with latent TB and those in the control group and gradually increased after effective TB treatment [36]. This mechanism is successful with other infecting agents that are Trp auxotrophs. On the other hand, Trp starvation can limit Mtb growth but fails as a killing mechanism, as Mtb is fully competent for its biosynthesis. Moreover, the production of kynurenines, including those generated by IDO, can have immunosuppressive effects: they can inhibit the activation and function of immune cells, leading to immune tolerance, which could allow chronic bacterial infections to persist and evade immune clearance, leading to Mtb persisting in a latent state [35]. Inhibitors of the Trp biosynthetic pathway can then function as potential antibiotics by disrupting Trp production in Mtb and synergizing with the host immune system to inhibit microbial growth and hinder the establishment and progression of the infection. Additionally, the lack of an equivalent biosynthetic pathway in mammals reduces the likelihood of host toxicity, making these inhibitors even more promising as potential anti-TB agents. This has inspired intense research to identify novel inhibitors of this pathway that could serve as potential antibiotics, many of which have already been reviewed [33].
The first inhibitor of this biosynthetic pathway was identified 10 years ago while studying the mycobacterial genetic requirements to endure the CD4 response (the CD4 “counteractome”) [6]. This seminal study demonstrated for the first time that mycobacteria express genes involved in gluconeogenesis and Trp biosynthesis as a response to immune system stimulation in immunocompetent mice. To validate the target, some anthranilate analogues were tested against Mtb in the presence and absence of Trp. Two compounds, 2-amino-5-fluorobenzoic acid (5-FABA) and 2-amino-6-fluorobenzoic acid (6-FABA) (Figure 4), showed an MIC of 5 µM without Trp, while the addition of Trp to the medium rescued Mtb growth.
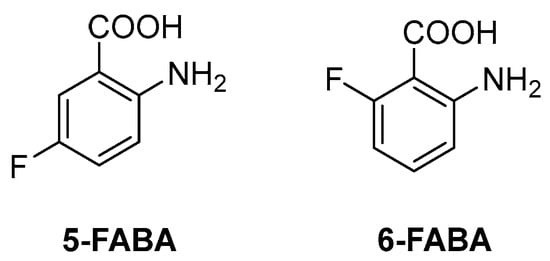
Figure 4.
Chemical structures of 5-FABA and 6-FABA.
When tested in vivo on a murine model of TB infection, the administration of 6-FABA and its ethyl ester resulted in a significant reduction in the bacterial load in infected mice spleens (10-fold reduction relative to the control) [6], suggesting that the alteration of Trp biosynthesis by an anthranilate-like compound synergizes with the host immune response to Mtb infection in vivo. 6-FABA, the only well-characterized compound, was thought to inhibit either the formation of anthranilate by TrpE or its modification by TrpD. Subsequent work suggested that the toxic mechanism of 6-FABA occurs downstream, potentially inhibiting a subsequent step or forming fluoro-Trp that is then incorporated into polypeptides, causing global protein stress [37]. Inspired by these findings, our research group developed a class of anthranilate-like compounds (Figure 5) to improve the activity and reduce the cytotoxicity of 6-FABA [38]. Our strategy was to replace the carboxylic moiety of 6-FABA with different bioisosters, including hydroxamates (3–4), oxadiazoles and tetrazoles (5–6), amides (7–18), hydrazides (19–39), aryl hydrazide hydrochlorides (40–41), and trifluoromethyl amines (42–44) (Figure 5).
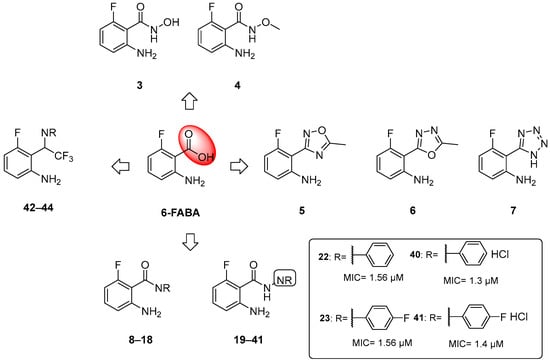
Figure 5.
Chemical structures of 6-FABA analogues and MICs against Mtb H37Rv of most active compounds.
The MICs against Mtb H37Rv ranged from 0.625 to >50 µM, with some analogues showing sub-micromolar activities and low cytotoxicities in Vero cells. We observed that the replacement of the carboxylic moiety with a hydrazide (compounds 19–41) led to a significant improvement in both activity and cytotoxicity relative to the parent compound 6-FABA, showing an MIC between 0.625 and >50 µM. Among them, 18 out of 23 compounds showed an MIC < 9.4 µM in Middlebrook 7H9/DPPC/casitone/Tx. Moreover, the preparation of hydrochloride salts (40 and 41) of hydrazides 22 and 23 provided an improvement in activity. Compounds 22, 23, and 40 (Figure 5) were tested against bacille Calmette–Guerin (BCG) growth in the presence of Trp and its main biosynthesis precursors, chorismate, anthranilate, and indole, to demonstrate that their growth inhibitory effects were due to their alteration of the Trp biosynthesis pathway. The ability of anthranilate, indole, and Trp to rescue BCG from growth inhibition by these hydrazides suggests that they exert a pleiotropic effect on Trp biosynthesis. By generating 6-FABA-resistant mutants and evaluating the activity of 40 against these resistors, it was also demonstrated that there is cross-resistance between compound 40 and 6-FABA, which is in agreement with the speculation that 6-FABA and hydrazides act by inhibiting the same target. Furthermore, whole genome sequencing (WGS) of resistors against 40 revealed that seven of the nine resistant mutants had mutations in the Trp pathway genes trpE, trpD, trpC, and trpB, confirming that 6-FABA hydrazide analogues exert their anti-mycobacterial activity by interfering with mycobacterial Trp biosynthesis. The lack of a confined mutational spot implies that these mutations modulate the metabolic flux of the biosynthesis pathway rather than directly affecting the binding of compound 40 to a specific enzymatic target. To further investigate the interaction between compound 40 and these genes, we achieved inducible repression by applying CRISPR interference. While the knockdown of trpD and trpB desensitized BCG to 40, trpC and trpE repression had a negligible effect on BCG’s sensitivity. This suggests that these compounds cause functional Trp depletion in mycobacteria by acting as substrate analogues, leading to the production of fluorinated Trp. However, the exact molecular mechanism requires further investigation.
In a study conducted by Naz et al. in 2021 [32], a ligand-based pharmacophore modeling approach was employed to identify potent inhibitors against the α-subunit of TrpAB. A virtual screening of drug-like molecules from the ZINC database was performed using a pharmacophore model generated starting from the structures of previously known TrpAB inhibitors and ligands. The best matches were subjected to molecular docking studies against the crystal structure of the α-subunit of TrpAB from Mtb. Five hits were then selected on the basis of the best fit value, binding score, binding interaction pattern with catalytically important amino acids in the active pocket, and physicochemical property analysis. These hits were further evaluated for their anti-TB activity through a whole cell-based assay, which led to the identification of a new inhibitor named ZINC09150898 (Figure 6). It was tested against Mtb and showed good antibacterial activity at concentrations up to 27.6 μM, achieving complete growth inhibition (100%) of mycobacteria at 115 μM. To gain insights into the stability of the identified inhibitor in the active pocket of TrpA, molecular dynamics (MD) simulations were performed. Various analyses were conducted to assess the binding mode stability of the inhibitor, whose binding score was determined to be −32.07 kcal/mol. Van der Waals interactions were demonstrated to play a crucial role in the retention of the inhibitor within the protein pocket through a molecular mechanics–Poisson–Boltzmann surface area (MM-PBSA) analysis. The authors also identified Gly69, Ser70, and Asp68 as key residues for the binding by performing site-directed mutagenesis; changing these residues into alanine (Ala) resulted, in fact, in a reduction in the binding affinity of the inhibitor.
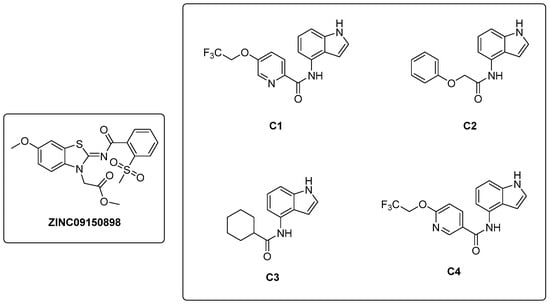
Figure 6.
Chemical structures of ZINC09150898 and indole carboxamides C1–C4.
In 2021, Libardo et al. [39] identified a series of indole-4-carboxamides with potent antitubercular activity. Representative members of the series (Figure 6) were selected to understand their antimycobacterial properties and how they inhibit the growth of Mtb. Compounds C1–C4 exhibited potent activities, inhibiting Mtb growth at low mM concentrations. Additionally, compound C1 was non-toxic to J774 macrophages and showed a reduction in intracellular Mtb titer in a murine macrophage model of infection. The mechanism of action was determined through the generation of C1-resistant mutants. Interestingly, indole-4-carboxamides act as prodrugs: AmiC-mediated hydrolysis converts them to 4-aminoindole (4-AI), an antimetabolite, which is then metabolically incorporated by Trp synthase to form cytotoxic 4-aminotryptophan. Mtb developed resistance to indole 4-carboxamides through three distinct mechanisms: (i) a decrease in drug metabolism, which reduces the conversion of the prodrug to the active cytotoxic compound; (ii) a specific mutation in trpE that makes the enzyme resistant to feedback inhibition, determining an increased biosynthetic flux in Trp that compensates for the cytotoxic effects of 4-aminotryptophan; and (iii) an in situ enzymatic attenuation of TrpAB, further reducing the incorporation of 4-AI into Trp biosynthesis, a genuine resistance mechanism in mycobacteria. This study unveils a very complex mechanism of metabolic flux regulation in mycobacteria, which can escape killing by toxic false metabolite flux either through a loss of feedback allosteric regulation, enhancing the flux of intermediates, or through enzymatic attenuation, which reduces the concentration of toxic metabolites. The medicinal chemistry optimization of 4-AI is essential to obtain a suitable candidate for in vivo studies, obtain further insights into its mechanism of action, and overcome resistance [40].
2.2. Inhibitors of Branched Chain Aminoacid (BCAA) Biosynthesis
Valine (Val), leucine (Leu), and isoleucine (Ile) share the first part of their biosynthesis (Figure 7). This biosynthetic pathway exists in plants, fungi, and bacteria but lacks a homolog in mammals, making the enzymes of this pathway attractive targets for drug development. The first enzyme in the pathway is acetohydroxyacid synthase (AHAS) encoded by ilvB1 for the largest domain and ilvN for the minor subunit. IlvB1 mutants are auxotrophic, and the resulting depletion of BCAAs leads to Mtb death [41]. The second promising target is a ketoacid reductoisomerase (KARI), a bifunctional enzyme that first catalyzes a methyl migration, requiring Mg2+ for its activity, and then reduces in the presence of nicotinamide adenine dinucleotide phosphate (NADPH). Finally, the gene ilvD encodes for dihydroxyacid dehydratase, which catalyzes one of the middle steps of Val and Ile biosynthesis. The ilvD mutant is auxotrophic as well, inspiring the development of inhibitors [7,42].
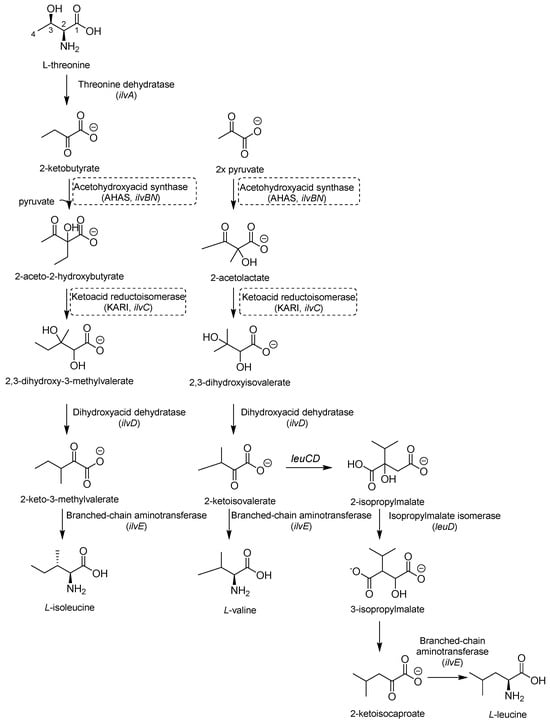
Figure 7.
A schematic representation of BCAA biosynthesis. The most relevant targets are depicted in dotted boxes.
2.2.1. AHAS Inhibitors
AHAS comprises a catalytic (IlvB1) and a regulatory subunit (IlvN) and catalyzes the condensation of two pyruvate molecules to acetolactate and pyruvate with 2-ketobutyrate to form 2-aceto-2-hydroxybutirate in the second step of BCAA biosynthesis (Figure 7). It is considered a biologically safe target for common herbicides, such as sulfonylurea sulfometuron methyl (SMM) and triazolopyrimidine sulfonamides. Interestingly, high doses of SMM were efficacious in a murine model of TB infection [43], disclosing the therapeutic potential of this target and inspiring the design of several analogues with improved activities [44,45,46].
Chlorflavonin (CF) (Figure 8) is an interesting hit of a new class of AHAS inhibitors that are structurally unrelated to the above-mentioned molecules. It was purified from an extract of Moringa stenopetala and displayed good antimycobacterial activity (MIC90 = 1.56 µM), no cytotoxicity at 100 µM against the two human cell lines THP-1 and MRC-5, a high selectivity index (SI) (IC50/MIC90 ≥ 64), and intracellular activity in infected macrophages. Mode of action and resistance mechanism experiments revealed that CF inhibits AHAS enzymatic activity by binding the ILvB1 catalytic subunit, causing a combined auxotrophic effect on Val, Ile, Leu, and pantothenic acid. The inhibitory effect is completely reversed through medium supplementation with BCAAs and pantothenic acid, suggesting that there are no relevant off-target effects for antibacterial activity. Docking studies predicted that CF could interact with the catalytic subunit IlvB1 through a hydrogen bond and a salt bridge with Lys 197 and π−π interactions between the phenyl moiety and Trp 516. Chlorine, which seemed to be essential for the activity, pointed toward a subunit pocket defined by Leu 65. Target engagement was confirmed by in vitro inhibition studies with recombinant ILvB1, which confirmed the direct interaction of CF with its target [47]. Recently, Berger and co-workers developed a five-step synthetic route to prepare CF and synthesized a small library of analogues by modifying the B ring of the scaffold (Figure 8). Unfortunately, any change in the B ring led to a loss of activity of the derivatives [48].
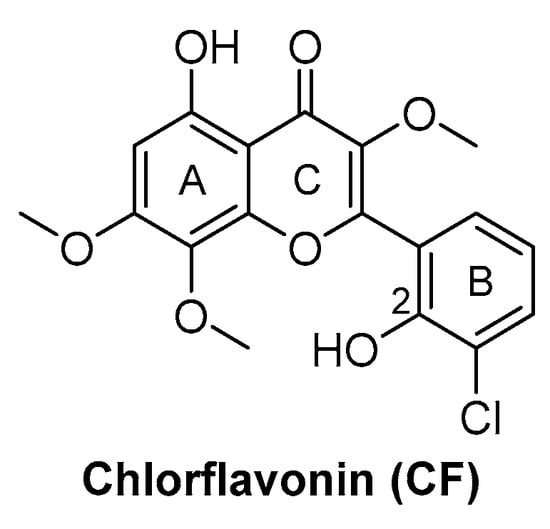
Figure 8.
CF chemical structure.
2.2.2. KARI Inhibitors
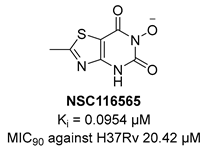
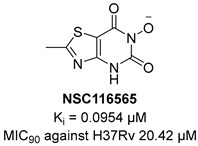
KARI catalyzes the transfer of a methyl group to generate the intermediate 3-hydroxy-3methyl-2-ketobutyrate and its reduction to R-2,3-dihydroxyisovalerate in the third step of BCAA biosynthesis (Figure 7). It is a bifunctional enzyme which requires Mg2+ for the methylation and NADH or NADPH for the reduction and has recently gained considerable attention as an innovative target in anti-TB drug discovery [49]. Lin and co-workers identified a promising compound (NSC116565, Ki = 0.0954 μM, Table 1) through a screening of the National Cancer Institute-Development Therapeutics Program (NCI-DTP) library. Kinetic, calorimetric, and structural studies showed that NSC116565 is a potent, competitive, and time-dependent inhibitor, which can bind the enzyme both in the presence and absence of NAPDH with KD values of 2.0 and 2.8 µM, respectively. To confirm that it acts as a KARI inhibitor, its biological activity was investigated with and without BCAA supplementation in the medium culture. In the absence of BCAAs, NSC116565 was able to inhibit the growth of Mtb H37Rv with an MIC50 of 6.06 µM and an MIC90 of 20.42 µM, while its MIC90 was >30 µM upon the addition of 1 mM of BCAA. NSC116565 did not display cytotoxicity in CD2F1 mice at doses of up to 300 mg/kg, confirming that it is a promising hit for further structural optimization [49,50].
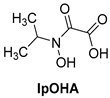
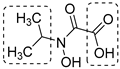
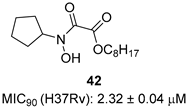
N-hydroxy-N-isopropyloxamate (IpOHA) (Table 1) is a herbicide that inhibits KARI in the nanomolar range. This compound acts as a transition-state analogue and is a time-dependent inhibitor of the enzyme. Twenty-two IpOHA analogues showed improved activities against the target, but none of them were active against Mtb [51]. This lack of activity was likely due to the hydrophilicity of this class of compounds, which limits their permeability through the cell wall. To improve lipophilicity, five prodrugs (42 a–e) of the most active analogue were prepared by esterifying the carboxylate group with alcohol functions of different lengths. Esterification with octanol, dodecanol, and decanol generated compounds with MIC90 values in the range of 2–3 μM against H37Rv strains. The addition of BCAAs to the media reversed the activity (MIC90 > 30 μM), confirming that these compounds act by inhibiting the BCAA pathway [52].
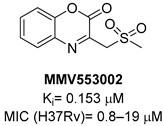
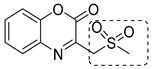
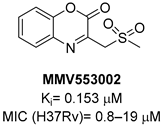
A screening of the Medicines for Malaria Venture Pathogen Box (MMVPB) against Mtb-KARI produced a potential hit (MMV553002, Table 1), with a Ki value of 0.153μM, an MIC between 0.8 and 19 μM, and no cytotoxicity against human HepG2 cells [51]. Kinetic studies revealed that this compound is rapidly hydrolyzed to 3-(methylsulfonyl)-2-oxopropanoic acid (4-EP) and 2-aminophenol (2-AP). Inhibition and crystallographic studies revealed that 4-EP is a time-dependent inhibitor and strongly interacts with Mg2+ ions inside the active site of the enzyme, similar to IpOHA. However, its charge and hydrophilic character prevent it from crossing the cell wall, and the anti-TB activity of MMV553002 is only due to its metabolite, the 2-amino-phenol. Four analogues were synthesized, but none of them were more active than the parent compound [51].
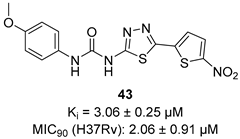
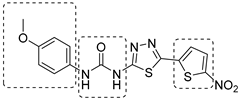
In recent years, Krishna et al. identified a new hit (compound 43, Table 1) acting against Mtb-KARI through a virtual screening of an in-house database. This thiadiazine derivative was able to inhibit Mtb-H37Rv growth with an MIC of 2.06 μM. To improve the inhibitory activity of the hit, 22 analogues were prepared by modifying the thiophen ring with bioisoster furan and by introducing various benzene-substituted rings. The ureidic moiety, the electron withdrawing groups on the thiophen, and the thiadiazine core were left unchanged as they seemed to be essential for the activity. The most promising compound was 44 (Table 1), which also showed encouraging activities in infected macrophages and in a nutrient starvation model. However, further studies are needed to clarify its mechanism of action and to determine whether KARI is the only target [53,54].

Table 1.
Chemical structures, SAR plan, and most advanced KARI inhibitors.
Table 1.
Chemical structures, SAR plan, and most advanced KARI inhibitors.
| Hit/Scaffold | SAR Plan from Hit | Most Advanced Analogue | Refs. |
|---|---|---|---|
 | - |  | [50] |
 |  |  | [52] |
 |  |  | [51] |
 |  |  | [53,54] |
2.3. Inhibitors of Arginine (Arg) Biosynthesis
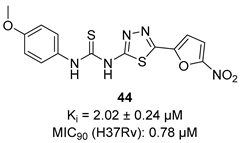
Arg biosynthesis (Figure 9) consists of eight different steps, each catalyzed by a different enzyme (argA-D, argF-H, and ArgJ) [55]. Like other microorganisms and cancer cells, the upregulation of de novo Arg biosynthesis is also an early response to oxidative stress. Unlike other amino acids, Arg deprivation leads to rapid sterilization. It has been demonstrated that Arg-mediated cell death in Mtb is caused by reactive-oxygen species (ROS)-mediated oxidative damage. Indeed, experiments with auxotrophs showed that Arg starvation and the accumulation of upstream metabolites caused a complex metabolic disruption, which ultimately led to rapid sterilization through the depletion of antioxidant thiols and ROS-mediated oxidative damage [8]. Its critical sterilization role and the absence of human homologues for many of the enzymes of Arg biosynthesis make the Arg biosynthetic pathway one of the most attractive targets for anti-TB drug development [55].
Figure 9.
A schematic representation of Arg biosynthesis. The most relevant targets are depicted in dotted boxes.
2.3.1. ArgA Inhibitors
ArgA is the first enzyme in the Arg biosynthetic pathway and catalyzes the transfer of an acetyl group from acetyl coenzyme A (AcCoA) to L-Glu. It is classified as an N-acetylglutamate synthase (NAGS) and belongs to the GCN5-related N-acetyl transferase superfamily [56]. ArgA is regulated by negative feedback through the binding of L-Arg to the active site [57].
Through the screening of the National Institutes of Health (NIH) Diversity set and Pathogen box, Khurana et al. identified a symmetric dicationic 2,5-bis (2-chloro-4-guanidinophenyl) furan (MMV688179, Figure 10) that inhibits Mtb growth in vitro with an MIC99 of 1.56 µM. This hit kills Mtb in a dose-dependent manner. As suggested by the MD simulation, it seems to strongly bind the L-Arg binding pocket, acting as an allosteric ArgA inhibitor.
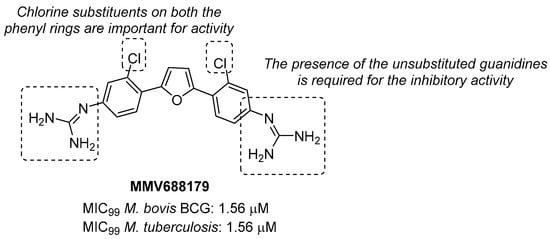
Figure 10.
Chemical structure, antimycobacterial activity, and preliminary SAR of MMV688179.
The proposed mechanism of action is the cell damage induced by ROS production in Arg starvation conditions, which leads to DNA damage and then cell death. The oral administration of MMV688179 only slightly inhibited mycobacterial growth after two weeks of treatment, and no antimycobacterial activity was observed in the lungs and spleen at 4 weeks post-infection. This lack of efficacy could be due to poor pharmacokinetic (PK) properties, and new analogues of MMV688179 were synthesized to expand the SAR of this new class of inhibitors. While shifting the chlorine atom to a different position did not impact the activity, the replacement of the two chlorine atoms with bromines lowered the potency, confirming that chlorine is the most appropriate halogen to guarantee antimycobacterial activity. Moreover, the freedom grades of the terminal guanidine groups seem to be required for essential interactions in the active site; indeed, both substituents on the guanidines and bulky substituents at position 3 on the phenyl rings reduce the activity as they might hamper the interactions between the nitrogen atoms and the active site (Figure 10) [58]. Unfortunately, none of the synthesized compounds were more active than the parent compound.
