
Single-Cell Transcriptomic and Targeted Genomic Profiling Adjusted for Inflammation and Therapy Bias Reveal CRTAM and PLCB1 as Novel Hub Genes for Anti-Tumor Necrosis Factor Alpha Therapy Response in Crohn’s Disease

 , , , ,
, , , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Enrolled Subjects
2.2. Preparation of Cells
2.3. scRNA-Seq Experiment and Library Preparation
2.4. scRNA-Seq Data Analysis
2.5. Differential Gene Expression Testing
2.6. Gene Ontology Analysis
2.7. Genomic Profiling
2.8. Replication and Meta-Analysis
2.9. Mendelian Randomization and Oligogenic Risk Score
3. Results
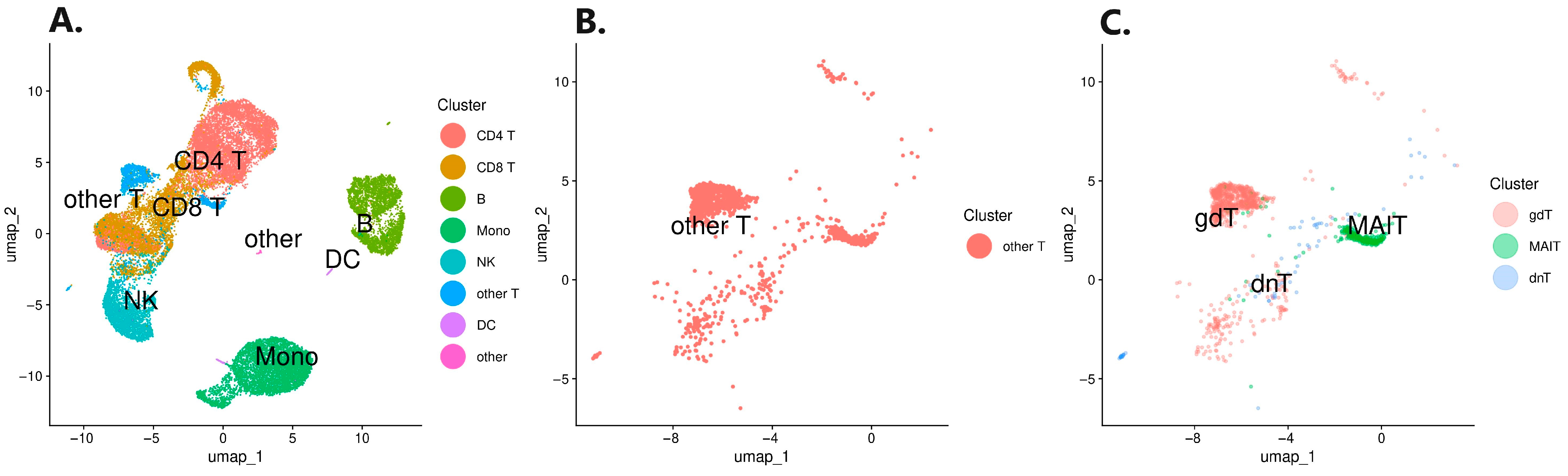
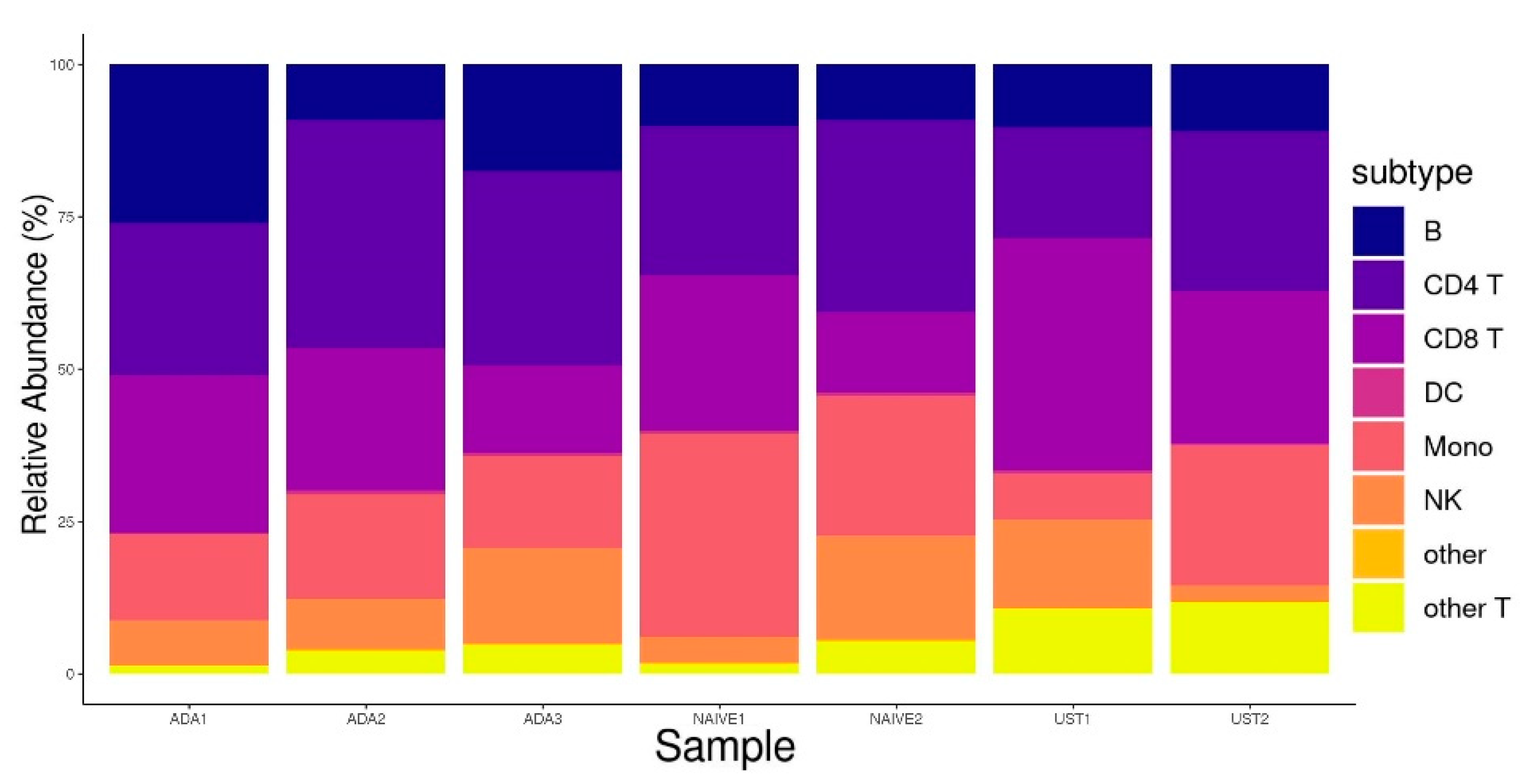
3.1. Single-Cell RNA Sequencing Analysis
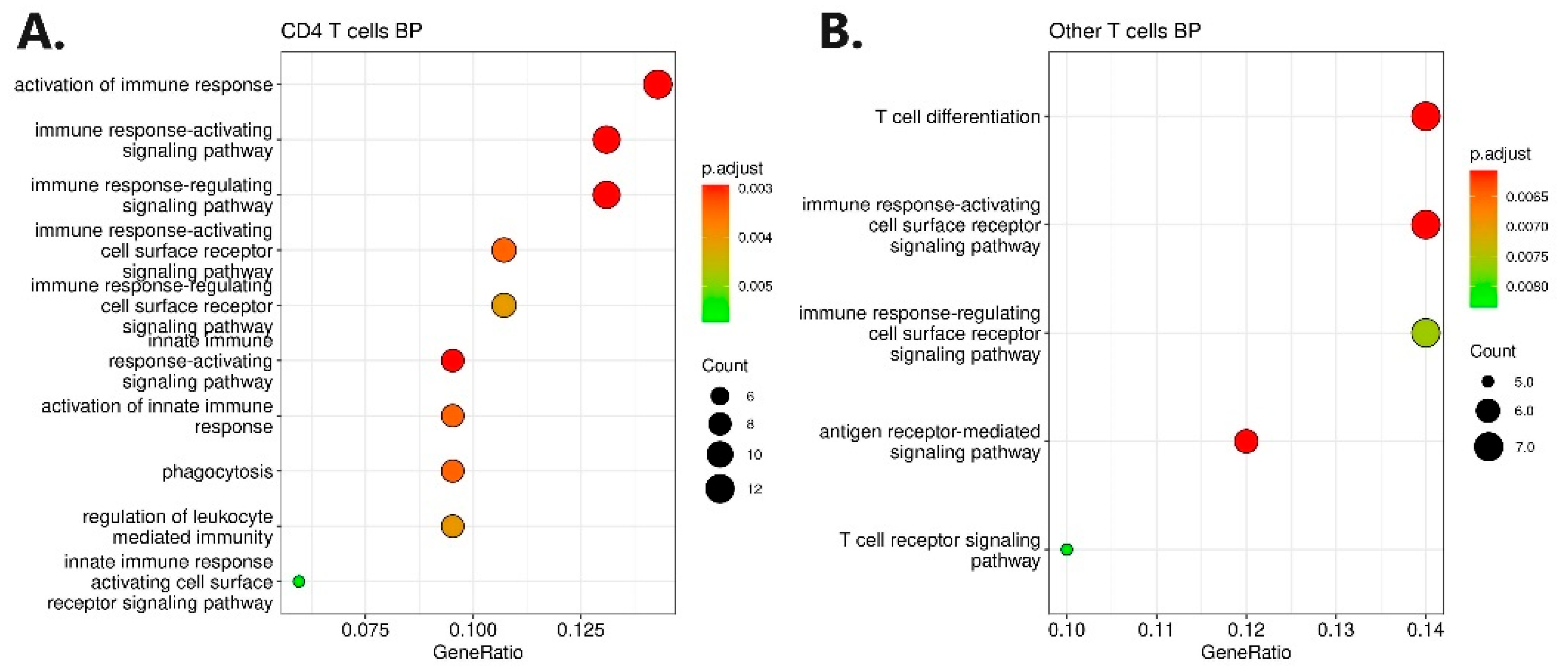
3.2. Gene Ontology Analysis
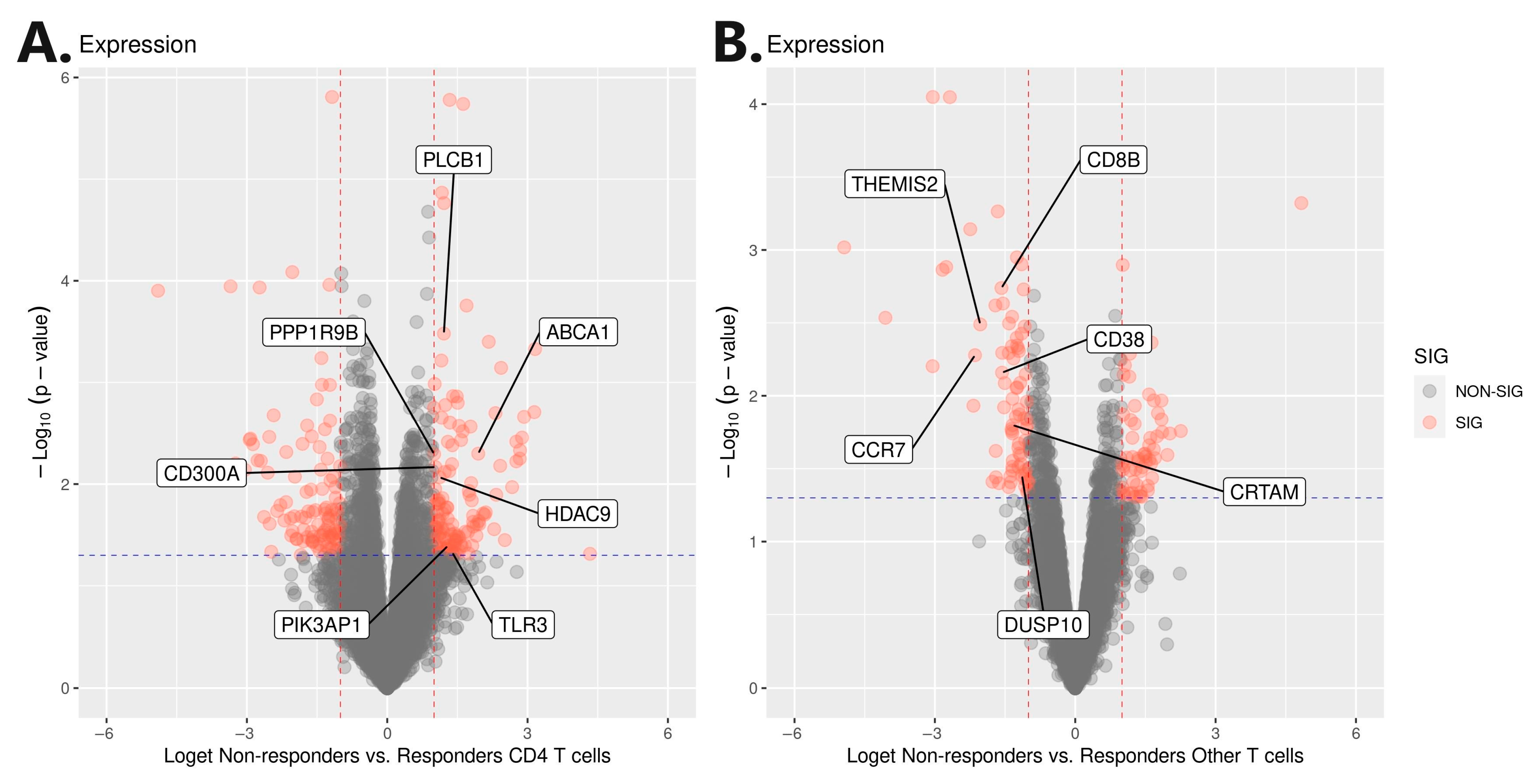
3.3. Genomic Profiling and Association Analysis
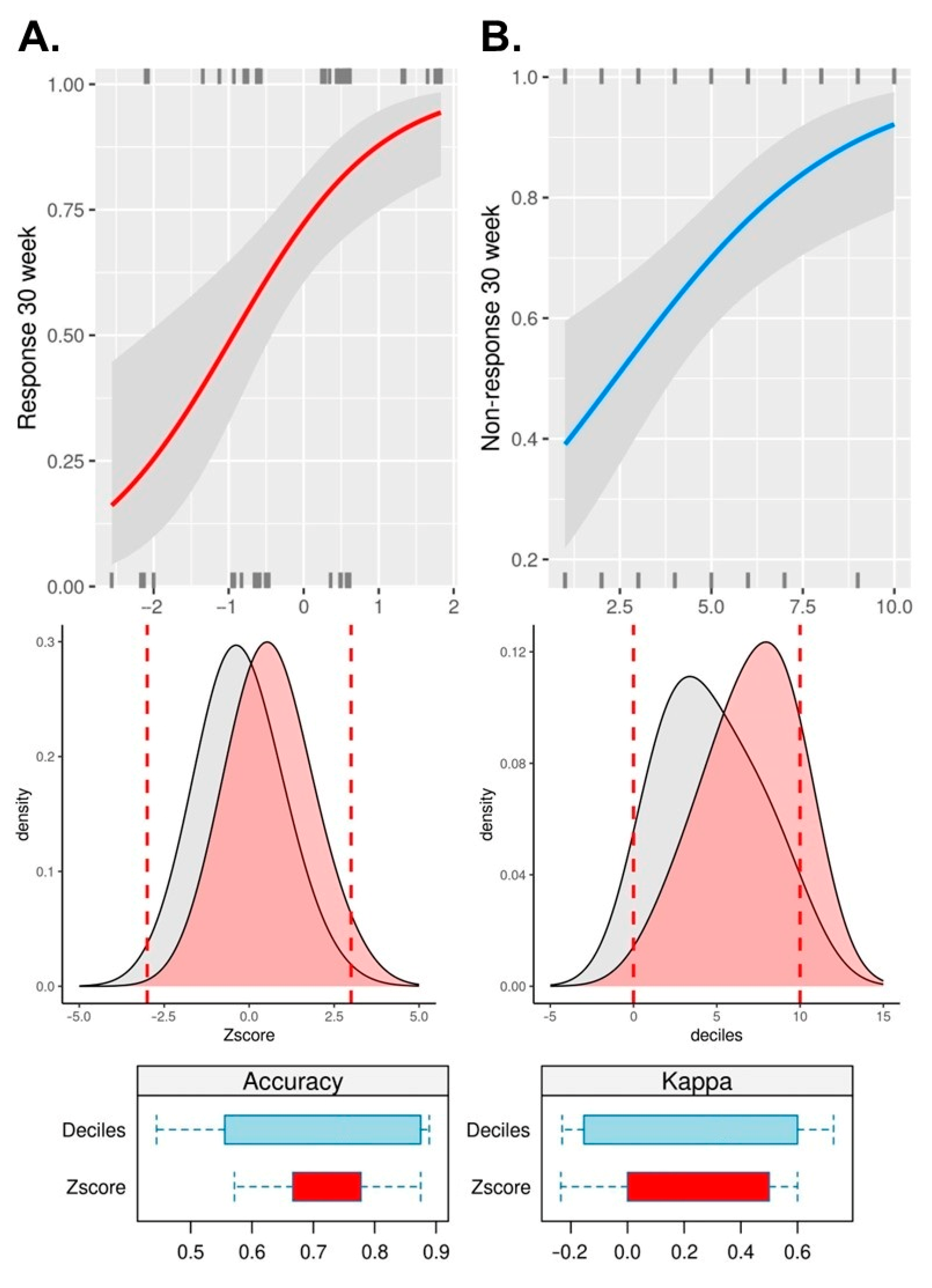
3.4. Replication, Meta-Analysis, Mendelian Randomization, and Oligogenic Risk Score
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 54.e42; quiz e30. [Google Scholar] [CrossRef]
- Rosenstiel, P.; Sina, C.; Franke, A.; Schreiber, S. Towards a molecular risk map--recent advances on the etiology of inflammatory bowel disease. Semin. Immunol. 2009, 21, 334–345. [Google Scholar] [CrossRef]
- Wehkamp, J.; Gotz, M.; Herrlinger, K.; Steurer, W.; Stange, E.F. Inflammatory Bowel Disease. Dtsch. Arztebl. Int. 2016, 113, 72–82. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Duchmann, R.; May, E.; Heike, M.; Knolle, P.; Neurath, M.; Meyer zum Buschenfelde, K.H. T cell specificity and cross reactivity towards enterobacteria, bacteroides, bifidobacterium, and antigens from resident intestinal flora in humans. Gut 1999, 44, 812–818. [Google Scholar] [CrossRef]
- Moussata, D.; Goetz, M.; Gloeckner, A.; Kerner, M.; Campbell, B.; Hoffman, A.; Biesterfeld, S.; Flourie, B.; Saurin, J.C.; Galle, P.R.; et al. Confocal laser endomicroscopy is a new imaging modality for recognition of intramucosal bacteria in inflammatory bowel disease in vivo. Gut 2011, 60, 26–33. [Google Scholar] [CrossRef]
- Wehkamp, J.; Fellermann, K.; Herrlinger, K.R.; Bevins, C.L.; Stange, E.F. Mechanisms of disease: Defensins in gastrointestinal diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 406–415. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Liu, T.C.; Li, D.; Towfic, F.; Modiano, N.; Winter, R.; Haritunians, T.; Taylor, K.D.; Dhall, D.; Targan, S.R.; et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 2014, 146, 200–209. [Google Scholar] [CrossRef]
- Zundler, S.; Neurath, M.F. Immunopathogenesis of inflammatory bowel diseases: Functional role of T cells and T cell homing. Clin. Exp. Rheumatol. 2015, 33, S19–S28. [Google Scholar]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Danese, S.; Fiorino, G.; Reinisch, W. Review article: Causative factors and the clinical management of patients with Crohn’s disease who lose response to anti-TNF-alpha therapy. Aliment. Pharmacol. Ther. 2011, 34, 1–10. [Google Scholar] [CrossRef]
- Van Deventer, S.J. Tumour necrosis factor and Crohn’s disease. Gut 1997, 40, 443–448. [Google Scholar] [CrossRef]
