Abstract
Current therapies targeting the human epidermal growth factor receptor (HER) family, including monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKIs), are limited by drug resistance and systemic toxicities. Antibody–drug conjugates (ADCs) are one of the most rapidly expanding classes of anti-cancer therapeutics with 13 presently approved by the FDA. Importantly, ADCs represent a promising therapeutic option with the potential to overcome traditional HER-targeted therapy resistance by delivering highly potent cytotoxins specifically to HER-overexpressing cancer cells and exerting both mAb- and payload-mediated antitumor efficacy. The clinical utility of HER-targeted ADCs is exemplified by the immense success of HER2-targeted ADCs including trastuzumab emtansine and trastuzumab deruxtecan. Still, strategies to improve upon existing HER2-targeted ADCs as well as the development of ADCs against other HER family members, particularly EGFR and HER3, are of great interest. To date, no HER4-targeting ADCs have been reported. In this review, we extensively detail clinical-stage EGFR-, HER2-, and HER3-targeting monospecific ADCs as well as novel clinical and pre-clinical bispecific ADCs (bsADCs) directed against this receptor family. We close by discussing nascent trends in the development of HER-targeting ADCs, including novel ADC payloads and HER ligand-targeted ADCs.
1. Introduction
The human epidermal growth factor receptor (HER) family is an important class of receptor tyrosine kinases (RTKs) consisting of the epidermal growth factor receptor (ErbB1/EGFR), ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4. The HER family signals through a variety of substrates with integral roles in normal cellular processes including proliferation, differentiation, development, migration, adhesion, and others [1]. HER family members are frequently hyperactivated through mutation or overexpression in human cancers [2]. In the context of cancer, the HER family promotes uncontrolled cell growth, tumor progression, and metastasis. Consequently, there have been extensive efforts to target this family therapeutically, with the most successful avenues being monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKIs) against EGFR and HER2. Yet, intrinsic and acquired drug resistance are frequently observed in response to these therapies, limiting their efficacy to particular patient subsets and emphasizing the need for novel therapeutic approaches [3,4]. Recently, antibody–drug conjugates (ADCs) have emerged as a rapidly growing class of anti-cancer therapeutics to target HER family members and other RTKs [5]. Overall, 2 of 13 FDA-approved ADCs target HER2 and more than 50 of the 100 plus ADCs in clinical trials are against RTKs, including EGFR, HER2, HER3, c-MET, AXL, ROR1/2, EPHA3/5, FGFR3, and IGF-1R [6]. In this review, we provide a comprehensive evaluation of mono- and bispecific ADCs targeting each of the HER family members and emergent trends in HER-targeting ADCs, including novel payloads and HER ligand-targeting ADCs.
1.1. HER Activation and Signaling Mechanisms
Prior to ligand binding, EGFR, HER3, and HER4 monomers exist on the cell surface in an autoinhibitory state in which their dimerization arms are inaccessible to other cognate receptors [7,8]. The dimerization arm of HER2, rather, is constitutively exposed, deeming HER2 the preferred dimerization partner for all other HER monomers [9]. Notably, HER2 does not form homodimers in response to ligands but is able to form homodimers under states of HER2 overexpression [10]. Ligand binding to EGFR, HER3, or HER4 promotes a conformational change that exposes the receptor dimerization arm to interact with HER2 or a second ligand-bound receptor. Additionally, an increasing number of reports have described the existence of pre-formed, inactive EGFR dimers that, upon ligand binding, undergo a conformational change to reorient and activate the intracellular kinase domain [11]. There are seven HER family ligands including heparin-binding EGF (HB-EGF), amphiregulin (AREG), epiregulin (EREG), epigen (EPGN), neuregulin (NRG1-4), betacellulin (BTC), and transforming growth factor alpha (TGFɑ). EGF, TGFɑ, AREG, and EPGN exclusively bind EGFR; HB-EGF, EREG, and BTC are able to bind EGFR or HER4; NRG1/2 can bind HER3 and HER4; and NRG3/4 bind HER4 [12]. HER2 has no known natural ligand [13]. Following ligand binding, the activating kinase domain of one monomer phosphorylates the receiving kinase domain of the other monomer, termed trans-autophosphorylation [14]. Notably, while HER3 heterodimers are functionally active, HER3 homodimers possess minimal autophosphorylation because of mutations in its kinase domain [15]. Phosphorylated tyrosine residues in the C-terminal tail serve as docking sites for Src homology 2 (SH2) or phosphotyrosine-binding (PTB) domain-containing adaptor proteins that potentiate RAS/MAPK, PI3K/AKT, and PLCγ/PKC pathways, among others [16]. Importantly, different heterodimers, ligands, and phosphotyrosine residues confer bias toward different signaling pathways with varying levels of activation [17,18].
1.2. The HER Family in Cancer
HER family signaling is frequently dysregulated in cancers. For example, EGFR point mutations such as L858R and the T790M “gatekeeper” mutation, as well as exon 19 in-frame deletions, are all commonly observed in lung adenocarcinomas [19,20]. Additionally, deletion of exons 2–7 in the EGFR extracellular domain (ECD) results in a constitutively active “EGFRvIII” mutant frequently observed in glioblastoma multiforme (GBM) [21]. EGFR amplification is also broadly observed across cancer types, including 60–80% of colorectal cancers (CRCs) [22,23,24,25]. HER2 overexpression, found in 15–25% of breast cancers (BCs) and 10–30% of gastric cancers (GCs), is a common mechanism to promote cancer progression via the formation of ligand-independent, constitutively active HER2 homodimers [26,27]. HER3 is overexpressed in 50–70% of BCs, where HER2/HER3 heterodimers serve as functional tumor drivers [15,28,29]. The roles of HER4 in cancer are less well-studied, with both oncogenic and tumor-suppressive functions having been documented [30,31,32,33,34,35,36]. Taken together, the implications of the HER family in mediating cancer progression have prompted the development of several therapeutics against this family.
1.3. HER-Targeted TKIs and mAbs: Successes and Challenges
TKIs were the first therapeutic agents designed to target the HER family. TKIs compete for the ATP binding site in the receptor catalytic domain, inhibiting receptor activation and downstream signaling [37]. The first EGFR-targeting TKI, gefitinib, was approved in 2003 for non-small cell lung cancer (NSCLC), followed by erlotinib for NSCLC and pancreatic cancer in 2004 and 2005, respectively [38,39,40]. Notably, despite both exhibiting reversible binding to the EGFR tyrosine kinase domain, erlotinib is able to bind to both active and inactive EGFR conformations, whereas gefitinib only binds the active EGFR conformation [41]. While these first-generation EGFR TKIs are initially efficacious, nearly all patients develop resistance due to acquired mutations in EGFR (i.e., T790M), downstream mutations in KRAS/BRAF/PIK3CA, and alternative RTK activation, such as mesenchymal–epithelial transition factor (c-MET) [4,42]. Because of resistance mutations that allow evasion of first-generation TKIs, osimertinib, which irreversibly binds to residue C797 in the EGFR ATP binding pocket and demonstrates efficacy against T790M-harboring tumors, was approved in 2015 [43,44]. Yet, mechanisms of osimertinib resistance, such as EGFR C797S mutation, are still observed [45]. Other FDA-approved HER-targeting TKIs, including afatinib (EGFR/HER2/HER4), dacomitinib (pan-HER), lapatinib (EGFR/HER2), and neratinib (EGFR/HER2), are subject to similar resistance mechanisms [46,47,48,49].
MAbs are another class of therapeutics that bind HER monomer ECDs [50,51]. HER-targeting mAbs may exert cytostatic or cytotoxic effects through multiple mechanisms that include inhibiting ligand binding, receptor dimerization, and downstream signaling or promoting receptor internalization and degradation. MAbs may also engage Fcγ receptors on immune cells, resulting in antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) [52]. There are currently six FDA-approved mAbs targeting EGFR or HER2 and ongoing phase I/II clinical trials for HER3-targeting mAbs (NCT05057013, NCT04383210, NCT05203601, NCT05910827) [53,54,55,56]. EGFR-targeting mAbs are approved for various oncologic indications and all share similar binding epitopes in the EGFR ECDIII as follows: cetuximab (mouse/human chimeric mAb approved for metastatic CRC and head and neck squamous cell carcinoma (HNSCC)), panitumumab (human mAb approved for metastatic CRC), and necitumumab (human mAb approved for NSCLC). HER2-targeting mAbs are also approved for various oncologic indications, though they target slightly different epitopes as follows: trastuzumab (humanized mAb approved for BC, GC, and gastroesophageal junction adenocarcinoma; binds HER2 ECDII), pertuzumab (humanized mAb approved for BC; binds HER2 ECDII separate from trastuzumab), and margetuximab (mouse/human chimeric Fc-modified mAb approved for BC; binds same HER2 ECDII as trastuzumab) [57,58]. Yet, mAbs are still subject to the aforementioned mechanisms of resistance as TKIs as well as impaired ADCC/ADCP and dysregulated receptor degradation [59,60]. Example resistance mechanisms are depicted in Figure 1. Bispecific antibodies (bsAbs) that target combinations of EGFR, HER2, and HER3 or other surface receptors such as hepatocyte growth factor receptor (c-MET) and programmed cell death protein 1 (PD-1) have been developed to circumvent some of these resistance mechanisms, though resistance to these modalities is also not uncommon. For example, analysis of patient responses to amivantamab, an EGFR/c-MET bsAb approved for NSCLC, revealed resistance mechanisms including c-MET/EGFR amplification and PIK3CA mutations [61]. Considering the pervasive resistance mechanisms associated with current HER-targeting therapies, there is still a need for alternative strategies to target this family.
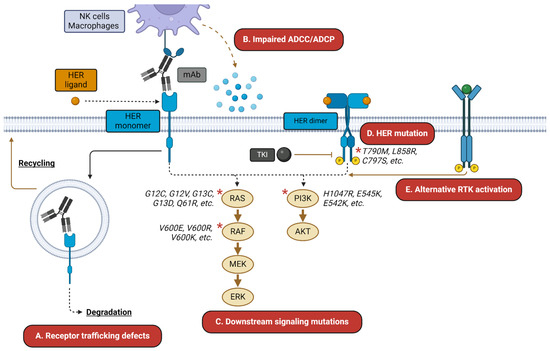
Figure 1.
HER-targeted therapy resistance mechanisms. HER-targeted mAbs and TKIs are subject to a wide variety of resistance mechanisms. These may include (A) altered trafficking of the mAb–receptor complex away from degradation and toward recycling; (B) impaired ADCC/ADCP-mediated killing; (C) resistance mutations in downstream signaling mediators; (D) mutations in the receptor itself; and (E) activation of alternative receptors, such as c-MET. Asterisks indicate commonly mutated signaling mediators.
1.4. Antibody–Drug Conjugates: The Anti-Cancer Biological Missiles
ADCs are a rapidly expanding class of anti-cancer therapeutics with 13 currently FDA-approved, including six for solid tumors and seven for hematological malignancies, and over 100 more in clinical trials [6]. ADCs consist of a mAb backbone conjugated to potent cytotoxins via a cleavable or non-cleavable linker. Cleavable linkers are more common among approved and preclinical ADCs and are cleaved by a variety of different mechanisms including lysosomal protease activity (e.g., cathepsin B), tumor-associated acidic/reducing conditions, or other enzymatic cleavage (i.e., beta-glucuronidase, sulfatases, phosphatases) [62]. ADCs with non-cleavable linkers, rather, require lysosomal degradation of the entire ADC for payload release. ADCs target surface-expressed, tumor-enriched antigens, enabling specific delivery of payloads to cancer cells and sparing normal tissue. The therapeutic index (TI) of ADC payloads is, in theory, enhanced compared with traditional chemotherapies by restricting their action to cancer cells. Among approved ADCs, traditional payload classes include DNA-damaging agents (i.e., calicheamicins and pyrrolobenzodiazepines (PBDs)); microtubule-disrupting auristatins and maytansanoids (i.e., monomethyl auristatin E (MMAE), monomethyl auristatin F (MMAF), DM1, and DM4); and topoisomerase I inhibitors (i.e., SN38 and DXd) [63]. While topoisomerase I inhibitors are the most predominant payload class in clinical trials, there is an increasing amount of alternative payloads being examined including topoisomerase II and RNA polymerase II inhibitors, immune agonists, proteolysis-targeting activating chimeras (PROTACs), and TKIs [64]. ADC payload is the most significant determinant of clinical toxicity profiles because of non-specific, normal tissue uptake [65]. Common treatment-related adverse events (TRAEs) include peripheral neuropathy and hematological malignancies for MMAE ADCs, ocular toxicity for DM4 and MMAF ADCs, thrombocytopenia and liver toxicity for DM1 ADCs, and hematological and gastrointestinal malignancies for PBD ADCs [66]. In response, several dose optimization and management strategies such as capping of treatment duration, fractionated dosing, treatment response-guided dose adjustments, and randomized dose-finding studies have all been employed [65]. Furthermore, innovations in ADC conjugation techniques have been made to improve TI and tolerability. Compared with traditional stochastic cysteine- or lysine-based conjugation strategies that result in heterogeneous pools of ADCs with varying DARs, site-specific and tag-free enzymatic conjugation methods have recently emerged as a promising strategy to generate homogenous ADC pools with improved pharmacodynamic and pharmacokinetic properties [67,68]. Upon binding its target antigen, ADCs are internalized and trafficked to the lysosome, resulting in linker cleavage, drug release, and antigen degradation [69]. Additionally, many ADC payloads are non-polar and diffuse into neighboring tumor cells to exert target-independent cytotoxicity, termed the “bystander effect,” though there are notable exceptions to this (MMAF and DM1) [70]. Furthermore, ADCs retain the therapeutic effects of the parent mAb. Therefore, the anti-tumor mechanisms of RTK-targeting ADCs may include (1) neutralization of downstream signaling; (2) Fcγ receptor-mediated immune cell engagement; (3) payload-mediated cytotoxicity; and (4) antigen degradation (Figure 2). Importantly, since ADC payloads exert cytotoxic effects independent of mAb and TKI cell-killing mechanisms, ADCs may be an effective option to overcome traditional resistance mechanisms of HER-targeted therapies.
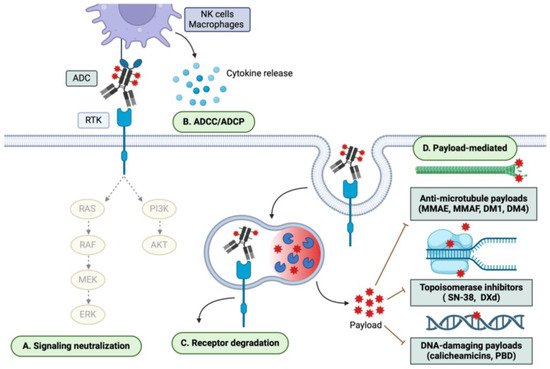
Figure 2.
ADC mechanisms of cytotoxicity. ADCs exert both mAb- and payload-mediated effects. Therefore, the mechanisms of ADC cytotoxicity may include (A) neutralization of downstream signaling pathways; (B) ADCC/ADCP; (C) degradation of the target receptor; and (D) payload-mediated cytotoxicity, which may be mediated through anti-microtubule, topoisomerase-inhibiting, and DNA-damaging mechanisms, among others.
A wide variety of novel ADC targets are being examined in clinical trials including RTKs, G protein-coupled receptors, glycoproteins, adhesion receptors, and immune checkpoint proteins [71,72,73,74,75,76]. The rapid growth of HER2-targeting ADCs exemplifies the potential for clinical success of HER-targeting ADCs. HER2 is the most popular ADC target, with 2 HER2-targeting ADCs having received FDA approval and 26 more in the clinical pipeline [6]. The two next most targeted RTKs lag far behind HER2, with seven c-MET- and four EGFR-targeted ADCs currently in clinical trials. The remainder of this review focuses on selected approved and clinically relevant HER-targeting monospecific and bispecific ADCs (bsADCs), as summarized in Table 1 and Table 2, respectively. Notably, no HER4-targeting ADCs have been reported to date.