2.3.2. ArgB Inhibitors
ArgB is an acetylglutamate kinase that catalyzes the second step of Arg biosynthesis (Figure 9). This enzyme holds great potential to be a good target for the development of new antitubercular drugs because it lacks a homologue in humans and is essential for Mtb growth and survival [8]. Still, it remains largely unexplored. In 2021, its ligandability was explored through a fragment-based approach. The screening of a library of 960 fragments yielded two structurally related positive hits: NMR711 and NMR446 (Figure 11). Crystallographic data demonstrate that both fragments tightly bind to an interfacial allosteric site in ArgB through π–π interactions and few hydrogen bonds, with both trifluoromethyl moieties occupying the same position in the enzyme pocket. NMR711 and NMR446 have IC50 values of 366 and 707 µM, respectively, against ArgB. Isothermal Titration Calorimetry (ITC) experiments confirmed the binding (KD = 7.7 and 23 µM, respectively), and Nuclear Magnetic Resonance (NMR) competition assays clarified that these derivatives do not compete with other ArgB natural ligands such as ATP, N-acetyl glutamate (NAG), and L-Arg. The MICs against Mtb H37Rv, ΔargB-c, and the MDR strain V2475 were 90–180 µM and 117–234 µM for NMR711 and >200 µM and >200 µM for NMR446. However, only NMR446 was inactive after adding L-Arg to the media, proving to be on-target. This study offers a proof of concept of the potential of this target and provides two interesting fragment hits for further fragment growth and medicinal chemistry campaigns. However, the intrinsic hydrophobic nature of the interfacial binding site of ArgB, as well as the lack of hydrogen bonds and polar contacts, strongly limits fragment development and drug discovery programs, and the potential of this class of inhibitors remains largely unexplored [59].
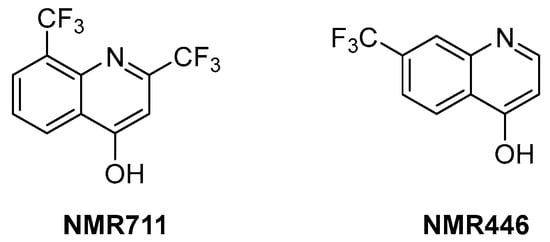
Figure 11.
Chemical structures of NMR711 and NMR446 fragments.
2.3.3. ArgJ Inhibitors
ArgJ encodes for an ornithine acetyltransferase (OAT) involved in the acetyl recycling in Arg biosynthesis and catalyzes the transfer of an acetyl group from N-acetylornithine (NAORN) to L-Glu to form N-acetyl L-Glu (Figure 9) [60]. This attractive target lacks a homologue in humans and is essential for mycobacterial survival [61]. However, its druggability has remained unexplored for years. A possible explanation could be that developing substrate analogue inhibitors could be accompanied by off-target effects since many substrates of the Arg pathway are common to host cellular pathways. To avoid off-target effects and explore the potential of this target, Mishra and coworkers rationalized an inhibitor of the allosteric site, which is less evolutionary conserved and should ensure selectivity. To avoid safety issues, a library of 1556 Food and Drug Administration (FDA)-approved drugs was selected for virtual screening against ArgJ. To provide a proper characterization of ArgJ for drug targeting, this study combined in silico and extensive biochemical studies as well as functional in vitro strategies. The most promising hit of this screening was Pranlukast (PRK, Figure 12), an antagonist of the cysteinyl leukotriene receptor that is effective for the treatment of asthma. This drug acted as an allosteric modulator by binding to a novel pocket in Mtb ArgJ, inhibiting the activity in a non-competitive manner. It markedly inhibited the growth of Mtb H37Rv (MIC90 = 10 µM) and was also effective against Mtb-infected monocytic THP-1 cells without detrimental effects on host cell survival. Moreover, it reduced Mtb-mediated apoptosis in macrophages and showed a synergistic effect with standard-of-care anti-TB drugs. This drug has a dual mechanism of action: Firstly, the Arg biosynthesis inhibition mediates the Mtb killing mechanism, as confirmed by Arg supplementation experiments. Secondly, it enables the efficient intra-macrophage elimination of mycobacteria by targeting the 5-lipooxigenase pathway, which facilitates Mtb survival and growth in host macrophages. Encouragingly, PRK also induced a significant decrease in the granuloma size in a chronic murine model of TB infection and showed a remarkable additive effect in combination with rifampicin (RIF), confirming its potential in combination therapies [41,62]. Moreover, PK studies revealed that the plasma concentration after single and multiple doses achieves levels close to the IC50 range, suggesting a translational value [63].
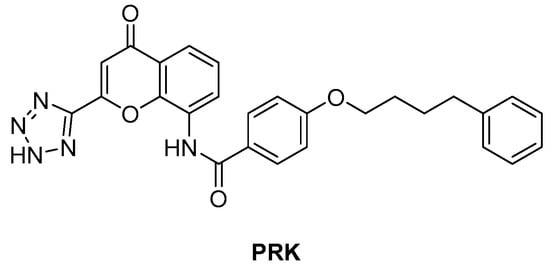
Figure 12.
Chemical structure of PRK.
2.4. Inhibitors of Gln Biosynthesis
The Gln biosynthetic pathway is essential for both the production of L-Gln and L-Glu and nitrogen metabolism. Moreover, it is implicated in host defense against TB, serving as a carbon and nitrogen source for M1 macrophage polarization. Gln synthetase (GS or GlnA1) is an ATP-dependent enzyme that catalyzes [64] the reaction between the ammonium ion and L-Glu to form L-Gln, Adenosine Diphosphate (ADP), phosphate, and L-Glu (Figure 13). Mtb has four different GS genes, but only GlnA1 is essential for both in vitro and in vivo survival [65]. The main advantage of targeting GlnA1 is that its metabolites are involved in the formation of a poly-L-Glu/Gln structure, a constituent of the cell wall of mycobacteria [66,67]. Thus, the inhibition of this enzyme can affect both amino acid biosynthesis and the integrity of the cell wall, leading to the death of mycobacteria [68,69,70]. GlnA1 is a dodecameric enzyme, and the active site is formed by two cones connected at their narrow ends. When ATP binds to the enzyme, in the presence of two or three metal ions (Mg2+ or Mn2+), it passes from a relaxed state to a taut (active) state, also known as closed conformation [66,69]. In general, the mechanism allows for the entry of L-Glu and ammonium ion from one side, while ATP passes via the opposite side of the cone. The first step is the phosphorylation of L-Glu, followed by the nucleophilic attack of the ammonium ion to generate the amide moiety of L-Gln (Figure 13).
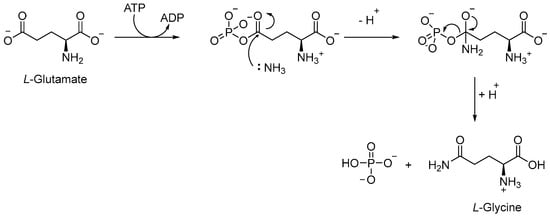
Figure 13.
Conversion of L-Glu to L-Gln, catalyzed by GlnA1.
An early seminal study demonstrated the druggability of this target by showing that the known epileptogenic agent, methionine sulfoximine (MSO) (Figure 14), which possesses direct antibacterial activity on a solid medium (MIC = 50 µM), can reduce the bacterial burden in a guinea pig model, and it protects animals from weight loss and has synergistic activity with isoniazid (INH) in vivo. However, the high frequency of spontaneous resistant mutants to MSO has dampened the enthusiasm in developing analogues against this target. Additionally, the amino acid binding site in GS exhibits a structure that is highly conserved in humans. For this reason, the ATP binding site has become a more attractive site for targeting for the development of new anti-TB drugs with reduced off-target effects [71,72,73]. Most recently, Dilebo et al. optimized a novel series of 4-pyridylamino and 4-(ethynylpyridine) quinazolines (Figure 15). All of these derivatives were subjected to in vitro anti-Mtb assays, cytotoxicity studies, and docking studies to predict their binding modes to the target enzyme. The 4-(ethynylpyridine) quinazolines’ MIC90 values were generally low (0.72 µM < MIC90 < 23 µM). The best activity was related to the presence of a methoxyl, which can probably engage hydrogen bonds with the target. The best compound of this series showed a very good SI and could serve as a useful starting point for further chemical optimization to expand this series. However, further metabolomic experiments and genetic and biochemical validation are needed to confirm the target [74].
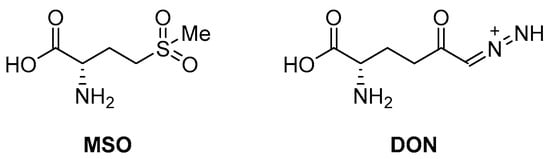
Figure 14.
Chemical structures of L-Gln antagonists MSO and 6-diazo-5-oxo-l-norleucine (DON).
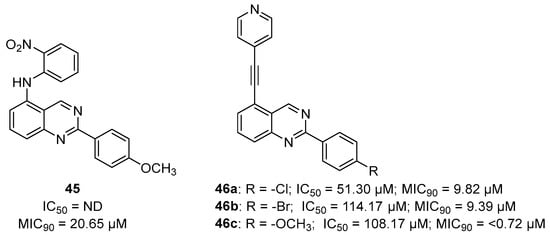
Figure 15.
Chemical structures and antimycobacterial activities of 4-pyridylamino and 4-(ethynylpyridine) quinazolines against Mtb H37Rv.
Target-based high-throughput screening (HTS) of the MMVPB against GlnA1 revealed that Lisitinib (LIN, Figure 16), a clinical-stage kinase inhibitor targeting insulin-like growth factor 1 and insulin receptor (IGF1R/IR), is a potent GlnA1 inhibitor (IC50 = 1 µM). Kinetic enzyme assays showed that LIN acts as an ATP-competitive inhibitor, and it was hypothesized that this mechanism of action is due to the imidazopyrazine moiety, which mimics the adenine of the natural ligand ATP quite well and establishes π-π interactions with the benzene of Phe232 of GlnA1. GlnA1 inhibition results in weak direct antimycobacterial activity, probably due to permeability issues, which require further chemical optimization. However, an analysis of the Mtb-infected THP-1 macrophages cell responses showed that LIN treatment enabled the intracellular killing of drug-sensitive and -resistant Mtb in a dose-dependent manner through autophagy activation [75]. Moreover, Phase I studies showed that it has excellent properties in terms of PK and metabolism, achieving plasma concentrations that are well correlated to the IC50 value [76]. Therefore, this early hit is worthy of chemical optimization to overcome permeability issues, increase direct antimycobacterial activity, and develop a novel class of anti-TB host-directed therapy (HDT) dual targeting inhibitors [75]. Even though GlnA1 has been extensively studied as a direct anti-TB drug target [68,69,77,78], HDT is the most emerging role in Gln metabolism inhibition. It is well-known that Gln metabolism antagonism is also a strategy to improve T-cell immunity effectors in a tumor microenvironment, but the mechanism in Mtb has only been disclosed recently. JHU083 (Figure 16), a prodrug developed to improve the therapeutic index of the Gln antagonist DON (Figure 14), has promising anticancer activity in preclinical models and acts by enhancing T-cell activity through metabolic reprogramming. A very recent study demonstrated that JHU-083 has a dual antibacterial and host-directed effect against TB [79]. Despite showing modest direct anti-TB activity, JHU-1083 treatment significantly reduced TB infection in murine models (1.9 log10 bacteria reduction after 5 weeks of treatment), leading to an improved anti-Mtb immune response in all treated animals. Its therapeutic efficacy requires an intact immune system and was completely abrogated in immunocompromised mice, supporting the hypothesis that the immunomodulatory effect, more than the direct antibacterial effect, was the predominant mechanism of its therapeutic efficacy. Moreover, the metabolic fluxes of the immunologically relevant amino acids Trp and Arg are altered following JHU-1083 treatment, which is consistent with an enhanced host immune response. This study also sheds light on an additional role of GlnA1, which could be exploited as a virulence factor to increase Gln metabolism in the granuloma, leading to an immunosuppressive environment (reduced T-cell function, decreased citrulline and NO production, and immunosuppressive myeloid accumulation), which promotes disease progression.
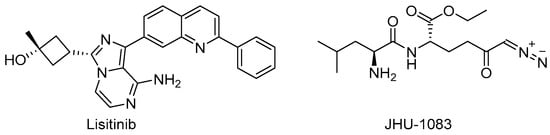
Figure 16.
Chemical structures of LIN and JHU-1083.
2.5. Inhibitors of Ser Biosynthesis
Ser biosynthesis is a crucial metabolic pathway that enables Mtb to synthesize L-Ser. This amino acid is required for Mtb growth and survival, and it is involved in various cellular processes such as nucleotide synthesis, redox balance, and cell wall biosynthesis [36,80,81]. The first step of this pathway is the NAD+-mediated oxidation of D-3-phosphoglycerate to 3-phosphohydroxypyruvate, catalyzed by the enzyme phosphoglycerate dehydrogenase (PGDH or SerA1) [81,82] (Figure 17). Next, phosphoserine aminotransferase (PSAT or SerC) transforms phosphohydroxypyruvate into L-3 phosphoserine using glutamate as an amino donor [80,81]. Finally, L-3 phosphoserine undergoes a dephosphorylation step mediated by the enzyme phosphoserine phosphatase (PSP or SerB2), leading to the formation of L-Ser. This step is essential for generating free Ser molecules that can be exploited by mycobacteria for various metabolic processes [81,82]. L-Ser is also a substrate for the biosynthesis of other important metabolites, such as glycine (Gly), an important precursor for the synthesis of nucleotides and peptidoglycan, and phosphatidylserine (PS), a major component of the bacterial cell membrane contributing to the maintenance and integrity of the bacterial cell envelope. Therefore, gaining a clear understanding of the intricacies of this pathway may provide insights into many potential targets for the development of novel therapeutic strategies against tuberculosis [36].
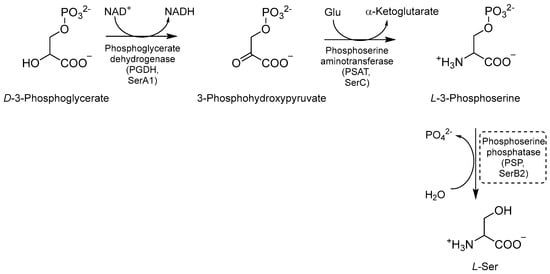
Figure 17.
A schematic representation of Ser biosynthesis. The most relevant target is shown in a dotted box.
Even though these three enzymes are essential for Mtb growth and survival [61], no inhibitors of SerA and SerC have been reported so far, and only a few inhibitors on SerB have been developed over the past five years. Indeed, the development of compounds targeting this pathway is limited by the existence of human orthologues, which share very similar catalytic pockets. Moreover, the lack of a thorough structural characterization hinders the structure-based design and the drug discovery process.
Mtb-SerB2 is predominantly secreted in the cytosol of infected macrophages (specifically in THP-1 cells), where it triggers a rearrangement of the cytoskeleton, creating a necrotic environment. These conditions promote the growth of Mtb and contribute to intra-macrophagic survival. Mtb-SerB2 is then considered an invasive secreted virulence factor employed by Mtb to evade the host immune system. Its phosphatase activity can alter the immune response by interacting with cofilin, nuclear factor-kappa B (NFkB), and P38 and by inhibiting the expression of interleukin 8 (IL-8), an important immune mediator [81,83]. In terms of structure, SerB2 possesses a core domain similar to the Rossman fold, and it is composed of α and β elements. This core domain comprises four loops that encompass all three motifs that are essential for PSP activity. Additionally, SerB2 includes a cap domain that acts as a closure for the active site, enabling specific recognition of the substrate. The catalytic core of SerB2 exhibits the characteristic features that are commonly observed in HAD (haloacid dehalogenase) phosphatases [83]. Mtb-SerB2 possesses two regulatory N-terminal Aspartate kinase, Chorismate mutase, and TyrA (ACT) amino acid-binding domains. These domains serve as conserved allosteric regions responsible for the regulation of enzymatic activity when exposed to high levels of reaction products. Notably, human PSP only comprises the phosphatase domain, making the ACT domains potential targets for the development of small inhibitors that could selectively inhibit Mtb-SerB2 [81]. Most of the known inhibitors of SerB2 target the catalytic binding site and were identified through the target-based screening of commercial libraries [84,85] (Table 2).
Table 2.
Chemical structures, SAR plan, and most advanced SerB inhibitors.
Table 2.
Chemical structures, SAR plan, and most advanced SerB inhibitors.
| Hit/Scaffold | SAR Plan from Hit | Most Advanced Analogue | Refs. |
|---|---|---|---|
 | - |  | [84] |
 | - |  | [85] |
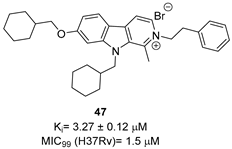 |  | 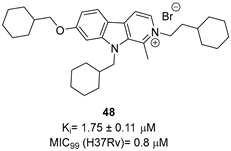 | [86] |
To increase chemical diversity, Pierson et al. [86] tested an in-house chemical library with a great range of novel chemical scaffolds. Interestingly, three hits were harmine-derived with bulky aliphatic or aromatic substituents and shared a 2,7,9-trisubstituted pattern, indicating the crucial involvement of the positive charge carried by the N2 atom on their activity. Compound 48 (Table 2) was synthesized to investigate the importance of aromatic substituents and was found to be the most potent. This confirmed the importance of flexible and hydrophobic substituents and suggested that aromatic residues are not involved in interactions within the binding site. Indeed, the three substituents on the β-carboline core occupy distinct pockets within the active site, primarily composed of hydrophobic residues such as Leu, Val, and Ile. These inhibitors are likely to directly interact with the active site of the enzyme, causing the disruption of substrate binding [83]. Based on this work, a recent study reported an innovative virtual screening-based approach to select fragment-sized harmine-derived compounds and chelators of the catalytic magnesium of Mtb-SerB2. This work offered more insights into the necessary constituents for the interactions between the enzyme and inhibitors and provided interesting fragments for future fragment growth and drug discovery efforts [87]. The same research group recently published a study dissecting the complex oligomeric behavior of this protein. A combination of biochemical and biophysical experiments demonstrated that this enzyme exists in different oligomeric forms of different activity and has unique morpheein behavior, which is different from other mycobacteria orthologs. Apart from providing a deeper understanding of the mechanism of control of this protein, this study may also be the base for allosteric drug discovery and for future structure-based drug designs of inhibitors targeting oligomeric interfaces with the aim of increasing selectivity and avoiding concomitant human ortholog inhibition [88].
2.6. Inhibitors of Proline (Pro) Biosynthesis
Pro biosynthesis starts with the phosphorylation of Glu to γ-glutamyl phosphate, which is catalyzed by the enzyme γ-glutamyl kinase (GK). γ-glutamyl phosphate reductase (GPR) then reduces γ-glutamyl phosphate to Glu-γ-semialdehyde (GSA), which undergoes cyclization to form Δ1-pyrroline-5-carboxylate (P5C). In the final step, P5C is further reduced to Pro by the enzyme P5C reductase (P5CR) using either NADH or NADPH as cofactors (Figure 18) [89,90].
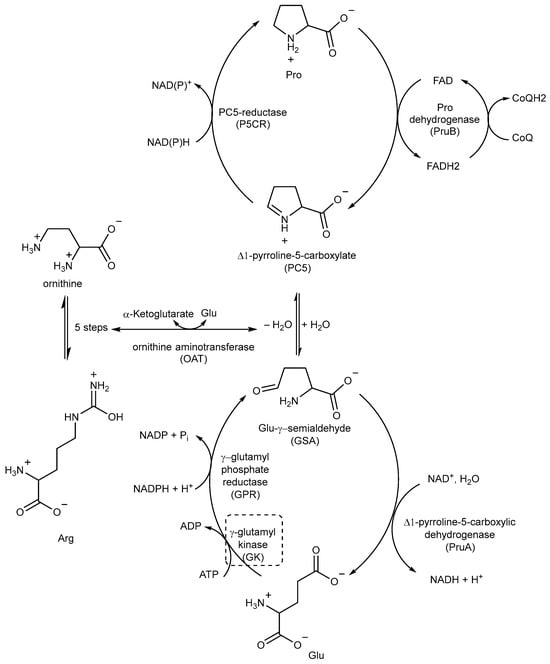
Figure 18.
A schematic representation of the Pro metabolic pathway. The most relevant target is shown in a dotted box.
P5C, the precursor of Pro, can also be obtained from ornithine through ornithine aminotransferase (OAT) (Figure 18). This important connection between Pro and Arg metabolism highlights the importance of Arg as an important source of nitrogen and carbon in Mtb, contributing to the overall metabolic pathways involved in nitrogen and carbon metabolism [41,89].
Pro catabolism is also a crucial factor in the ability of Mtb to persist within the host. The conversion of Pro to Glu is regulated by two specific enzymes: Pro dehydrogenase (PruB), which catalyzes Pro oxidation to Δ1-pyrroline-5-carboxylic acid (P5C) using a flavin cofactor, and Δ1-pyrroline-5-carboxylic dehydrogenase (PruA), which converts the tautomeric form of P5C (Glu-γ-semialdehyde) into Glu using NAD+ as a cofactor. These enzymatic reactions play key roles in the Pro metabolic pathway of Mtb, contribute to its ability to persist and thrive in the host environment, and protect it from methylglyoxal, a toxic and electrophilic metabolic intermediate [91,92]. However, even though studies could help in designing inhibitors, the potential of these enzymes as TB drug targets remains unexplored.
The first study on GK, as a potential drug target, only appeared in 2019 while studying the mechanism of action of Z0930 and Z0933 (Figure 19), two compounds synthesized in the hit-to-lead optimization of a [3,2-c]quinoline scaffold, identified by the whole-cell screening of a diversity-oriented in-house library [93]. GK consists of two distinct domains: the main catalytic domain, AAK, which is responsible for binding and interacting with the substrate(s), and the PUA domain. Its catalytic activity can be regulated by the presence of L-Pro, which can act as a feedback inhibitor [93]. WGS of Z0930- and Z0933-resistant mutants revealed mutations within the Rv2439c gene, encoding for GK. Intriguingly, kinetic and biochemical studies demonstrated that these compounds enhance GK catalytic activity in a dose-dependent manner. Moreover, their activation kinetics do not follow a competitive pattern with ATP, indicating the existence of a separate allosteric regulatory site in GK, which is distinct from the ATP binding site [93]. Upregulated Pro production may eliminate Mtb both in vitro and in macrophages through the generation of ROS, disrupting cellular redox homeostasis and leading to cell death. A preliminary SAR analysis suggested that the heterocyclic framework is essential for GK activation. In particular, the quinoline core, which is shared by both compounds, is crucial for the binding of these compounds to GK [93]. This groundbreaking study not only discovered and unveiled the potential of a new drug target, advancing the knowledge and the importance of the Pro biosynthetic pathway, but also highlighted enzyme activation as a new opportunity in TB drug discovery. Inspired by this work, a more recent study identified two pyrroloquinolines, compounds 49 and 50 (Figure 19, with promising efficacies against Mtb (MIC = 4.1 and 4.2 μM, respectively)) [94]. Compound 50 binds to an allosteric pocket adjacent to the catalytic site responsible for L-Glu binding, whereas inhibitor 49 specifically interacts with a pocket in proximity to the ATP adenosine group recognition site. In silico studies predicted that compound 49 could hamper ATP binding by inducing long-distance conformational alterations in the ATP binding site. On the other hand, compound 50 does not influence ATP recognition and could cause conformational modifications in the L-Glu catalytic site, thereby preventing the anchoring of L-Glu which is necessary for catalysis. The discovery of these promising structural frameworks paves the way for the development of allosteric inhibitors targeting the GK enzyme and further supports the therapeutic potential of this biological target in the pursuit of novel anti-TB drugs [94].