- Colombel, J.F.; Sandborn, W.J.; Rutgeerts, P.; Enns, R.; Hanauer, S.B.; Panaccione, R.; Schreiber, S.; Byczkowski, D.; Li, J.; Kent, J.D.; et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: The CHARM trial. Gastroenterology 2007, 132, 52–65. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239. [Google Scholar] [CrossRef]
- Sands, B.E.; Anderson, F.H.; Bernstein, C.N.; Chey, W.Y.; Feagan, B.G.; Fedorak, R.N.; Kamm, M.A.; Korzenik, J.R.; Lashner, B.A.; Onken, J.E.; et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N. Engl. J. Med. 2004, 350, 876–885. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef]
- Schoultz, I.; Keita, A.V. Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease-Focusing on Intestinal Barrier Function. Cells 2019, 8, 193. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Feagan, B.G.; Panes, J.; Ferrante, M.; Kaser, A.; D‘Haens, G.R.; Sandborn, W.J.; Louis, E.; Neurath, M.F.; Franchimont, D.; Dewit, O.; et al. Risankizumab in patients with moderate to severe Crohn’s disease: An open-label extension study. Lancet Gastroenterol. Hepatol. 2018, 3, 671–680. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Loftus, E.V., Jr.; Danese, S.; Colombel, J.F.; Toruner, M.; Jonaitis, L.; Abhyankar, B.; Chen, J.; Rogers, R.; et al. Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1215–1226. [Google Scholar] [CrossRef]
- Singh, S.; Murad, M.H.; Fumery, M.; Sedano, R.; Jairath, V.; Panaccione, R.; Sandborn, W.J.; Ma, C. Comparative efficacy and safety of biologic therapies for moderate-to-severe Crohn’s disease: A systematic review and network meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 1002–1014. [Google Scholar] [CrossRef]
- Restellini, S.; Chao, C.Y.; Lakatos, P.L.; Aruljothy, A.; Aziz, H.; Kherad, O.; Bitton, A.; Wild, G.; Afif, W.; Bessissow, T. Therapeutic Drug Monitoring Guides the Management of Crohn’s Patients with Secondary Loss of Response to Adalimumab. Inflamm. Bowel Dis. 2018, 24, 1531–1538. [Google Scholar] [CrossRef]
- Dezelak, M.; Repnik, K.; Koder, S.; Ferkolj, I.; Potocnik, U. A Prospective Pharmacogenomic Study of Crohn’s Disease Patients during Routine Therapy with Anti-TNF-alpha Drug Adalimumab: Contribution of ATG5, NFKB1, and CRP Genes to Pharmacodynamic Variability. OMICS 2016, 20, 296–309. [Google Scholar] [CrossRef]
- Koder, S.; Repnik, K.; Ferkolj, I.; Pernat, C.; Skok, P.; Weersma, R.K.; Potocnik, U. Genetic polymorphism in ATG16L1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics 2015, 16, 191–204. [Google Scholar] [CrossRef]
- Repnik, K.; Koder, S.; Skok, P.; Ferkolj, I.; Potocnik, U. Transferrin Level Before Treatment and Genetic Polymorphism in HFE Gene as Predictive Markers for Response to Adalimumab in Crohn’s Disease Patients. Biochem. Genet. 2016, 54, 476–486. [Google Scholar] [CrossRef]
- Barber, G.E.; Yajnik, V.; Khalili, H.; Giallourakis, C.; Garber, J.; Xavier, R.; Ananthakrishnan, A.N. Genetic Markers Predict Primary Non-Response and Durable Response To Anti-TNF Biologic Therapies in Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1816–1822. [Google Scholar] [CrossRef]
- Yoon, S.M.; Haritunians, T.; Chhina, S.; Liu, Z.; Yang, S.; Landers, C.; Li, D.; Ye, B.D.; Shih, D.; Vasiliauskas, E.A.; et al. Colonic Phenotypes Are Associated with Poorer Response to Anti-TNF Therapies in Patients with IBD. Inflamm. Bowel Dis. 2017, 23, 1382–1393. [Google Scholar] [CrossRef]