Table 1.
HER-targeting monospecific ADCs.
Table 2.
HER-targeting bsADCs.
2. Monospecific ADCs Targeting the ErbB Receptor Family
2.1. EGFR-Targeting ADCs
As previously discussed, EGFR amplification and oncogenic mutations are commonly implicated in tumorigenesis, disease progression, and drug resistance. While there are several FDA-approved EGFR-targeting mAbs and TKIs, their efficacy is limited. EGFR-targeting ADCs have yet to receive FDA approval, though there are several currently under clinical investigation (Table 1) and in preclinical development.
2.1.1. Depatuxizumab Mafodotin (ABT-414)
Depatuxizumab mafodotin, or ABT-414, developed by Abbvie, consists of an ABT-806 mAb backbone conjugated via interchain cysteines to the microtubule-inhibiting payload MMAF via a non-cleavable maleimidocaproyl (mc) linker with an average drug-to-antibody ratio (DAR) of 3.8 [77,78]. Importantly, ABT-806 exhibits nearly 10-fold greater affinity for overexpressed, tumor-associated EGFR versus WT-EGFR. Phase I clinical trials (NCT01472003) of the ABT-806 mAb showed it was well-tolerated with significant brain uptake and minimal cutaneous toxicity, deeming it a rational choice for ADC development for brain tumors [79]. Preclinically, ABT-414 promoted tumor growth inhibition (TGI) and regression superior to ABT-806 in several EGFR-overexpressing xenografts [78]. In particular, ABT-414 was highly effective in GBM xenografts (2–10 mg/kg) and strongly synergized with the DNA-alkylating agent temozolomide (TMZ) and fractionated radiation at a low ADC dose (1 mg/kg). ABT-414 was assessed in phase I trials for EGFR-amplified, recurrent GBM (NCT01800695), wherein the most commonly observed TRAEs were reversible and ocular (i.e., blurred vision and dry eye), as is common with the MMAF payload [80,168]. Importantly, ABT-414 demonstrated reasonable efficacy with a median progression-free survival (PFS) of 2.1 months. ABT-414 was further evaluated in phase II trials at 1.25 mg/kg in adult participants with recurrent GBM with or without TMZ versus TMZ/lomustine monotherapies (NCT02343406). Combination ABT-414 and TMZ treatment compared with TMZ/lomustine monotherapy increased median PFS and overall survival (OS), warranting the investigation of ABT-414 in combination with standard chemo-irradiation and TMZ in phase III trials for newly diagnosed EGFR-amplified GBM patients (NCT02573324).
Unfortunately, ABT-414 phase III trials were discontinued for futility purposes with one potential explanation being limited tumor and blood–brain barrier (BBB) penetration [81]. Regarding tumor penetration, preclinical and clinical data showed that ABT-414 efficacy and intratumoral concentration were inversely correlated with tumor size (NCT01800695) [82]. Furthermore, while the ABT-806 mAb showed significant brain uptake in clinical studies, post-phase III trial ABT-414 studies demonstrated that GBM patient-derived xenografts (PDXs) intrinsically sensitive to ABT-414 as subcutaneous tumors were significantly less responsive when engrafted as orthotopic, intracranial tumors (2/7 responders). Furthermore, sensitivity was directly correlated with ABT-414 brain tumor uptake, suggesting ABT-414 BBB penetration may hinder efficacy [83]. Consistently, ABT-414 direct intratumoral injection restored PDX sensitivity. While the study did not examine ABT-806 uptake alongside ABT-414, it would be intriguing to evaluate how the addition of the linker–payload moiety affects biodistribution, BBB penetration, and, therefore, ADC efficacy. Beyond strategies to enhance BBB/tumor penetration, higher affinity mAbs with bystander effect–competent payloads may represent more effective approaches to GBM-indicated ADCs. Accordingly, additional ADCs were developed by Abbvie utilizing an affinity-matured ABT-806 (AM1) backbone as follows: losatuxizumab vedotin, or ABBV-221 (MMAE payload), and serclutamab talirine, or ABBV-321 (PBD payload) [84,85,87]. Unfortunately, phase I trials of ABBV-221 were halted because of the high incidence of infusion reactions, and ABBV-321 phase I trials were closed because of a lack of efficacy (NCT02365662, NCT03234712) [86,87].
2.1.2. MRG003
MRG003, developed by Miracogen, is an EGFR-targeting ADC consisting of an MMAE payload and a cleavable valine–citrulline (vc) linker. Phase I trials for MRG003 were completed for patients with EGFR-positive tumors (NCT04868344), and reasonable objective response rates (ORRs) were observed for HNSCC (40%) and nasopharyngeal carcinoma (NPC; 44%), although not for CRC patients (0%) [88]. Inefficacy in CRCs may be due to the relative insensitivity of these tumors to microtubule-destabilizing agents [169]. Thirty-one percent of patients reported ≥ grade 3 treatment-related AEs (TRAEs), including hyponatremia, leukocytopenia, and neutropenia. As observed with ABT-414, MRG003 exhibited less skin toxicity than EGFR mAbs cetuximab or panitumumab. Several phase II trials examining the efficacy of MRG003 monotherapy are underway for solid cancers (NCT04868162, NCT04838548, NCT05188209, NCT04838964, NCT05126719) as well as in combination with pucotenlimab or HX-008, a PD-1-targeting mAb (NCT05688605).
2.1.3. HLX42
HLX42, developed by Henlius, is a next-generation EGFR-targeting ADC with a novel topoisomerase I inhibitor and a DAR of 8 [89]. HLX42 integrates a linker that is cleaved in the tumor microenvironment (TME), circumventing the requirement for ADC internalization. Preclinical studies of HLX42 demonstrated its superior efficacy over an anti-EGFR-GGFG-DXd ADC in lung cancer xenografts (8 mg/kg), supporting the idea that the novel HLX42 linker and topoisomerase payload may confer improved efficacy over validated linker–payloads on FDA-approved ADCs (i.e., GGFG-DXd on trastuzumab deruxtecan). Pilot toxicity studies determined the highest non-severely toxic dose (HNSTD) in nonhuman primates as 20 mg/kg. HLX42 recently began phase I trials for advanced and metastatic solid tumors (NCT06210815).
While an EGFR-targeted ADC has yet to receive FDA approval, candidates like ABT-414 and MRG003 have demonstrated reasonable safety profiles in late-stage clinical trials, a major concern for therapeutic targeting of EGFR due to its ubiquitous expression. Despite ABT-414 inefficacy in phase III trials, MRG003 is the next most-progressed EGFR-targeting ADC and is being evaluated in a variety of EGFR-expressing tumors.
2.2. HER2-Targeting ADCs
ADCs targeting HER2 have been rather prolific in the clinical setting, and one of these agents, trastuzumab deruxtecan, recently received tumor-agnostic approval for HER2-positive (IHC3+) tumors [170]. Here, we briefly review recent clinical advances for approved HER2 ADCs and discuss HER2-targeted ADCs in phase III clinical development (Table 1). Additional information on earlier-stage HER2 ADCs may be found elsewhere [171].
2.2.1. Trastuzumab Emtansine (T-DM1)
Trastuzumab emtansine or T-DM1 is a HER2 ADC developed by Genentech in collaboration with ImmunoGen that comprises a trastuzumab backbone conjugated to DM1 via a nonreducible thioether linker (DAR = 3.5) [90,91]. T-DM1 was approved in 2013 for HER2-positive metastatic BC (mBC) based on the EMILIA trial, wherein T-DM1 significantly improved PFS and OS compared with lapatinib plus the antimetabolite capecitabine [92]. T-DM1 was subsequently approved in 2019 as adjuvant therapy for the treatment of HER2-positive early BC in patients with residual invasive disease following taxane (mitotic inhibitor)-based chemotherapy and trastuzumab treatment [93]. This approval was based on results from the KATHERINE trial where three years post-treatment, patients receiving T-DM1 experienced improved rates of invasive disease-free survival (88.3%) versus those on trastuzumab (77%). Common TRAEs associated with T-DM1 include nausea, fatigue, thrombocytopenia, and diarrhea [94]. Multiple trials are ongoing to increase T-DM1 efficacy and expand its utility. The ATEMPT 2.0 phase II trial (NCT04893109), for example, is examining T-DM1 in stage I HER2-positive BC versus the combination of paclitaxel and trastuzumab. Other trials are examining T-DM1 combination with HER2 TKI, tucatinib (NCT03975647); PD-1 mAb, pembrolizumab (NCT03032107); and PD-0332991, a cyclin-dependent kinase 4/6 (CDK4/6) inhibitor (NCT01976169).
2.2.2. Trastuzumab Deruxtecan (T-DXd)
Similar to T-DM1, trastuzumab deruxtecan, or T-DXd, co-developed by Daiichi Sankyo and AstraZeneca, is composed of a trastuzumab backbone. Deruxtecan refers to the linker-drug entity of a cathepsin-cleavable tetrapeptide (GGFG) linker and exatecan derivative DXd payload, which functions via topoisomerase I inhibition (DAR = 8) [95,96]. T-DXd was approved for HER2-positive mBC in 2019 following results from the DESTINY-Breast01 trial, where patients previously treated with T-DM1 achieved a PFS of 16.4 months and an estimated OS of 93.9% after 5.4 mg/kg T-DXd treatment (NCT03248492) [97]. Common ≥ grade 3 TRAEs include decreased neutrophil count, anemia, and nausea, and 13.6% of patients in the DESTINY-Breast01 trial also experienced interstitial lung disease (ILD) thought to be mediated by Fcγ receptor-mediated uptake on alveolar macrophages [98]. DESTINY-Breast03, which directly assessed T-DXd versus T-DM1 in HER2-positive mBC, found that patients receiving T-DXd experienced significantly improved median PFS compared with T-DM1 with similar rates of ≥ grade 3 TRAEs [99]. Furthermore, in patients with brain metastases, T-DXd promoted an intracranial ORR of 65.7% versus 34.3% for T-DM1. Similarly, the DESTINY-Breast05 trial is directly comparing the head-to-head efficacy of T-DXd versus T-DM1 in HER2-positive BC patients with residual disease following neoadjuvant therapy (NCT04622319). Recent clinical approval of T-DXd in HER2-low tumors, a majority subset of advanced BCs without adequate treatments, has significantly altered BC treatment and the ADC paradigm [172]. The DESTINY-Breast04 phase III trial evaluated the efficacy of T-DXd in previously treated, HER2-low (IHC 1+ or ICH2+/ISH−) mBCs (NCT03734029) and showed that T-DXd significantly improved median PFS (9.9 months) versus physician’s choice (5.5 months). T-DXd was therefore approved for unresectable or metastatic, previously treated HER2-low BC. The DESTINY-Breast06 trial aims to further expand this by evaluating hormone receptor-positive (HR+) HER2-low or HER2-ultralow (IHC > 0, <1+) patients (NCT04494425). Importantly, primary results from this trial showed that T-DXd conferred a clinically relevant increase in PFS for both HER2-low and HER2-ultralow patients versus physician’s choice of chemotherapy with no unexpected changes in safety (13.2 months versus 8.1 months and 13.2 months versus 8.3 months for HER2-low and HER2-ultralow, respectively) [100]. This may signal an important paradigm shift in the determinants of ADC efficacy beyond antigen abundance [173].
While T-DM1 is solely approved for BC, the DESTINY-Lung02 (NCT04644237) and DESTINY-Gastric01 (NCT03329690) trials prompted T-DXd approval for HER2-positive NSCLC and GCs. Furthermore, in April 2024, T-DXd also received tumor-agnostic approval in metastatic HER2-positive (IHC3+) solid tumors following results from the DESTINY-PanTumor02, DESTINY-CRC02, and DESTINY-Lung01 trials (NCT04482309; NCT04744831; NCT03505710) [101,102,170]. Like T-DM1, ongoing trials are evaluating the efficacy of T-DXd as a frontline treatment and in combination with other agents. For example, the DESTINY-Breast09 phase III trial (NCT04784715) is evaluating T-DXd as a first-line treatment in HER2-positive BC, either as monotherapy or in combination with pertuzumab. The SATEEN trial is examining the efficacy of sacituzumab govitecan, an FDA-approved TROP2-targeting ADC, in HER2-positive BCs following progression on T-DXd (NCT06100874). Additional trials are evaluating the efficacy of T-DXd with immune therapies and TKIs, such as nivolumab and tucatinib (NCT05480384; NCT02614794).
2.2.3. Disitamab Vedotin (RC48)
Disitamab vedotin, or RC48, developed by Remegen Biosciences and Yantai Rongchang Biological Engineering, is a hertuzumab-based HER2 ADC that consists of a cleavable mc-vc-PABC linker and MMAE payload (DAR = 4). Phase I trials of RC48 in HER2-positive advanced solid cancers demonstrated reasonable efficacy (ORR = 21% and disease control rate (DCR) = 49.1%) with a well-tolerated safety profile (NCT02881190). Most common ≥ grade 3 TRAEs included neutropenia, leukopenia, and hypoesthesia [103]. There is an ongoing phase III trial assessing RC48 efficacy in advanced GC (2.5 mg/kg Q2W) versus physician’s choice (NCT04714190) as well as numerous other trials in HER2-expressing cancers (NCT05996952, NCT05955209, NCT05980481, NCT0513715, NCT04329429). RC48 is also being examined in combination with immune therapies including PD-1 mAbs as well as the PD1xCTLA4 bsAb, cadonilimab (NCT06178601, NCT05912816, NCT05979740, NCT05726175).
2.2.4. MRG002
MRG002, developed by Shanghai Miracogen Inc., consists of the parent mAb MAB802 and MMAE payload conjugated to interchain cysteines via a protease-cleavable vc linker (DAR = 3.8). MAB802 shares the same amino acid sequence as trastuzumab but is hyper-fucosylated to dampen ADCC activity and reduce the potential incidence of immune-related AEs. Importantly, preclinical evaluation of MRG002 in GC and BC PDX models demonstrated equivalent or superior efficacy over T-DM1 [104]. GLP toxicity studies in nonhuman primates determined the HNSTD as 6 mg/kg (Q3Wx4), and the major toxicities were reversible hematotoxicity and myelotoxicity. MRG002 is now undergoing several clinical trials (phases I through III) in HER2-positive and HER2-low cancers (NCT04941339, NCT04839510, NCT04742153).
2.2.5. DP303c
Similar to MRG002 and RC48, DP303c, developed by CPSC ZhongQi Pharmaceutical Technology Co., Ltd., is a HER2-targeting, MMAE-conjugated ADC. DP303c incorporates a cleavable NH2-PEG3-vc linker via site-specific, microbial transglutaminase-mediated conjugation at Q295 in the mAb Fc region (DAR = 2). Compared with T-DM1, DP303c demonstrated equivalent or superior antitumor efficacy in GC and BC cell line xenografts (0.3–15 mg/kg similar to MRG002). This may be attributed in part to the fact that MMAE can exert a bystander effect, whereas DM1 cannot. Nonhuman primate studies found the HNSTD of DP303c to be 20 mg/kg (Q3Wx5), and major TRAEs were minimal to moderate lymphocyte depletion and lung inflammation, both of which were reversible [105]. DP303c has now progressed to phase III trials where it is being evaluated against T-DM1 in HER2-positive BC (NCT06313086).
2.2.6. FS-1502
FS-1502 is a HER2-targeting ADC developed by Iksuda Biotherapeutics consisting of a cleavable β-glucuronide linker and MMAF payload. FS-1502 demonstrated reasonable antitumor activity in phase I clinical trials in HER2-expressing advanced solid tumors with an ORR of 66.7% (4/6) in patients receiving a dose greater than 1 mg/kg, and the recommended phase II dose (RP2D) was 2.3 mg/kg Q3W (NCT03944499). Grade 3 TRAEs were observed in 24/67 of patients, the most common being hypokalemia and decreased platelet counts [106]. FS-1502 is now being evaluated against T-DM1 (3.6 mg/kg) in a phase III trial in locally advanced or mBC (NCT05755048).