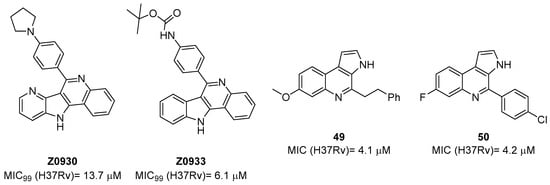
Figure 19.
Chemical structures of Z0930, Z0933, 49, and 50.
2.7. Inhibitors of Asparagine (Asn) Metabolism
Ans and Asp are among the major sources of nitrogen for Mtb. The mycobacterium mainly exploits Ans from host tissues through the transporter AnsP1 and the paralogue AnsP2 (Rv0346c), which are involved in Asp and Ans uptake in the mycobacterial phagosome [9,95]. Even though AnsP2 is not responsible for virulence, ansP2 expression is strongly induced in the lungs of TB patients, which could reflect the importance of this transporter. Ans catabolism supports acid stress resistance and intracellular survival. The only enzyme of the asparaginase family encoded by the Mtb genome is AnsA, which is secreted in the mycobacterial cytosol and outside the mycobacterial envelope and is responsible for the hydrolysis of Ans giving Asp and ammonia (NH3). NH3 is spontaneously protonated in the phagosomal lumen, reacting with protons transported by V-ATPase to form ammonium ions. This allows for phagosomal pH buffering and represents an essential strategy to resist acid stress both in vitro and inside macrophages [96,97]. Indeed, an Mtb mutant lacking the encoding gene ansA shows impaired nitrogen incorporation from Asn and has attenuated virulence in interferon gamma (IFN-γ)-activated macrophages and in mice [9]. Nonetheless, compared to other metabolic enzymes involved in nitrogen metabolism, those belonging to Ans metabolism are largely unexplored [95]. The only study highlighting the potential role of asparaginase as a potential drug target reports a three-dimensional structural model via the SWISS-MODEL server (http://swissmodel.expasy.org/) using P. horikoshi L.-asparaginase as a structure template (percentage of sequence identity equal to 27%). A comparison with asparaginase of other pathogens and the human one revealed a significant difference between them, suggesting that it could be a good and safe target. Potential inhibitors were selected from the Traditional Chinese Medicine (TCM) database, the ZINC database, and the FDA-approved drug database and predicted through a virtual screening against AnsA. Positive hits showed satisfactory activities against M. smegmatis, but the results have not yet been confirmed in Mtb [98].
2.8. Inhibitors of the Asp Metabolic Pathway
The Asp metabolic pathway (Figure 20) exists in plants, fungi, archaea, and microbes, but not in mammals. It produces Met (Met), threonine (Thr), isoleucine (Ile), lysine (Lys), S-adenosyl-L-methionine (SAM), and diaminopimelate (DAP), a cell wall constituent that is important for the virulence of Mtb [99]. The disruption of the Asp pathway leads to a complex imbalance of metabolic fluxes, which is counteracted by Mtb through different compensatory mechanisms. Thr, homoserine, and Met auxotrophies exhibited a rapid cell death phenotype, confirming the vulnerability of this pathway and that both Met and Thr are required for Mtb survival. Thr starvation leads to an accumulation of Lys and homoserine metabolic intermediates. Indeed, Asp kinase (AK), the first enzyme of the pathway (refs. [10,11]) is controlled by Thr allosteric feedback, but not by Met or Lys. Instead of the ubiquitous Lys-AK feedback loop regulation, Mtb regulates Lys accumulation through two unique mechanisms, Lys degradation and export. Moreover, conditional knockdowns of thrA and metX revealed that these two branch point enzymes are required for late-stage infection. This demonstrates that the inability to scavenge Met, Thr, and homoserine from the host affects both the establishment and late-stage persistence of the infection, which is an essential feature for future drug discovery efforts. Therefore, due to its branched nature and multifaceted bactericidal mechanism, along with its importance in both acute and chronic infections, the Asp pathway offers a wide and still unexplored target space for anti-TB drug discovery [10].
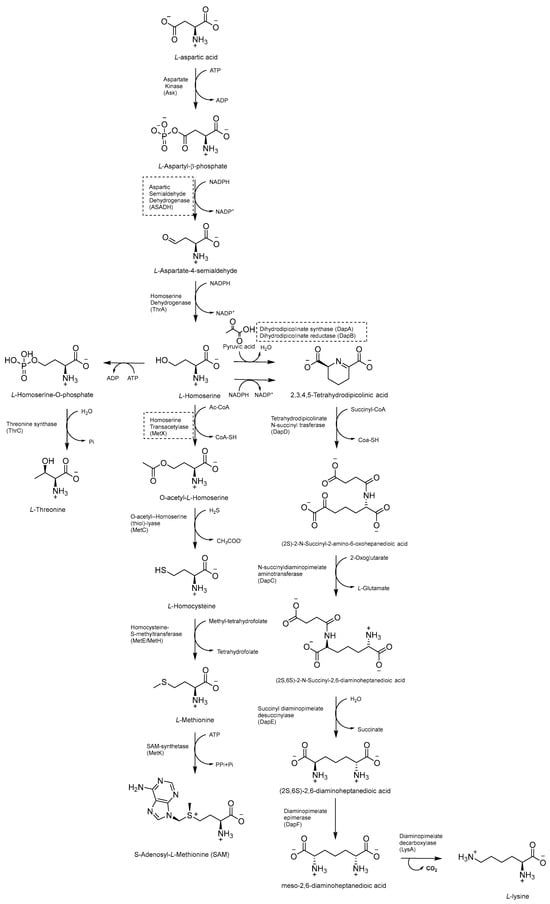
Figure 20.
Schematic representation of Asp metabolic pathway.
2.8.1. Aspartic Acid Semialdehyde Dehydrogenase (ASADH) Inhibitors
ASADH is a critical branch point for the biosynthesis of Lys, Thr, Met, and isoleucine [99]. Even though several inhibitors were identified through in silico approaches [100,101], the first study validating the efficacy of ASADH inhibitors in an experimental model only came out in 2021. Wang and coworkers identified IMB-XMA0038 (Figure 21) through the HTS of 150,000 compounds using a surface plasmon resonance (SPR) assay as a secondary screening method for hit validation and a molecular docking analysis to predict interaction with the target [102]. The further characterization of in vitro IMB-XMA0038 activity revealed that this compound is not only active against drug-sensitive strains (MIC = 1.7 µM), but also against clinical isolates (MICs = 1.7–3.4 µM) and dormant bacteria [103]. In addition, it showed a clear synergistic effect when combined with INH, RIF, bedaquiline (BDQ), moxifloxacin (MFX), and delamanid (DLM) because it is likely to improve drug penetration, which ultimately inhibits the synthesis of Mtb cell walls. The post-antibiotic effect (PAE) was also studied using Mtb H37Rv cells grown to the midlogarithmic phase (OD600 = 0.5). The PAE is an important pharmacodynamic indicator, resulting in a delayed resumption of bacterial growth following the removal of antibiotics from the cell culture. In this case, IMB-XMA0038 acted in a dose-dependent manner, like INH. Finally, it reduced the bacterial load by 1.67 log10 in a murine model of acute Mtb H37Rv infection, confirming its potential as an anti-TB hit for further hit-to-lead optimization. Further studies are also needed to elucidate its mechanism of action and investigate its effects on cell wall integrity [103].
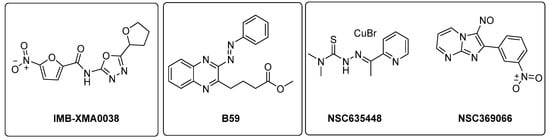
Figure 21.
Chemical structures of IMB-XMA0038, B59, NSC635448, and NSC369066.
2.8.2. Inhibitors of Lysine (Lys) Metabolic Branch
The Lys metabolic branch, also known as the diaminopimelate DAP pathway, is absent in mammals and is crucial for producing other important metabolites, such as dipicolinate and DAP, which are essential for the cross-linking of peptidoglycan polymers in bacterial cell wall synthesis [104,105]. It involves several enzymatic steps (Figure 20), starting from aldol condensation catalyzed by the enzyme dihydrodipicolinate synthase (DapA) to convert homoserine and pyruvate to dihydrodipicolinate. The latter is reduced by dihydrodipicolinate reductase (DapB) to tetrahydrodipicolinate, which, in turn, is converted to meso-DAP. Meso-DAP is then converted to Lys by diaminopimelate decarboxylase (LysA), a pyridoxal 5′-phosphate (PLP)-dependent enzyme [106] (Figure 20). The genes of this pathway are essential for Mtb growth, and some of them have already been investigated as targets for potential inhibitors [107,108,109,110]. The importance of dihydrodipicolinate reductase (DapB), encoded by dapB, has only been recently elucidated [104]. This enzyme uses NADH/NADPH as a cofactor to reduce dihydrodipicolinate to tetradihydrodipicolinate. It is composed of four identical subunits, each of them comprising two domains connected through a flexible region. The N-terminal domain binds the cofactor, while the C-terminal domain is responsible for substrate or inhibitor binding. An antisense dapB knockdown mutant strain exhibited growth defects and a reduced ability to infect macrophages, proving the importance of DapB in mycobacterial survival and virulence and its potential as a drug target. A virtual screening of 95 k molecules from an NCI database, followed by docking studies, identified 60 potential DapB inhibitors. The hit of this series was quinoxaline derivative B59 (Figure 21), showing good enzymatic activity (32.9 µM), a reasonable MIC (59.8 µM), and negligible cytotoxicity against three different cell lines. This offered a proof of concept that DapB is a potential drug target and that B59 is worth investigating with further hit-to-lead optimization studies to enhance its antimycobacterial activity and improve its cell permeability and drug-like properties [104].
2.8.3. Inhibitors of Met Metabolic Branch
The essential branch point of the Asp pathway for Met biosynthesis is the conversion of homoserine to O-acetyl-L-homoserine (OAHS) catalyzed by the enzyme homoserine transacetylase (MetX) [10] (Figure 20). MetX exploits acetyl-CoA as a cofactor and has a conserved catalytic triad comprising Ser157, His350, and Asp320 and Ser157 as a nucleophile. The catalyzed reaction follows a ping-pong mechanism: once activated, Ser157 reacts with acetyl-CoA, producing the acetyl-enzyme intermediate and CoA. Following the dissociation of CoA, it binds L-homoserine, and then the γ hydroxyl of L-homoserine attacks the acetyl–enzyme intermediate complex, generating an OAHS product and free enzyme [111].
Met is an important factor for translational initiation and is the precursor of SAM, a cofactor involved in biochemical reactions and regulatory mechanisms. Both Met and SAM are involved in essential roles in Mtb, such as the control of the eukaryotic cell cycle, autophagy, and differentiation of human pluripotent stem cells. Moreover, SAM is a cofactor for one-carbon metabolism, and it is responsible for the methylation of DNA, RNA, proteins, and lipids by SAM-dependent methyltransferases [11]. The deletion of MetX (encoded by the Rv3341 gene) generates auxotrophic mutants that are unable to establish a productive infection both in immunocompetent and immunocompromised mice. Compared to other amino acids, this auxotrophy has a strong bactericidal effect, which rapidly leads to mycobacterial death. This indicates that Mtb is completely dependent on this pathway for its survival and suggests a multitarget cell death mechanism. Consistent with their composite biological roles, the blockade of Met and SAM production causes pleiotropic effects, including the inhibition of several methyl-transferase-dependent processes, a stalling of translation initiation, the toxic accumulation of Lys, and a pervasive metabolic shutdown. Therefore, the Met metabolic branch provides an excellent drug target space. Recent structural studies aiming to elucidate the druggability of MetX have revealed the existence of druggable sites, confirming that this is an excellent candidate for structure-based small-molecule drug discovery [111]. The first HTS assay for identifying MetX inhibitors was developed in 2022 by Chaudhary and coworkers, who screened a library of 2334 compounds of the NCI-DTP library and identified two primary hits (NSC635448 and NSC369066) that are active against MetX in a dose dependent manner with MIC values of 3.125 µM and 6.25 µM, respectively (Figure 21). Docking studies revealed that compound NSC635448 forms a hydrogen bond with Tr61 residue, while NSC369066 is involved in the hydrogen bond with Leu60. NSC369066 seems to be the best compound, also fitting in the binding pocket for interactions with Arg227, Lys272, and Arg276 residues, which are located near the active site. NSC369066 was the most potent, with killing levels comparable to INH, which were partially restored after Met supplementation. Unfortunately, this first-in-class MetX inhibitor suffers from high cytotoxicity (5 µM) and multitarget killing activity, and further effort is required to improve its antimycobacterial activity and its SI before considering it as a promising candidate for further hit-to-lead optimization [112].
2.9. Inhibitors of Cysteine (Cys) Biosynthesis
Cys is required for mycobacterial growth and is involved in mycothiol biosynthesis, which is necessary to maintain redox homeostasis in dormant models. Cys contributes to the repair of the iron–sulfur center containing proteins, which is damaged by ROS and reactive nitrogen intermediate (RNI) species. There are three distinct pathways for de novo Cys biosynthesis. Each of them is characterized by a different PLP-dependent Cys synthase: (i) CysK1, a bona fide PLP-dependent O-acetylserine sulfhydrylase encoded by the gene rv2334, which converts O-acetylserine and sulfide to Cys; (ii) CysK2, a sulfocysteine synthase encoded by the gene rv0848, which converts O-Phospho-L-Ser into S-sulfocysteine and then into L-Cys; and (iii) CysM, a unique sulfhydrylase for Mtb encoded by the gene rv1336 [113] (Figure 22).
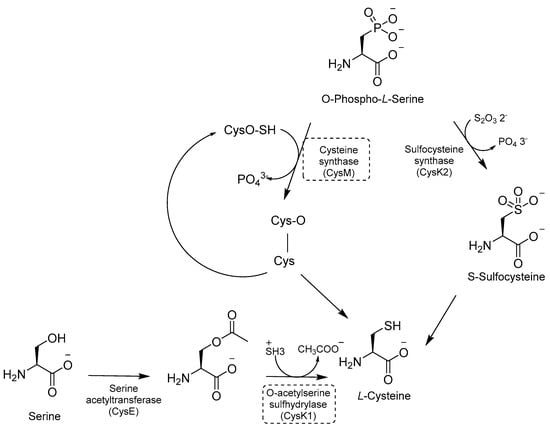
Figure 22.
A schematic representation of Cys biosynthesis. The most relevant targets are shown in dotted boxes.
Inhibitors developed in the past decade have provided valuable tools to investigate the role of the Cys pathway in Mtb (Table 3) [114,115,116,117]. However, in vivo efficacies remain to be tested, along with the need for clear target validation. Therefore, none of them have further progressed, and research in this field has been stalled in the past five years. Moreover, the existence of three different pathways is intricated, and further gene knock-out and animal models are needed to demonstrate their essentiality for Mtb survival [113].
Besides the de novo biosynthetic pathway, Cys can also be produced by a less known and poorly characterized reverse transsulfuration pathway, which is the only source of glutathione and Cys in eukaryotes. Cystathionine β-synthase (MtbCbs) has recently been characterized and appears to be an important regulator of sulfur metabolism in Mtb, but its druggability has not yet been validated [118].
Table 3.
Chemical structures, SAR plan, and most active inhibitors of Cys biosynthesis.
Table 3.
Chemical structures, SAR plan, and most active inhibitors of Cys biosynthesis.
| Target | Hit/Scaffold | SAR Plan from Hit | Most Advanced Analogue | Refs. |
|---|---|---|---|---|
| CysK1 | 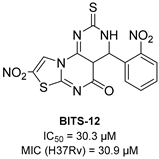 | 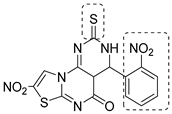 | 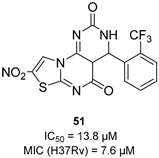 | [114] |
| CysK1 | 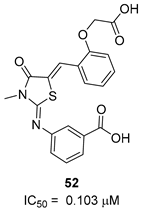 |  |  | [115] |
| CysM |  |  | 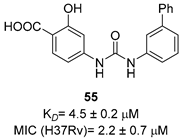 | [116,117] |
2.10. Inhibitors of Histidine (His) Biosynthesis
Besides its proteogenic and catalytic functions, His is also an important regulator of several cellular processes, such as cellular pH maintenance and metal chelation. While bacteria and plants possess enzymes for de novo biosynthesis, it is an essential amino acid for humans. Even though enzymes of this biosynthetic pathway have been explored for decades as potential targets for TB treatment [119], the importance of His in an in vivo setting was only elucidated recently [120]. His is one of the most abundant amino acids in the host intracellular milieu. Nonetheless, His auxotrophs fail to infect immunocompetent mice, as His restriction is a host adaptive immunity mechanism to contain Mtb infection. When Mtb infects macrophages, a CD4 T-cell-mediated mechanism induces an enhanced production of IFN-γ, which activates downstream signaling to upregulate His ammonia-lyase (HAL) and His decarboxylase (HDC), two host catabolizing enzymes. Similar to Trp, this mechanism reduces His availability to Mtb, which counteracts the immune response by activating the de novo His biosynthetic pathway. His biosynthesis is then an immune evasion strategy and is vital for Mtb to persist in the host microenvironment. Therefore, inhibitors of this pathway can synergize with the host in clearing the infection and can lead to rapid bacterial clearance.
The His pathway starts from phosphorybosylpyrophosphate (PRPP), which is converted to His through ten enzymatic reactions [119]. Many enzymes of this pathway have been biochemically and structurally characterized to guide the rational development of specific inhibitors based on a structure-based approach [121,122,123,124,125,126]. However, only few of them were validated through target-specific inhibitors. One of the most well-characterized targets is an ATP-phosphorybosyltransferase (ATP-PRTase, HisG), which catalyzes Mg2+-dependent nucleophilic substitution of PRPP using ATP to produce phosphoribosyl-ATP and inorganic pyrophosphate (Figure 23A).
Figure 23.
(A) A schematic representation of the first step of His biosynthesis, catalyzed by HisG; (B) the chemical structures and antimycobacterial activities of compounds 56 and 57.
With the His pathway being metabolically expensive (10 enzymatic reactions and 41 ATP molecules consumed), the activity of HisG is allosterically regulated by His through negative feedback [122,127]. Transposon insertion experiments [128] and knockout studies [129] showed that it is essential for in vivo growth, highlighting its potential as a drug target. In 2008, a virtual screening of more than 500,000 molecules identified diverse potential inhibitors [127]. Interestingly, several of the hits contain a nitrobenzothiazole fragment, opening the way to new possibilities for drug design and hit optimization against this target. Inspired by these findings, Dhameliya and coworkers have explored the potential of this scaffold in the past decade [130,131]. Hit optimization studies allowed for the identification of two new chemotypes, benzo[d]thiazole-2-carboxamides and benzo[d]thiazole-2-carbanilides, which are devoid of the potential mutagenicity of the nitro group and match with the topological features of PR-ATP, the natural ligand of PRPP transferase. An extensive SAR study led to 56 and 57, the most potent HisG inhibitors reported to date, with EC50 values of 20 ± 2.2 and 14 ± 1.8 µM, respectively, good antimycobacterial activities (2.2 and 2.7 µM, respectively), and non-cytotoxic behavior (Figure 23B). A His complementation assay confirmed that these compounds acted on His biosynthesis. Biochemical and biophysical studies revealed competitive inhibition towards ATP and also revealed that they induced a significant change in the HisG secondary structure. These data were corroborated by docking and molecular dynamics simulations, suggesting that they form a stable complex with the target and bind to the catalytic cleft at the interface between Domain I and Domain II.
Looking at the future, two interesting targets of recent characterization are imidazoleglycerolphosphate dehydratase (IGPD) and histidinol phosphate phosphatase (HolPase), catalyzing the sixth and the eight steps of the pathway, respectively [124,126].
2.11. Inhibitors of Ala Biosynthesis
Ala is a substrate for protein synthesis and an important component for the synthesis of cell wall peptidoglycan. It is synthesized by Ala dehydrogenase (Ald), a NADH-dependent enzyme which catalyzes the reductive amination of pyruvate and its reverse reaction [13]. While ald knockout strains are not auxotroph for Ala [12], the absence of Ald affects mycobacterial growth under anaerobic conditions [13]. This suggests that Mtb could possess an alternative Ala biosynthetic pathway and that Ald could be involved in mycobacterial survival under oxygen-limiting conditions [13,132,133]. The most important role of Ald is the regulation of redox homeostasis. Hypoxic and starvation conditions encountered by mycobacteria in the granuloma, as well as an altered function of the respiratory chain, cause a shift of the NAD+/NADH balance toward a reduced state, which promotes the Ald-mediated pyruvate conversion to Ala and concomitant NADH oxidation, increasing the Ala cellular levels [13]. Moreover, Ald activity is regulated by Ala availability, which, under anaerobic conditions, strongly upregulates ald. Differently from other biosynthetic pathways, the main aim of the application of Ald inhibitors is to disrupt the redox balance in nonreplicating mycobacteria rather than to target Ala biosynthesis to induce starvation. The availability of its crystal structure served as the structural framework for many virtual screenings against this target [134,135]. The most recent study identified a series of adenosine-based inhibitors using a versatile label-free assay [136]. A first round of screening identified an N6-methyladenosine inhibitor, which occupies the NAD binding site of Ald. This inhibitor was co-crystallized and optimized through a rational design based on the crystal structure. Since the N6-methyl moiety extends and interacts with a hydrophobic groove structure, a series of N6-alkyl bulkier analogues was synthesized to evaluate hydrophobicity contributions and increase affinity by extending hydrophobic interactions. These structural changes substantially improved the affinity for the target, increasing the affinity by a factor of ten (N-6-isobutyl derivative Ki= 80 µM). This study describes a very versatile screening platform, providing an efficient drug discovery strategy which could serve to identify several hits against this target. However, it is limited by the absence of activity confirmation in Mtb.
3. Conclusions
Although it is responsible for a preventable and curable disease, Mtb remains among the world’s top infectious killers, causing over one million deaths annually. Significant concern arises from the emergence of MDR and XDR strains, fueled by the length of anti-TB treatment and HIV comorbidity. Indeed, the rise of drug resistance has significantly increased treatment failure rates and the need for long and far more toxic and costly therapies [137]. To shorten treatment duration, an ideal anti-TB drug should be amenable to oral administration, safe in association, less toxic, and more effective and have an innovative mechanism of action. In recent decades, a renewed interest in TB drug discovery and the combined efforts of academia and industry have significantly fueled the TB drug pipeline, providing a positive perspective of the future of TB drug discovery. Even though this pipeline contains novel chemical scaffolds and a variety of targets, there are still some redundancies, and drugs acting with novel mechanisms of action remain unrepresented. Until recently, Mtb metabolism has been seldom studied from a drug discovery perspective due to the belief that Mtb can scavenge amino acids from the host and subsequently reverse the activity. More recently, several studies with amino acid auxotrophs [5] have reconsidered this hypothesis, proving that subverting metabolic restrictions is an essential mechanism to escape host immune surveillance and that unbalanced metabolic fluxes are bactericidal during different stages of infection. Therefore, altering amino acid metabolism and host responses critical for amino acid starvation could provide new avenues for TB drug discovery [6]. However, not all metabolic pathways are suitable for targeting, as some of them are produced by multiple enzymes. Moreover, information regarding amino acid transporters is limited, which hampers our understanding of competition mechanisms and the overall link between host and mycobacteria metabolism [138]. Therefore, it is crucial to understand which pathways are essential, which enzymes are the most important, and whether these enzymes are druggable [5]. In the past five years, the multifaceted bactericidal mechanism of amino acid starvation has gained considerable attention, and a better understanding of amino acid biosynthetic pathways has paved the way for target-based screenings against defined protein targets of the pathways. Many inhibitors of key enzymes of several amino acid biosynthetic pathways have been discovered, proving their chemical vulnerability and providing optimism for future advances [95]. While several studies have reported nanomolar enzymatic inhibitions, there is still a lack of cell and animal experiments. Many studies are still at the level of in silico prediction or biochemical experiments, with only a few demonstrating documented activity against whole-cell Mtb. For many of these potential inhibitors, crossing the complex Mtb cell envelope and avoiding efflux expulsion to engage their targets could be extremely challenging [55]. As frequently occurs with novel pathways, research has focused more on validating targets and discovering new chemical scaffolds despite optimizing existing hits. Yet many hits have been discovered over the past five years, most of them still suffer from low bactericidal activity and a lack of in vivo validation, and extensive improvements in their drug-like properties are required. Among these, Trp inhibitors stand out as the most advanced and have significantly contributed to demonstrating the feasibility of this approach for future TB drug discovery. Future efforts should be directed at prioritizing the chemical optimization of these hits to enrich the TB drug pipeline with high-quality leads.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Global Tuberculosis Report. 2023. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023 (accessed on 27 February 2024).