- Arijs, I.; De Hertogh, G.; Lemaire, K.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; Cleynen, I.; Van Assche, G.; Vermeire, S.; et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS ONE 2009, 4, e7984. [Google Scholar] [CrossRef]
- Gorenjak, M.; Repnik, K.; Jezernik, G.; Jurgec, S.; Skok, P.; Potocnik, U. Genetic prediction profile for adalimumab response in Slovenian Crohn’s disease patients. Z. Gastroenterol. 2019, 57, 1218–1225. [Google Scholar] [CrossRef]
- Gorenjak, M.; Zupin, M.; Jezernik, G.; Skok, P.; Potocnik, U. Omics data integration identifies ELOVL7 and MMD gene regions as novel loci for adalimumab response in patients with Crohn’s disease. Sci. Rep. 2021, 11, 5449. [Google Scholar] [CrossRef]
- Schmitt, H.; Billmeier, U.; Dieterich, W.; Rath, T.; Sonnewald, S.; Reid, S.; Hirschmann, S.; Hildner, K.; Waldner, M.J.; Mudter, J.; et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 2019, 68, 814–828. [Google Scholar] [CrossRef]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef]
- Gole, B.; Potocnik, U. Pre-Treatment Biomarkers of Anti-Tumour Necrosis Factor Therapy Response in Crohn’s Disease-A Systematic Review and Gene Ontology Analysis. Cells 2019, 8, 515. [Google Scholar] [CrossRef]
- Bai, X.; Liu, W.; Chen, H.; Zuo, T.; Wu, X. Immune Cell Landscaping Reveals Distinct Immune Signatures of Inflammatory Bowel Disease. Front. Immunol. 2022, 13, 861790. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Kwak, M.S.; Hwang, C.I.; Cha, J.M.; Jeon, J.W.; Yoon, J.Y.; Park, S.B. Single-Cell Network-Based Drug Repositioning for Discovery of Therapies against Anti-Tumour Necrosis Factor-Resistant Crohn’s Disease. Int. J. Mol. Sci. 2023, 24, 14099. [Google Scholar] [CrossRef]
- Wu, Y.; Gettler, K.; Kars, M.E.; Giri, M.; Li, D.; Bayrak, C.S.; Zhang, P.; Jain, A.; Maffucci, P.; Sabic, K.; et al. Identifying high-impact variants and genes in exomes of Ashkenazi Jewish inflammatory bowel disease patients. Nat. Commun. 2023, 14, 2256. [Google Scholar] [CrossRef]
- Gole, B.; Pernat, C.; Jezernik, G.; Potocnik, U. The expression IL1B correlates negatively with the clinical response to adalimumab in Crohn’s disease patients: An ex vivo approach using peripheral blood mononuclear cells. Life Sci. 2023, 326, 121822. [Google Scholar] [CrossRef]
- Harvey, R.F.; Bradshaw, J.M. A simple index of Crohn’s-disease activity. Lancet 1980, 1, 514. [Google Scholar] [CrossRef]
- Panaccione, R.; Loftus, E.V., Jr.; Binion, D.; McHugh, K.; Alam, S.; Chen, N.; Guerette, B.; Mulani, P.; Chao, J. Efficacy and safety of adalimumab in Canadian patients with moderate to severe Crohn’s disease: Results of the Adalimumab in Canadian SubjeCts with ModErate to Severe Crohn’s DiseaSe (ACCESS) trial. Can. J. Gastroenterol. 2011, 25, 419–425. [Google Scholar] [CrossRef]
- Hlavaty, T.; Persoons, P.; Vermeire, S.; Ferrante, M.; Pierik, M.; Van Assche, G.; Rutgeerts, P. Evaluation of short-term responsiveness and cutoff values of inflammatory bowel disease questionnaire in Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 199–204. [Google Scholar] [CrossRef]
- Guyatt, G.; Mitchell, A.; Irvine, E.J.; Singer, J.; Williams, N.; Goodacre, R.; Tompkins, C. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology 1989, 96, 804–810. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 11 June 2022).