2.2.7. Trastuzumab Duocarmazine (SYD985)
Trastuzumab duocarmazine, or SYD985, developed by Byondis, is composed of a trastuzumab backbone conjugated to the duocarmycin prodrug seco-duocarmycin-hydroxybenzamide-azaindole (seco-DUBA) via a cleavable vc linker (DAR = 2.8). Preclinical evaluation of SYD985 showed broader efficacy versus T-DM1 in BC PDX models, particularly in HER2-low models. A phase I dose-escalation study for SYD985 (NCT02277717) in several HER2-expressing cancers determined the RP2D at 1.2 mg/kg and the most common TRAEs included fatigue and dry eye with reasonable anti-tumor activity across HER2-positive and HER2-low tumors. SYD985 was assessed in the phase III TULIP trial versus physician’s choice in pretreated HER2-positive mBC and significantly improved PFS (7.0 months vs. 4.9 months) (NCT03262935) [107]. Presently, the FDA has issued a complete response letter for the Biological License Application of SYD985, suspending the approval decision for SYD985 in HER2-positive unresectable locally advanced or mBC [108].
2.2.8. Emergent Clinical-Stage HER2-Targeting ADCs
In addition to the previously discussed phase III/approved HER2-targeted ADCs, there are several more clinical assets that have shown promising preliminary safety and efficacy. A166 and ZV0203, for example, are two HER2-targeted ADCs conjugated to a duostatin-5 (MMAF derivative) payload that have demonstrated efficacy in patients heavily pretreated with other HER2-targeted therapies, including T-DM1 and T-DXd, in phase I/II trials (NCT03602079; NCT05346328; NCT05423977) [109,110,111,112,113]. Similarly, ARX788, which consists of an MMAF payload conjugated via unnatural para-acetylphenylalanine amino acid conjugation, is being examined in the phase II ACE-Breast-03 clinical trial for HER2-positive mBC patients previously treated with T-DXd (NCT04829604) [114,115,116].
2.3. HER3-Targeting ADCs
In addition to EGFR and HER2, HER3-targeting ADCs have also been evaluated in the clinical setting (Table 1). HER3 is frequently upregulated in multiple cancer subtypes and correlates with poor disease prognosis [174,175,176]. Despite favorable safety profiles, the clinical progress of HER3-targeted mAbs has been stalled by a lack of objective responses [177,178,179,180]. Yet, HER3-targeted ADCs may be a more favorable option because of increased antitumor effects conferred by payload-mediated cytotoxicity.
2.3.1. Patritumab Deruxtecan (U3-1402, HER3-DXd)
Patritumab deruxtecan, or U3-1402/HER3-DXd, developed by Daichii Sankyo in collaboration with Amgen, comprises the parent mAb patritumab conjugated to the cleavable GGFG tetrapeptide linker and topoisomerase I inhibitor DXd used in T-DXd [117]. The parent patritumab mAb competitively inhibits NRG1/2 binding to the HER3 ECD [118] and demonstrated reasonable safety and antitumor efficacy in phase I/II trials, although phase III trials were terminated because of a failure to meet pre-defined efficacy criteria, rationalizing the development of a patritumab-based ADC, HER3-DXd (NCT02134015; NCT01957280; NCT01211483) [118,119,177]. Preclinically, HER3-DXd exhibited durable responses and favorable toxicity profiles in lung, colorectal, breast, gastric, and melanoma cell line xenografts and PDX models (3–10 mg/kg) with negligible activity in HER3-low models [117,120,121]. Encouraging preclinical results prompted the phase I/II trial investigating HER3-DXd efficacy in heavily pretreated HER3-expressing mBC patients (NCT02980341) [122]. At lower doses (4.8 mg/kg), HER3-DXd exhibited a tolerable toxicity profile, with most common ≥ grade 3 TRAEs including decreased neutrophil, white blood cell, or platelet counts and anemia. Furthermore, HER-DXd achieved a median OS of 14.6 months in HR+/HER2− and triple-negative BC (TNBC) subtypes and 19.5% in HER2-positive BCs [123]. The ICARUS-BREAST01 phase II trial (NCT04965766), which evaluated HER3-DXd in heavily pretreated, HER3-unselected mBC patients, observed a DCR of 82.1% (40/56) [124]. The SOLTI TOT-HER3 (NCT0461528) study evaluated HER3-DXd in treatment-naïve patients with HR+/HER2− BC and reported meaningful clinical response following a single HER3-DXd dose (6.4 mg/kg) [125,126].
In addition to BC, clinical trials for HER3-DXd were also initiated in NSCLC. In a phase I dose escalation trial for EGFR TKI-pretreated metastatic NSCLC, HER3-Dxd (5.6 mg/kg Q3W) was well tolerated with a 39% ORR and median PFS of 8.2% (NCT03260491) [127]. In the dose expansion arm, HER3-Dxd (5.6 mg/kg) promoted a clinically meaningful response irrespective of RAS/PIK3CA/HER2 mutations, EGFR mutation, or c-MET amplification [128]. In the recently concluded phase II HERTHENA-Lung01 (NCT04619004) study, HER3-DXd dramatically improved ORR in EGFR-mutant NSCLC patients, including activity against brain metastases [129]. These data support the ongoing phase III HERTHENA-Lung02 (NCT05338970) trial evaluating HER3-Dxd versus standard-of-care chemotherapy in EGFR-mutated progressed NSCLC [130]. Yet, it is worth noting that ~60% of EGFR-mutated NSCLC patients do not respond to HER3-DXd because of low expression of HER3 [131]. Furthermore, HER3-DXd has been shown to be ineffective in other cancer subtypes, as indicated by the termination of a phase II trial examining HER3-DXd in advanced or metastatic CRC (NCT04479436).
2.3.2. AMT-562
AMT-562, developed by Multitude Therapeutics, was generated by conjugating HER3-targeting mAb Ab562 to exatecan via a self-immolating PABC spacer [132]. Ab562 has moderate HER3 affinity, minimizing the potential for on-target toxicity and improving tumor penetration. AMT-562 also improved TGI in CRC cell line xenografts compared with HER3-DXd and Ab562-GGFG-DXd, demonstrating improved tumor-killing capacity conferred by Ab562 over patritumab [132]. Consistently, AMT-562 (5–10 mg/kg) demonstrated improved antitumor activity over HER3-DXd in several HER3-low xenograft models (5–10 mg/kg) and durable responses in HER3-Dxd insensitive CRC PDXs. AMT-562 also strongly synergized with VEGF- or EGFR-targeting mAbs bevacizumab and cetuximab, respectively, in CRC PDXs and with osimertinib or KRAS G12C inhibitor sotorasib in NSCLC PDX models [132]. Safety studies of AMT-562 in nonhuman primates determined the HNSTD as 30 mg/kg, with primarily gastrointestinal-related toxicities that were reversible following 1-week treatment withdrawal [132]. Taken together, these preclinical findings suggest that AMT-562 may be a clinically viable option for HER3-low patients and cancers including CRC. AMT-562 is now under evaluation in phase I clinical trials (NCT06199908).
2.3.3. DB-1310
DB-1310, by Duality Biologics, is a HER3-targeting ADC produced by conjugating GenScript’s humanized 3F8 antibody via a cleavable maleimide tetrapeptide-based linker to the proprietary topoisomerase I inhibitor P1021 (DAR = 8) [133]. DB-1310 binds a distinct epitope from that of HER3-DXd with improved internalization and lysosomal trafficking [133]. Consistently, DB-1310 demonstrated increased TGI compared with HER3-DXd in lung, prostate, breast, and colon cancer cell line xenografts and PDXs (1–10 mg/kg) [133]. Similar to AMT-562, DB-1310 was also shown to synergize with osimertinib in EGFR-mutant NSCLC cell line xenografts and dramatically suppressed tumor growth in an osimertinib-resistant NSCLC PDX model [133]. Thus, DB-1310 may represent a viable clinical therapeutic option either in combination with or following patient progression on osimertinib. DB-1310 was well-tolerated in nonhuman primates (Q3Wx3) at an HNSTD of 45 mg/kg [133]. Lung and thymic toxicities were the primary observed TRAEs. Based on preclinical evaluation, a phase I/IIa trial of DB-1310 is underway in advanced/metastatic solid tumors (NCT05785741).
2.3.4. YL202/BNT326
YL202/BNT326 is a HER3-targeting ADC developed by MediLink Therapeutics consisting of a cleavable tripeptide linker and a novel topoisomerase I inhibitor, YL0010014 (DAR = 8) [134]. Uniquely, YL-202 incorporates a TME-cleavable linker utilizing MediLink’s TMALIN platform. Pre-clinical evaluation of YL202 demonstrated dose-dependent antitumor activity in cancer cell line xenografts and PDXs of several cancer types and revealed a tolerable safety profile in nonhuman primates [135]. YL202 was subsequently evaluated in a phase I trial (NCT05653752) for NSCLC and HR+/HER2− mBC. Commonly observed TRAEs included anemia and hematopoietic toxicities associated with topoisomerase I inhibition. YL202 is now being evaluated in two separate phase II trials for several tumor types (NCT06107686; NCT06439771).
3. Bispecific ADCs Targeting the HER Family
While single-target ADCs have experienced great clinical success, their efficacy is limited by two main factors including drug resistance and TRAEs. ADC drug resistance is mediated through several mechanisms including upregulation of drug efflux pumps, antigen downregulation, and alterations in ADC processing [181]. For example, HER2 ADCs are often routed for membrane recycling rather than lysosomal degradation, resulting in premature release into the extracellular space [66,182,183]. Example resistance mechanisms are depicted in Figure 3. TRAEs are largely, though not exclusively, driven by ADC payloads, such as neutropenia for topoisomerase I inhibitors and hepatotoxicity and thrombocytopenia for maytansine derivatives [65]. One emerging strategy to overcome drug resistance and reduce TRAEs is the development of bsADCs that simultaneously target two surface antigens. Notably, while all bsADCs target two cognate antigens or antigen epitopes, their formats are incredibly diverse. Some commonly employed bsADC formats are depicted in Figure 4. Compared with monospecific ADCs, bsADCs may increase efficacy via enhanced internalization and lysosomal trafficking, more effective targeting of tumor heterogeneity, and direct targeting of drug resistance-mediating receptors [184]. Clinical-stage and select emergent pre-clinical bsADCs consisting of at least one EGFR/HER2/HER3 targeting arm are reviewed below and summarized in Table 2.
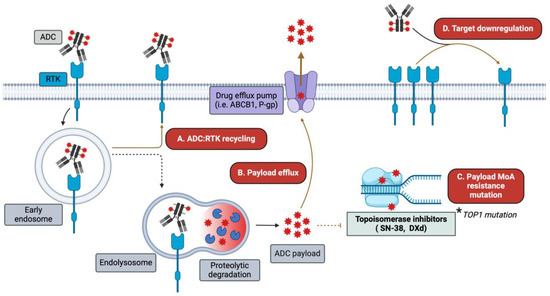
Figure 3.
Mechanisms of ADC resistance. While ADCs circumvent some of the resistance mechanisms encountered by traditional HER-targeting mAbs and TKIs, alternative resistance mechanisms emerge. Among these are (A) increased ADC–receptor recycling/routing away from degradation pathways; (B) upregulation of drug efflux transporters (e.g., ABCB1 and P-gp); (C) mutations in the machinery involved in payload-mediated cytotoxicity, such as TOP1; and (D) target antigen downregulation following ADC treatment.
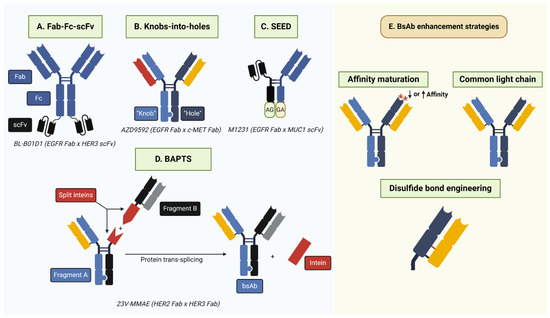
Figure 4.
BsAb generation strategies. (A) Fab-Fc-scFv involves the fusion of Fab and scFv antibody fragments targeting separate antigens. Notably, the scFv region may be attached to regions other than those depicted above, such as the Fab N-terminus. (B) Knobs-into-holes require the incorporation of complementary “knob” (T366W) and “hole” (T366S; L368A; Y407V) mutations into the antibody CH3 region to facilitate heterodimerization. BsAbs may have common or different light chains. (C) Strand-exchange engineered domain (SEED) relies on the generation of CH3 domains with alternating IgG/IGA domains (“GA”/“AG”) that facilitate heterodimerization. SEED technology is also amenable to the incorporation of alternative antibody formats, such as scFv, as depicted above. (D) Bispecific Antibody by Protein Trans-Splicing (BAPTS) generates two antibody fragments fused to the split intein Npu DnaE. Following protein-splicing, the full-length intein is removed and the full-length bsAb is generated. (E) Common strategies to enhance bsAb efficacy, selectivity, and production include affinity maturation to alter antibody affinity for a target antigen and the incorporation of a common light chain or engineered cysteine/disulfide bond to eliminate heavy–light chain mispairing. Asterisks depict amino acid mutations in the complementarity-determining region.
3.1. EGFR-Targeting bsADCs
3.1.1. BL-B01D1 (EGFR x HER3)
BL-B01D1, developed by Systimmune, Inc. and Sichuan Baili Pharmaceutical Co., Ltd., is an EGFR x HER3 bsADC consisting of a cathepsin-cleavable tetrapeptide linker and a topoisomerase I-inhibiting camptothecin derivative payload, ED-04 (DAR = 8) [136]. BL-B01D1 consists of a biparatopic anti-EGFR Fab arm fused to two anti-HER3 single-chain fragment variables (scFv) by glycine–serine linkage, generating a tetravalent bsAb backbone [62]. Preclinical evaluation in lung, colorectal, and pancreatic cancer cell line xenografts demonstrated that BL-B01D1 improved cytotoxicity over EGFR or HER3 monospecific ADCs. Phase I trials for BL-B01D1 in locally advanced or metastatic solid tumors (NCT05194982) determined an RP2D of 2.5 mg/kg (D1D8Q3W). Commonly observed ≥ grade 3 TRAEs were predominantly hematopoietic and gastrointestinal as are commonly implicated with other topoisomerase inhibitor ADCs, such as SN38-loaded TROP2 ADC and sacituzumab govitecan [137,185]. Furthermore, 3% of patients (5/195) experienced serious AEs resulting in treatment discontinuation, and only one patient experienced ILD. Importantly, BL-B01D1 demonstrated promising antitumor activity with a DCR of 89%, ORR of 34%, and median PFS of 5.7 months [138]. BL-B01D1 is now being tested in phase I-III trials for different tumor indications (NCT05983432; NCT05880706; NCT06118333; etc.). Several trials are also examining the efficacy of BL-B01D1 in combination with a PD-1xCTLA4 bsAb (SI-B003) (NCT05990803; NCT05924841; NCT06006169). Of note, BCG019 is another EGFR x HER3 bsADC currently in preclinical development by Biocytogen conjugated to either MMAE or the novel topoisomerase I inhibitor BCPT02, although its characterization thus far is limited [139].
3.1.2. M1231 (EGFR x MUC1)
M1231, a joint venture between Merck and Sutro Biopharma, is a bsADC targeting EGFR and mucin 1 (MUC1), which are highly co-expressed in cancers such as NSCLC, esophageal squamous cell carcinoma, HNSCC, TNBC, and ovarian cancer [140]. M1231 consists of an anti-MUC1 scFv and anti-EGFR Fab domain engineered by a SEED (strand-exchange engineered domain) antibody scaffold based on complementary IgG/IgA domains in the antibody CH3 Fc regions that promote heterodimerization [186]. M1231 consists of a cleavable vc-PABA linker conjugated via site-specific unnatural amino acids to SC209, a hemiasterlin derivative that disrupts microtubule dynamics [62]. M1231 improved internalization, lysosomal trafficking, and antitumor efficacy compared with monospecific EGFR or MUC1 bivalent antibodies, and pharmacokinetic (PK) studies in nonhuman primates determined an efficacious dose prediction range of 2.4–4.3 mg/kg Q3W [141]. M1231 has now completed phase I clinical trials (NCT04695847) for EGFR-expressing solid tumors, although results are not yet available nor are there ongoing phase II trials. Biocytogen has also reported the preclinical development of BSA01, another EGFR x MUC1 bsADC conjugated to MMAE [142,143].