- Pecoraro, C.; Carbone, D.; Parrino, B.; Cascioferro, S.; Diana, P. Recent Developments in the Inhibition of Bacterial Adhesion as Promising Anti-Virulence Strategy. Int. J. Mol. Sci. 2023, 24, 4872. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.S.; Thompson, A.M.; Castagnolo, D.; Denny, W.A.; Dos Santos, J.L. Tuberculosis Drug Discovery: Challenges and New Horizons. J. Med. Chem. 2022, 65, 7489–7531. [Google Scholar] [CrossRef] [PubMed]
- Wellington, S.; Hung, D.T. The Expanding Diversity of Mycobacterium tuberculosis Drug Targets. ACS Infect. Dis. 2018, 4, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Berney, M.; Berney-Meyer, L. Mycobacterium tuberculosis in the Face of Host-Imposed Nutrient Limitation. Microbiol. Spectr. 2017, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Reddy, M.C.; Ioerger, T.R.; Rothchild, A.C.; Dartois, V.; Schuster, B.M.; Trauner, A.; Wallis, D.; Galaviz, S.; Huttenhower, C.; et al. Tryptophan Biosynthesis Protects Mycobacteria from CD4 T-Cell-Mediated Killing. Cell 2013, 155, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Awasthy, D.; Gaonkar, S.; Shandil, R.K.; Yadav, R.; Bharath, S.; Marcel, N.; Subbulakshmi, V.; Sharma, U. Inactivation of the ilvB1 Gene in Mycobacterium tuberculosis Leads to Branched-Chain Amino Acid Auxotrophy and Attenuation of Virulence in Mice. Microbiology 2009, 155, 2978–2987. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; van Tonder, A.J.; Vilchèze, C.; Mendes, V.; Thomas, S.E.; Malek, A.; Chen, B.; Chen, M.; Kim, J.; Blundell, T.L.; et al. Arginine-Deprivation-Induced Oxidative Damage Sterilizes Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Gouzy, A.; Poquet, Y.; Neyrolles, O. Nitrogen Metabolism in Mycobacterium tuberculosis Physiology and Virulence. Nat. Rev. Microbiol. 2014, 12, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Hasenoehrl, E.J.; Rae Sajorda, D.; Berney-Meyer, L.; Johnson, S.; Tufariello, J.M.; Fuhrer, T.; Cook, G.M.; Jacobs, W.R.; Berney, M. Derailing the Aspartate Pathway of Mycobacterium tuberculosis to Eradicate Persistent Infection. Nat. Commun. 2019, 10, 4215. [Google Scholar] [CrossRef]
- Berney, M.; Berney-Meyer, L.; Wong, K.-W.; Chen, B.; Chen, M.; Kim, J.; Wang, J.; Harris, D.; Parkhill, J.; Chan, J.; et al. Essential Roles of Methionine and S-Adenosylmethionine in the Autarkic Lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 10008–10013. [Google Scholar] [CrossRef]
- Agapova, A.; Serafini, A.; Petridis, M.; Hunt, D.M.; Garza-Garcia, A.; Sohaskey, C.D.; de Carvalho, L.P.S. Flexible Nitrogen Utilisation by the Metabolic Generalist Pathogen Mycobacterium tuberculosis. Elife 2019, 8, e41129. [Google Scholar] [CrossRef]
- Jeong, J.-A.; Oh, J.-I. Alanine Dehydrogenases in Mycobacteria. J. Microbiol. 2019, 57, 81–92. [Google Scholar] [CrossRef]
- Nunes, J.E.S.; Duque, M.A.; de Freitas, T.F.; Galina, L.; Timmers, L.F.S.M.; Bizarro, C.V.; Machado, P.; Basso, L.A.; Ducati, R.G. Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs. Molecules 2020, 25, 1259. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. The Common Aromatic Amino Acid Biosynthesis Pathway Is Essential in Mycobacterium tuberculosis. Microbiology 2002, 148, 3069–3077. [Google Scholar] [CrossRef]
- Galina, L.; Hopf, F.S.M.; Abbadi, B.L.; de Moura Sperotto, N.D.; Czeczot, A.M.; Duque-Villegas, M.A.; Perello, M.A.; Matter, L.B.; de Souza, E.V.; Parish, T.; et al. Evaluation of 3-Deoxy-D-Arabino-Heptulosonate 7-Phosphate Synthase (DAHPS) as a Vulnerable Target in Mycobacterium tuberculosis. Microbiol. Spectr. 2022, 10, e0072822. [Google Scholar] [CrossRef]
- Rajput, V.S.; Mehra, R.; Kumar, S.; Nargotra, A.; Singh, P.P.; Khan, I.A. Screening of Antitubercular Compound Library Identifies Novel Shikimate Kinase Inhibitors of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2016, 100, 5415–5426. [Google Scholar] [CrossRef]
- Miranda, P.H.d.S.; Lourenço, E.M.G.; Morais, A.M.S.; de Oliveira, P.I.C.; Silverio, P.S.d.S.N.; Jordão, A.K.; Barbosa, E.G. Molecular Modeling of a Series of Dehydroquinate Dehydratase Type II Inhibitors of Mycobacterium tuberculosis and Design of New Binders. Mol. Divers. 2021, 25, 1–12. [Google Scholar] [CrossRef]
- Freitas de Freitas, T.; Roth, C.D.; Abbadi, B.L.; Hopf, F.S.M.; Perelló, M.A.; de Matos Czeczot, A.; de Souza, E.V.; Borsoi, A.F.; Machado, P.; Bizarro, C.V.; et al. Identification of Potential Inhibitors of Mycobacterium tuberculosis Shikimate Kinase: Molecular Docking, in Silico Toxicity and in Vitro Experiments. J. Comput. Aided Mol. Des. 2023, 37, 117–128. [Google Scholar] [CrossRef]
- Rajput, V.S.; Runthala, A.; Khan, I.A. Shikimate Kinase Inhibitors: An Update on Promising Strategy against Mycobacterium tuberculosis. Curr. Drug Targets 2023, 24, 388–405. [Google Scholar] [CrossRef]
- Deng, Q.; Meng, J.; Liu, Y.; Guan, Y.; Xiao, C. IMB-SD62, a Triazolothiadiazoles Derivative with Promising Action against Tuberculosis. Tuberculosis 2018, 112, 37–44. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, X.; Li, D.; Lin, Y.; You, X.; Jiang, J.; Xu, Y.; Jiang, W.; Si, S. IMB-T130 Targets 3-Dehydroquinate Synthase and Inhibits Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 17439. [Google Scholar] [CrossRef]
- Carbone, D.; Pecoraro, C.; Scianò, F.; Catania, V.; Schillaci, D.; Manachini, B.; Cascioferro, S.; Diana, P.; Parrino, B. Novel [1,3,4]Thiadiazole[3,2-a]Pyrimidin-5-Ones as Promising Biofilm Dispersal Agents against Relevant Gram-Positive and Gram-Negative Pathogens. Mar. Drugs 2024, 22, 133. [Google Scholar] [CrossRef]
- Simithy, J.; Fuanta, N.R.; Alturki, M.; Hobrath, J.V.; Wahba, A.E.; Pina, I.; Rath, J.; Hamann, M.T.; DeRuiter, J.; Goodwin, D.C.; et al. Slow-Binding Inhibition of Mycobacterium tuberculosis Shikimate Kinase by Manzamine Alkaloids. Biochemistry 2018, 57, 4923–4933. [Google Scholar] [CrossRef]
- Rahul Reddy, M.B.; Krishnasamy, S.K.; Kathiravan, M.K. Identification of Novel Scaffold Using Ligand and Structure Based Approach Targeting Shikimate Kinase. Bioorg. Chem. 2020, 102, 104083. [Google Scholar] [CrossRef]
- Kawamoto, S.; Hori, C.; Taniguchi, H.; Okubo, S.; Aoki, S. Identification of Novel Antimicrobial Compounds Targeting Mycobacterium tuberculosis Shikimate Kinase Using in Silico Hierarchical Structure-Based Drug Screening. Tuberculosis 2023, 141, 102362. [Google Scholar] [CrossRef]
- Dadlani, V.G.; Chhabhaiya, H.; Somani, R.R.; Tripathi, P.K. Synthesis, Molecular Docking, and Biological Evaluation of Novel 1,2,4-Triazole-Isatin Derivatives as Potential Mycobacterium tuberculosis Shikimate Kinase Inhibitors. Chem. Biol. Drug Des. 2022, 100, 230–244. [Google Scholar] [CrossRef]
- Duque-Villegas, M.A.; Abbadi, B.L.; Romero, P.R.; Matter, L.B.; Galina, L.; Dalberto, P.F.; Rodrigues-Junior, V.d.S.; Ducati, R.G.; Roth, C.D.; Rambo, R.S.; et al. EPSP Synthase-Depleted Cells Are Aromatic Amino Acid Auxotrophs in Mycobacterium Smegmatis. Microbiol. Spectr. 2021, 9, e0000921. [Google Scholar] [CrossRef]
- Khanapur, M.; Alvala, M.; Prabhakar, M.; Shiva Kumar, K.; Edwin, R.K.; Sri Saranya, P.S.V.K.; Patel, R.K.; Bulusu, G.; Misra, P.; Pal, M. Mycobacterium tuberculosis Chorismate Mutase: A Potential Target for TB. Bioorg. Med. Chem. 2017, 25, 1725–1736. [Google Scholar] [CrossRef]
- Shukla, S.; Nishanth Rao, R.; Bhuktar, H.; Edwin, R.K.; Jamma, T.; Medishetti, R.; Banerjee, S.; Giliyaru, V.B.; Shenoy, G.G.; Oruganti, S.; et al. Wang Resin Catalysed Sonochemical Synthesis of Pyrazolo[4,3-d]Pyrimidinones and 2,3-Dihydroquinazolin-4(1H)-Ones: Identification of Chorismate Mutase Inhibitors Having Effects on Mycobacterium tuberculosis Cell Viability. Bioorg. Chem. 2023, 134, 106452. [Google Scholar] [CrossRef]