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef]
- Lu, S.; Li, J.; Song, C.; Shen, K.; Tseng, G.C. Biomarker detection in the integration of multiple multi-class genomic studies. Bioinformatics 2010, 26, 333–340. [Google Scholar] [CrossRef]
- Wang, X.; Kang, D.D.; Shen, K.; Song, C.; Lu, S.; Chang, L.C.; Liao, S.G.; Huo, Z.; Tang, S.; Ding, Y.; et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics 2012, 28, 2534–2536. [Google Scholar] [CrossRef]
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsström, B.; Edfors, F.; Odeberg, J.; et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carre, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef]
- Squair, J.W.; Gautier, M.; Kathe, C.; Anderson, M.A.; James, N.D.; Hutson, T.H.; Hudelle, R.; Qaiser, T.; Matson, K.J.E.; Barraud, Q.; et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [Google Scholar] [CrossRef]
- Zimmerman, K.D.; Espeland, M.A.; Langefeld, C.D. A practical solution to pseudoreplication bias in single-cell studies. Nat. Commun. 2021, 12, 738. [Google Scholar] [CrossRef]
- Junttila, S.; Smolander, J.; Elo, L.L. Benchmarking methods for detecting differential states between conditions from multi-subject single-cell RNA-seq data. Brief. Bioinform. 2022, 23, bbac286. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, A.P.; Zondervan, K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2010, 5, 1564–1573. [Google Scholar] [CrossRef]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Abecasis, G.R.; Hinds, D.A. minimac2: Faster genotype imputation. Bioinformatics 2015, 31, 782–784. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.R.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef]
- Kerimov, N.; Hayhurst, J.D.; Peikova, K.; Manning, J.R.; Walter, P.; Kolberg, L.; Samovica, M.; Sakthivel, M.P.; Kuzmin, I.; Trevanion, S.J.; et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 2021, 53, 1290–1299. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Ha, B.; Altay, G.; Greenbaum, J.A.; McVicker, G.; et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 2018, 175, 1701–1715.e16. [Google Scholar] [CrossRef]
- Buil, A.; Brown, A.A.; Lappalainen, T.; Vinuela, A.; Davies, M.N.; Zheng, H.F.; Richards, J.B.; Glass, D.; Small, K.S.; Durbin, R.; et al. Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat. Genet. 2015, 47, 88–91. [Google Scholar] [CrossRef]
- Chen, L.; Ge, B.; Casale, F.P.; Vasquez, L.; Kwan, T.; Garrido-Martin, D.; Watt, S.; Yan, Y.; Kundu, K.; Ecker, S.; et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 2016, 167, 1398–1414.e24. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Daperno, M.; D‘Haens, G.; Van Assche, G.; Baert, F.; Bulois, P.; Maunoury, V.; Sostegni, R.; Rocca, R.; Pera, A.; Gevers, A.; et al. Development and validation of a new, simplified endoscopic activity score for Crohn’s disease: The SES-CD. Gastrointest. Endosc. 2004, 60, 505–512. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Prada, C.; Lima, D.; Nakaya, H. MetaVolcanoR: Gene Expression Meta-analysis Visualization Tool. 2020. Available online: https://github.com/csbl-usp/MetaVolcanoR (accessed on 11 June 2022).