3.1.3. AZD9592 (EGFR x c-MET)
AZD9592, developed by AstraZeneca, is an EGFR x c-MET bsADC consisting of a proprietary topoisomerase I inhibitor, AZ14170132, conjugated to an EGFR x c-MET bsAb backbone via a cleavable peptide linker [144]. The EGFR x c-MET bsAb was constructed utilizing a DuetMab monovalent bispecific IgG platform that uses knobs-into-holes technology to promote heavy chain dimerization and, furthermore, reduces heavy and light chain mispairing via the introduction of an engineered disulfide bond in one of the CH1-CL interfaces. Importantly, the bsAb backbone demonstrates a higher binding affinity for c-MET so as to reduce EGFR-mediated toxicity [144]. Intriguingly, AZD9592 in vitro efficacy was most robust when both EGFR and c-MET were engaged, suggesting that bispecific targeting of EGFR and c-MET may increase tumor selectivity and reduce on-target toxicities [144]. AZD9592 showed efficacy in EGFR-mutated NSCLC and HNSCC PDX models (2 mg/kg) and in EGFR, ALK, and c-MET TKI-refractory PDX models [145,146]. Nonhuman primate safety studies revealed a well-tolerated safety profile consistent with other topoisomerase I inhibitors [144]. The ongoing EGRET trial (NCT05647122) is the first-in-human phase I study evaluating AZD9592 monotherapy and in combination with osimertinib in advanced/metastatic NSCLC and HNSCC [147].
3.1.4. VBC101-F11 (EGFR x c-MET)
VBC101-F11 is another EGFR x c-MET bsADC developed by VelaVigo. Uniquely, VBC101-F11 is designed in a nanobody-based format (90kDa) that incorporates a low-affinity EGFR-targeting arm coupled to a high-affinity biparatopic c-MET arm and demonstrates superior cytotoxicity over the monospecific c-MET ADC ABBV-399 currently in phase III trials [148]. The nanobody format of VBC101-F11 also confers increased tumor penetration and accumulation compared with ABBV-399 and AZD9592 [148]. MMAE-conjugated VBC101-F11 displayed superior TGI over ABBV-399 in lung cancer cell line xenografts (3 mg/kg), and conjugation to a novel, undisclosed DNA replication inhibitor resulted in similar efficacy to AZD9592 in EGFR-low/c-MET-high and EGFR-/c-MET-moderate models [148]. The preclinical efficacy of VBC101-F11 supports its development in clinical trials, although phase I trials have not yet been initiated.
3.1.5. DM001 (EGFR x TROP2)
DM001 is an EGFR x TROP2 bsADC developed by Biocytogen utilizing their RenLite antibody production platform that utilizes knobs-into-holes technology to facilitate heavy chain heterodimerization and incorporates a common light chain [149]. Notably, TROP2 ADCs such as FDA-approved sacituzumab govitecan (SN38 payload) and phase III datopotamab deruxtecan (DXd payload), have shown efficacy in HR+/HER2− mBC and TNBC or metastatic urothelial cancer, respectively (NCT05104866) [187]. EGFR and TROP2 are co-expressed in many solid tumors, and co-targeting these receptors may improve tumor selectivity compared with EGFR or TROP2 monospecific ADCs [149]. The DM001 EGFR x TROP2 bsAb was generated, which demonstrated comparable internalization to its parental EGFR or TROP2 monospecific parental mAbs. DM001 was conjugated to MMAE via a protease-cleavable linker to generate the DM001 bsADC, which showed more potent cell-killing capacity in EGFR/TROP2-positive cells than single-target positive cells and showed potent anti-tumor activity in lung and pancreatic cell line xenografts and PDXs [149]. Furthermore, the DM001 bsADC demonstrated superior efficacy over its parental monospecific EGFR- and TROP2 ADCs in lung and pancreatic cancer PDXs. At this point, Biocytogen has reported the submission of an Investigational New Drug application for DM001.
3.2. HER2-Targeting bsADCs
3.2.1. Zanidatamab Zovodotin (ZW49; HER2 Biparatopic)
Zanidatamab zovodotin, or ZW49, developed by Zymeworks, is one of three biparatopic HER2-targeted ADCs to reach clinical trials. ZW49 consists of a zanidatamab (ZW25) mAb composed of an anti-HER2 ECDII Fab fragment and anti-HER2 ECDIV scFv fragment conjugated to a proprietary auristatin microtubule-inhibiting payload, ZD02044, via a cleavable dipeptide linker [150,151]. ZW25 induces receptor crosslinking and surface clustering, thereby enhancing internalization, lysosomal delivery, and receptor degradation [152]. Indeed, ZW49 internalized more rapidly than a monospecific trastuzumab-ZD02044 HER2 ADC. ZW49 also promoted tumor regression in HER2-high (3 mg/kg) and HER2-low (6 mg/kg) BC xenografts as well as GC PDXs (6 mg/kg) [151,153]. ZW49 was tolerable in a nonhuman primate toxicology study with no TRAEs observed up to the maximum dose tested (Q2Wx3; up to 12 mg/kg). A phase I dose escalation study for ZW49 for locally advanced or metastatic HER2-expressing cancers was initiated, with preliminary results determining a recommended dose as 2.5 mg/kg Q3W (ORR = 28%; DCR = 72%) (NCT03821233) [154]. Furthermore, 9% of patients had ≥ grade 3 TRAEs, including a grade 4 infusion reaction. Presently, plans for ZW49 phase II trials have been halted by Zymeworks pending further assessment of the clinical landscape [155].
3.2.2. MEDI4276 (HER2 Biparatopic)
Like ZW49, MEDI4276 developed by AstraZeneca also targets HER2 ECDs II and IV. ZW49 is composed of the trastuzumab scFv and HER2-directed mAb 39S and is conjugated to a tubulysin variant, AZ13599185, via an mc protease-cleavable peptide-based linker attached utilizing site-specific (S239C and S442C) cysteine conjugation (DAR = 4) [156,157]. MEDI4276 also incorporates an L234F mutation in the Fc region to impair FcγR binding, potentially limiting FcγR-mediated toxicities like T-DXd- and T-DM1-associated ILD [98,188]. Importantly, MEDI4276 internalized faster with greater lysosomal trafficking and HER2 degradation versus monospecific HER2 mAbs. Furthermore, MEDI4276 promoted regression in T-DM1-resistant BC and GC xenografts and was tolerable in nonhuman primates with a safety profile similar to other microtubule-inhibiting agents (HNSTD = 1 mg/kg) [158]. These results motivated a phase I/II study of MEDI4276 in patients with HER2-expressing BCs or GCs (NCT02576548). However, ORR was only 9.4% and MEDI4276 was poorly tolerated, with 75% of patients experiencing one or more ≥ grade 3 TRAE at the maximum-tolerated dose of 0.75 mg/kg [158]. Five patients experienced TRAEs leading to discontinuation, including hepatotoxicity and peripheral neuropathy, which may potentially be attributed to on-target effects due to enhanced internalization and/or ECDII inhibition.
3.2.3. JSKN003 (HER2 Biparatopic)
Developed by Alphamab Oncology, JSKN003 is the third HER2 biparatopic ADC in clinical trials and, like ZW49 and MEDI4276, targets HER2 ECDs II and IV. JSKN003 consists of a KN026 bsAb backbone conjugated to a topoisomerase I inhibitor via a dibenzocyclooctyne tetrapeptide linker by a proprietary glycan-specific conjugation strategy (DAR = 4) [159,160]. Notably, JSKN003 demonstrated more rapid internalization than T-DXd in GC and pancreatic cancer cell lines and promoted TGI in HER2-positive GC and BC cell line xenografts [159]. A first-in-human phase I study for JSKN003 (NCT05494918) was conducted in HER2-expressing patients, and interim analyses showed a well-tolerated safety profile with promising antitumor activity, particularly in HER2-amplified tumors (ORR = 71.4% for HER2 IHC 3+ patients; ORR = 55.6% and 37.5% for IHC 1+ and 2+, respectively). The maximum tolerated dose has not yet been reached after dosing up to 8.4 mg/kg [161]. JSKN003 is now under evaluation in phase I/II (NCT06226766; NCT05744427) tolerability and dose-escalation studies as well as phase III trials (NCT06079983) for HER2-low mBC.
3.2.4. 23V-MMAE (HER2 x HER3)
To date, one preclinical HER2 x HER3 bsADC, 23V-MMAE developed by researchers at Shanghai Jiao Tong University, has been reported. 23V-MMAE utilizes bispecific antibody by protein trans-splicing (BAPTS) to join the anti-HER2 antibody sequence from trastuzumab and the anti-HER3 antibody sequence from DL11, an EGFR x HER3 bsAb [162]. The BAPTS platform joins two separately generated antibody fragments via autocatalytic protein trans-splicing of split inteins at the antibody hinge region and eliminates heavy or light chain mispairing [189]. Following protein trans-splicing, there is the insertion of a five-amino acid residue “CFNAS” sequence in the hinge region containing a cysteine with high oxidation efficacy, and V205C mutations were introduced into each antibody light chain for site-specific cysteine conjugation to an mc-vc-PAB-MMAE linker–payload, thus generating the 23V-MMAE bsADC (DAR = 2.89). The binding affinity of 23V-MMAE for HER2 and HER3 was comparable to that of parental anti-HER2 2V-MMAE and anti-HER3 3V-MMAE monospecific ADCs in BC cells and exhibited slightly improved internalization. Importantly, 23V-MMAE promoted more substantial TGI in T-DM1-resistant cells compared with 2V-MMAE or 3V-MMAE monotherapy. In BC cell line xenografts, TGI and the survival of 23V-MMAE-treated mice were comparable to 2V-MMAE and 3V-MMAE combination therapy (3 mg/kg) and 2V-MMAE monotherapy (10 mg/kg), although 3V-MMAE monotherapy had no effect. To date, 23V-MMAE is still in the preclinical stages, though this work provides proof of concept that HER2 x HER3 bsADCs may be more effective than monospecific ADCs.
3.2.5. YH012 (HER2 x TROP2)
Additional HER2 bsADCs are being developed to enhance tumor specificity by co-targeting other non-RTK tumor-associated antigens such as TROP2, which is co-expressed with HER2 in various cancers [163]. YH012 is a HER2 x TROP2 developed by Biocytogen comprising an MMAE payload and protease-cleavable vc linker (DAR = 4). Importantly, the YH012 bsAb backbone demonstrated improved binding avidity and internalization compared with parental monospecific HER2 and TROP2 mAbs [163]. YH012 was also shown to be more selective for HER2+/TROP2+ cells rather than single target-positive cells, thereby enhancing tumor specificity and potentially reducing on-target toxicity compared with monospecific HER2 or TROP2 ADCs. Furthermore, YH012 induced potent TGI in NSCLC and GC cell line xenografts and HER2-low GC and CRC PDXs [163,164]. Nonhuman primate safety studies are still needed to determine the safety profile of YH012.
3.2.6. BIO-201 (HER2 x TROP2)
BIO-201, a HER2 x TROP2 bsADC developed by BiOneCure Therapeutics, is conjugated to an undisclosed topoisomerase I inhibitor via a cleavable linker [165]. Preclinical characterization indicated that BIO-201 showed similar binding and internalization compared to parental HER2/TROP2 mAbs and promoted TGI in HER2- or TROP2-positive xenograft models similar to T-Dxd [165]. Notably, the efficacy of BIO-201 in HER2- or TROP2-positive xenografts may indicate broader clinical utility over traditional monospecific ADCs targeting HER2 or TROP2. Presently, BIO-201 is still in the preclinical phases of development.
3.3. HER3-Targeting bsADCs
3.3.1. BCG022 (HER3 x c-MET)
BCG022, developed by Biocytogen, targets HER3 and c-MET, which are over-expressed in multiple solid tumors and mediate resistance to EGFR TKIs [180,190,191,192]. Similar to DM001, the BCG022 bsAb directed against HER3 and c-MET was generated using the RenLite production platform and showed superior internalization over HER3 or the c-MET parental mAbs. BCG022 was conjugated to either vc-MMAE or a BLD1102 linker-payload, which consists of a novel topoisomerase I inhibitor, BCPT02, and a highly hydrophilic protease-cleavable linker [166]. Both bsADCs induced robust tumor regression in NSCLC, GC, and pancreatic cancer xenografts, demonstrating potential clinical utility for BCG022-based bsADCs, although additional safety studies are warranted [166].
3.3.2. DM002 (HER3 x MUC1)
DM002 developed by Biocytogen is a preclinical bsADC that simultaneously targets HER3 and MUC1, which are co-expressed in lung, gastric, breast, and pancreatic cancers. As with other Biocytogen-developed bsADCs (i.e., DM001 and BCG022), the DM002 bsAb was generated utilizing the RenLite production platform utilizing knobs-into-holes technology and conjugated to MMAE via a protease-cleavable linker [167]. Importantly, DM002 recognizes the juxtamembrane domain of MUC1 as high levels of MUC1 may induce auto-proteolysis and render MUC1 N-terminal-targeting antibodies ineffective. Preliminary characterization showed the DM002 bsADC induced marked antitumor effects in lung cancer, BC, GC, and pancreatic cancer cell line xenografts and PDXs [167]. Yet, as with the aforementioned Biocytogen bsADCs, DM002 is still in preclinical stages and warrants additional safety studies prior to entering clinical trials.
4. Emergent Trends
While ADCs have emerged as a prominent force in the field of anti-cancer therapeutics, the efficacy of FDA-approved ADCs is limited by TRAEs, underscoring the importance of novel strategies to improve tolerability and expand their therapeutic index. As previously outlined, bsADCs are one strategy that may hold significant potential to build on the success of monospecific ADCs. ADC combination therapies with agents such as TKIs, mAbs, chemotherapy, and immune checkpoint therapies are another potential strategy to improve ADC efficacy, as previously described. Other novel approaches toward advancing ADC development include novel payloads and ligand-targeting ADCs, which we briefly detail below as they pertain to HER family-targeted ADCs. These emergent trends are summarized in Figure 5.
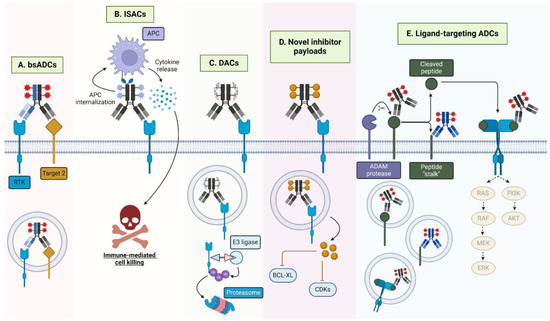
Figure 5.
Emergent HER-targeting antibody-conjugate modalities. (A) BsADCs engage two tumor-associated antigens, such as two RTKs. The development of bsADCs that engage a rapidly internalizing receptor is another common motif for bsADCs. (B) ISACs, loaded with immune agonists, such as TLR7/8 agonists, engage the FcγR on antigen-presenting cells (APCs), mediating internalization, immune activation, and cytokine release, resulting in target cell death. (C) DACs are loaded with protein degraders such as PROTACs or molecular glues that, upon internalization, mediate ubiquitination and subsequent proteasomal degradation of the degrader-targeted protein (i.e., BRD4, GSPT1). (D) ADCs with novel inhibitors, such as BCL-XL and CDK inhibitors function by specifically targeting these cytotoxins to target-expressing cells. (E) Ligand-targeted ADCs may recognize full-length surface-bound ligands, solubilized ligands, or novel ligand epitopes remaining following ligand proteolytic cleavage. Furthermore, ligand-targeted ADCs may have the added benefit of signaling neutralization through the associated ligand’s receptor.