- Dahmer, B.R.; Ethur, E.M.; Timmers, L.F.S.M. Discovery of New Inhibitors of Mycobacterium tuberculosis EPSP Synthase—A Computational Study. J. Mol. Graph. Model. 2023, 121, 108404. [Google Scholar] [CrossRef]
- Naz, S.; Farooq, U.; Khan, S.; Sarwar, R.; Mabkhot, Y.N.; Saeed, M.; Alsayari, A.; Muhsinah, A.B.; Ul-Haq, Z. Pharmacophore Model-Based Virtual Screening, Docking, Biological Evaluation and Molecular Dynamics Simulations for Inhibitors Discovery against α-Tryptophan Synthase from Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2021, 39, 610–620. [Google Scholar] [CrossRef]
- Consalvi, S.; Scarpecci, C.; Biava, M.; Poce, G. Mycobacterial Tryptophan Biosynthesis: A Promising Target for Tuberculosis Drug Development? Bioorg. Med. Chem. Lett. 2019, 29, 126731. [Google Scholar] [CrossRef] [PubMed]
- Adu-Gyamfi, C.G.; Savulescu, D.; George, J.A.; Suchard, M.S. Indoleamine 2, 3-Dioxygenase-Mediated Tryptophan Catabolism: A Leading Star or Supporting Act in the Tuberculosis and HIV Pas-de-Deux? Front. Cell Infect. Microbiol. 2019, 9, 372. [Google Scholar] [CrossRef]
- Lott, J.S. The Tryptophan Biosynthetic Pathway Is Essential for Mycobacterium tuberculosis to Cause Disease. Biochem. Soc. Trans. 2020, 48, 2029–2037. [Google Scholar] [CrossRef]
- Amalia, F.; Syamsunarno, M.R.A.A.; Triatin, R.D.; Fatimah, S.N.; Chaidir, L.; Achmad, T.H. The Role of Amino Acids in Tuberculosis Infection: A Literature Review. Metabolites 2022, 12, 933. [Google Scholar] [CrossRef]
- Nurul Islam, M.; Hitchings, R.; Kumar, S.; Fontes, F.L.; Lott, J.S.; Kruh-Garcia, N.A.; Crick, D.C. Mechanism of Fluorinated Anthranilate-Induced Growth Inhibition in Mycobacterium tuberculosis. ACS Infect. Dis. 2019, 5, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Venditti, G.; Zhu, J.; Boshoff, H.I.; Arora, K.; De Logu, A.; Ioerger, T.R.; Rubin, E.J.; Biava, M.; Poce, G. 6-Fluorophenylbenzohydrazides Inhibit Mycobacterium tuberculosis Growth through Alteration of Tryptophan Biosynthesis. Eur. J. Med. Chem. 2021, 226, 113843. [Google Scholar] [CrossRef] [PubMed]
- Libardo, M.D.J.; Duncombe, C.J.; Green, S.R.; Wyatt, P.G.; Thompson, S.; Ray, P.C.; Ioerger, T.R.; Oh, S.; Goodwin, M.B.; Boshoff, H.I.M.; et al. Resistance of Mycobacterium tuberculosis to Indole 4-Carboxamides Occurs through Alterations in Drug Metabolism and Tryptophan Biosynthesis. Cell Chem. Biol. 2021, 28, 1180–1191. [Google Scholar] [CrossRef]
- Jackson, M. An Antitubercular Prodrug Leaves Mycobacterium tuberculosis Facing a Difficult Choice, Poisoning or Starvation? Cell Chem. Biol. 2021, 28, 1101–1103. [Google Scholar] [CrossRef]
- Yelamanchi, S.D.; Surolia, A. Targeting Amino Acid Metabolism of Mycobacterium tuberculosis for Developing Inhibitors to Curtail Its Survival. IUBMB Life 2021, 73, 643–658. [Google Scholar] [CrossRef]
- Amorim Franco, T.M.; Hegde, S.; Blanchard, J.S. Chemical Mechanism of the Branched-Chain Aminotransferase IlvE from Mycobacterium tuberculosis. Biochemistry 2016, 55, 6295–6303. [Google Scholar] [CrossRef]
- Grandoni, J.A.; Marta, P.T.; Schloss, J.V. Inhibitors of Branched-Chain Amino Acid Biosynthesis as Potential Antituberculosis Agents. J. Antimicrob. Chemother. 1998, 42, 475–482. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, X.; Cui, C.; Dong, M.; Jiang, H.; Li, Z.; Liu, Z.; Zhu, W.; Wang, J.-G. Discovery of Novel Acetohydroxyacid Synthase Inhibitors as Active Agents against Mycobacterium tuberculosis by Virtual Screening and Bioassay. J. Chem. Inf. Model. 2013, 53, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Kale, M.; Raichurkar, A.; Bhaskar, B.; Prahlad, D.; Balganesh, M.; Nandan, S.; Shahul Hameed, P. Design and Synthesis of Triazolopyrimidine Acylsulfonamides as Novel Anti-Mycobacterial Leads Acting through Inhibition of Acetohydroxyacid Synthase. Bioorg. Med. Chem. Lett. 2014, 24, 2222–2225. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Baig, I.A.; Sun, H.-J.; Cui, C.-J.; Guo, R.; Jung, I.-P.; Wang, D.; Dong, M.; Yoon, M.-Y.; Wang, J.-G. Synthesis, Crystal Structure and Biological Evaluation of Substituted Quinazolinone Benzoates as Novel Antituberculosis Agents Targeting Acetohydroxyacid Synthase. Eur. J. Med. Chem. 2015, 94, 298–305. [Google Scholar] [CrossRef]
- Rehberg, N.; Akone, H.S.; Ioerger, T.R.; Erlenkamp, G.; Daletos, G.; Gohlke, H.; Proksch, P.; Kalscheuer, R. Chlorflavonin Targets Acetohydroxyacid Synthase Catalytic Subunit IlvB1 for Synergistic Killing of Mycobacterium tuberculosis. ACS Infect. Dis. 2018, 4, 123–134. [Google Scholar] [CrossRef]
- Berger, A.; Knak, T.; Kiffe-Delf, A.-L.; Mudrovcic, K.; Singh, V.; Njoroge, M.; Burckhardt, B.B.; Gopalswamy, M.; Lungerich, B.; Ackermann, L.; et al. Total Synthesis of the Antimycobacterial Natural Product Chlorflavonin and Analogs via a Late-Stage Ruthenium(II)-Catalyzed Ortho-C(Sp2)-H-Hydroxylation. Pharmaceuticals 2022, 15, 984. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Wang, S.; Carr, R.; Trembleau, L.; Deng, H. Characterization of a Class II Ketol-Acid Reductoisomerase from Mycobacterium tuberculosis. RSC Adv. 2022, 12, 10540–10544. [Google Scholar] [CrossRef]
- Lin, X.; Kurz, J.L.; Patel, K.M.; Wun, S.J.; Hussein, W.M.; Lonhienne, T.; West, N.P.; McGeary, R.P.; Schenk, G.; Guddat, L.W. Discovery of a Pyrimidinedione Derivative with Potent Inhibitory Activity against Mycobacterium tuberculosis Ketol-Acid Reductoisomerase. Chemistry 2021, 27, 3130–3141. [Google Scholar] [CrossRef]
- Bayaraa, T.; Kurz, J.L.; Patel, K.M.; Hussein, W.M.; Bilyj, J.K.; West, N.P.; Schenk, G.; McGeary, R.P.; Guddat, L.W. Discovery, Synthesis and Evaluation of a Ketol-Acid Reductoisomerase Inhibitor. Chemistry 2020, 26, 8958–8968. [Google Scholar] [CrossRef]
- Kandale, A.; Patel, K.; Hussein, W.M.; Wun, S.J.; Zheng, S.; Tan, L.; West, N.P.; Schenk, G.; Guddat, L.W.; McGeary, R.P. Analogues of the Herbicide, N-Hydroxy-N-Isopropyloxamate, Inhibit Mycobacterium tuberculosis Ketol-Acid Reductoisomerase and Their Prodrugs Are Promising Anti-TB Drug Leads. J. Med. Chem. 2021, 64, 1670–1684. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.S.; Zheng, S.; Rekha, E.M.; Guddat, L.W.; Sriram, D. Discovery and Evaluation of Novel Mycobacterium tuberculosis Ketol-Acid Reductoisomerase Inhibitors as Therapeutic Drug Leads. J. Comput. Aided Mol. Des. 2019, 33, 357–366. [Google Scholar] [CrossRef]
- Krishna, V.S.; Zheng, S.; Rekha, E.M.; Nallangi, R.; Sai Prasad, D.V.; George, S.E.; Guddat, L.W.; Sriram, D. Design and Development of ((4-Methoxyphenyl)Carbamoyl) (5-(5-Nitrothiophen-2-Yl)-1,3,4-Thiadiazol-2-Yl)Amide Analogues as Mycobacterium tuberculosis Ketol-Acid Reductoisomerase Inhibitors. Eur. J. Med. Chem. 2020, 193, 112178. [Google Scholar] [CrossRef]
- Mizrahi, V.; Warner, D.F. Death of Mycobacterium tuberculosis by L-Arginine Starvation. Proc. Natl. Acad. Sci. USA 2018, 115, 9658–9660. [Google Scholar] [CrossRef]
- Yang, X.; Wu, L.; Ran, Y.; Xu, A.; Zhang, B.; Yang, X.; Zhang, R.; Rao, Z.; Li, J. Crystal Structure of L-Glutamate N-Acetyltransferase ArgA from Mycobacterium tuberculosis. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2017, 1865, 1800–1807. [Google Scholar] [CrossRef]
- Das, U.; Singh, E.; Dharavath, S.; Tiruttani Subhramanyam, U.K.; Pal, R.K.; Vijayan, R.; Menon, S.; Kumar, S.; Gourinath, S.; Srinivasan, A. Structural Insights into the Substrate Binding Mechanism of Novel ArgA from Mycobacterium tuberculosis. Int. J. Biol. Macromol. 2019, 125, 970–978. [Google Scholar] [CrossRef]