- Aterido, A.; Palau, N.; Domenech, E.; Nos Mateu, P.; Gutierrez, A.; Gomollon, F.; Mendoza, J.L.; Garcia-Planella, E.; Barreiro-de Acosta, M.; Munoz, F.; et al. Genetic association between CD96 locus and immunogenicity to anti-TNF therapy in Crohn’s disease. Pharmacogenom. J. 2019, 19, 547–555. [Google Scholar] [CrossRef]
- Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- Yavorska, O.O.; Burgess, S. MendelianRandomization: An R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 2017, 46, 1734–1739. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef]
- Collister, J.A.; Liu, X.; Clifton, L. Calculating Polygenic Risk Scores (PRS) in UK Biobank: A Practical Guide for Epidemiologists. Front. Genet. 2022, 13, 818574. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Autissier, P.; Soulas, C.; Burdo, T.H.; Williams, K.C. Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry A 2010, 77, 410–419. [Google Scholar] [CrossRef]
- Hernandez-Pacheco, N.; Gorenjak, M.; Jurgec, S.; Corrales, A.; Jorgensen, A.; Karimi, L.; Vijverberg, S.J.; Berce, V.; Schieck, M.; Acosta-Herrera, M.; et al. Combined analysis of transcriptomic and genetic data for the identification of loci involved in glucocorticosteroid response in asthma. Allergy 2020, 76, 1238–1243. [Google Scholar] [CrossRef]
- Gorenjak, M.; Jezernik, G.; Krusic, M.; Skok, P.; Potocnik, U. Identification of Novel Loci Involved in Adalimumab Response in Crohn’s Disease Patients Using Integration of Genome Profiling and Isoform-Level Immune-Cell Deconvoluted Transcriptome Profiling of Colon Tissue. Pharmaceutics 2022, 14, 1893. [Google Scholar] [CrossRef]
- Dahl, E.F.; Wu, S.C.; Healy, C.L.; Harsch, B.A.; Shearer, G.C.; O’Connell, T.D. Subcellular compartmentalization of proximal Galpha(q)-receptor signaling produces unique hypertrophic phenotypes in adult cardiac myocytes. J. Biol. Chem. 2018, 293, 8734–8749. [Google Scholar] [CrossRef]
- Desprairies, C.; Valence, S.; Maurey, H.; Helal, S.I.; Weckhuysen, S.; Soliman, H.; Mefford, H.C.; Spentchian, M.; Heron, D.; Leguern, E.; et al. Three novel patients with epileptic encephalopathy due to biallelic mutations in the PLCB1 gene. Clin. Genet. 2020, 97, 477–482. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, Z.; Zhao, W.; Wang, L.; Jiang, J.; Gu, L.; Li, M.; Jiang, L.; Bi, Y. PLCB1 Enhances Cell Migration and Invasion in Gastric Cancer Via Regulating Actin Cytoskeletal Remodeling and Epithelial-Mesenchymal Transition. Biochem. Genet. 2023, 61, 2618–2632. [Google Scholar] [CrossRef]
- Ratti, S.; Marvi, M.V.; Mongiorgi, S.; Obeng, E.O.; Rusciano, I.; Ramazzotti, G.; Morandi, L.; Asioli, S.; Zoli, M.; Mazzatenta, D.; et al. Impact of phospholipase C beta1 in glioblastoma: A study on the main mechanisms of tumor aggressiveness. Cell Mol. Life Sci. 2022, 79, 195. [Google Scholar] [CrossRef]
- Lu, G.; Chang, J.T.; Liu, Z.; Chen, Y.; Li, M.; Zhu, J.J. Phospholipase C Beta 1: A Candidate Signature Gene for Proneural Subtype High-Grade Glioma. Mol. Neurobiol. 2016, 53, 6511–6525. [Google Scholar] [CrossRef]
- Sengelaub, C.A.; Navrazhina, K.; Ross, J.B.; Halberg, N.; Tavazoie, S.F. PTPRN2 and PLCbeta1 promote metastatic breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J. 2016, 35, 62–76. [Google Scholar] [CrossRef]
- Xu, Z.W.; Liu, N.N.; Wang, X.Y.; Ding, B.C.; Zhang, H.F.; Li, Y.; Sun, W.Y.; Wei, W. Effect of PLC-beta1/CaM signaling pathway mediated by AT1R on the occurrence and development of hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 587. [Google Scholar] [CrossRef]
- Jia, W.H.; Zhang, B.; Matsuo, K.; Shin, A.; Xiang, Y.B.; Jee, S.H.; Kim, D.H.; Ren, Z.; Cai, Q.; Long, J.; et al. Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat. Genet. 2013, 45, 191–196. [Google Scholar] [CrossRef]
- Radhakrishna, U.; Ratnamala, U.; Jhala, D.D.; Uppala, L.V.; Vedangi, A.; Saiyed, N.; Patel, M.; Vadsaria, N.; Shah, S.R.; Rawal, R.M.; et al. Hidradenitis suppurativa associated telomere-methylome dysregulations in blood. J. Eur. Acad. Dermatol. Venereol. 2023, 38, 393–403. [Google Scholar] [CrossRef]
- Janse, I.C.; Koldijk, M.J.; Spekhorst, L.M.; Vila, A.V.; Weersma, R.K.; Dijkstra, G.; Horvath, B. Identification of Clinical and Genetic Parameters Associated with Hidradenitis Suppurativa in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 106–113. [Google Scholar] [CrossRef]
- Ha, S.D.; Lewin, N.; Li, S.S.C.; Kim, S.O. HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells. Cells 2021, 10, 1101. [Google Scholar] [CrossRef]
- Roger, T.; Lugrin, J.; Le Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef]
- Usami, M.; Kishimoto, K.; Ohata, A.; Miyoshi, M.; Aoyama, M.; Fueda, Y.; Kotani, J. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr. Res. 2008, 28, 321–328. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Jezernik, G.; Potocnik, U. Comprehensive genetic study of fatty acids helps explain the role of noncoding inflammatory bowel disease associated SNPs and fatty acid metabolism in disease pathogenesis. Prostaglandins Leukot. Essent. Fatty Acids 2018, 130, 1–10. [Google Scholar] [CrossRef]