4.1. ADCs Incorporating Novel Payloads
The majority of approved ADC payloads are either microtubule-inhibiting agents, DNA-damaging agents, or topoisomerase I inhibitors. However, the realm of alternative ADC payloads is rapidly expanding to include immune agonists, proteolysis-targeting activating chimeras (PROTACs), and BCL-XL and CDK inhibitors. Detailed below are several clinical and pre-clinical HER-targeting ADCs incorporating such novel payloads including toll-like receptor agonists and bromodomain-containing protein 4 (BRD4) PROTACs, among others.
4.1.1. Immune-Stimulating Antibody Conjugates (ISACs)
HER2-targeting ADCs SBT6050, NJH395, and BDC-1001 are loaded with toll-like receptor 7/8 (TLR7/8) agonists that induce immune activation, also referred to as immune-stimulating antibody conjugates (ISACs). SBT6050 underwent phase I trials as a monotherapy and in combination with PD-1 mAbs pembrolizumab or cemiplimab (NCT04460456) and demonstrated a tolerable safety profile at 0.6 mg/kg Q2W according to interim analyses [193]. However, it appears that since these studies, SBT6050 development has been halted based on limited efficacy and cytokine-related TRAEs [194]. Similarly, NJH395 demonstrated severe cytokine-associated toxicities in phase I trials (NCT03696771) [195]. BDC-1001, consisting of a trastuzumab biosimilar backbone conjugated to a TLR7/8 agonist via a non-cleavable linker, is in phase I/II trials as monotherapy and in combination with nivolumab (NCT04278144) [196]. When dosed Q2W, BDC-1001 demonstrated a reasonable safety profile and efficacy up to 20 mg/kg, supporting further phase II expansion [197]. BDC-1001 recently began a separate phase II clinical trial in combination with pertuzumab (NCT05954143). Further studies are necessary to clarify the efficacy and safety of ISACs in the clinical setting across cancers.
4.1.2. Degrader-Antibody Conjugates (DACs)
PROTACs are heterobifunctional molecules consisting of an E3 ligase-targeting moiety and a ligand for a protein of interest tethered together via a chemical linker often based on simple alkyl or PEG chains [198]. Functionally, PROTACs bring a protein of interest into proximity with the E3 ligase, promoting protein ubiquitination and proteasomal degradation. Much like ADCs, PROTAC linker design (i.e., length and composition) is a critical determinant of PROTAC efficacy, influencing binding cooperativity and protein isoform specificity [199]. Yet, PROTACs face challenges including cell permeability and lack of tissue specificity [200]. The conjugation of PROTACs to mAbs targeting tumor-enriched antigens, termed degrader–antibody conjugates, or DACs, may then increase the internalization and specificity of PROTACs. The potential of HER-targeting DACs was demonstrated with the preclinical development of a trastuzumab-based DAC conjugated to a BRD4 PROTAC (DAR = 4) that promoted BRD4 degradation exclusively in HER2-positive cells [201]. Furthermore, ORM-5029 is another DAC developed by Orum Therapeutics that consists of a pertuzumab backbone conjugated to a degrader of the translational termination factor GSTP1 (SMol006) via a cleavable vc-PABC linker [202]. Functionally, then, ORM-5029 exerts antitumor effects via GSPT1 degradation specifically in HER2-expressing cells. Preclinically, ORM-5029 demonstrated 10–1000 fold increased potency in HER2-expressing cell lines versus SMol006, T-DM1, and/or T-DXd monotherapies as well as antitumor efficacy in BC cell line xenografts superior to T-DM1 and comparable to T-DXd [202]. ORM-5029 has now begun phase I first-in-class, first-in-human clinical trials for HER2-expressing tumors (NCT05511844) and represents an exciting area for future collaboration between the ADC and PROTAC fields [203].
4.1.3. ADCs Incorporating BCL-XL or CDK Inhibitors
Inhibitors of anti-apoptotic BLC-2 family members (i.e., BCL-2, BCL-XL) as well as CDK inhibitors have recently emerged as a potential ADC payload class. BCL-2 proteins play an important role in cancer by preventing apoptosis and promoting therapy resistance [204]. ABBV-637 is an EGFR-targeting ADC that consists of a BCL-XL inhibitor payload and has undergone phase I clinical trials in combination with osimertinib in EGFR-mutant NSCLC (NCT04721015). ABBV-637 was reported to show reasonable clinical activity (DCR = 73% or 65% for third- or second-line therapy, respectively) with a manageable safety profile, although phase II trials have yet to start [205]. Additionally, CDK inhibitors such as palbociclib, ribociclib, and ademaciclib have experienced great clinical success, though they are limited by normal tissue toxicity in animal models and patients [206,207,208]. To improve selectivity, an EGFR-targeting cetuximab-based ADC loaded with a selective CDK2/7/9 inhibitor, SNS-032, was recently developed and reported to inhibit tumor growth in EGFR-high TNBC xenografts [209]. Overall, novel payloads are an exciting avenue for ongoing and future ADC diversification.
4.2. Internalization-Enhancing bsADCs
For ADCs that rely on lysosomal degradation for payload release, internalization of the ADC–target complex is a crucial determinant of efficacy. Hence, there are ongoing efforts to increase the lysosomal trafficking of slow-internalizing receptors, such as members of the HER family, via the generation of bsADCs that target HER family members and other rapidly internalizing proteins. Though these types of bsADCs have yet to reach clinical evaluation, preclinical studies have shown potential translational promise.
Prolactin receptor (PRLR) has low expression in BC tumors and rapidly internalizes to the lysosome [210]. A HER2xPRLR bsADC with a DM1 payload (DAR = 3.3) and non-cleavable linker induced HER2 degradation and exhibited increased potency over parental ADC monotherapy and combination therapy in BC cells [210]. Similarly, the transmembrane amyloid precursor-like protein 2 (APLP2) is efficiently trafficked to lysosomes upon internalization [211]. A HER2xAPLP2 bsADC was developed via conjugation of trastuzumab to microtubule inhibitor dimethyl dolastatin 10 payload [211]. While the HER2xAPLP2 bsAb increased HER2 internalization and lysosomal trafficking versus trastuzumab, HER2xAPLP2 bsADC efficacy was inferior to a bivalent HER2 ADC in BC xenograft models, suggesting that HER2 bivalent engagement may be a more effective strategy for improved ADC efficacy.
Lysosome-associated membrane glycoprotein 3 (LAMP3) and sortilin-1 (SORT1) are protein transport regulators with roles in endocytosis. BsADCs targeting HER2 and LAMP3 or SORT1 were developed and conjugated to microtubule inhibitor duostatin-3 via a cleavable vc linker (DAR = 1) and DXd (DAR = 6.1), respectively [212,213]. Notably, the HER2xLAMP3 ADC utilized a low-affinity LAMP3 Fab to increase selectivity for cells expressing both targets as LAMP3 is ubiquitously expressed. Both bsAb backbones enhanced HER2 internalization in HER2-low cells, though not in HER2-high cells. Consistently, HER2xLAMP3 bsADC potency was similar to the bivalent HER2 ADC in HER2-high cells but showed enhanced TGI (8 mg/kg) compared with monospecific ADCs in HER2-moderate cells and xenografts [212]. Similarly, the HER2xSORT1 ADC promoted more marked TGI in HER2-low BC cell line xenografts (two weekly 10 mg/kg doses) compared with T-DXd and monospecific parental ADCs [213]. Thus, coupling HER family members with other rapidly internalizing proteins represents a promising avenue for enhanced ADC internalization and efficacy, particularly when HER family members are expressed at lower levels. At higher expression levels, though, bivalent HER engagement may be more effective.
4.3. HER Ligand-Targeted ADCs
Novel target antigens represent another opportunity to broaden ADC utility. Ligand-targeted ADCs are a unique subclass of ADCs directed against cell surface-tethered ligands enriched on tumor cells, such as inhibitory Notch ligand delta-like ligand 3 (DLL3) and Siglec-9 ligand galectin 3 binding protein (LGALS3BP) [214,215]. Within the HER family, some surface-tethered ligands are enriched on tumors and may make for suitable ADC targets [216,217,218,219]. For example, an AREG ADC, GMF-1A3-MMAE, is currently in preclinical investigation for endocrine-resistant BC [220]. GMF-1A3 recognizes the neo-epitope or transmembrane stalk of AREG that remains on the cell surface following its proteolytic cleavage. GMF-1A3-MMAE was successfully internalized to the lysosome and promoted tumor regression in BC cell line xenografts [220,221]. Similarly, our group recently reported EREG-targeting ADCs, including H231-EGC-qDuoDM-gluc, incorporating a stable tripeptide linker and self-immolative glucuronide-modified duocarmycin DM payload [222,223]. H231 recognizes the unprocessed proepiregulin and the mature, soluble EREG forms, thereby providing the additional benefit of EREG signaling neutralization mediated through EGFR and HER4. Furthermore, H231-EGC-qDuoDM-gluc promoted significant TGI in CRC cell line xenografts and PDX models. Importantly, targeting ligands that signal through multiple receptors may increase efficacy over traditional receptor-targeting ADCs.
5. Conclusions
It is evident that extensive work has been undertaken to develop safe and effective ADCs targeting members of the HER family. While HER2-directed T-DM1 and T-DXd are the only FDA-approved HER family-targeting ADCs, EGFR-targeting MRG003 and HER3-DXd in phase II and III trials, respectively, demonstrate potential clinical utility in targeting other HER family members. Importantly, while improving patient survival and disease control, HER-targeted ADCs have demonstrated mostly well-tolerated safety profiles, with most toxicity being driven by off-target, payload-mediated effects. MAb affinity maturation and Fc modifications, alterations in linker chemistry and stability, and payload diversification (i.e., tubulysin, eribulin, amanitin, and doxorubicin) have all been employed as attempts to improve the toxicities that are observed with ADC treatment and may further broaden their therapeutic indices [65,67]. Combination therapies with different immunotherapies, chemotherapies, TKIs, and mAbs may also represent a promising approach to improve the present success of ADCs. Furthermore, although not yet reported for ADCs, combination treatment regimens that target cancer-specific vulnerabilities (i.e., fasting to starve tumor cells of glucose and/or strategies to increase TME pH) may be worthwhile pursuits in improving ADC clinical efficacy. For example, bicarbonate improved response to several immunotherapies (anti-PD-1 and anti-CTLA4 mAbs and adoptive cell therapy) in melanoma xenograft models by increasing TME pH and counteracting immunosuppressive effects mediated by the acidic TME pH [224]. In addition to these strategies to improve current ADCs, there is a growing appreciation for determinants of efficacy that go beyond target expression, as exemplified by T-DXd efficacy in HER2-low and HER2-ultralow settings [173]. While the exact mechanisms are still unclear, these exciting developments seem to suggest that high antigen abundance is not required for ADC efficacy and may shed light on other important cell-killing mechanisms, such as the target-independent bystander effect. Lastly, while we have focused exclusively on HER-targeted ADCs in this review, it is worth noting that there are several other RTK-targeting ADCs in late-stage clinical trials, such as c-MET-targeting telisotuzumab vedotin and ROR1-targeting zilovertamab vedotin, both of which are currently being assessed in phase III trials (NCT04928846; NCT06093503; NCT05139017) [225,226]. Thus, as ADCs continue to expand rapidly in the anti-cancer therapeutic realm, the HER family and RTKs at large have vast potential to broaden ADC utility significantly and the portfolio of FDA-approved targets.