- Khurana, H.; Srivastava, M.; Chaudhary, D.; Gosain, T.P.; Kumari, R.; Bean, A.C.; Chugh, S.; Maiti, T.K.; Stephens, C.E.; Asthana, S.; et al. Identification of Diphenyl Furan Derivatives via High Throughput and Computational Studies as ArgA Inhibitors of Mycobacterium tuberculosis. Int. J. Biol. Macromol. 2021, 193, 1845–1858. [Google Scholar] [CrossRef]
- Gupta, P.; Thomas, S.E.; Zaidan, S.A.; Pasillas, M.A.; Cory-Wright, J.; Sebastián-Pérez, V.; Burgess, A.; Cattermole, E.; Meghir, C.; Abell, C.; et al. A Fragment-Based Approach to Assess the Ligandability of ArgB, ArgC, ArgD and ArgF in the L-Arginine Biosynthetic Pathway of Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3491–3506. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Cherney, M.M.; Garen, C.; Garen, G.; Niu, C.; Yuan, M.; James, M.N.G. The Molecular Structure of Ornithine Acetyltransferase from Mycobacterium tuberculosis Bound to Ornithine, a Competitive Inhibitor. J. Mol. Biol. 2010, 397, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Rubin, E.J. Genetic Requirements for Mycobacterial Survival during Infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef]
- Mishra, A.; Mamidi, A.S.; Rajmani, R.S.; Ray, A.; Roy, R.; Surolia, A. An Allosteric Inhibitor of Mycobacterium tuberculosis ArgJ: Implications to a Novel Combinatorial Therapy. EMBO Mol. Med. 2018, 10, e8038. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R.; Upward, J.W.; Georgiou, P.; Stelman, G.; Doyle, E.; Allen, E.; Wyld, P.; Dennis, M.J. The Single and Multiple Dose Pharmacokinetics of Pranlukast in Healthy Volunteers. Eur. J. Clin. Pharmacol. 1996, 51, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Koeken, V.A.C.M.; Lachmandas, E.; Riza, A.; Matzaraki, V.; Li, Y.; Kumar, V.; Oosting, M.; Joosten, L.A.B.; Netea, M.G.; van Crevel, R. Role of Glutamine Metabolism in Host Defense Against Mycobacterium tuberculosis Infection. J. Infect. Dis. 2019, 219, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Harth, G.; Maslesa-Galić, S.; Tullius, M.V.; Horwitz, M.A. All Four Mycobacterium tuberculosis glnA Genes Encode Glutamine Synthetase Activities but Only GlnA1 Is Abundantly Expressed and Essential for Bacterial Homeostasis. Mol. Microbiol. 2005, 58, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Dou, C.; Zhang, G.; Arnaout, T.; Cheng, W. The Pathogenic Mechanism of Mycobacterium tuberculosis: Implication for New Drug Development. Mol. Biomed. 2022, 3, 48. [Google Scholar] [CrossRef]
- Tripathi, D.; Kant, S.; Garg, R.; Bhatnagar, R. Low Expression Level of glnA1 Accounts for Absence of Cell Wall Associated Poly-l-Glutamate/Glutamine in Mycobacterium Smegmatis. Biochem. Biophys. Res. Commun. 2015, 458, 240–245. [Google Scholar] [CrossRef]
- Couturier, C.; Silve, S.; Morales, R.; Pessegue, B.; Llopart, S.; Nair, A.; Bauer, A.; Scheiper, B.; Pöverlein, C.; Ganzhorn, A.; et al. Nanomolar Inhibitors of Mycobacterium tuberculosis Glutamine Synthetase 1: Synthesis, Biological Evaluation and X-Ray Crystallographic Studies. Bioorg. Med. Chem. Lett. 2015, 25, 1455–1459. [Google Scholar] [CrossRef]
- Kosikowska, P.; Bochno, M.; Macegoniuk, K.; Forlani, G.; Kafarski, P.; Berlicki, Ł. Bisphosphonic Acids as Effective Inhibitors of Mycobacterium tuberculosis Glutamine Synthetase. J. Enzyme Inhib. Med. Chem. 2016, 31, 931–938. [Google Scholar] [CrossRef]
- Chanda, A.; Kalita, S.; Mishra, A.K.; Changkakoti, L.; Sarma, J.B.; Biswas, K.; Kakati, D.; Mohanta, Y.K.; Tanti, B.; Mahanta, S.; et al. Identification of Concomitant Inhibitors against Glutamine Synthetase and Isocitrate Lyase in Mycobacterium tuberculosis from Natural Sources. Biomed. Res. Int. 2022, 2022, 4661491. [Google Scholar] [CrossRef]
- Gising, J.; Nilsson, M.T.; Odell, L.R.; Yahiaoui, S.; Lindh, M.; Iyer, H.; Sinha, A.M.; Srinivasa, B.R.; Larhed, M.; Mowbray, S.L.; et al. Trisubstituted Imidazoles as Mycobacterium tuberculosis Glutamine Synthetase Inhibitors. J. Med. Chem. 2012, 55, 2894–2898. [Google Scholar] [CrossRef]
- De Rosa, M.; Gising, J.; Odell, L.R.; Larhed, M. Syntheses of New Tuberculosis Inhibitors Promoted by Microwave Irradiation. Upsala J. Med. Sci. 2014, 119, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, S.L.; Kathiravan, M.K.; Pandey, A.A.; Odell, L.R. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis. Molecules 2014, 19, 13161–13176. [Google Scholar] [CrossRef] [PubMed]
- Dilebo, K.B.; Gumede, N.J.; Nxumalo, W.; Matsebatlela, T.M.; Mangokoana, D.; Moraone, N.R.; Omondi, B.; Mampa, R.M. Synthesis, in Vitro Cytotoxic, Anti-Mycobacterium tuberculosis and Molecular Docking Studies of 4-Pyridylamino- and 4-(Ethynylpyridine)Quinazolines. J. Mol. Struct. 2021, 1243, 130824. [Google Scholar] [CrossRef]
- Wang, H.; Bi, J.; Zhang, Y.; Pan, M.; Guo, Q.; Xiao, G.; Cui, Y.; Hu, S.; Chan, C.K.; Yuan, Y.; et al. Human Kinase IGF1R/IR Inhibitor Linsitinib Controls the In Vitro and Intracellular Growth of Mycobacterium tuberculosis. ACS Infect. Dis. 2022, 8, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Kim, E.S.; Nava-Parada, P.; Alam, S.; Johnson, F.M.; Stephens, A.W.; Simantov, R.; Poondru, S.; Gedrich, R.; Lippman, S.M.; et al. Phase I Study of Intermittent Oral Dosing of the Insulin-like Growth Factor-1 and Insulin Receptors Inhibitor OSI-906 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Jose, G.; Suresha Kumara, T.H.; Sowmya, H.B.V.; Sriram, D.; Guru Row, T.N.; Hosamani, A.A.; More, S.S.; Janardhan, B.; Harish, B.G.; Telkar, S.; et al. Synthesis, Molecular Docking, Antimycobacterial and Antimicrobial Evaluation of New Pyrrolo[3,2-c]Pyridine Mannich Bases. Eur. J. Med. Chem. 2017, 131, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Theron, A.; Roth, R.L.; Hoppe, H.; Parkinson, C.; van der Westhuyzen, C.W.; Stoychev, S.; Wiid, I.; Pietersen, R.D.; Baker, B.; Kenyon, C.P. Differential Inhibition of Adenylylated and Deadenylylated Forms of M. tuberculosis Glutamine Synthetase as a Drug Discovery Platform. PLoS ONE 2017, 12, e0185068. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; Shen, J.; Lun, S.; Zhao, L.; Alt, J.; Koleske, B.; Leone, R.D.; Rais, R.; Powell, J.D.; Murphy, J.R.; et al. Glutamine Metabolism Inhibition Has Dual Immunomodulatory and Antibacterial Activities against Mycobacterium tuberculosis. Nat. Commun. 2023, 14, 7427. [Google Scholar] [CrossRef]
- Bai, G.; Schaak, D.D.; Smith, E.A.; McDonough, K.A. Dysregulation of Serine Biosynthesis Contributes to the Growth Defect of a Mycobacterium tuberculosis Crp Mutant. Mol. Microbiol. 2011, 82, 180–198. [Google Scholar] [CrossRef]
- Haufroid, M.; Wouters, J. Targeting the Serine Pathway: A Promising Approach against Tuberculosis? Pharmaceuticals 2019, 12, 66. [Google Scholar] [CrossRef]
- Grant, G.A. D-3-Phosphoglycerate Dehydrogenase. Front. Mol. Biosci. 2018, 5, 110. [Google Scholar] [CrossRef]
- Pierson, E.; Wouters, J. Biochemical Characterization of Phosphoserine Phosphatase SerB2 from Mycobacterium Marinum. Biochem. Biophys. Res. Commun. 2020, 530, 739–744. [Google Scholar] [CrossRef]
- Arora, G.; Tiwari, P.; Mandal, R.S.; Gupta, A.; Sharma, D.; Saha, S.; Singh, R. High Throughput Screen Identifies Small Molecule Inhibitors Specific for Mycobacterium tuberculosis Phosphoserine Phosphatase. J. Biol. Chem. 2014, 289, 25149–25165. [Google Scholar] [CrossRef]
- Shree, S.; Singh, A.K.; Saxena, R.; Kumar, H.; Agarwal, A.; Sharma, V.K.; Srivastava, K.; Srivastava, K.K.; Sanyal, S.; Ramachandran, R. The M. tuberculosis HAD Phosphatase (Rv3042c) Interacts with Host Proteins and Is Inhibited by Clofazimine. Cell Mol. Life Sci. 2016, 73, 3401–3417. [Google Scholar] [CrossRef] [PubMed]