- Sanford, J.A.; Zhang, L.J.; Williams, M.R.; Gangoiti, J.A.; Huang, C.M.; Gallo, R.L. Inhibition of HDAC8 and HDAC9 by microbial short-chain fatty acids breaks immune tolerance of the epidermis to TLR ligands. Sci. Immunol. 2016, 1, eaah4609. [Google Scholar] [CrossRef]
- Yang, L.; Wu, C.; Cui, Y.; Dong, S. Knockdown of histone deacetylase 9 attenuates sepsis-induced myocardial injury and inflammatory response. Exp. Anim. 2023, 72, 356–366. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, R.; Wu, L.; Jiang, J. Expression and function of Toll-like receptors in peripheral blood mononuclear cells in patients with ankylosing spondylitis. Mol. Med. Rep. 2019, 20, 3565–3572. [Google Scholar] [CrossRef]
- Starkhammar, M.; Kumlien Georen, S.; Dahlen, S.E.; Cardell, L.O.; Adner, M. TNFalpha-blockade stabilizes local airway hyperresponsiveness during TLR-induced exacerbations in murine model of asthma. Respir. Res. 2015, 16, 129. [Google Scholar] [CrossRef]
- Houser, M.C.; Caudle, W.M.; Chang, J.; Kannarkat, G.T.; Yang, Y.; Kelly, S.D.; Oliver, D.; Joers, V.; Shannon, K.M.; Keshavarzian, A.; et al. Experimental colitis promotes sustained, sex-dependent, T-cell-associated neuroinflammation and parkinsonian neuropathology. Acta Neuropathol. Commun. 2021, 9, 139. [Google Scholar] [CrossRef]
- Cole, D.K.; Laugel, B.; Clement, M.; Price, D.A.; Wooldridge, L.; Sewell, A.K. The molecular determinants of CD8 co-receptor function. Immunology 2012, 137, 139–148. [Google Scholar] [CrossRef]
- Rojas, M.; Rodriguez, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Zhu, C.; Li, Q.Z.; Ramirez-Santana, C.; Anaya, J.M. Autoimmunity is a hallmark of post-COVID syndrome. J. Transl. Med. 2022, 20, 129. [Google Scholar] [CrossRef]
- Liu, Y.; Ni, M.; Li, L.; Wang, J.; Tu, Z.; Zhou, H.; Zhang, S. A novel four-gene signature predicts immunotherapy response of patients with different cancers. J. Clin. Lab. Anal. 2022, 36, e24494. [Google Scholar] [CrossRef]
- Straube, J.; Bukhari, S.; Lerrer, S.; Winchester, R.J.; Gartshteyn, Y.; Henick, B.S.; Dragovich, M.A.; Mor, A. PD-1 signaling uncovers a pathogenic subset of T cells in inflammatory arthritis. Arthritis Res. Ther. 2024, 26, 32. [Google Scholar] [CrossRef]
- Halling, M.L.; Kjeldsen, J.; Knudsen, T.; Nielsen, J.; Hansen, L.K. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J. Gastroenterol. 2017, 23, 6137–6146. [Google Scholar] [CrossRef]
- Meisinger, C.; Freuer, D. Rheumatoid arthritis and inflammatory bowel disease: A bidirectional two-sample Mendelian randomization study. Semin. Arthritis Rheum. 2022, 55, 151992. [Google Scholar] [CrossRef]
- He, X.; Yin, J.; Yu, M.; Wang, H.; Qiu, J.; Wang, A.; Wu, X. Identification and Validation of Hub Genes for Predicting Treatment Targets and Immune Landscape in Rheumatoid Arthritis. Biomed. Res. Int. 2022, 2022, 8023779. [Google Scholar] [CrossRef]
- Meisinger, C.; Freuer, D. Causal Association Between Atopic Dermatitis and Inflammatory Bowel Disease: A 2-Sample Bidirectional Mendelian Randomization Study. Inflamm. Bowel Dis. 2022, 28, 1543–1548. [Google Scholar] [CrossRef]
- Chiesa Fuxench, Z.C.; Wan, J.; Wang, S.; Syed, M.N.; Shin, D.B.; Abuabara, K.; Gelfand, J.M. Risk of Inflammatory Bowel Disease in Patients With Atopic Dermatitis. JAMA Dermatol. 2023, 159, 1085–1092. [Google Scholar] [CrossRef]
- Bangert, C.; Rindler, K.; Krausgruber, T.; Alkon, N.; Thaler, F.M.; Kurz, H.; Ayub, T.; Demirtas, D.; Fortelny, N.; Vorstandlechner, V.; et al. Persistence of mature dendritic cells, T(H)2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Ralpha blockade. Sci. Immunol. 2021, 6, eabe2749. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Cortez, V.S.; Wang, Q.; McDonald, K.G.; Chai, J.N.; Di Luccia, B.; Gilfillan, S.; Hsieh, C.S.; Newberry, R.D.; Sibley, L.D.; et al. CRTAM Protects Against Intestinal Dysbiosis During Pathogenic Parasitic Infection by Enabling Th17 Maturation. Front. Immunol. 2019, 10, 1423. [Google Scholar] [CrossRef]
- Perez-Lopez, A.; Nuccio, S.P.; Ushach, I.; Edwards, R.A.; Pahu, R.; Silva, S.; Zlotnik, A.; Raffatellu, M. CRTAM Shapes the Gut Microbiota and Enhances the Severity of Infection. J. Immunol. 2019, 203, 532–543. [Google Scholar] [CrossRef]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Cortez, V.S.; Cervantes-Barragan, L.; Song, C.; Gilfillan, S.; McDonald, K.G.; Tussiwand, R.; Edelson, B.T.; Murakami, Y.; Murphy, K.M.; Newberry, R.D.; et al. CRTAM controls residency of gut CD4+CD8+ T cells in the steady state and maintenance of gut CD4+ Th17 during parasitic infection. J. Exp. Med. 2014, 211, 623–633. [Google Scholar] [CrossRef]
- Takeuchi, A.; Badr Mel, S.; Miyauchi, K.; Ishihara, C.; Onishi, R.; Guo, Z.; Sasaki, Y.; Ike, H.; Takumi, A.; Tsuji, N.M.; et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 2016, 213, 123–138. [Google Scholar] [CrossRef]