Author Contributions
Conceptualization, P.H. and K.S.C.; writing—original draft preparation, P.H., C.G., S.S., and J.J.; writing—review and editing, P.H., C.G., S.S., and K.S.C.; visualization, P.H.; supervision, K.S.C.; funding acquisition, K.S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from NCI (R01 CA226894, R21 CA270716, and R21CA282378) and the Cancer Prevention and Research Institute of Texas (CPRIT) RP210092 to K.S.C.; predoctoral fellowships of the Gulf Coast Consortia, on the Training Interdisciplinary Pharmacology Scientists Program (T32 GM139801) to P.H.; and a predoctoral fellowship in the Biomedical Informatics, Genomics and Translational Cancer Research Training Program (BIG-TCR) funded by CPRIT (RP210045) to S.S.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
All figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wieduwilt, M.J.; Moasser, M.M. The Epidermal Growth Factor Receptor Family: Biology Driving Targeted Therapeutics. Cell. Mol. Life Sci. CMLS 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in Cancer Cells Is Mediated by Loss of IGF-Binding Proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef]
- Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Flynn, P.; Suryaprakash, S.; Grossman, D.; Panier, V.; Wu, J. The Antibody–Drug Conjugate Landscape. Nat. Rev. Drug Discov. 2024. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions That Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Leahy, D.J. Structure of the Extracellular Region of HER3 Reveals an Interdomain Tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the Preferred Heterodimerization Partner of All ErbB Receptors, Is a Mediator of Lateral Signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF Receptor Ligands: Recent Advances. F1000Research 2016, 5, 2270. [Google Scholar] [CrossRef]
- Gutierrez, C.; Schiff, R. HER2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef]
- Honegger, A.M.; Kris, R.M.; Ullrich, A.; Schlessinger, J. Evidence That Autophosphorylation of Solubilized Receptors for Epidermal Growth Factor Is Mediated by Intermolecular Cross-Phosphorylation. Proc. Natl. Acad. Sci. USA 1989, 86, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the Biology of HER3 Receptor as a Therapeutic Target in Human Cancer. Acta Pharm. Sin. B 2018, 8, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Stacey, M.M.; Liu, B.A.; Pawson, T. Molecular Mechanisms of SH2- and PTB-Domain-Containing Proteins in Receptor Tyrosine Kinase Signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a008987. [Google Scholar] [CrossRef]
- Beerli, R.R.; Hynes, N.E. Epidermal Growth Factor-Related Peptides Activate Distinct Subsets of ErbB Receptors and Differ in Their Biological Activities. J. Biol. Chem. 1996, 271, 6071–6076. [Google Scholar] [CrossRef]
- Riese, D.J.; Bermingham, Y.; van Raaij, T.M.; Buckley, S.; Plowman, G.D.; Stern, D.F. Betacellulin Activates the Epidermal Growth Factor Receptor and erbB-4, and Induces Cellular Response Patterns Distinct from Those Stimulated by Epidermal Growth Factor or Neuregulin-Beta. Oncogene 1996, 12, 345–353. [Google Scholar]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal Growth Factor Receptor (EGFR) in Lung Cancer: An Overview and Update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- O’Leary, C.; Gasper, H.; Sahin, K.B.; Tang, M.; Kulasinghe, A.; Adams, M.N.; Richard, D.J.; O’Byrne, K.J. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals 2020, 13, 273. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational Up-Regulation of the EGFR by Tumor Hypoxia Provides a Nonmutational Explanation for Its Overexpression in Human Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Pabla, B.; Bissonnette, M.; Konda, V.J. Colon Cancer and the Epidermal Growth Factor Receptor: Current Treatment Paradigms, the Importance of Diet, and the Role of Chemoprevention. World J. Clin. Oncol. 2015, 6, 133–141. [Google Scholar] [CrossRef]
- Hynes, N.E.; Stern, D.F. The Biology of erbB-2/Neu/HER-2 and Its Role in Cancer. Biochim. Biophys. Acta 1994, 1198, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Amisha, F.; Malik, P.; Saluja, P.; Gautam, N.; Patel, T.H.; Roy, A.M.; Singh, S.R.K.; Malapati, S.J. A Comprehensive Review on the Role of Human Epidermal Growth Factor Receptor 2 (HER2) as a Biomarker in Extra-Mammary and Extra-Gastric Cancers. Onco 2023, 3, 96–124. [Google Scholar] [CrossRef]
- Alimandi, M.; Romano, A.; Curia, M.C.; Muraro, R.; Fedi, P.; Aaronson, S.A.; Di Fiore, P.P.; Kraus, M.H. Cooperative Signaling of ErbB3 and ErbB2 in Neoplastic Transformation and Human Mammary Carcinomas. Oncogene 1995, 10, 1813–1821. [Google Scholar] [PubMed]
- Majumder, A. HER3: Toward the Prognostic Significance, Therapeutic Potential, Current Challenges, and Future Therapeutics in Different Types of Cancer. Cells 2023, 12, 2517. [Google Scholar] [CrossRef]
- Jia, X.; Wang, H.; Li, Z.; Yan, J.; Guo, Y.; Zhao, W.; Gao, L.; Wang, B.; Jia, Y. HER4 Promotes the Progression of Colorectal Cancer by Promoting Epithelial-Mesenchymal Transition. Mol. Med. Rep. 2020, 21, 1779–1788. [Google Scholar] [CrossRef]
- Canfield, K.; Li, J.; Wilkins, O.M.; Morrison, M.M.; Ung, M.; Wells, W.; Williams, C.R.; Liby, K.T.; Vullhorst, D.; Buonanno, A.; et al. Receptor Tyrosine Kinase ERBB4 Mediates Acquired Resistance to ERBB2 Inhibitors in Breast Cancer Cells. Cell Cycle Georget. Tex 2015, 14, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.R.; Edelblum, K.L.; Mullane, M.T.; Liang, D.; Polk, D.B. The ErbB4 Growth Factor Receptor Is Required for Colon Epithelial Cell Survival in the Presence of TNF. Gastroenterology 2009, 136, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gong, L.; Qian, Z.; Song, G.; Liu, J. ERBB4 Promotes the Proliferation of Gastric Cancer Cells via the PI3K/Akt Signaling Pathway. Oncol. Rep. 2018, 39, 2892–2898. [Google Scholar] [CrossRef] [PubMed]
- Portier, B.P.; Minca, E.C.; Wang, Z.; Lanigan, C.; Gruver, A.M.; Downs-Kelly, E.; Budd, G.T.; Tubbs, R.R. HER4 Expression Status Correlates with Improved Outcome in Both Neoadjuvant and Adjuvant Trastuzumab Treated Invasive Breast Carcinoma. Oncotarget 2013, 4, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Gullick, W.J. C-erbB-4/HER4: Friend or Foe? J. Pathol. 2003, 200, 279–281. [Google Scholar] [CrossRef]
- Lucas, L.M.; Dwivedi, V.; Senfeld, J.I.; Cullum, R.L.; Mill, C.P.; Piazza, J.T.; Bryant, I.N.; Cook, L.J.; Miller, S.T.; Lott, J.H.; et al. The Yin and Yang of ERBB4: Tumor Suppressor and Oncoprotein. Pharmacol. Rev. 2022, 74, 18–47. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Haap, M.; Kopp, H.-G.; Lipp, H.-P. Tyrosine Kinase Inhibitors—A Review on Pharmacology, Metabolism and Side Effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa) Tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Erlotinib (Tarceva) Tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef]
- Starling, N.; Neoptolemos, J.; Cunningham, D. Role of Erlotinib in the Management of Pancreatic Cancer. Ther. Clin. Risk Manag. 2006, 2, 435–445. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448 Pt 3, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Soulières, D.; Melezínek, I.; Moecks, J.; Keil, L.; Mok, T.; Rosell, R.; Klughammer, B. Clinical Outcomes in Non-Small-Cell Lung Cancer Patients with EGFR Mutations: Pooled Analysis. J. Cell. Mol. Med. 2010, 14, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, E.; De Carlo, E.; Del Conte, A.; Stanzione, B.; Revelant, A.; Fassetta, K.; Spina, M.; Bearz, A. Acquired Resistance to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer: How Do We Overcome It? Int. J. Mol. Sci. 2022, 23, 6936. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Yang, J.C.-H.; Lee, C.K.; Kurata, T.; Kim, D.-W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation–Positive Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nakashima, C.; Komiya, K.; Kitera, K.; Hirai, M.; Kimura, S.; Aragane, N. Mechanisms of Acquired Resistance to Afatinib Clarified with Liquid Biopsy. PLoS ONE 2018, 13, e0209384. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018, 13, 727–731. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, V.; Raimondo, L.; Formisano, L.; Giuliano, M.; De Placido, S.; Rosa, R.; Bianco, R. Mechanisms of Lapatinib Resistance in HER2-Driven Breast Cancer. Cancer Treat. Rev. 2015, 41, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Eli, L.D.; Kavuri, S.M. Mechanisms of Neratinib Resistance in HER2-Mutant Metastatic Breast Cancer. Cancer Drug Resist. 2022, 5, 873–881. [Google Scholar] [CrossRef]
- Freeman, D.; Sun, J.; Bass, R.; Jung, K.; Ogbagabriel, S.; Elliott, G.; Radinsky, R. Panitumumab and Cetuximab Epitope Mapping and in Vitro Activity. J. Clin. Oncol. 2008, 26 (Suppl. S15), 14536. [Google Scholar] [CrossRef]
- Hao, Y.; Yu, X.; Bai, Y.; McBride, H.J.; Huang, X. Cryo-EM Structure of HER2-Trastuzumab-Pertuzumab Complex. PLoS ONE 2019, 14, e0216095. [Google Scholar] [CrossRef] [PubMed]
- Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.E.; Lyu, H.; Liu, B. HER3-Targeted Therapeutic Antibodies and Antibody–Drug Conjugates in Non-Small Cell Lung Cancer Refractory to EGFR-Tyrosine Kinase Inhibitors. Chin. Med. J. Pulm. Crit. Care Med. 2023, 1, 11–17. [Google Scholar] [CrossRef]
- Thakkar, D.; Paliwal, S.; Kar, S.; Gandhi, N.; Paszkiewicz, K.; Ingram, P.; Boyd-Kirkup, J. Abstract P197: An Anti-HER3 Antibody, HMBD-001, That Uniquely Binds to and Blocks the HER3 Heterodimerization Interface, Shows Superior Tumor Growth Inhibition in Biomarker-Defined Preclinical Cancer Models Including NRG1-Fusion Driven Cancers. Mol. Cancer Ther. 2021, 20 (Suppl. S12), P197. [Google Scholar] [CrossRef]
- Li, W.; Xie, C.; Zhu, X.; Tang, J.; Wang, L.; Lou, L. SIBP-03, a Novel Anti-HER3 Antibody, Exerts Antitumor Effects and Synergizes with EGFR- and HER2-Targeted Drugs. Acta Pharmacol. Sin. 2024, 45, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Thavaneswaran, S.; Chan, W.Y.; Asghari, R.; Grady, J.P.; Deegan, M.; Jansen, V.M.; Thomas, D.M. Clinical Response to Seribantumab, an Anti–Human Epidermal Growth Factor Receptor-3 Immunoglobulin 2 Monoclonal Antibody, in a Patient With Metastatic Pancreatic Ductal Adenocarcinoma Harboring an NRG1 Fusion. JCO Precis. Oncol. 2022, 6, e2200263. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-Q.; Zeng, L.-S.; Wang, L.-F.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Han, Z.-W.; Zhou, Y.; Huang, S.-L.; Wang, X.-W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-Positive Breast Cancer: Advances and Future Directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Torka, P.; Barth, M.; Ferdman, R.; Hernandez-Ilizaliturri, F.J. Mechanisms of Resistance to Monoclonal Antibodies (mAbs) in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 426–438. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular Mechanisms of Resistance to the EGFR Monoclonal Antibody Cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef]
- Wang, K.; Du, R.; Myall, N.J.; Lewis, W.E.; Uy, N.; Hong, L.; Skoulidis, F.; Byers, L.A.; Tsao, A.; Cascone, T.; et al. Real-World Efficacy and Safety of Amivantamab for EGFR-Mutant NSCLC. J. Thorac. Oncol. 2024, 19, 500–506. [Google Scholar] [CrossRef]
- Tsuchikama, K.; Anami, Y.; Ha, S.Y.Y.; Yamazaki, C.M. Exploring the next Generation of Antibody-Drug Conjugates. Nat. Rev. Clin. Oncol. 2024, 21, 203–223. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload Diversification: A Key Step in the Development of Antibody–Drug Conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Ricciuti, B.; Pradhan, S.M.; Tolaney, S.M. Optimizing the Safety of Antibody–Drug Conjugates for Patients with Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 558–576. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Matsuda, Y. Novel Formats of Antibody Conjugates: Recent Advances in Payload Diversity, Conjugation, and Linker Chemistry. Expert Opin. Biol. Ther. 2023, 23, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-Drug Conjugates: Recent Advances in Conjugation and Linker Chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- High, P.; Carmon, K.S. G Protein-Coupled Receptor-Targeting Antibody-Drug Conjugates: Current Status and Future Directions. Cancer Lett. 2023, 564, 216191. [Google Scholar] [CrossRef]
- Smith, R.A.; Zammit, D.J.; Damle, N.K.; Usansky, H.; Reddy, S.P.; Lin, J.-H.; Mistry, M.; Rao, N.S.; Denis, L.J.; Gupta, S. ASN004, A 5T4-Targeting scFv-Fc Antibody-Drug Conjugate with High Drug-to-Antibody Ratio, Induces Complete and Durable Tumor Regressions in Preclinical Models. Mol. Cancer Ther. 2021, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Francisco, L.E.; Chatterjee, T.; Liang, Z.; Subramanian, S.; Liu, Q.J.; Rowe, J.H.; Carmon, K.S. An Antibody-Drug Conjugate Targeting GPR56 Demonstrates Efficacy in Preclinical Models of Colorectal Cancer. Br. J. Cancer 2023, 128, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, Z.; Hu, C.; Zhang, S.; Zi, M.; Yuan, L.; Cheng, X. Targeting CLDN18.2 in Cancers of the Gastrointestinal Tract: New Drugs and New Indications. Front. Oncol. 2023, 13, 1132319. [Google Scholar] [CrossRef]
- Sau, S.; Petrovici, A.; Alsaab, H.O.; Bhise, K.; Iyer, A.K. PDL-1 Antibody Drug Conjugate for Selective Chemo-Guided Immune Modulation of Cancer. Cancers 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Reilly, E.B.; Phillips, A.C.; Buchanan, F.G.; Kingsbury, G.; Zhang, Y.; Meulbroek, J.A.; Cole, T.B.; DeVries, P.J.; Falls, H.D.; Beam, C.; et al. Characterization of ABT-806, a Humanized Tumor-Specific Anti-EGFR Monoclonal Antibody. Mol. Cancer Ther. 2015, 14, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; DeVries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol. Cancer Ther. 2016, 15, 661–669. [Google Scholar] [CrossRef]
- Gan, H.K.; Burge, M.; Solomon, B.; Lee, S.T.; Holen, K.D.; Zhang, Y.; Ciprotti, M.; Lee, F.T.; Munasinghe, W.; Fischer, J.; et al. A Phase 1 and Biodistribution Study of ABT-806i, an 111In-Radiolabeled Conjugate of the Tumor-Specific Anti-EGFR Antibody ABT-806. J. Nucl. Med. 2021, 62, 787–794. [Google Scholar] [CrossRef]
- van den Bent, M.; Gan, H.K.; Lassman, A.B.; Kumthekar, P.; Merrell, R.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Efficacy of Depatuxizumab Mafodotin (ABT-414) Monotherapy in Patients with EGFR-Amplified, Recurrent Glioblastoma: Results from a Multi-Center, International Study. Cancer Chemother. Pharmacol. 2017, 80, 1209–1217. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; Vogelbaum, M.A.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab Mafodotin in EGFR-Amplified Newly Diagnosed Glioblastoma: A Phase III Randomized Clinical Trial. Neuro-Oncology 2023, 25, 339–350. [Google Scholar] [CrossRef]
- Gan, H.K.; Parakh, S.; Lassman, A.B.; Seow, A.; Lau, E.; Lee, S.T.; Ameratunga, M.; Perchyonok, Y.; Cao, D.; Burvenich, I.J.G.; et al. Tumor Volumes as a Predictor of Response to the Anti-EGFR Antibody Drug Conjugate Depatuxizumab Mafadotin. Neuro-Oncol. Adv. 2021, 3, vdab102. [Google Scholar] [CrossRef]
- Marin, B.-M.; Porath, K.A.; Jain, S.; Kim, M.; Conage-Pough, J.E.; Oh, J.-H.; Miller, C.L.; Talele, S.; Kitange, G.J.; Tian, S.; et al. Heterogeneous Delivery across the Blood-Brain Barrier Limits the Efficacy of an EGFR-Targeting Antibody Drug Conjugate in Glioblastoma. Neuro-Oncology 2021, 23, 2042–2053. [Google Scholar] [CrossRef]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Falls, H.D.; Mitten, M.J.; DeVries, P.J.; Benatuil, L.; Hsieh, C.-M.; Meulbroek, J.A.; Panchal, S.C.; et al. Characterization of ABBV-221, a Tumor-Selective EGFR-Targeting Antibody Drug Conjugate. Mol. Cancer Ther. 2018, 17, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Falls, H.D.; Mitten, M.J.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Gao, W.; Palma, J.P.; Cao, D.; Chia, P.-L.; et al. Targeting Multiple EGFR-Expressing Tumors with a Highly Potent Tumor-Selective Antibody-Drug Conjugate. Mol. Cancer Ther. 2020, 19, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Calvo, E.; Moreno, V.; Juric, D.; Shapiro, G.I.; Vanderwal, C.A.; Hu, B.; Gifford, M.; Barch, D.; Roberts-Rapp, L.; et al. A Phase 1 Study Evaluating Safety and Pharmacokinetics of Losatuxizumab Vedotin (ABBV-221), an Anti-EGFR Antibody-Drug Conjugate Carrying Monomethyl Auristatin E, in Patients with Solid Tumors Likely to Overexpress EGFR. Investig. New Drugs 2020, 38, 1483–1494. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Papadopoulos, K.P.; Strickler, J.H.; Lassman, A.B.; Waqar, S.N.; Chae, Y.K.; Patel, J.D.; Shacham-Shmueli, E.; Kelly, K.; Khasraw, M.; et al. Phase I Study of Anti-Epidermal Growth Factor Receptor Antibody-Drug Conjugate Serclutamab Talirine: Safety, Pharmacokinetics, and Antitumor Activity in Advanced Glioblastoma. Neuro-Oncol. Adv. 2023, 5, vdac183. [Google Scholar] [CrossRef]
- Qiu, M.-Z.; Zhang, Y.; Guo, Y.; Guo, W.; Nian, W.; Liao, W.; Xu, Z.; Zhang, W.; Zhao, H.-Y.; Wei, X.; et al. Evaluation of Safety of Treatment With Anti–Epidermal Growth Factor Receptor Antibody Drug Conjugate MRG003 in Patients With Advanced Solid Tumors. JAMA Oncol. 2022, 8, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Liu, R.; Song, G.; Song, H.; Jiang, J.; Jia, C.; Huang, X.; Yuan, X.; Yang, W.-J.; Wang, X.; et al. 683P Preclinical Evaluation of HLX42, a Novel EGFR-Targeting ADC, for Cetuximab or TKI Resistant Cancer. Ann. Oncol. 2023, 34, S477–S478. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V.J. Ado-Trastuzumab Emtansine (T-DM1): An Antibody-Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab Emtansine: Mechanisms of Action and Drug Resistance. Breast Cancer Res. BCR 2014, 16, 209. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Wedam, S.; Fashoyin-Aje, L.; Gao, X.; Bloomquist, E.; Tang, S.; Sridhara, R.; Goldberg, K.B.; King-Kallimanis, B.L.; Theoret, M.R.; Ibrahim, A.; et al. FDA Approval Summary: Ado-Trastuzumab Emtansine for the Adjuvant Treatment of HER2-Positive Early Breast Cancer. Clin. Cancer Res. 2020, 26, 4180–4185. [Google Scholar] [CrossRef]
- Kowalczyk, L.; Bartsch, R.; Singer, C.F.; Farr, A. Adverse Events of Trastuzumab Emtansine (T-DM1) in the Treatment of HER2-Positive Breast Cancer Patients. Breast Care 2017, 12, 401–408. [Google Scholar] [CrossRef]
- Azar, I.; Alkassis, S.; Fukui, J.; Alsawah, F.; Fedak, K.; Al Hallak, M.N.; Sukari, A.; Nagasaka, M. Spotlight on Trastuzumab Deruxtecan (DS-8201,T-DXd) for HER2 Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer Targets Ther. 2021, 12, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Baba, E.; Fujitani, K.; Oki, E.; Fujii, S.; Yamaguchi, K. Discovery and Development of Trastuzumab Deruxtecan and Safety Management for Patients with HER2-Positive Gastric Cancer. Gastric Cancer 2021, 24, 780–789. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Kumagai, K.; Aida, T.; Tsuchiya, Y.; Kishino, Y.; Kai, K.; Mori, K. Interstitial Pneumonitis Related to Trastuzumab Deruxtecan, a Human Epidermal Growth Factor Receptor 2-Targeting Ab-Drug Conjugate, in Monkeys. Cancer Sci. 2020, 111, 4636–4645. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Hegg, R.; Chung, W.-P.; Im, S.-A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine in Patients with HER2-Positive Metastatic Breast Cancer: Updated Results from DESTINY-Breast03, a Randomised, Open-Label, Phase 3 Trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Curigliano, G.; Hu, X.; Dent, R.A.; Yonemori, K.; Barrios, C.H.; O’Shaughnessy, J.; Wildiers, H.; Zhang, Q.; Im, S.-A.; Saura, C.; et al. Trastuzumab Deruxtecan (T-DXd) vs Physician’s Choice of Chemotherapy (TPC) in Patients (pts) with Hormone Receptor-positive (HR+), Human Epidermal Growth Factor Receptor 2 (HER2)-Low or HER2-Ultralow Metastatic Breast Cancer (mBC) with Prior Endocrine Therapy (ET): Primary Results from DESTINY-Breast06 (DB-06). Available online: https://meetings.asco.org/abstracts-presentations/238015 (accessed on 3 June 2024).