- Pierson, E.; Haufroid, M.; Gosain, T.P.; Chopra, P.; Singh, R.; Wouters, J. Identification and Repurposing of Trisubstituted Harmine Derivatives as Novel Inhibitors of Mycobacterium tuberculosis Phosphoserine Phosphatase. Molecules 2020, 25, 415. [Google Scholar] [CrossRef]
- Haufroid, M.; Volkov, A.N.; Wouters, J. Targeting the Phosphoserine Phosphatase MtSerB2 for Tuberculosis Drug Discovery, an Hybrid Knowledge Based/Fragment Based Approach. Eur. J. Med. Chem. 2023, 245, 114935. [Google Scholar] [CrossRef] [PubMed]
- Pierson, E.; De Pol, F.; Fillet, M.; Wouters, J. A Morpheein Equilibrium Regulates Catalysis in Phosphoserine Phosphatase SerB2 from Mycobacterium tuberculosis. Commun. Biol. 2023, 6, 1024. [Google Scholar] [CrossRef]
- Christgen, S.L.; Becker, D.F. Role of Proline in Pathogen and Host Interactions. Antioxid. Redox Signal 2019, 30, 683–709. [Google Scholar] [CrossRef] [PubMed]
- Fichman, Y.; Gerdes, S.Y.; Kovács, H.; Szabados, L.; Zilberstein, A.; Csonka, L.N. Evolution of Proline Biosynthesis: Enzymology, Bioinformatics, Genetics, and Transcriptional Regulation. Biol. Rev. Camb. Philos. Soc. 2015, 90, 1065–1099. [Google Scholar] [CrossRef]
- Lagautriere, T.; Bashiri, G.; Paterson, N.G.; Berney, M.; Cook, G.M.; Baker, E.N. Characterization of the Proline-Utilization Pathway in Mycobacterium tuberculosis through Structural and Functional Studies. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 968–980. [Google Scholar] [CrossRef]
- Kumar, S.; Sega, S.; Lynn-Barbe, J.K.; Harris, D.L.; Koehn, J.T.; Crans, D.C.; Crick, D.C. Proline Dehydrogenase and Pyrroline 5 Carboxylate Dehydrogenase from Mycobacterium tuberculosis: Evidence for Substrate Channeling. Pathogens 2023, 12, 1171. [Google Scholar] [CrossRef] [PubMed]
- Makafe, G.G.; Hussain, M.; Surineni, G.; Tan, Y.; Wong, N.-K.; Julius, M.; Liu, L.; Gift, C.; Jiang, H.; Tang, Y.; et al. Quinoline Derivatives Kill Mycobacterium tuberculosis by Activating Glutamate Kinase. Cell Chem. Biol. 2019, 26, 1187–1194.e5. [Google Scholar] [CrossRef]
- Panciera, M.; Lence, E.; Rodríguez, Á.; Gracia, B.; Aínsa, J.A.; Marco-Marín, C.; Rubio, V.; Duarte Correia, C.R.; González-Bello, C. Discovery of 3H-Pyrrolo[2,3-c]Quinolines with Activity against Mycobacterium tuberculosis by Allosteric Inhibition of the Glutamate-5-Kinase Enzyme. Eur. J. Med. Chem. 2022, 232, 114206. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, S.; Huang, Z.; Wang, L.; Raza, S.H.A.; Wang, Z. Nitrogen Metabolism in Mycobacteria: The Key Genes and Targeted Antimicrobials. Front. Microbiol. 2023, 14, 1149041. [Google Scholar] [CrossRef]
- Gouzy, A.; Poquet, Y.; Neyrolles, O. Amino Acid Capture and Utilization within the Mycobacterium tuberculosis Phagosome. Future Microbiol. 2014, 9, 631–637. [Google Scholar] [CrossRef]
- Gouzy, A.; Larrouy-Maumus, G.; Bottai, D.; Levillain, F.; Dumas, A.; Wallach, J.B.; Caire-Brandli, I.; de Chastellier, C.; Wu, T.-D.; Poincloux, R.; et al. Mycobacterium tuberculosis Exploits Asparagine to Assimilate Nitrogen and Resist Acid Stress during Infection. PLoS Pathog. 2014, 10, e1003928. [Google Scholar] [CrossRef]
- Kataria, A.; Singh, J.; Kundu, B. Identification and Validation of L-Asparaginase as a Potential Metabolic Target against Mycobacterium tuberculosis. J. Cell Biochem. 2019, 120, 143–154. [Google Scholar] [CrossRef]
- Meng, J.; Yang, Y.; Xiao, C.; Guan, Y.; Hao, X.; Deng, Q.; Lu, Z. Identification and Validation of Aspartic Acid Semialdehyde Dehydrogenase as a New Anti-Mycobacterium tuberculosis Target. Int. J. Mol. Sci. 2015, 16, 23572–23586. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Garg, P.; Bharatam, P.V. Shape-Based Virtual Screening, Docking, and Molecular Dynamics Simulations to Identify Mtb-ASADH Inhibitors. J. Biomol. Struct. Dyn. 2015, 33, 1082–1093. [Google Scholar] [CrossRef]
- Kumar, R.; Sethi, R.; Shah, P.; Roy, I.; Singh, I.P.; Bharatam, P.V.; Tewari, R.; Garg, P. Biological Evaluation of Small Molecule Inhibitors of Mtb-ASADH Enzyme. Lett. Drug Des. Discov. 2016, 13, 587–590. [Google Scholar] [CrossRef]
- Wang, X.; Yang, R.; Liu, S.; Guan, Y.; Xiao, C.; Li, C.; Meng, J.; Pang, Y.; Liu, Y. IMB-XMA0038, a New Inhibitor Targeting Aspartate-Semialdehyde Dehydrogenase of Mycobacterium tuberculosis. Emerg. Microbes Infect. 2021, 10, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Cao, W.; Liu, S.; Li, Q.; Sun, Y.; Liang, C.; Ren, W.; Liu, Y.; Meng, J.; Li, C. Evaluation of a Novel Inhibitor of Aspartate Semialdehyde Dehydrogenase as a Potent Antitubercular Agent against Mycobacterium tuberculosis. J. Antibiot. 2022, 75, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Angrish, N.; Lalwani, N.; Khare, G. In Silico Virtual Screening for the Identification of Novel Inhibitors against Dihydrodipicolinate Reductase (DapB) of Mycobacterium tuberculosis, a Key Enzyme of Diaminopimelate Pathway. Microbiol. Spectr. 2023, 11, e0135923. [Google Scholar] [CrossRef] [PubMed]
- Ayu Eka Pitaloka, D.; Izzati, A.; Rafa Amirah, S.; Abdan Syakuran, L.; Muhammad Irham, L.; Darumas Putri, A.; Adikusuma, W. Bioinformatics Analysis to Uncover the Potential Drug Targets Responsible for Mycobacterium tuberculosis Peptidoglycan and Lysine Biosynthesis. Bioinform. Biol. Insights 2023, 17, 11779322231171774. [Google Scholar] [CrossRef] [PubMed]
- Peverelli, M.G.; da Costa, T.P.S.; Kirby, N.; Perugini, M.A. Dimerization of Bacterial Diaminopimelate Decarboxylase Is Essential for Catalysis. J. Biol. Chem. 2016, 291, 9785. [Google Scholar] [CrossRef]
- Shrivastava, P.; Navratna, V.; Silla, Y.; Dewangan, R.P.; Pramanik, A.; Chaudhary, S.; Rayasam, G.; Kumar, A.; Gopal, B.; Ramachandran, S. Inhibition of Mycobacterium tuberculosis Dihydrodipicolinate Synthase by Alpha-Ketopimelic Acid and Its Other Structural Analogues. Sci. Rep. 2016, 6, 30827. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Tewari, R.; Raghava, G.P.S. Virtual Screening of Potential Drug-like Inhibitors against Lysine/DAP Pathway of Mycobacterium tuberculosis. BMC Bioinform. 2010, 11 (Suppl. 1), S53. [Google Scholar] [CrossRef] [PubMed]
- Bhargav, A.; Chaurasia, P.; Ivanisenko, N.; Ivanisenko, V.; Taneja, B.; Ramachandran, S. Screening and Identification of Novel Small Molecule Inhibitors against Mycobacterium tuberculosis Dihydrodipicolinate Synthase Enzyme Using In Silico and In Vitro Method. Preprints 2022, 2022050349. [Google Scholar] [CrossRef]
- Rehman, A.; Akhtar, S.; Siddiqui, M.H.; Sayeed, U.; Ahmad, S.S.; Arif, J.M.; Khan, M.K.A. Identification of Potential Leads against 4-Hydroxytetrahydrodipicolinate Synthase from Mycobacterium tuberculosis. Bioinformation 2016, 12, 400–407. [Google Scholar] [CrossRef]
- Sharma, S.; Jayasinghe, Y.P.; Mishra, N.K.; Orimoloye, M.O.; Wong, T.-Y.; Dalluge, J.J.; Ronning, D.R.; Aldrich, C.C. Structural and Functional Characterization of Mycobacterium tuberculosis Homoserine Transacetylase. ACS Infect. Dis. 2023, 9, 540–553. [Google Scholar] [CrossRef]
- Chaudhary, D.; Singh, A.; Marzuki, M.; Ghosh, A.; Kidwai, S.; Gosain, T.P.; Chawla, K.; Gupta, S.K.; Agarwal, N.; Saha, S.; et al. Identification of Small Molecules Targeting Homoserine Acetyl Transferase from Mycobacterium tuberculosis and Staphylococcus Aureus. Sci. Rep. 2022, 12, 13801. [Google Scholar] [CrossRef]
- Schnell, R.; Sriram, D.; Schneider, G. Pyridoxal-Phosphate Dependent Mycobacterial Cysteine Synthases: Structure, Mechanism and Potential as Drug Targets. Biochim. Biophys. Acta 2015, 1854, 1175–1183. [Google Scholar] [CrossRef]