- Li, H.; Tsokos, G.C. Double-negative T cells in autoimmune diseases. Curr. Opin. Rheumatol. 2021, 33, 163–172. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Data | Therapies Prior to Induction of Biologicals | Sampling Timepoints and Therapy Response Assessments | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nr | Age at Inclusion | Sex | 5-Amino-Salicylate | Cortico-Steroids | Azathioprine | T0 | A3 | A6 | U3 | U6 |
| 001 | 59 | Male | Yes | Yes | No | S | RE/NA | RE/NA S | NA | NA |
| 002 | 27 | Male | Yes | Yes | Yes | S | RE/RE | RE/RE S | NA | NA |
| 003 | 63 | Female | Yes | Yes | Yes | NA | RE/RE | RE/RE S | NA | NA |
| 004 | 39 | Male | Yes | Yes | No | NA | NR/NR | NA | RE/RE | RE/RE S |
| 005 | 43 | Male | Yes | Yes | Yes | NA | RE/RE | RE/RE | RE/NA | RE/NA S |
| Chr | Base Pair | dbSNP ID | A1 | Gene | Rank | Subset | OR | L95 | U95 | p Value | AdjP Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 17 | 50,042,849 | rs12150443 | G | PPP1R9B | 5 | 1 | 0.3 | 0.1 | 0.7 | 0.009 | 0.017 |
| 11 | 122,774,340 | rs60999716 | T | CRTAM | 5.3 | 1 | 0.2 | 0.1 | 0.6 | 0.004 | 0.004 |
| 4 | 186,049,848 | rs13123257 | T | TLR3 | 6.3 | 1 | 4.8 | 1.7 | 13.6 | 0.003 | 0.021 |
| 4 | 186,100,161 | rs9312342 | C | TLR3 | 7.1 | 1 | 3.7 | 1.5 | 9.1 | 0.005 | 0.028 |
| 1 | 27,877,578 | rs12140013 | C | THEMIS2 | 8 | 1 | 3.9 | 1.4 | 10.8 | 0.008 | 0.008 |
| 9 | 104,789,257 | rs2297406 | T | ABCA1 | 8.1 | 1 | 0.1 | 0.0 | 0.3 | 0.001 | 0.001 |
| 2 | 86,942,294 | rs201118660 | T | CD8B | 8.4 | 2 | 16.0 | 2.1 | 123.0 | 0.008 | 0.008 |
| 9 | 104,982,380 | rs4743784 | A | ABCA1 | 9.2 | 2 | 0.2 | 0.1 | 0.6 | 0.002 | 0.004 |
| 17 | 50,222,983 | rs67288212 | A | PPP1R9B | 10 | 2 | 16.9 | 2.2 | 131.9 | 0.007 | 0.014 |
| 17 | 40,458,620 | rs10305315 | C | CCR7 | 10.7 | 2 | 12.2 | 2.1 | 72.7 | 0.006 | 0.006 |
| 20 | 8,147,472 | rs2327025 | T | PLCB1 | 10.9 | 2 | 5.2 | 1.5 | 17.6 | 0.008 | 0.008 |
| 7 | 18,770,935 | rs62446605 | A | HDAC9 | 11.6 | 2 | 4.1 | 1.5 | 11.8 | 0.008 | 0.023 |
| 17 | 74,541,040 | rs1532800 | C | CD300A | 12.4 | 3 | 3.8 | 1.4 | 10.2 | 0.010 | 0.010 |
| 4 | 15,679,295 | rs11722854 | G | CD38 | 13.8 | 3 | 0.3 | 0.1 | 0.7 | 0.006 | 0.019 |
| 1 | 221,839,608 | rs4579763 | A | DUSP10 | 14.5 | 3 | 5.8 | 1.5 | 21.9 | 0.009 | 0.009 |
| 10 | 96,773,725 | rs2861627 | A | PIK3AP1 | 15.1 | 3 | 13.2 | 2.1 | 83.0 | 0.006 | 0.006 |
| 4 | 186,098,752 | rs12645085 | T | TLR3 | 15.8 | 3 | 0.3 | 0.1 | 0.7 | 0.006 | 0.037 |
| 4 | 186,028,258 | rs62335289 | G | TLR3 | 16.6 | 3 | 11.5 | 2.0 | 67.8 | 0.007 | 0.040 |
| 4 | 15,857,239 | rs10001128 | A | CD38 | 17.2 | 6 | 3.8 | 1.5 | 10.1 | 0.007 | 0.020 |
| 4 | 186,077,934 | rs6811484 | G | TLR3 | 17.2 | 6 | 4.6 | 1.8 | 12.0 | 0.002 | 0.010 |
| 4 | 15,722,169 | rs10018756 | T | CD38 | 17.7 | 6 | 0.2 | 0.1 | 0.6 | 0.005 | 0.015 |
| 4 | 186,063,851 | rs35114430 | G | TLR3 | 19.3 | 6 | 0.2 | 0.1 | 0.6 | 0.006 | 0.034 |
| 7 | 18,580,458 | rs1012658 | C | HDAC9 | 19.5 | 6 | 3.3 | 1.4 | 7.8 | 0.007 | 0.020 |
| 7 | 18,729,677 | rs35242513 | G | HDAC9 | 20.3 | 6 | 5.1 | 1.6 | 15.7 | 0.005 | 0.014 |
| dbSNP ID | Gene | Tissue | NES | SE | p Value |
|---|---|---|---|---|---|
| rs12150443 | PPP1R9B | Th2 memory | −0.13 | 0.04 | 0.0020 |
| rs60999716 | CRTAM | CD8+ T naïve | −0.058 | 0.034 | 0.0912 * |
| rs13123257 | TLR3 | T cells | 0.18 | 0.09 | 0.0427 |
| rs9312342 | TLR3 | T cells | 0.28 | 0.069 | 0.0001 |
| rs12140013 | THEMIS2 | Th1-17 memory | −0.5 | 0.17 | 0.0044 |
| rs2297406 | ABCA1 | T cells | −0.17 | 0.077 | 0.0275 |
| rs201118660 | CD8B | Tfh memory | 1.33 | 0.65 | 0.0457 |
| rs4743784 | ABCA1 | CD4+ T cell | −0.26 | 0.096 | 0.0072 |
| rs67288212 | PPP1R9B | Monocytes | 0.075 | 0.031 | 0.0155 |
| rs10305315 | CCR7 | Blood | −0.46 | 0.18 | 0.0115 |
| rs2327025 | PLCB1 | Th1 memory | −0.26 | 0.094 | 0.0076 |
| rs62446605 | HDAC9 | NA | NA | NA | NA |
| rs1532800 | CD300A | Th1-17_memory | 0.17 | 0.071 | 0.0219 |
| rs11722854 | CD38 | T cells | −0.074 | 0.039 | 0.0562 * |
| rs4579763 | DUSP10 | B cell naïve | 0.19 | 0.068 | 0.0056 |
| rs2861627 | PIK3AP1 | Th2 memory | −0.37 | 0.18 | 0.0490 |
| rs12645085 | TLR3 | T cells | −0.13 | 0.071 | 0.0646 * |
| rs62335289 | TLR3 | T cells | 0.51 | 0.21 | 0.0145 |
| rs10001128 | CD38 | Blood | 0.1 | 0.045 | 0.0251 |
| rs6811484 | TLR3 | Blood | 0.11 | 0.037 | 0.0040 |
| rs10018756 | CD38 | Monocytes | 0.2 | 0.077 | 0.0105 |
| rs35114430 | TLR3 | Macrophage naïve | −0.21 | 0.13 | 0.1072 |
| rs1012658 | HDAC9 | NA | NA | NA | NA |
| rs35242513 | HDAC9 | NA | NA | NA | NA |
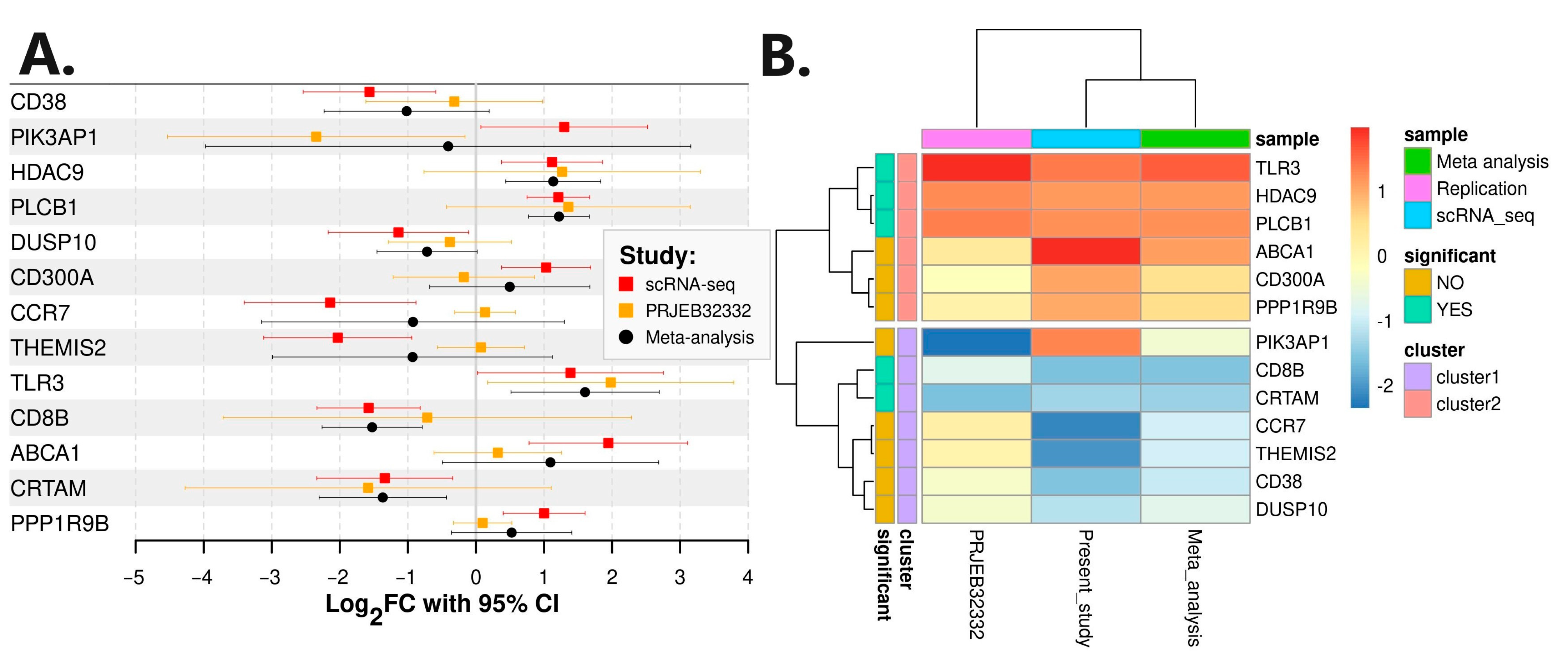
| Gene | scRNA-Seq Cohort | Replication Cohort | Meta-Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log2FC | L95 | U95 | p Value | Log2FC | L95 | U95 | p Value | Log2FC | L95 | U95 | p Value | |
| PLCB1 | 1.21 | 0.75 | 1.67 | 0.0003 | 1.36 | −0.43 | 3.15 | 0.1307 | 1.22 | 0.77 | 1.67 | 8.43 × 10−8 |
| PPP1R9B | 1.00 | 0.40 | 1.60 | 0.0051 | 0.10 | −0.33 | 0.53 | 0.6402 | 0.53 | −0.36 | 1.41 | 2.44 × 10−1 |