- Smit, E.F.; Felip, E.; Uprety, D.; Nagasaka, M.; Nakagawa, K.; Rodríguez, L.P.-A.; Pacheco, J.M.; Li, B.T.; Planchard, D.; Baik, C.; et al. Trastuzumab Deruxtecan in Patients with Metastatic Non-Small-Cell Lung Cancer (DESTINY-Lung01): Primary Results of the HER2-Overexpressing Cohorts from a Single-Arm, Phase 2 Trial. Lancet Oncol. 2024, 25, 439–454. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Fam-Trastuzumab Deruxtecan-Nxki for Unresectable or Metastatic HER2-Positive Solid Tumors. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-unresectable-or-metastatic-her2 (accessed on 30 June 2024).
- Xu, Y.; Wang, Y.; Gong, J.; Zhang, X.; Peng, Z.; Sheng, X.; Mao, C.; Fan, Q.; Bai, Y.; Ba, Y.; et al. Phase I Study of the Recombinant Humanized Anti-HER2 Monoclonal Antibody-MMAE Conjugate RC48-ADC in Patients with HER2-Positive Advanced Solid Tumors. Gastric Cancer 2021, 24, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, X.; Xu, Z.; Li, L.; Liu, W.; Dai, Z.; Zhao, Z.; Xiao, L.; Li, H.; Hu, C. Preclinical Evaluation of MRG002, a Novel HER2-Targeting Antibody-Drug Conjugate with Potent Antitumor Activity against HER2-Positive Solid Tumors. Antib. Ther. 2021, 4, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Yuan, C.; Cao, W.; Ge, W.; Zhang, D.; Dan, M.; Zhao, Q.; Liu, B.; Yao, B. An Innovative Site-Specific Anti-HER2 Antibody-Drug Conjugate with High Homogeneity and Improved Therapeutic Index. OncoTargets Ther. 2022, 15, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Y.; Tong, Z.; Liu, Y.; Wang, X.; Yan, M.; Chang, J.; Wang, S.; Du, C.; Li, L.; et al. FS-1502 in HER2-positive advanced breast cancer: Results from an open-label, phase 1 study. J. Clin. Oncol. 2023, 41, 3044. [Google Scholar] [CrossRef]
- Aftimos, P.G.; Turner, N.; O’Shaughnessy, J.; van den Tweel, E.; Oesterholt, M.; Escrivá-de-Romaní, S.; Tueux, N.Q.; Tan, T.J.Y.; Lim, J.; Ladoire, S.; et al. 386MO Trastuzumab Duocarmazine versus Physician’s Choice Therapy in Pre-Treated HER2-Positive Metastatic Breast Cancer: Final Results of the Phase III TULIP Trial. Ann. Oncol. 2023, 34, S340–S341. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Issues Complete Response Letter for Byondis’ [Vic-]Trastuzumab Duocarmazine. Available online: https://www.byondis.com/media/press-releases/us-food-and-drug-administration-issues-complete-response-letter-for-vic-trastuzumab-duocarmazine (accessed on 26 May 2024).
- Hu, X.; Zhang, J.; Liu, R.; Gao, S.; Qing, Y.; Yi, S.; Yuan, J.; Chen, H.; Fan, B.; Zheng, H.; et al. Phase I Study of A166 in Patients with HER2-Expressing Locally Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2021, 39 (Suppl. S15), 1024. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; Liu, R.; Gao, S.; Wu, J.; Wang, Y.; Hao, Y.; Ge, J.; Qing, Y.; Yi, S.; et al. Updated Results and Biomarker Analyses from the Phase I Trial of A166 in Patients with HER2-Expressing Locally Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2022, 40 (Suppl. S16), 1037. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, R.; Gao, S.; Li, W.; Chen, Y.; Meng, Y.; Liu, C.; Jin, W.; Wu, J.; Wang, Y.; et al. Phase I Study of A166, an Antibody–drug Conjugate in Advanced HER2-Expressing Solid Tumours. Npj Breast Cancer 2023, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Huang, Y.; Lu, Y.; Rao, X.; Wang, F.; Huang, Y.; Huang, X. 438P Phase I Study of ZV0203, a First in Class Pertuzumab ADC, in Patients with HER2+ Advanced Solid Tumors. Ann. Oncol. 2023, 34, S365. [Google Scholar] [CrossRef]
- Miao, Z.; Lin, F.; Rao, X.; Huang, Y.; Wang, F.; Lu, Y.; Huang, X.; Huang, Y. Phase 1 study of ZV0203, a first in class pertuzumab ADC, in patients with HER2 positive advanced solid tumors. J. Clin. Oncol. 2024, 42, e15004. [Google Scholar] [CrossRef]
- Barok, M.; Le Joncour, V.; Martins, A.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H. ARX788, a Novel Anti-HER2 Antibody-Drug Conjugate, Shows Anti-Tumor Effects in Preclinical Models of Trastuzumab Emtansine-Resistant HER2-Positive Breast Cancer and Gastric Cancer. Cancer Lett. 2020, 473, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-Specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-Low and T-DM1-Resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, D.; Shen, W.; Xiao, Q.; Gu, Y.; O’Shaughnessy, J.; Hu, X. Phase I Trial of a Novel Anti-HER2 Antibody-Drug Conjugate, ARX788, for the Treatment of HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2022, 28, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Yonesaka, K.; Takegawa, N.; Watanabe, S.; Haratani, K.; Kawakami, H.; Sakai, K.; Chiba, Y.; Maeda, N.; Kagari, T.; Hirotani, K.; et al. An HER3-Targeting Antibody-Drug Conjugate Incorporating a DNA Topoisomerase I Inhibitor U3-1402 Conquers EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC. Oncogene 2019, 38, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Jänne, P.A.; Oliveira, M.; Rizvi, N.; Malburg, L.; Keedy, V.; Yee, L.; Copigneaux, C.; Hettmann, T.; Wu, C.-Y.; et al. Phase I Study of U3-1287, a Fully Human Anti-HER3 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 3078–3087. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.J.; Ogbagabriel, S.; Bready, J.; Sun, J.-R.; Radinsky, R.; Hettmann, T. Abstract A182: U3–1287 (AMG 888), a Fully Human Anti-HER3 mAb, Demonstrates in Vitro and in Vivo Efficacy in the FaDu Model of Human Squamous Cell Carcinoma of the Head and Neck (SCCHN). Mol. Cancer Ther. 2011, 10 (Suppl. S11), A182. [Google Scholar] [CrossRef]
- Koganemaru, S.; Kuboki, Y.; Koga, Y.; Kojima, T.; Yamauchi, M.; Maeda, N.; Kagari, T.; Hirotani, K.; Yasunaga, M.; Matsumura, Y.; et al. U3-1402, a Novel HER3-Targeting Antibody-Drug Conjugate, for the Treatment of Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody-Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Masuda, N.; Mukohara, T.; Takahashi, S.; Nakayama, T.; Inoue, K.; Iwata, H.; Toyama, T.; Yamamoto, Y.; Hansra, D.M.; et al. Results from the Phase 1/2 Study of Patritumab Deruxtecan, a HER3-Directed Antibody-Drug Conjugate (ADC), in Patients with HER3-Expressing Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 1002. [Google Scholar] [CrossRef]
- Krop, I.E.; Masuda, N.; Mukohara, T.; Takahashi, S.; Nakayama, T.; Inoue, K.; Iwata, H.; Yamamoto, Y.; Alvarez, R.H.; Toyama, T.; et al. Patritumab Deruxtecan (HER3-DXd), a Human Epidermal Growth Factor Receptor 3–Directed Antibody-Drug Conjugate, in Patients With Previously Treated Human Epidermal Growth Factor Receptor 3–Expressing Metastatic Breast Cancer: A Multicenter, Phase I/II Trial. J. Clin. Oncol. 2023, 41, 5550–5560. [Google Scholar] [CrossRef]
- Pistilli, B.; Ibrahimi, N.; Lacroix-Triki, M.; D’Hondt, V.; Vicier, C.; Frenel, J.-S.; Dalenc, F.; Bachelot, T.; Benderra, M.-A.; Loirat, D.; et al. 189O A Phase II Study of Patritumab Deruxtecan (HER3-DXd), in Patients (Pts) with Advanced Breast Cancer (ABC), with Biomarker Analysis to Characterize Response to Therapy (ICARUS-BREAST01). ESMO Open 2023, 8, 101378. [Google Scholar] [CrossRef]
- Oliveira, M.; Falato, C.; Cejalvo, J.M.; Vila, M.M.; Tolosa, P.; Salvador-Bofill, F.J.; Cruz, J.; Arumi, M.; Luna, A.M.; Guerra, J.A.; et al. Patritumab Deruxtecan in Untreated Hormone Receptor-Positive/HER2-Negative Early Breast Cancer: Final Results from Part A of the Window-of-Opportunity SOLTI TOT-HER3 Pre-Operative Study. Ann. Oncol. 2023, 34, 670–680. [Google Scholar] [CrossRef]
- Pascual, T.; Oliveira, M.; Ciruelos, E.; Bellet Ezquerra, M.; Saura, C.; Gavilá, J.; Pernas, S.; Muñoz, M.; Vidal, M.J.; Margelí Vila, M.; et al. SOLTI-1805 TOT-HER3 Study Concept: A Window-of-Opportunity Trial of Patritumab Deruxtecan, a HER3 Directed Antibody Drug Conjugate, in Patients With Early Breast Cancer. Front. Oncol. 2021, 11, 638482. [Google Scholar] [CrossRef]
- Steuer, C.E.; Hayashi, H.; Su, W.-C.; Nishio, M.; Johnson, M.L.; Kim, D.-W.; Koczywas, M.; Felip, E.; Gold, K.A.; Murakami, H.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in Advanced/Metastatic Non-Small Cell Lung Cancer (NSCLC) without EGFR-Activating Mutations. J. Clin. Oncol. 2022, 40 (Suppl. S16), 9017. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Dosunmu, O.; Shastry, M.; Finney, L.; Sellami, D.B.; Sternberg, D.W.; Wright-Browne, V.; Toppmeyer, D.; Gwin, W.R.; Thaddeus, J.T.; et al. A Phase 2 Study of HER3-DXd in Patients (Pts) with Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2023, 41 (Suppl. S16), 1004. [Google Scholar] [CrossRef]
- Yu, H.A.; Goto, Y.; Hayashi, H.; Felip, E.; Yang, J.C.-H.; Reck, M.; Yoh, K.; Lee, S.-H.; Paz-Ares, L.; Besse, B.; et al. HERTHENA-Lung01, a Phase II Trial of Patritumab Deruxtecan (HER3-DXd) in Epidermal Growth Factor Receptor-Mutated Non-Small-Cell Lung Cancer After Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy and Platinum-Based Chemotherapy. J. Clin. Oncol. 2023, 41, 5363–5375. [Google Scholar] [CrossRef]
- Mok, T.; Jänne, P.A.; Nishio, M.; Novello, S.; Reck, M.; Steuer, C.; Wu, Y.-L.; Fougeray, R.; Fan, P.-D.; Meng, J.; et al. HERTHENA-Lung02: Phase III Study of Patritumab Deruxtecan in Advanced EGFR-Mutated NSCLC after a Third-Generation EGFR TKI. Future Oncol. 2024, 20, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Baik, C.S.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated (EGFRm) Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2021, 39 (Suppl. S15), 9007. [Google Scholar] [CrossRef]
- Weng, W.; Meng, T.; Pu, J.; Ma, L.; Shen, Y.; Wang, Z.; Pan, R.; Wang, M.; Chen, C.; Wang, L.; et al. AMT-562, a Novel HER3-Targeting Antibody-Drug Conjugate, Demonstrates a Potential to Broaden Therapeutic Opportunities for HER3-Expressing Tumors. Mol. Cancer Ther. 2023, 22, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, J.; Qu, C.; Luo, L.; Li, B.; Zhang, Y.; Zhu, Z.; Qiu, Y.; Hua, H. DB-1310, an ADC Comprised of a Novel Anti-HER3 Antibody Conjugated to a DNA Topoisomerase I Inhibitor, Is Highly Effective for the Treatment of HER3-Positive Solid Tumors. J. Transl. Med. 2024, 22, 362. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, X.; Liu, J.; Huang, Z.; Lv, D.; Zhang, Y.; Wang, X.; Chaudhry, A.; Amin, H.; Damodaran, S.; et al. YL202/BNT326, a HER3-Targeted ADC, in Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer and Breast Cancer: Preliminary Results from a First-in-Human Phase I Trial. J. Clin. Oncol. 2024, 42 (Suppl. S16), 3034. [Google Scholar] [CrossRef]
- Xu, J.; Zong, Q.; Zhu, L.; Liu, Q.; Stann, S.; Cai, J. Abstract 563: Preclinical Development of YL202, a Novel HER3-Targeting Antibody-Drug Conjugate (ADC) with Novel DNA Topoisomerase I Inhibitor for Treatment of Solid Tumors. Cancer Res. 2023, 83 (Suppl. S7), 563. [Google Scholar] [CrossRef]
- Wan, W.; Zhao, S.; Zhuo, S.; Zhang, Y.; Chen, L.; Li, G.; Renshaw, B.; Khalili, J.S.; Xiao, S.; Zhu, Y. Abstract 2642: BL-B01D1, a Novel EGFR×HER3-Targeting ADC, Demonstrates Robust Anti-Tumor Efficacy in Preclinical Evaluation. Cancer Res. 2023, 83 (Suppl. S7), 2642. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, Y.; Zhao, Y.; Fang, W.; Zhao, H.; Huang, Y.; Yang, Y.; Chen, L.; Hou, X.; Zou, W.; et al. BL-B01D1, a First-in-Class EGFRxHER3 Bispecific Antibody-Drug Conjugate (ADC), in Patients with Locally Advanced or Metastatic Solid Tumor: Results from a First-in-Human Phase 1 Study. J. Clin. Oncol. 2023, 41 (Suppl. S16), 3001. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, Y.; Zhao, Y.; Zhao, S.; Xue, J.; Yang, Y.; Fang, W.; Guo, Y.; Han, Y.; Yang, K.; et al. BL-B01D1, a First-in-Class EGFR–HER3 Bispecific Antibody–Drug Conjugate, in Patients with Locally Advanced or Metastatic Solid Tumours: A First-in-Human, Open-Label, Multicentre, Phase 1 Study. Lancet Oncol. 2024, 25, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shang, C.; An, G.; Guo, C.; An, W.; Yang, Y. A Novel EGFR×HER3-Targeting Bispecific Antibody Drug-Conjugate, BCG019, Demonstrates Robust Anti-Tumor Efficacy in Preclinical Evaluation. Available online: https://www.abstractsonline.com/pp8/#!/20272/presentation/6184 (accessed on 26 May 2024).