- Jean Kumar, V.U.; Poyraz, Ö.; Saxena, S.; Schnell, R.; Yogeeswari, P.; Schneider, G.; Sriram, D. Discovery of Novel Inhibitors Targeting the Mycobacterium tuberculosis O-Acetylserine Sulfhydrylase (CysK1) Using Virtual High-Throughput Screening. Bioorg. Med. Chem. Lett. 2013, 23, 1182–1186. [Google Scholar] [CrossRef]
- Poyraz, O.; Jeankumar, V.U.; Saxena, S.; Schnell, R.; Haraldsson, M.; Yogeeswari, P.; Sriram, D.; Schneider, G. Structure-Guided Design of Novel Thiazolidine Inhibitors of O-Acetyl Serine Sulfhydrylase from Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 6457–6466. [Google Scholar] [CrossRef]
- Brunner, K.; Maric, S.; Reshma, R.S.; Almqvist, H.; Seashore-Ludlow, B.; Gustavsson, A.-L.; Poyraz, Ö.; Yogeeswari, P.; Lundbäck, T.; Vallin, M.; et al. Inhibitors of the Cysteine Synthase CysM with Antibacterial Potency against Dormant Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 6848–6859. [Google Scholar] [CrossRef]
- Brunner, K.; Steiner, E.M.; Reshma, R.S.; Sriram, D.; Schnell, R.; Schneider, G. Profiling of in Vitro Activities of Urea-Based Inhibitors against Cysteine Synthases from Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2017, 27, 4582–4587. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.; Pramanick, I.; Biswas, R.; Ps, S.; Sreedharan, S.; Singh, S.; Rajmani, R.S.; Laxman, S.; Dutta, S.; Singh, A. S-Adenosylmethionine-Responsive Cystathionine β-Synthase Modulates Sulfur Metabolism and Redox Balance in Mycobacterium tuberculosis. Sci. Adv. 2022, 8, eabo0097. [Google Scholar] [CrossRef]
- Lunardi, J.; Nunes, J.E.S.; Bizarro, C.V.; Basso, L.A.; Santos, D.S.; Machado, P. Targeting the Histidine Pathway in Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2013, 13, 2866–2884. [Google Scholar] [CrossRef]
- Dwivedy, A.; Ashraf, A.; Jha, B.; Kumar, D.; Agarwal, N.; Biswal, B.K. De Novo Histidine Biosynthesis Protects Mycobacterium tuberculosis from Host IFN-γ Mediated Histidine Starvation. Commun. Biol. 2021, 4, 410. [Google Scholar] [CrossRef]
- Ahangar, M.S.; Vyas, R.; Nasir, N.; Biswal, B.K. Structures of Native, Substrate-Bound and Inhibited Forms of Mycobacterium tuberculosis Imidazoleglycerol-Phosphate Dehydratase. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2461–2467. [Google Scholar] [CrossRef]
- Pacholarz, K.J.; Burnley, R.J.; Jowitt, T.A.; Ordsmith, V.; Pisco, J.P.; Porrini, M.; Larrouy-Maumus, G.; Garlish, R.A.; Taylor, R.J.; de Carvalho, L.P.S.; et al. Hybrid Mass Spectrometry Approaches to Determine How L-Histidine Feedback Regulates the Enzyzme MtATP-Phosphoribosyltransferase. Structure 2017, 25, 730–738.e4. [Google Scholar] [CrossRef]
- Donini, S.; Garavaglia, S.; Ferraris, D.M.; Miggiano, R.; Mori, S.; Shibayama, K.; Rizzi, M. Biochemical and Structural Investigations on Phosphoribosylpyrophosphate Synthetase from Mycobacterium Smegmatis. PLoS ONE 2017, 12, e0175815. [Google Scholar] [CrossRef]
- Jha, B.; Kumar, D.; Sharma, A.; Dwivedy, A.; Singh, R.; Biswal, B.K. Identification and Structural Characterization of a Histidinol Phosphate Phosphatase from Mycobacterium tuberculosis. J. Biol. Chem. 2018, 293, 10102–10118. [Google Scholar] [CrossRef]
- Lunardi, J.; Martinelli, L.K.B.; Raupp, A.S.; Nunes, J.E.S.; Rostirolla, D.C.; Timmers, L.F.S.M.; Villela, A.D.; Pissinate, K.; Limberger, J.; de Souza, O.N.; et al. Mycobacterium tuberculosis Histidinol Dehydrogenase: Biochemical Characterization and Inhibition Studies. RSC Adv. 2016, 6, 28406–28418. [Google Scholar] [CrossRef]
- Kumar, U.; Mehta, P.; Kumar, S.; Jain, A.; Guleria, A.; Kumar, R.V.; Misra, R.; Kumar, D. Circulatory Histidine Levels as Predictive Indicators of Disease Activity in Takayasu Arteritis. Anal. Sci. Adv. 2021, 2, 527–535. [Google Scholar] [CrossRef]
- Cho, Y.; Ioerger, T.R.; Sacchettini, J.C. Discovery of Novel Nitrobenzothiazole Inhibitors for M. tuberculosis ATP Phosphoribosyl Transferase (HisG) through Virtual Screening. J. Med. Chem. 2008, 51, 5984–5992. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Comprehensive Identification of Conditionally Essential Genes in Mycobacteria. Proc. Natl. Acad. Sci. USA 2001, 98, 12712–12717. [Google Scholar] [CrossRef]
- Parish, T. Starvation Survival Response of Mycobacterium tuberculosis. J. Bacteriol. 2003, 185, 6702–6706. [Google Scholar] [CrossRef]
- Dhameliya, T.M.; Tiwari, R.; Banerjee, A.; Pancholia, S.; Sriram, D.; Panda, D.; Chakraborti, A.K. Benzo[d]Thiazole-2-Carbanilides as New Anti-TB Chemotypes: Design, Synthesis, Biological Evaluation, and Structure-Activity Relationship. Eur. J. Med. Chem. 2018, 155, 364–380. [Google Scholar] [CrossRef]
- Dhameliya, T.M.; Tiwari, R.; Banerjee, A.; Pancholia, S.; Sriram, D.; Panda, D.; Chakraborti, A.K. Benzo[d]Thiazole-2-Carboxamides as New Antituberculosis Chemotypes Inhibiting Mycobacterial ATP Phosphoribosyl Transferase. Future Med. Chem. 2022, 14, 1847–1864. [Google Scholar] [CrossRef]
- Hutter, B.; Dick, T. Increased Alanine Dehydrogenase Activity during Dormancy in Mycobacterium Smegmatis. FEMS Microbiol. Lett. 1998, 167, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Giffin, M.M.; Shi, L.; Gennaro, M.L.; Sohaskey, C.D. Role of Alanine Dehydrogenase of Mycobacterium tuberculosis during Recovery from Hypoxic Nonreplicating Persistence. PLoS ONE 2016, 11, e0155522. [Google Scholar] [CrossRef] [PubMed]
- Reshma, R.S.; Saxena, S.; Bobesh, K.A.; Jeankumar, V.U.; Gunda, S.; Yogeeswari, P.; Sriram, D. Design and Development of New Class of Mycobacterium tuberculosis L-Alanine Dehydrogenase Inhibitors. Bioorg. Med. Chem. 2016, 24, 4499–4508. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Samala, G.; Sridevi, J.P.; Devi, P.B.; Yogeeswari, P.; Sriram, D. Design and Development of Novel Mycobacterium tuberculosis L-Alanine Dehydrogenase Inhibitors. Eur. J. Med. Chem. 2015, 92, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-B.; Bacik, J.-P.; Wu, R.; Jha, R.K.; Hebron, M.; Triandafillou, C.; McCown, J.E.; Baek, N.-I.; Kim, J.H.; Kim, Y.J.; et al. Label-Free Affinity Screening, Design and Synthesis of Inhibitors Targeting the Mycobacterium tuberculosis L-Alanine Dehydrogenase. PLoS ONE 2022, 17, e0277670. [Google Scholar] [CrossRef]
- Dartois, V.A.; Rubin, E.J. Anti-Tuberculosis Treatment Strategies and Drug Development: Challenges and Priorities. Nat. Rev. Microbiol. 2022, 20, 685–701. [Google Scholar] [CrossRef]
- Borah, K.; Beyß, M.; Theorell, A.; Wu, H.; Basu, P.; Mendum, T.A.; Nöh, K.; Beste, D.J.V.; McFadden, J. Intracellular Mycobacterium tuberculosis Exploits Multiple Host Nitrogen Sources during Growth in Human Macrophages. Cell Rep. 2019, 29, 3580–3591.e4. [Google Scholar] [CrossRef]
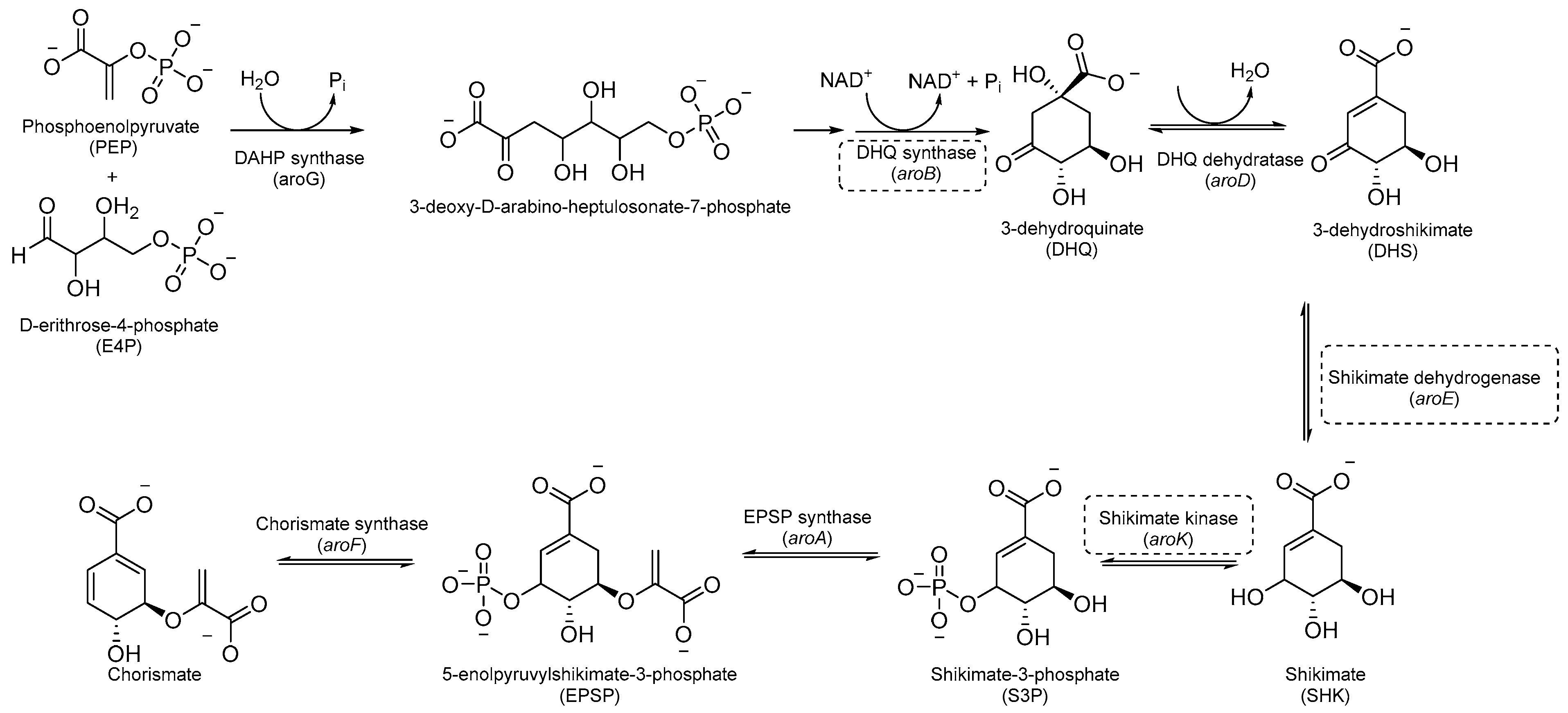
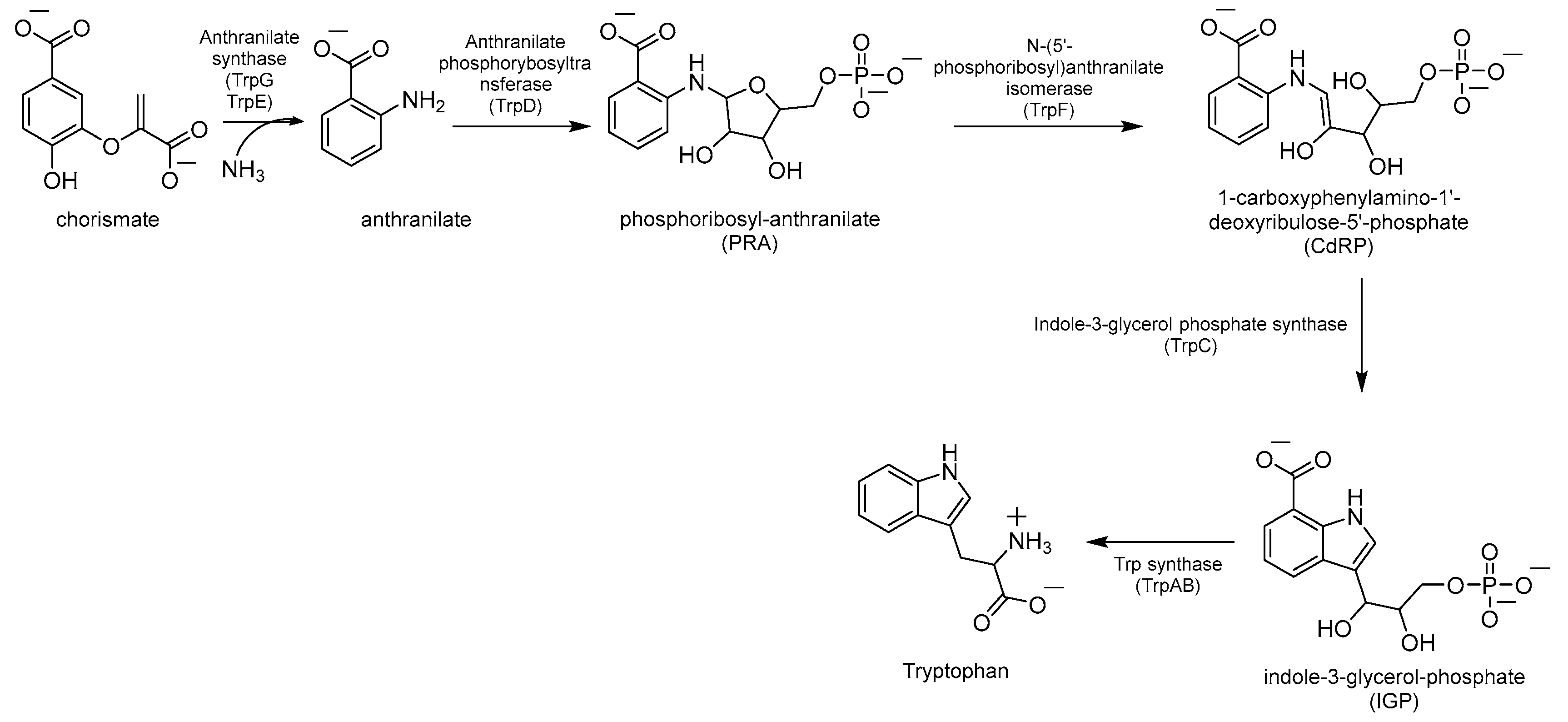
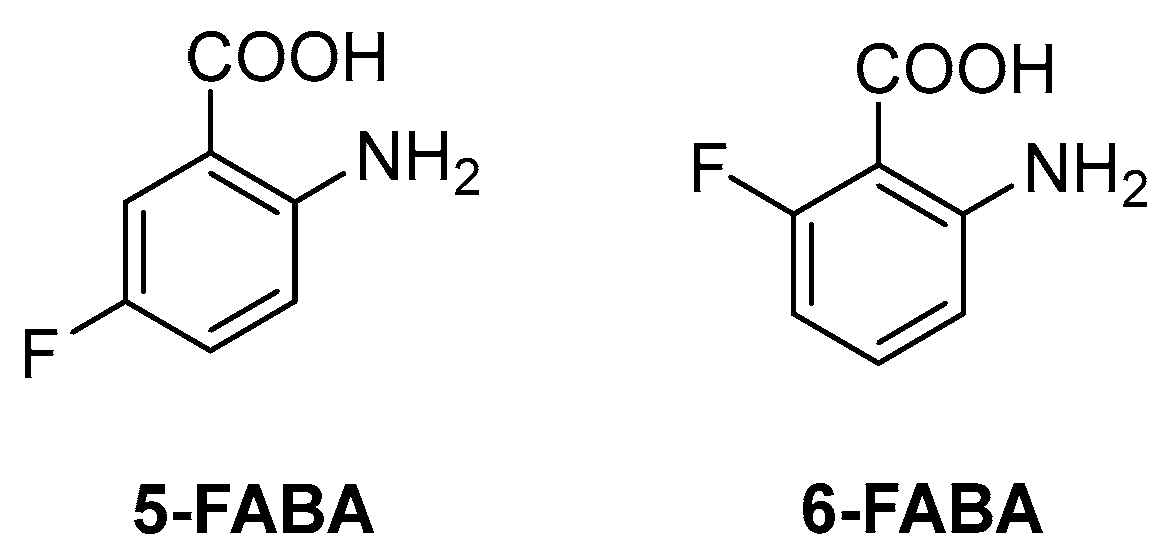
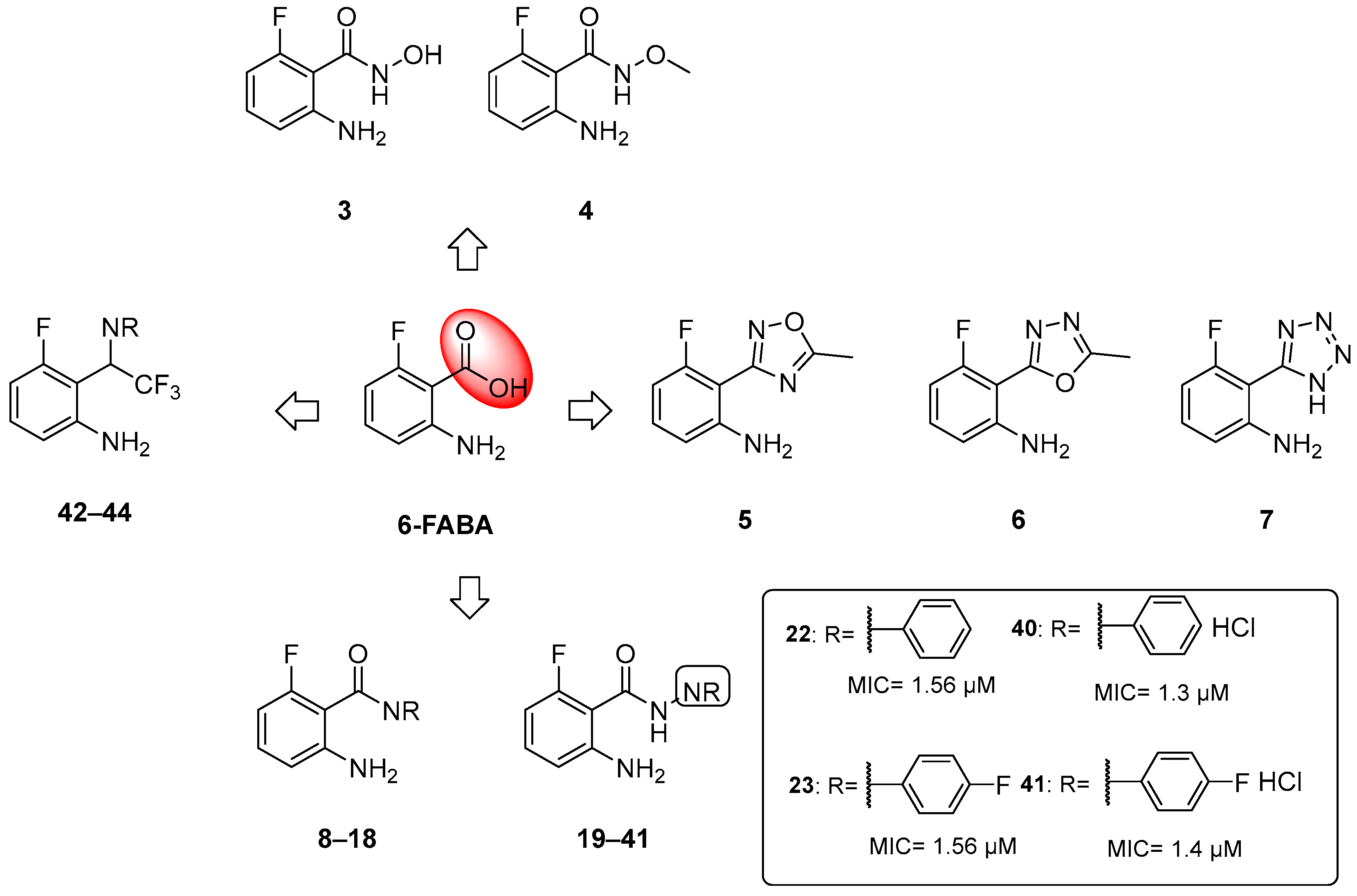
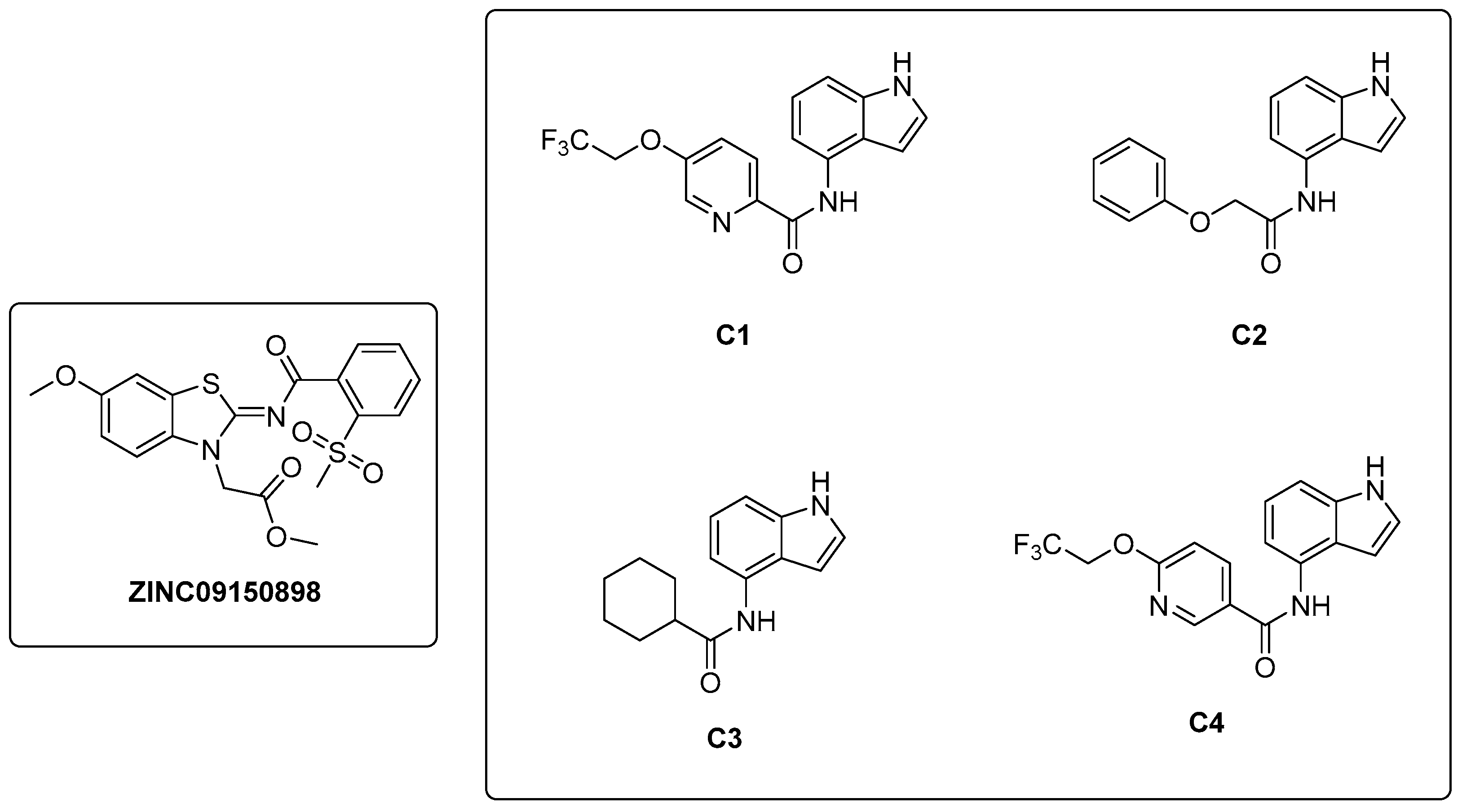
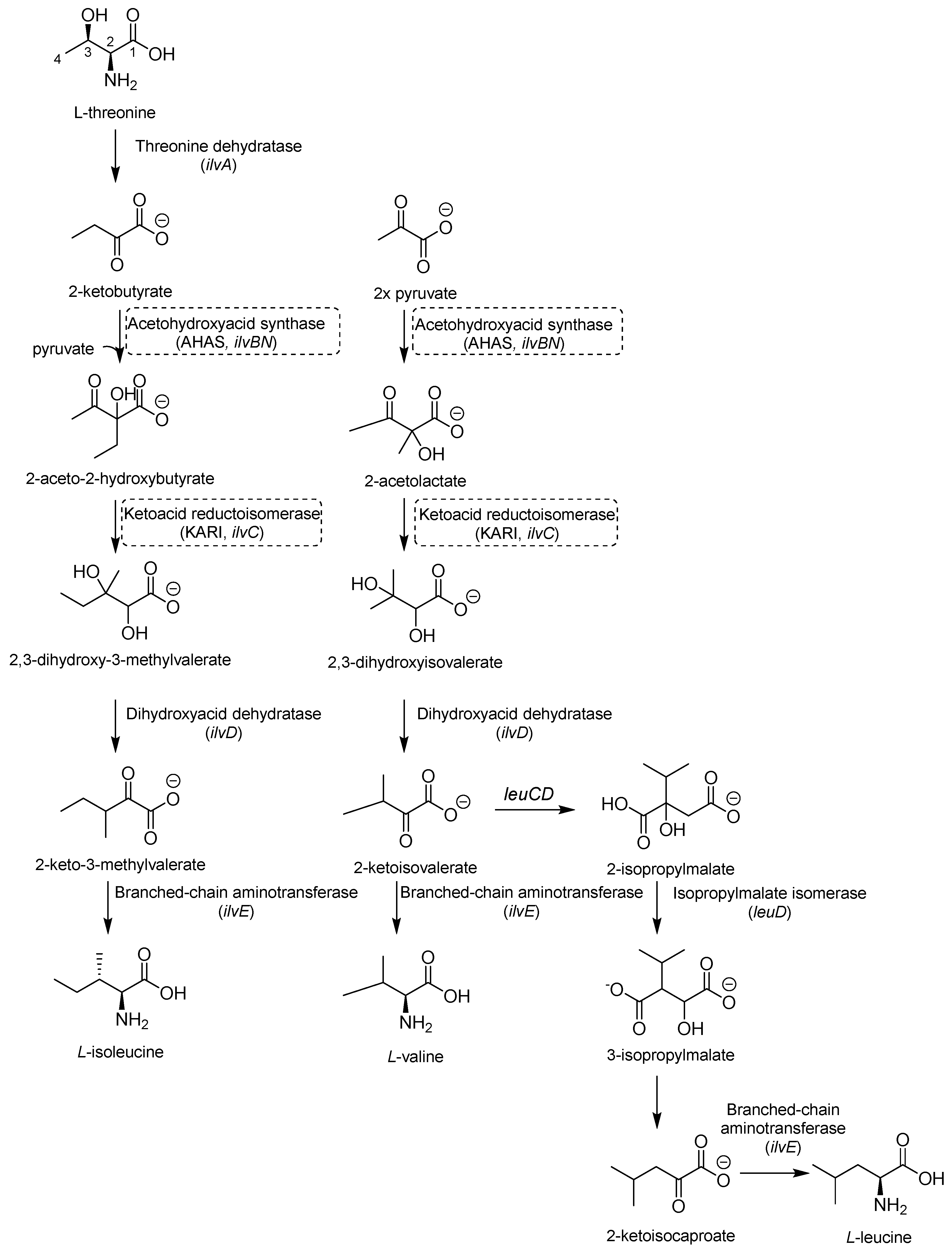
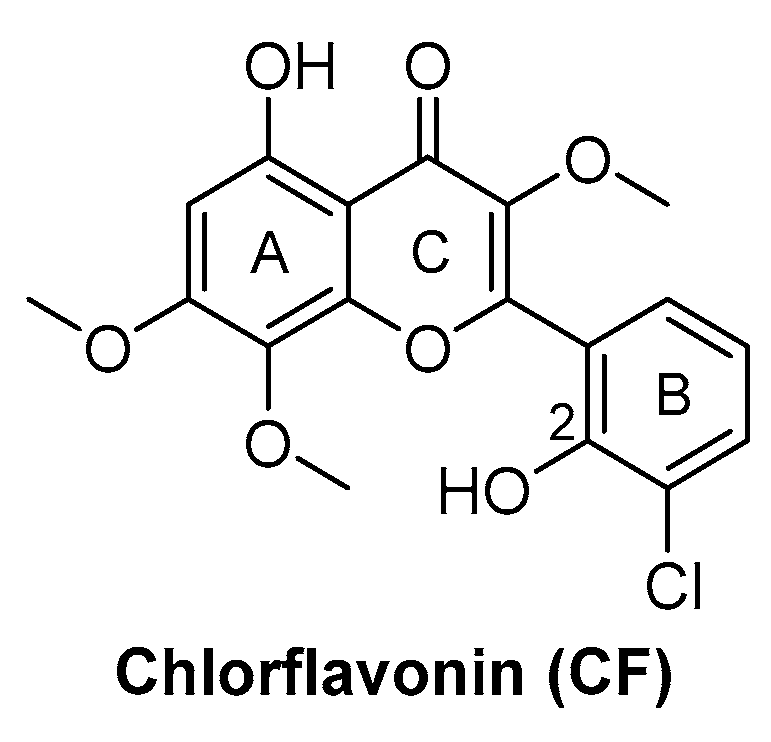
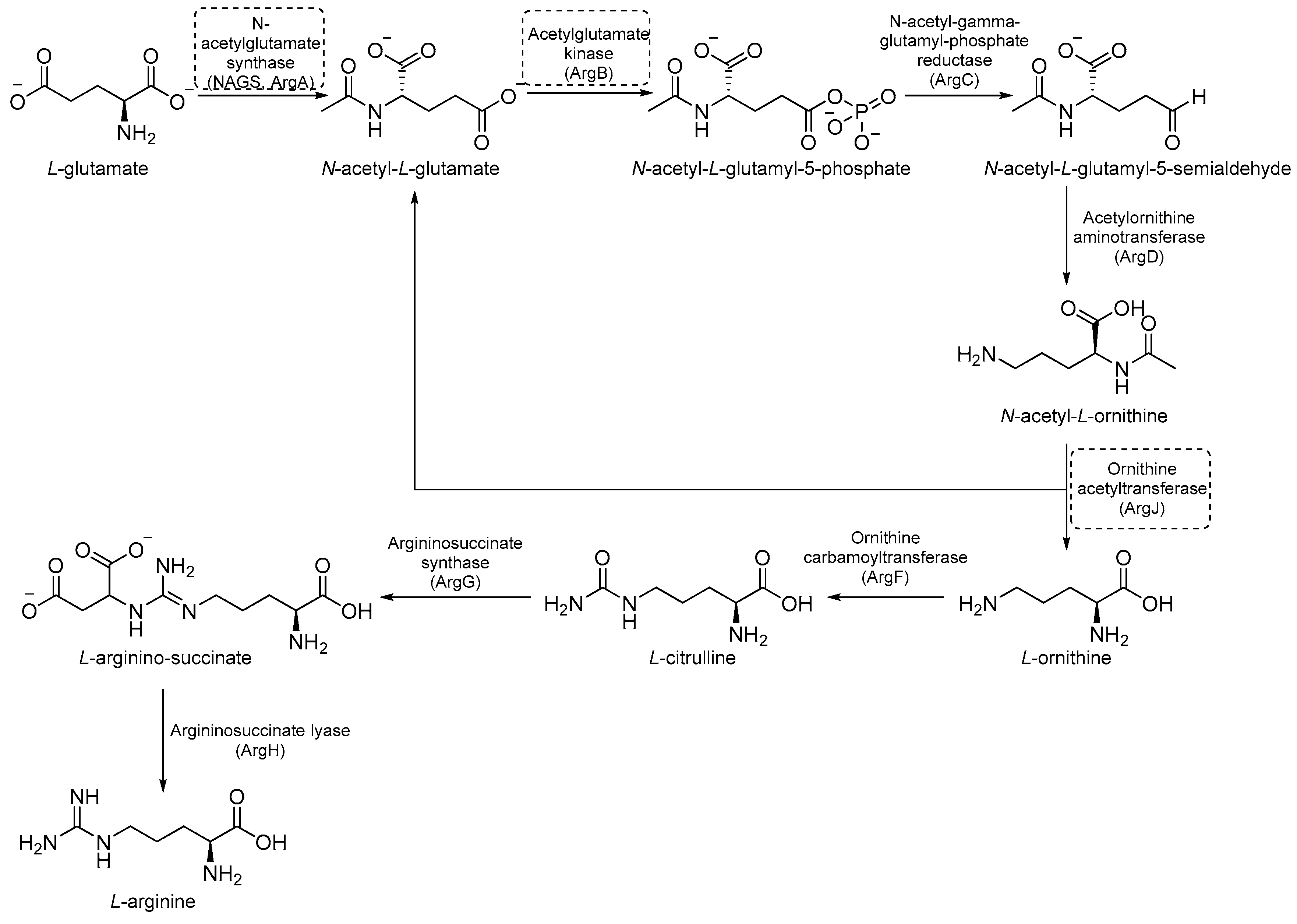
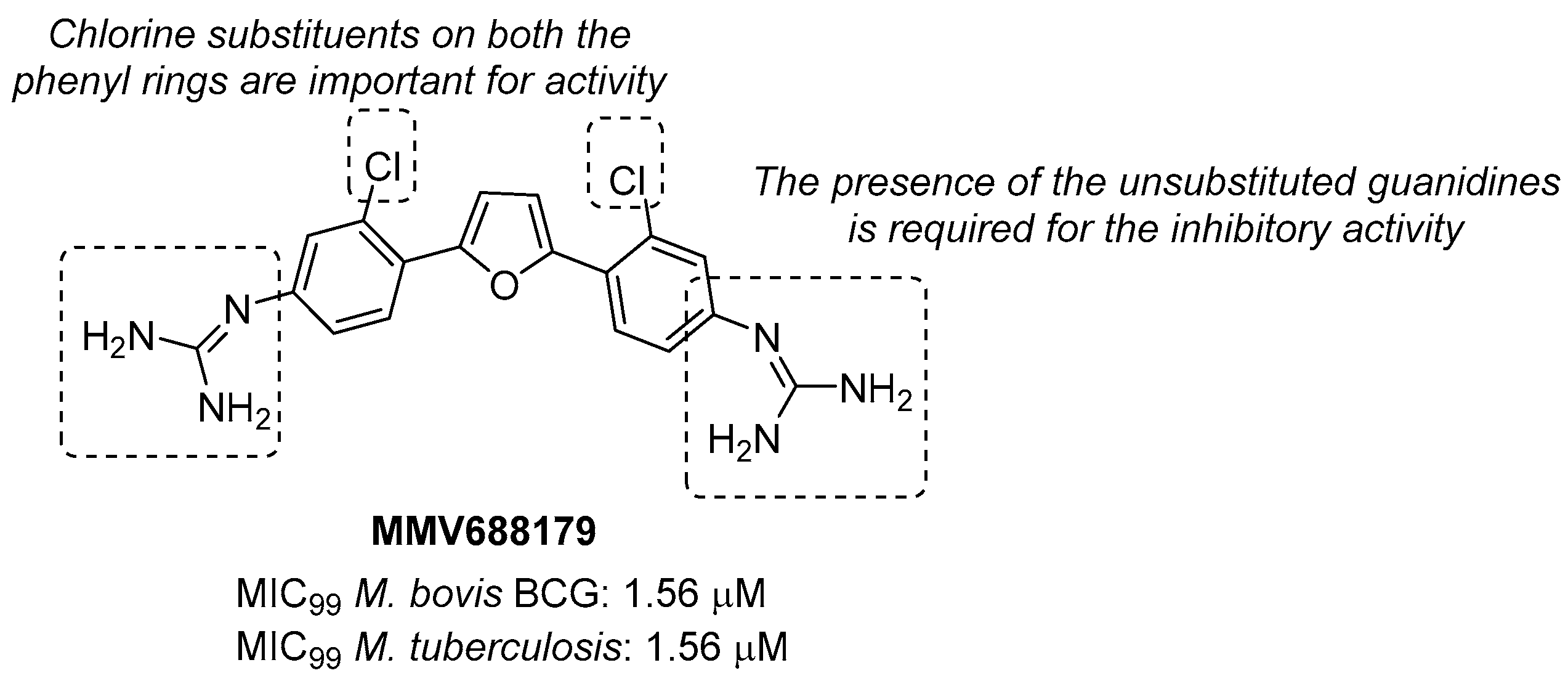
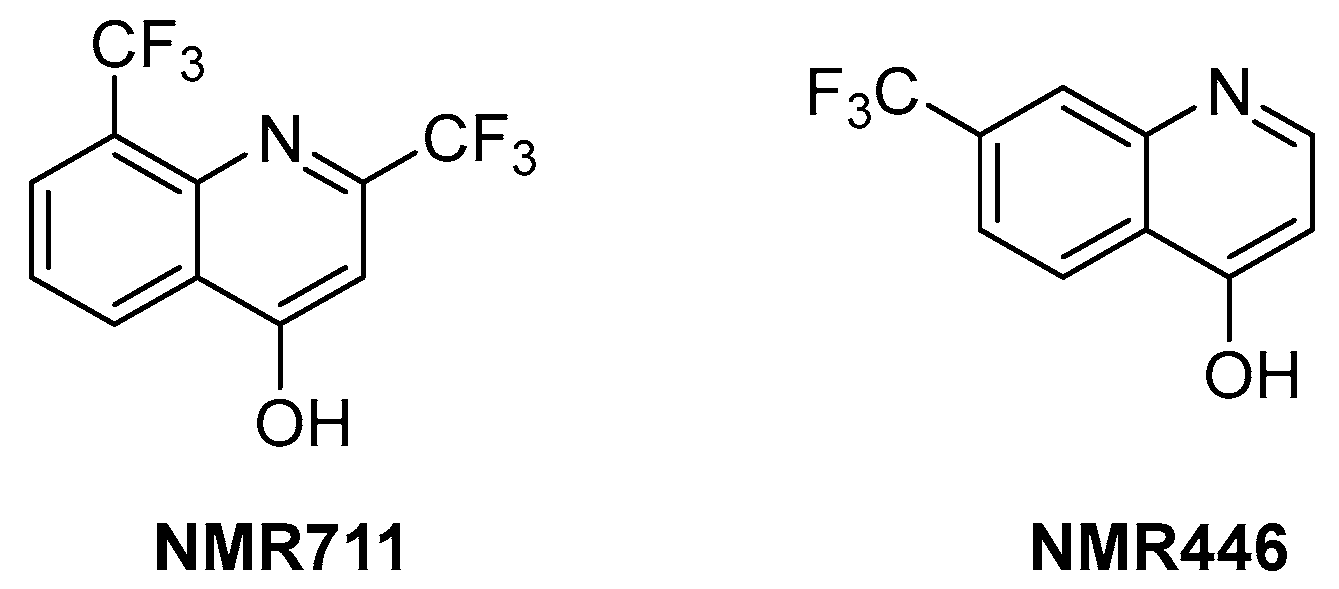
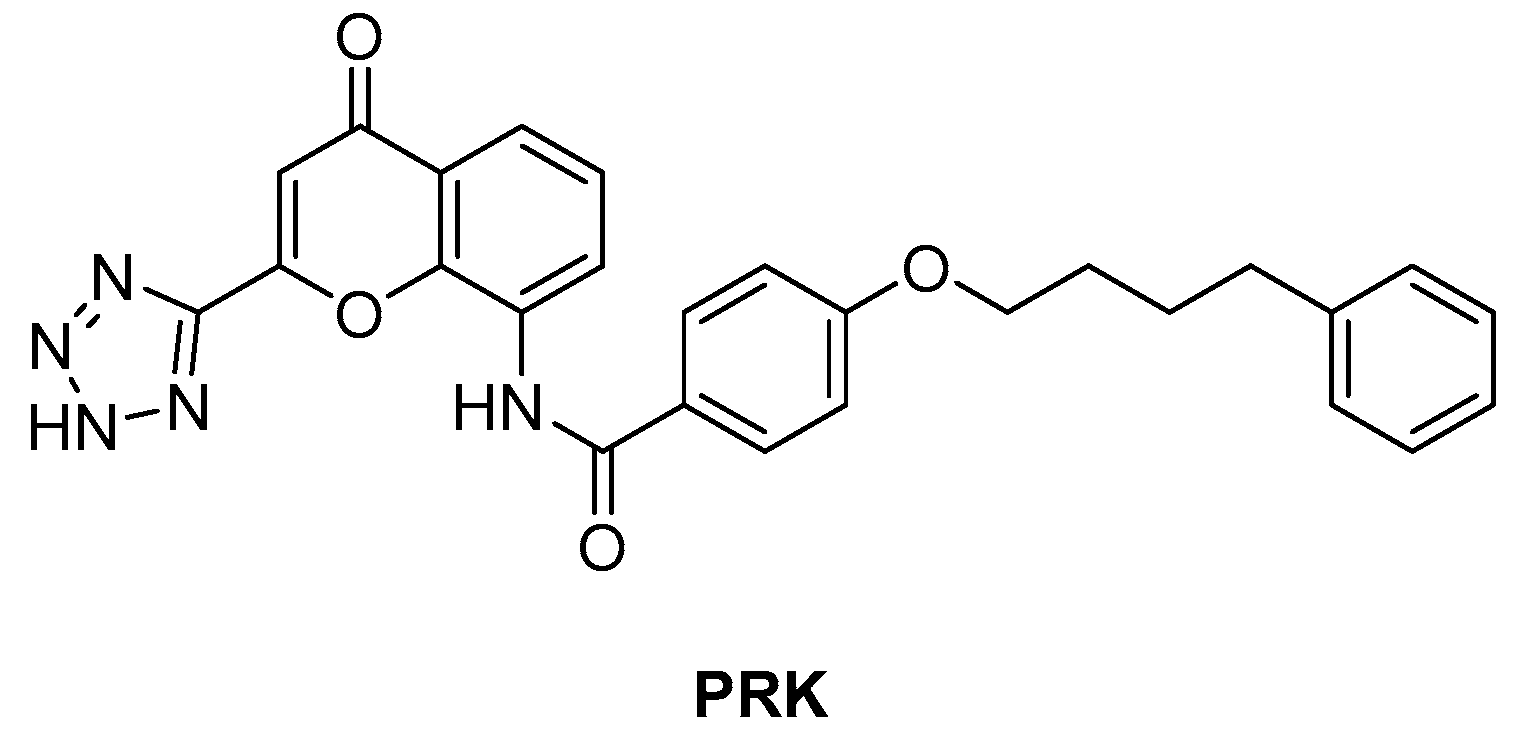
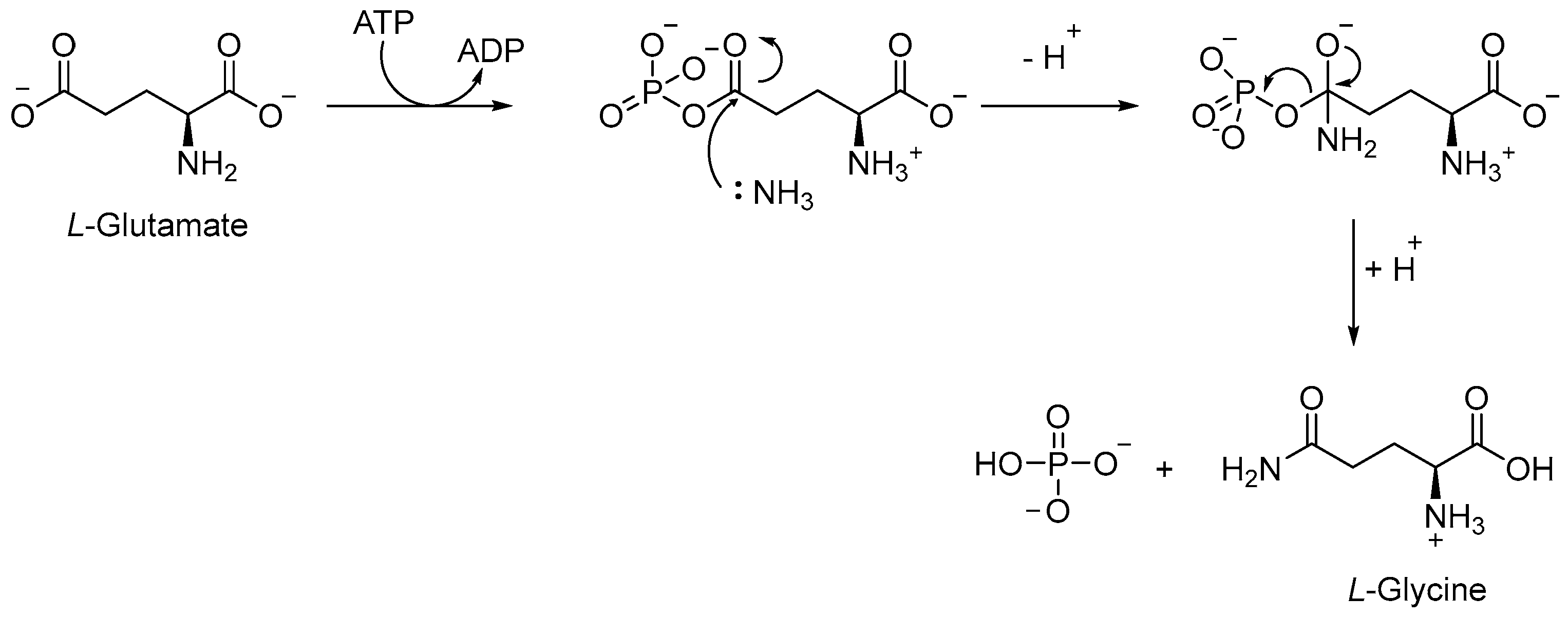
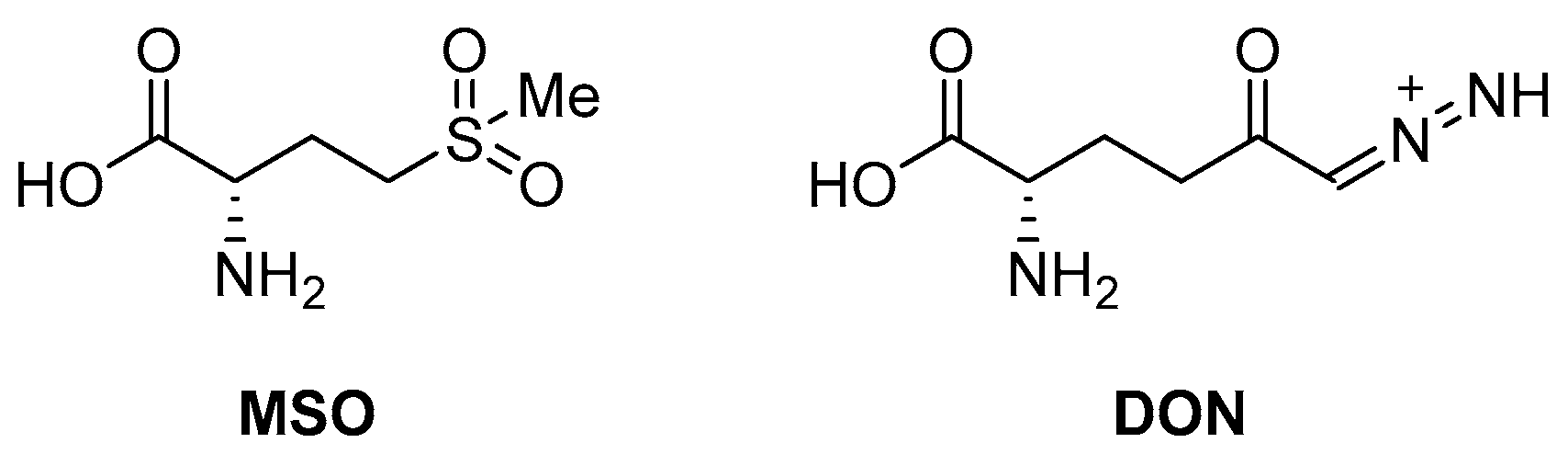
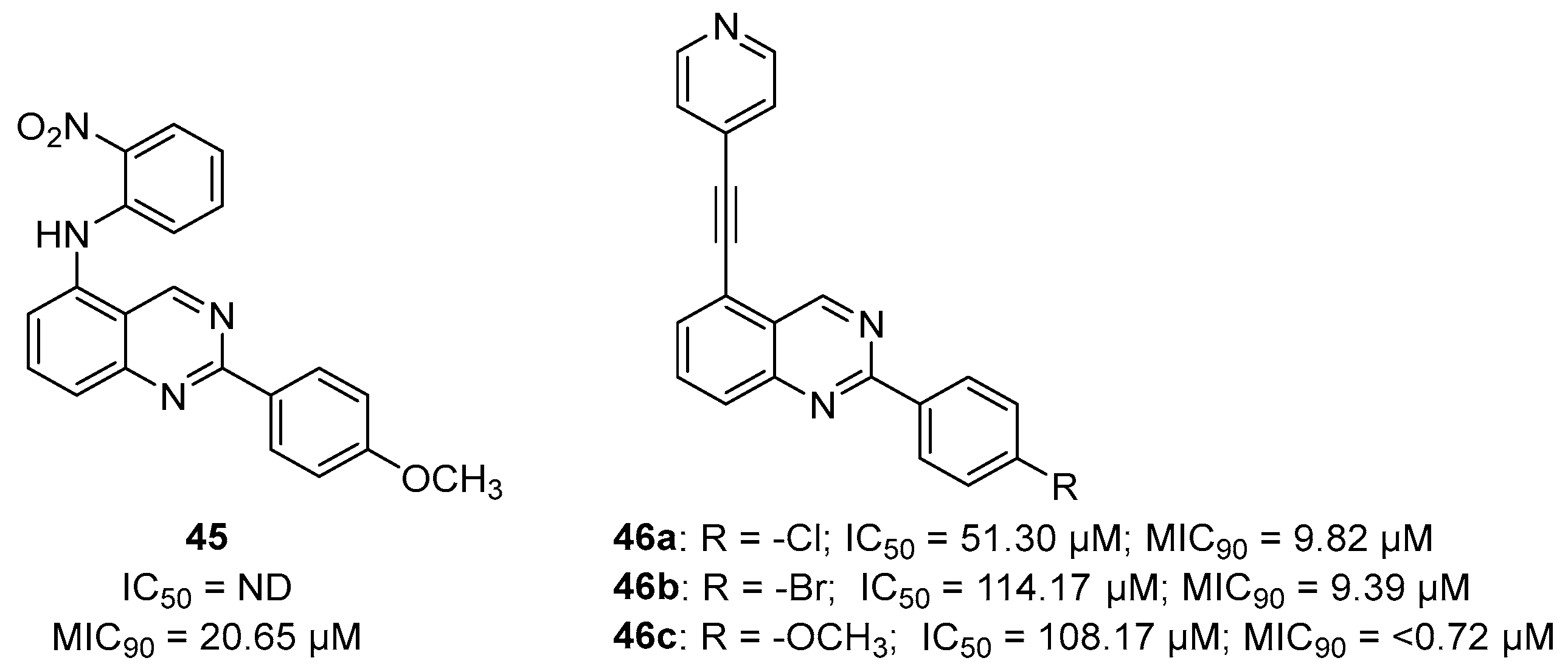
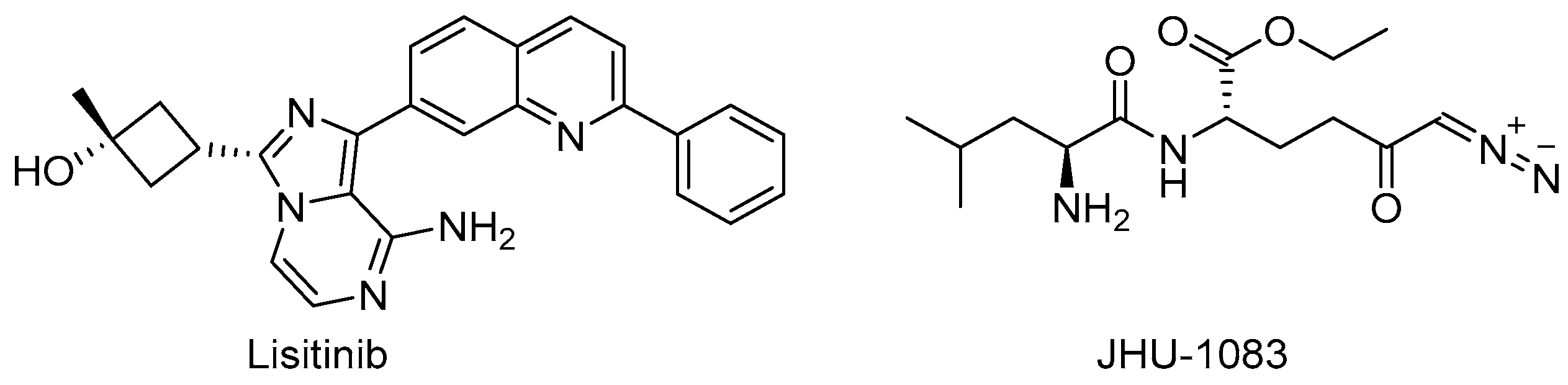
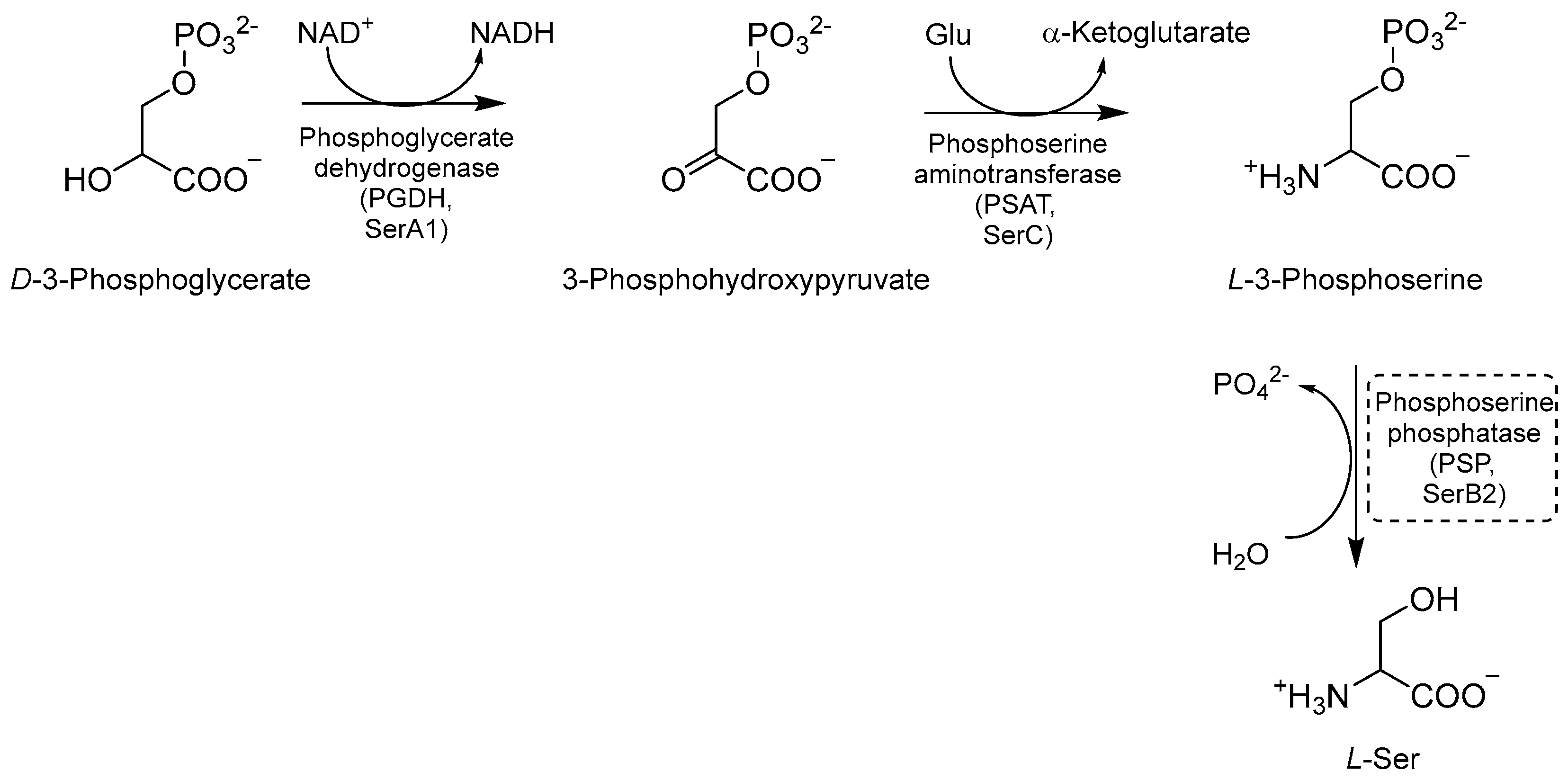
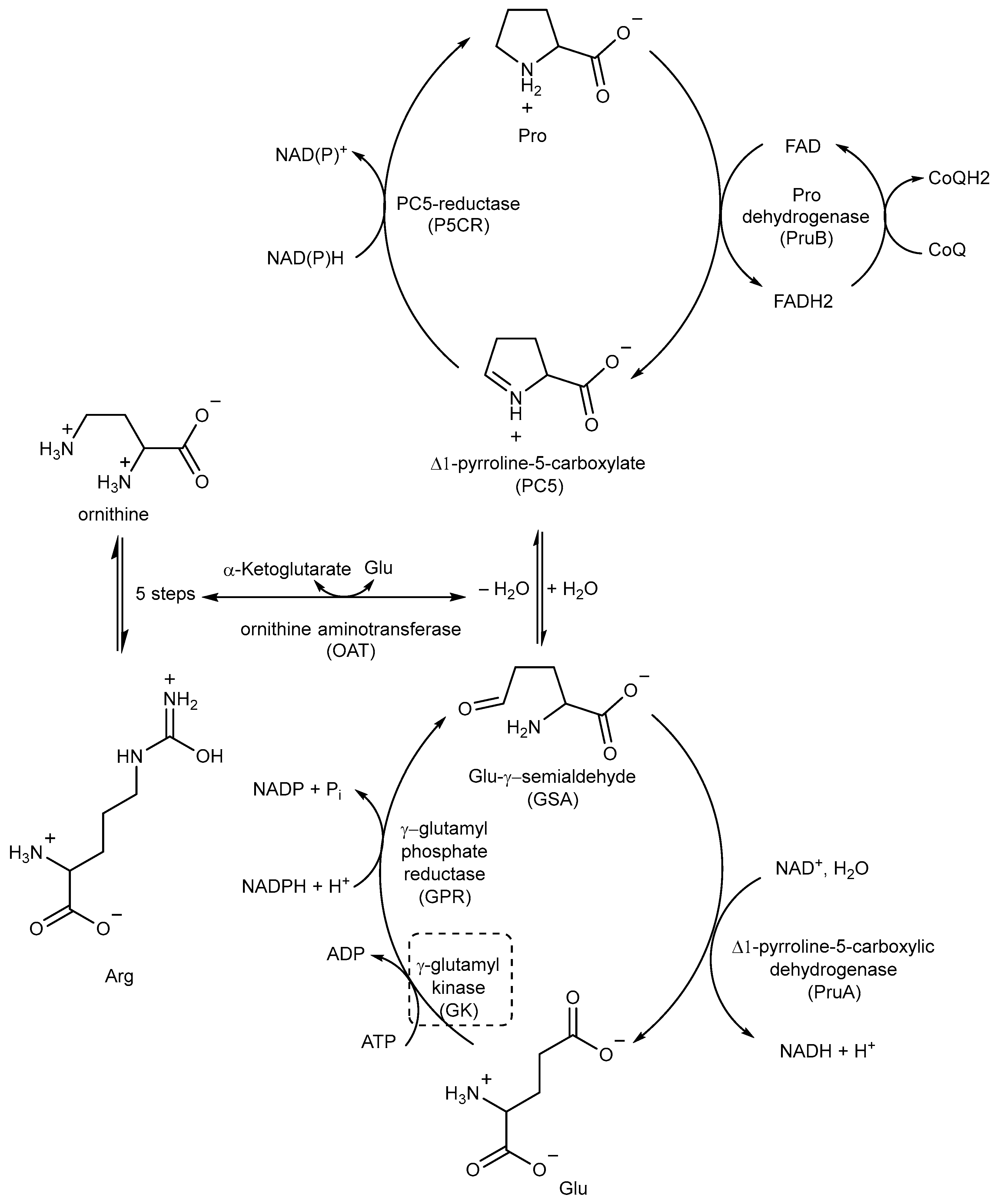
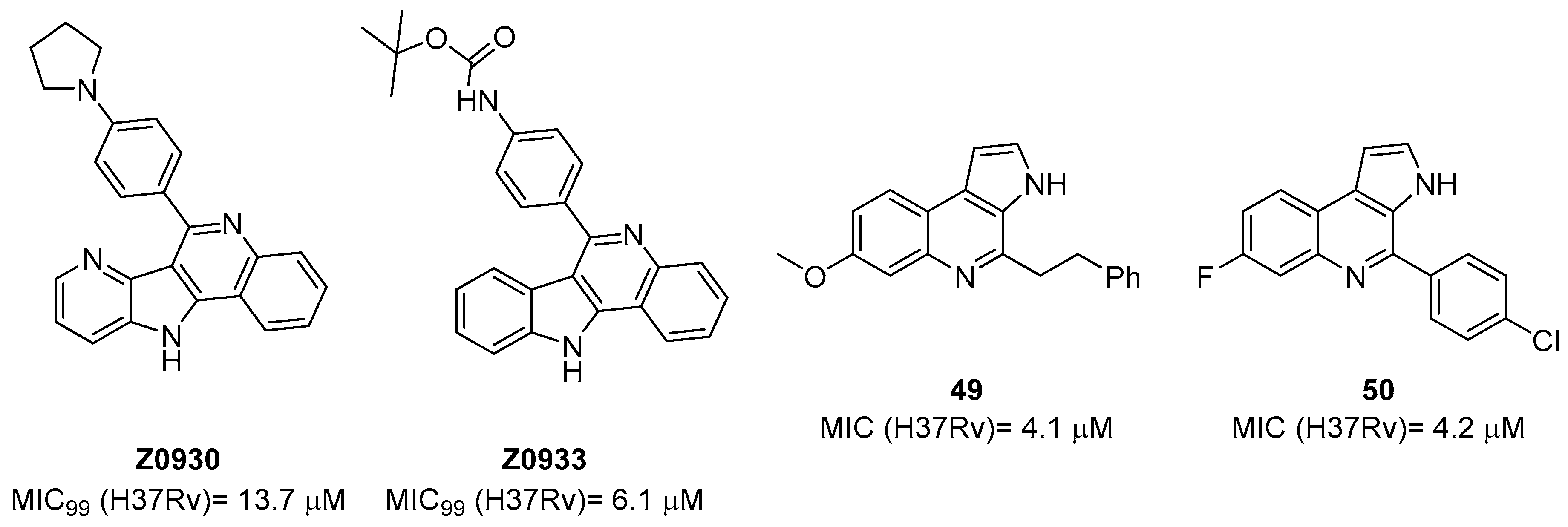
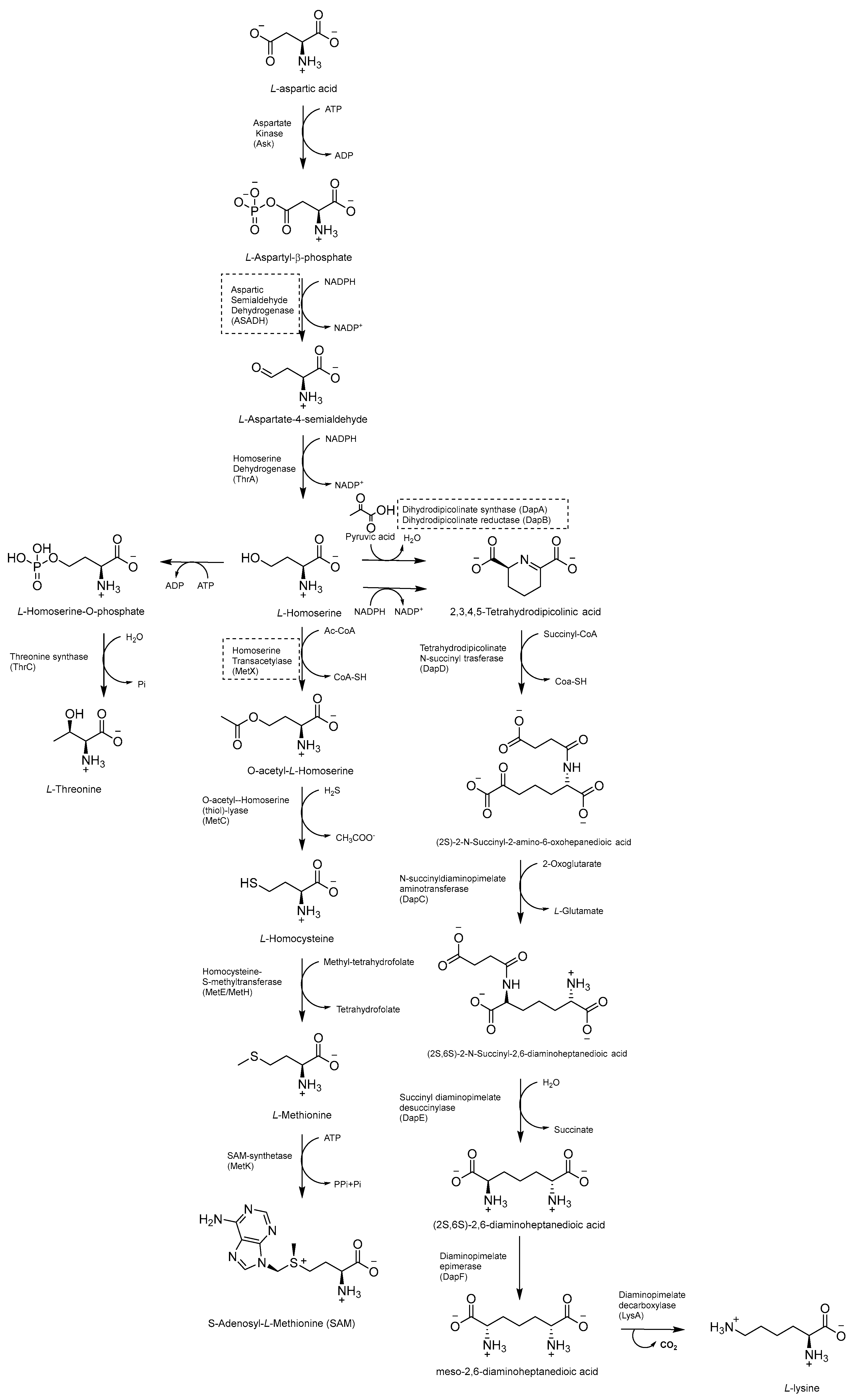
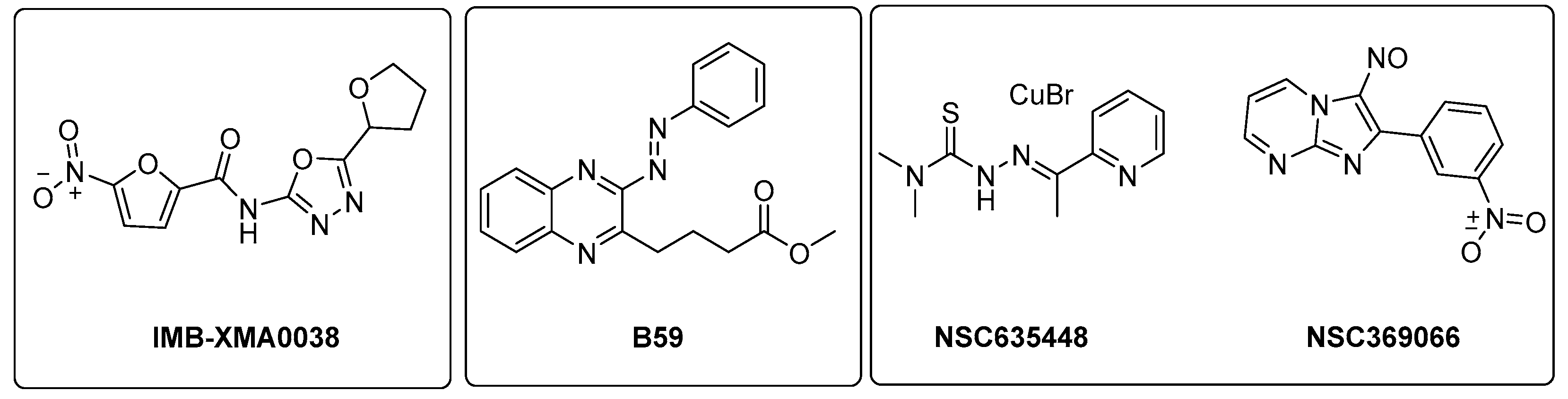
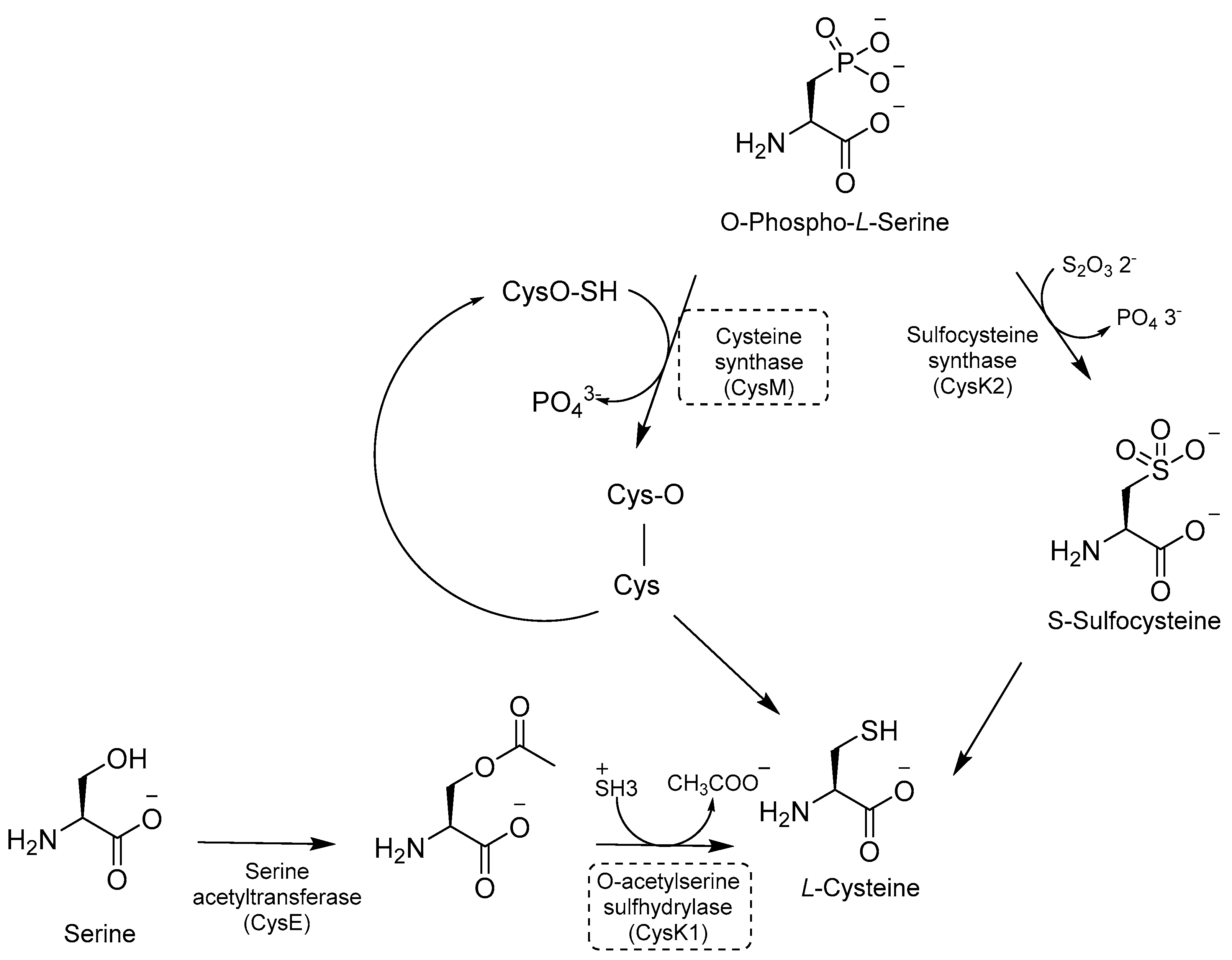
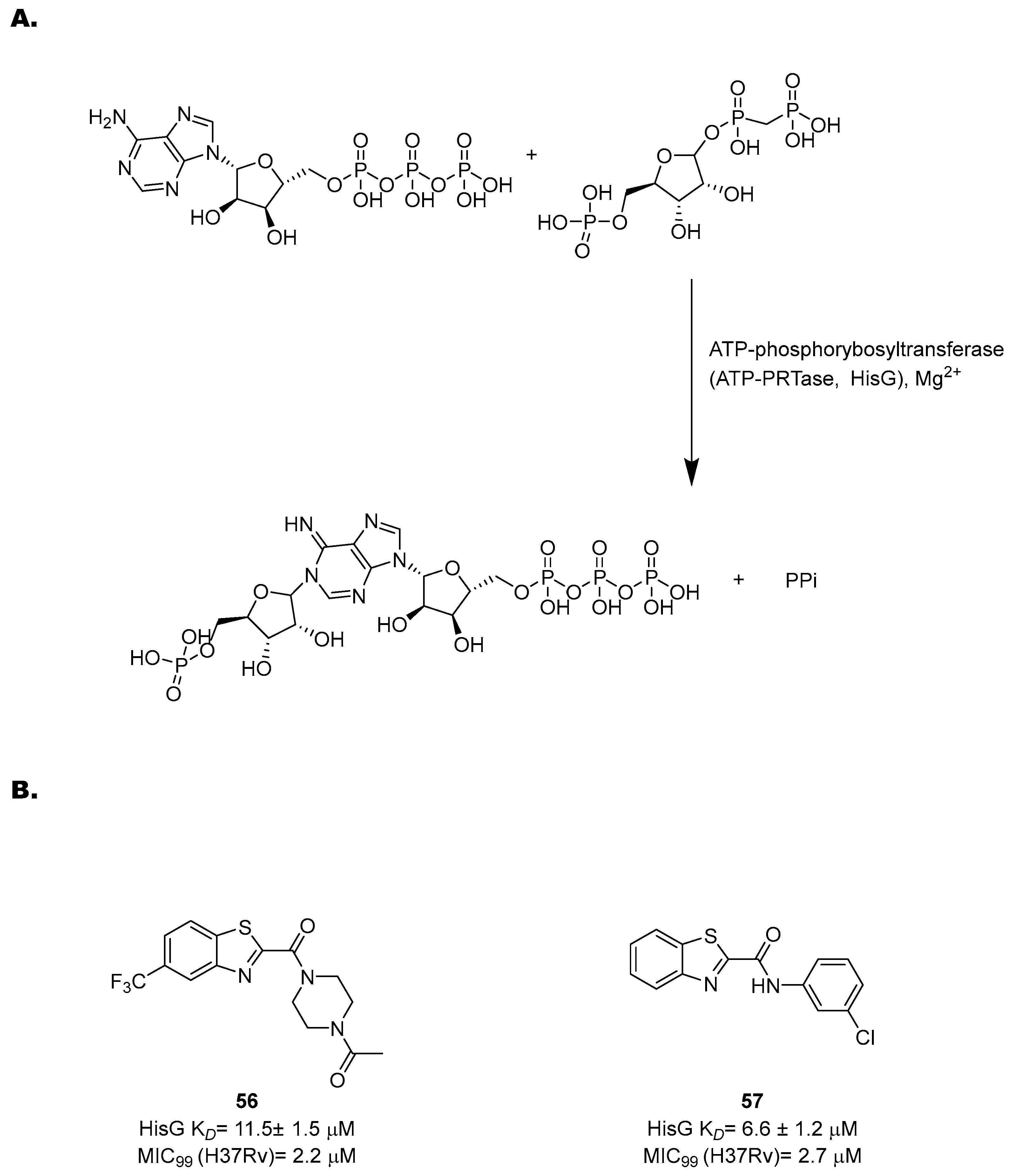
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).