| CD300A | 1.03 | 0.38 | 1.69 | 0.0068 | −0.18 | −1.22 | 0.86 | 0.7278 | 0.50 | −0.68 | 1.67 | 4.08 × 10−1 |
| HDAC9 | 1.12 | 0.38 | 1.86 | 0.0086 | 1.27 | −0.76 | 3.30 | 0.2111 | 1.14 | 0.44 | 1.83 | 1.41 × 10−3 |
| ABCA1 | 1.95 | 0.78 | 3.11 | 0.0050 | 0.32 | −0.62 | 1.26 | 0.4872 | 1.10 | −0.49 | 2.68 | 1.77 × 10−1 |
| PIK3AP1 | 1.30 | 0.07 | 2.52 | 0.0404 | −2.35 | −4.53 | −0.16 | 0.0363 | −0.41 | −3.97 | 3.16 | 8.23 × 10−1 |
| TLR3 | 1.39 | 0.02 | 2.75 | 0.0469 | 1.98 | 0.17 | 3.79 | 0.0331 | 1.60 | 0.51 | 2.69 | 3.91 × 10−3 |
| CD8B | −1.58 | −2.34 | −0.82 | 0.0018 | −0.72 | −3.72 | 2.28 | 0.6281 | −1.53 | −2.26 | −0.79 | 4.85 × 10−5 |
| CRTAM | −1.34 | −2.34 | −0.34 | 0.0159 | −1.58 | −4.27 | 1.11 | 0.2370 | −1.37 | −2.31 | −0.43 | 4.15 × 10−3 |
| CD38 | −1.56 | −2.54 | −0.59 | 0.0069 | −0.32 | −1.62 | 0.98 | 0.6190 | −1.02 | −2.23 | 0.19 | 9.97 × 10−2 |
| THEMIS2 | −2.03 | −3.12 | −0.94 | 0.0032 | 0.07 | −0.57 | 0.71 | 0.8162 | −0.93 | −2.99 | 1.13 | 3.76 × 10−1 |
| DUSP10 | −1.14 | −2.17 | −0.11 | 0.0351 | −0.38 | −1.29 | 0.52 | 0.3926 | −0.72 | −1.45 | 0.02 | 5.58 × 10−2 |
| CCR7 | −2.14 | −3.40 | −0.88 | 0.0053 | 0.13 | −0.31 | 0.58 | 0.5422 | −0.92 | −3.15 | 1.30 | 4.15 × 10−1 |
| scRNA-Seq COHORT | Replication Cohort | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| dbSNP ID | Gene | A1 | OR | AdjP Value | dbSNP ID | D′ | A1 | p Value | OR | CI95 |
| rs60999716 | CRTAM | T | 4.12 | 0.0043 | rs10892897 | 1 | T | 0.01781 | 3.42 | 1.22–9.569 |
| / | / | / | / | / | rs10892893 | 0.9397 | T | 0.03554 | 3.02 | 1.091–8.383 |
| / | / | / | / | / | rs10892894 | 0.9354 | T | 0.00923 | 3.85 | 1.374–10.77 |
| rs2327025 | PLCB1 | T | 5.21 | 0.0079 | rs2327025 | NA | T | 0.01166 | 4.71 | 1.339–16.58 |
| rs62446605 | HDAC9 | A | 4.13 | 0.0233 | rs212671 | 0.5312 | G | 0.03246 | 2.93 | 1.053–8.131 |
| rs4579763 | DUSP10 | A | 5.83 | 0.0091 | rs6673674 | 1 | T | 0.04575 | 3.05 | 1.062–8.748 |
| rs35242513 | HDAC9 | G | 5.09 | 0.0144 | rs212671 | 0.6932 | G | 0.03246 | 2.93 | 1.053–8.131 |
| scRNA-Seq Cohort | Replication Cohort | Meta-Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| dbSNP ID | OR | L95 | U95 | dbSNP ID | OR | L95 | U95 | OR | L95 | U95 | p Value |
| rs60999716 | 4.12 | 1.56 | 10.89 | rs10892897 | 3.42 | 1.22 | 9.57 | 3.77 | 1.86 | 7.65 | 0.0002 |
| / | / | / | / | rs10892893 | 3.024 | 1.091 | 8.38 | 3.56 | 1.76 | 7.19 | 0.0004 |
| / | / | / | / | rs10892894 | 3.846 | 1.374 | 10.77 | 3.99 | 1.97 | 8.09 | 0.0001 |
| rs2327025 | 5.21 | 1.54 | 17.59 | rs2327025 | 4.71 | 1.34 | 16.58 | 4.96 | 2.07 | 11.90 | 0.0003 |
| rs62446605 | 4.13 | 1.45 | 11.75 | rs212671 | 2.93 | 1.05 | 8.13 | 3.46 | 1.67 | 7.19 | 0.0009 |
| rs4579763 | 5.83 | 1.55 | 21.93 | rs6673674 | 3.05 | 1.06 | 8.75 | 3.92 | 1.72 | 8.94 | 0.0012 |
| rs35242513 | 5.09 | 1.64 | 15.75 | rs212671 | 2.93 | 1.05 | 8.13 | 3.76 | 1.76 | 8.01 | 0.0006 |
| dbSNP ID | Gene | Tissue | NES | SE | p Value |
|---|---|---|---|---|---|
| rs10892893 | CRTAM | Blood | −0.079 | 0.039 | 0.0427 |
| rs10892897 | CRTAM | T cells | 0.13 | 0.072 | 0.0646 |
| rs10892894 | CRTAM | T cells | 0.15 | 0.073 | 0.0417 |
| rs212671 | HDAC9 | NA | NA | NA | NA |
| rs6673674 | DUSP10 | Blood | 0.03 | 0.014 | 0.0339 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorenjak, M.; Gole, B.; Goričan, L.; Jezernik, G.; Prosenc Zmrzljak, U.; Pernat, C.; Skok, P.; Potočnik, U. Single-Cell Transcriptomic and Targeted Genomic Profiling Adjusted for Inflammation and Therapy Bias Reveal CRTAM and PLCB1 as Novel Hub Genes for Anti-Tumor Necrosis Factor Alpha Therapy Response in Crohn’s Disease. Pharmaceutics 2024, 16, 835. https://doi.org/10.3390/pharmaceutics16060835
Gorenjak M, Gole B, Goričan L, Jezernik G, Prosenc Zmrzljak U, Pernat C, Skok P, Potočnik U. Single-Cell Transcriptomic and Targeted Genomic Profiling Adjusted for Inflammation and Therapy Bias Reveal CRTAM and PLCB1 as Novel Hub Genes for Anti-Tumor Necrosis Factor Alpha Therapy Response in Crohn’s Disease. Pharmaceutics. 2024; 16(6):835. https://doi.org/10.3390/pharmaceutics16060835
Chicago/Turabian StyleGorenjak, Mario, Boris Gole, Larisa Goričan, Gregor Jezernik, Uršula Prosenc Zmrzljak, Cvetka Pernat, Pavel Skok, and Uroš Potočnik. 2024. "Single-Cell Transcriptomic and Targeted Genomic Profiling Adjusted for Inflammation and Therapy Bias Reveal CRTAM and PLCB1 as Novel Hub Genes for Anti-Tumor Necrosis Factor Alpha Therapy Response in Crohn’s Disease" Pharmaceutics 16, no. 6: 835. https://doi.org/10.3390/pharmaceutics16060835
APA StyleGorenjak, M., Gole, B., Goričan, L., Jezernik, G., Prosenc Zmrzljak, U., Pernat, C., Skok, P., & Potočnik, U. (2024). Single-Cell Transcriptomic and Targeted Genomic Profiling Adjusted for Inflammation and Therapy Bias Reveal CRTAM and PLCB1 as Novel Hub Genes for Anti-Tumor Necrosis Factor Alpha Therapy Response in Crohn’s Disease. Pharmaceutics, 16(6), 835. https://doi.org/10.3390/pharmaceutics16060835