- Knuehl, C.; Toleikis, L.; Dotterweich, J.; Ma, J.; Kumar, S.; Ross, E.; Wilm, C.; Schmitt, M.; Grote, H.J.; Amendt, C. Abstract 5284: M1231 Is a Bispecific Anti-MUC1xEGFR Antibody-Drug Conjugate Designed to Treat Solid Tumors with MUC1 and EGFR Co-Expression. Cancer Res. 2022, 82 (Suppl. S12), 5284. [Google Scholar] [CrossRef]
- Zutshi, A.; Neuteboom, B.; Kumar, S.; Sloot, W.; Knuehl, C.; Dotterweich, J.; Ma, J.; Amendt, C.; Venkatakrishnan, K.; Park, T.; et al. Abstract 5423: Translational PK/PD/Efficacy Modeling and Efficacious Human Dose Prediction for a First-in-Class MUC1-EGFR (M1231) Bispecific Antibody Drug Conjugate. Cancer Res. 2022, 82 (Suppl. S12), 5423. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, A.; Zhang, J.; Liu, Y.; Li, H.; Li, X.; An, G.; An, W.F.; Guo, C.; et al. 1164 BSA01, a Bispecific Antibody-Drug Conjugate Targeting EGFR and Membrane-Bound MUC-1-C, Exhibits Anti-Tumor Efficacy In Vivo. J. Immunother. Cancer 2023, 11 (Suppl. S1). [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, A.; Zhang, J.; Liu, Y.; Li, H.; Li, X.; An, G.; Hui, L.; An, F.; et al. Abstract 6325: A Novel EGFR x MUC1 Bispecific Antibody-Drug Conjugate, BSA01, Targets MUC1 Transmembrane Cleavage Products and Improves Tumor Selectivity. Cancer Res. 2023, 83 (Suppl. S7), 6325. [Google Scholar] [CrossRef]
- Comer, F.; Mazor, Y.; Hurt, E.; Yang, C.; Fleming, R.; Shandilya, H.; Vijayakrishnan, B.; Sterba, M.; Chen, R.; Rosfjord, E.; et al. Abstract 5736: AZD9592: An EGFR-cMET Bispecific Antibody-Drug Conjugate (ADC) Targeting Key Oncogenic Drivers in Non-Small-Cell Lung Cancer (NSCLC) and Beyond. Cancer Res. 2023, 83 (Suppl. S7), 5736. [Google Scholar] [CrossRef]
- McGrath, L.; Zheng, Y.; Christ, S.; Sachs, C.C.; Khelifa, S.; Windmüller, C.; Sweet, S.; Kim, Y.J.; Sutton, D.; Sulikowski, M.; et al. Abstract 5737: Evaluation of the Relationship between Target Expression and in Vivo Anti-Tumor Efficacy of AZD9592, an EGFR/c-MET Targeted Bispecific Antibody Drug Conjugate. Cancer Res. 2023, 83 (Suppl. S7), 5737. [Google Scholar] [CrossRef]
- McGrath, L.; Rosfjord, E.; Christ, S.; Zheng, Y.; Floch, N.; Sachs, C.C.; Cotoi, C.; Batelli, S.; Cantu, E.; Makaretz, S.; et al. P1.12-04 In Vivo Efficacy of AZD9592, an EGFR-cMET Bispecific ADC, in a Broad Panel of NSCLC Patient-Derived Xenograft Models. J. Thorac. Oncol. 2023, 18, S208. [Google Scholar] [CrossRef]
- Aggarwal, C.; Azzoli, C.G.; Spira, A.I.; Solomon, B.J.; Le, X.; Rolfo, C.; Planchard, D.; Felip, E.; Wu, Y.-L.; Ahn, M.-J.; et al. EGRET: A First-in-Human Study of the Novel Antibody-Drug Conjugate (ADC) AZD9592 as Monotherapy or Combined with Other Anticancer Agents in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS3156. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Ma, L.; Xu, M.; Yin, Q. Abstract LB043: VBC101-F11: An Innovative EGFR/cMet Bispecific Antibody Drug Conjugate (ADC) Targeting Key Oncogenic Drivers in Solid Tumors. Cancer Res. 2024, 84 (Suppl. S7), LB043. [Google Scholar] [CrossRef]
- Li, Z.; Shang, C.; Guan, X.; An, G.; Guo, Y.; Zhang, E.; Lin, Q.; Yang, Y. Abstract LB215: A First-in-Class Anti-TROP2/EGFR Bispecific Antibody-Drug Conjugate, DM001, Exhibits Potent Anti-Tumor Efficacy. Cancer Res. 2023, 83 (Suppl. S8), LB215. [Google Scholar] [CrossRef]
- Weisser, N.E.; Sanches, M.; Escobar-Cabrera, E.; O’Toole, J.; Whalen, E.; Chan, P.W.Y.; Wickman, G.; Abraham, L.; Choi, K.; Harbourne, B.; et al. An Anti-HER2 Biparatopic Antibody That Induces Unique HER2 Clustering and Complement-Dependent Cytotoxicity. Nat. Commun. 2023, 14, 1394. [Google Scholar] [CrossRef]
- Hamblett, K.; Barnscher, S.; Davies, R.; Hammond, P.; Hernandez, A.; Wickman, G.; Fung, V.; Ding, T.; Garnett, G.; Galey, A.; et al. Abstract P6-17-13: ZW49, a HER2 Targeted Biparatopic Antibody Drug Conjugate for the Treatment of HER2 Expressing Cancers. Cancer Res. 2019, 79 (Suppl. S4), P6-17-13. [Google Scholar] [CrossRef]
- Moody, P.R.; Sayers, E.J.; Magnusson, J.P.; Alexander, C.; Borri, P.; Watson, P.; Jones, A.T. Receptor Crosslinking: A General Method to Trigger Internalization and Lysosomal Targeting of Therapeutic Receptor:Ligand Complexes. Mol. Ther. 2015, 23, 1888–1898. [Google Scholar] [CrossRef]
- Barnscher, S.D.; Rojas, A.H.; Hamblett, K.J.; Escalante, N. Abstract 2633: Zanidatamab Zovodotin (ZW49) Induces Hallmarks of Immunogenic Cell Death and Is Active in Patient-Derived Xenograft Models of Gastric Cancer. Cancer Res. 2023, 83 (Suppl. S7), 2633. [Google Scholar] [CrossRef]
- Jhaveri, K.; Han, H.; Dotan, E.; Oh, D.-Y.; Ferrario, C.; Tolcher, A.; Lee, K.-W.; Liao, C.-Y.; Kang, Y.-K.; Kim, Y.H.; et al. 460MO Preliminary Results from a Phase I Study Using the Bispecific, Human Epidermal Growth Factor 2 (HER2)-Targeting Antibody-Drug Conjugate (ADC) Zanidatamab Zovodotin (ZW49) in Solid Cancers. Ann. Oncol. 2022, 33, S749–S750. [Google Scholar] [CrossRef]
- Waldron, J. Zymeworks Halts Plans for Phase 2 ADC Trial to Mull ‘Evolving Clinical Landscape’. Available online: https://www.fiercebiotech.com/biotech/zymeworks-halts-plans-phase-2-adc-trial-mull-evolving-clinical-landscape (accessed on 26 June 2024).
- Oganesyan, V.; Peng, L.; Bee, J.S.; Li, J.; Perry, S.R.; Comer, F.; Xu, L.; Cook, K.; Senthil, K.; Clarke, L.; et al. Structural Insights into the Mechanism of Action of a Biparatopic Anti-HER2 Antibody. J. Biol. Chem. 2018, 293, 8439–8448. [Google Scholar] [CrossRef]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef]
- Pegram, M.D.; Hamilton, E.P.; Tan, A.R.; Storniolo, A.M.; Balic, K.; Rosenbaum, A.I.; Liang, M.; He, P.; Marshall, S.; Scheuber, A.; et al. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody–Drug Conjugate MEDI4276 in Patients with HER2-Positive Advanced Breast or Gastric Cancer. Mol. Cancer Ther. 2021, 20, 1442–1453. [Google Scholar] [CrossRef]
- Wang, P.; Guo, K.; Peng, J.; Sun, J.; Xu, T. JSKN003, A NOVEL BIPARATOPIC ANTI-HER2 ANTIBODY-DRUG CONJUGATE, EXHIBITS POTENT ANTITUMOR EFFICACY. Antib. Ther. 2023, 6 (Suppl. S1), tbad014.009. [Google Scholar] [CrossRef]
- Park, J.J.; Gao, B.; Beecroft, C.; Wilkinson, K.J.; Parsonson, A.; Zhang, K.; Yan, X.; Wang, N. Safety and efficacy of JSKN003 in patients with advanced/metastatic solid tumors: A first-in-human, dose-escalation, multicenter, open-label, phase I study. J. Clin. Oncol. 2024, 42, 3038. [Google Scholar] [CrossRef]
- Beecroft, C.; Gao, B.; Park, J.; Wilkinson, K.; Zhang, K.; Yan, X.; Lv, Y. Abstract CT179: Safety and Efficacy of JSKN003 in Patients with Advanced/Metastatic Solid Tumors: A First-in-Human, Dose-Escalation, Multicenter, Open-Label, Phase I Study. Cancer Res. 2024, 84 (Suppl. S7), CT179. [Google Scholar] [CrossRef]
- Schaefer, G.; Haber, L.; Crocker, L.M.; Shia, S.; Shao, L.; Dowbenko, D.; Totpal, K.; Wong, A.; Lee, C.V.; Stawicki, S.; et al. A Two-in-One Antibody against HER3 and EGFR Has Superior Inhibitory Activity Compared with Monospecific Antibodies. Cancer Cell 2011, 20, 472–486. [Google Scholar] [CrossRef]
- Shang, C.; An, G.; Guo, Y.; Zhang, E.; Lin, Q.; Yang, Y. Abstract 2977: A First-in-Class Anti-HER2/TROP2 Bispecific Antibody-Drug Conjugate (YH012) Exhibits Potent Anti-Tumor Efficacy. Cancer Res. 2023, 83 (Suppl. S7), 2977. [Google Scholar] [CrossRef]
- Shang, C.; Yang, L.; Zhang, J.; Han, Y.; Li, Z.; Han, Z.; Li, J.; Meng, Y.; An, G.; Yang, H.; et al. Abstract 4256: YH012, a Novel Bispecific Anti-HER2 and TROP2 Antibody-Drug Conjugate, Exhibits Potent Antitumor Efficacy. Cancer Res. 2022, 82 (Suppl. S12), 4256. [Google Scholar] [CrossRef]
- Zhong, H.; Xiong, Y.; Huang, H.; Pan, Y.; Cao, L.; Liu, S.; Wang, N.; Sun, B.; Liu, D.; Yuan, W.; et al. Preclinical Antitumor Activity of an Anti-HER2/Trop-2 Bispecific Antibody-Drug Conjugate with a New DNA Topoisomerase I Inhibitor. J. Clin. Oncol. 2023, 41 (Suppl. S16), e15013. [Google Scholar] [CrossRef]
- Li, Z.; Shang, C.; Han, Z.; An, G.; Guo, C.; An, W.F.; Yang, Y. Abstract 2617: BCG022, a Novel HER3 × MET Bispecific Antibody-Drug Conjugate (bsADC), Demonstrates Promising Anti-Tumor Efficacy. Cancer Res. 2024, 84 (Suppl. S6), 2617. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, N.; An, G.; Zhang, E.; Lin, Q.; Yang, Y. Abstract LB214: A First-in-Class Bispecific Antibody-Drug Conjugate (DM002) Targeting HER3 and the Juxtamembrane Domain of MUC1. Cancer Res. 2023, 83 (Suppl. S8), LB214. [Google Scholar] [CrossRef]
- Wahab, A.; Rafae, A.; Mushtaq, K.; Masood, A.; Ehsan, H.; Khakwani, M.; Khan, A. Ocular Toxicity of Belantamab Mafodotin, an Oncological Perspective of Management in Relapsed and Refractory Multiple Myeloma. Front. Oncol. 2021, 11, 678634. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W. Antibody Drug Conjugates: Lessons from 20 Years of Clinical Experience. Ann. Oncol. 2016, 27, 2168–2172. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Makker, V.; Oaknin, A.; Oh, D.-Y.; Banerjee, S.; González-Martín, A.; Jung, K.H.; Ługowska, I.; Manso, L.; Manzano, A.; et al. Efficacy and Safety of Trastuzumab Deruxtecan in Patients With HER2-Expressing Solid Tumors: Primary Results From the DESTINY-PanTumor02 Phase II Trial. J. Clin. Oncol. 2024, 42, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.K.; Manore, S.G.; Regua, A.T.; Lo, H.-W. Antibody-Drug Conjugates for the Treatment of HER2-Positive Breast Cancer. Genes 2022, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Chic, N.; Brasó-Maristany, F.; Paré, L.; Pascual, T.; Conte, B.; Martínez-Sáez, O.; Adamo, B.; Vidal, M.; Barnadas, E.; et al. Clinical, Pathological, and PAM50 Gene Expression Features of HER2-Low Breast Cancer. NPJ Breast Cancer 2021, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Gandini, S.; Nicolò, E.; Trillo, P.; Giugliano, F.; Zagami, P.; Vivanet, G.; Bellerba, F.; Trapani, D.; Marra, A.; et al. Evolution of Low HER2 Expression between Early and Advanced-Stage Breast Cancer. Eur. J. Cancer 2022, 163, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Inokuchi, M.; Takagi, Y.; Yamada, H.; Kojima, K.; Kumagai, J.; Kawano, T.; Sugihara, K. High Expression of HER3 Is Associated with a Decreased Survival in Gastric Cancer. Clin. Cancer Res. 2008, 14, 7843–7849. [Google Scholar] [CrossRef]
- Reschke, M.; Mihic-Probst, D.; van der Horst, E.H.; Knyazev, P.; Wild, P.J.; Hutterer, M.; Meyer, S.; Dummer, R.; Moch, H.; Ullrich, A. HER3 Is a Determinant for Poor Prognosis in Melanoma. Clin. Cancer Res. 2008, 14, 5188–5197. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, R.; Yan, H.; Zhao, P.; Wu, L.; Wang, H.; Li, T.; Cao, B. Prognostic Significance of HER3 in Patients with Malignant Solid Tumors. Oncotarget 2017, 8, 67140–67151. [Google Scholar] [CrossRef]
- Mendell, J.; Freeman, D.J.; Feng, W.; Hettmann, T.; Schneider, M.; Blum, S.; Ruhe, J.; Bange, J.; Nakamaru, K.; Chen, S.; et al. Clinical Translation and Validation of a Predictive Biomarker for Patritumab, an Anti-Human Epidermal Growth Factor Receptor 3 (HER3) Monoclonal Antibody, in Patients With Advanced Non-Small Cell Lung Cancer. eBioMedicine 2015, 2, 264–271. [Google Scholar] [CrossRef]
- Meulendijks, D.; Jacob, W.; Voest, E.E.; Mau-Sorensen, M.; Martinez-Garcia, M.; Taus, A.; Fleitas, T.; Cervantes, A.; Lolkema, M.P.; Langenberg, M.H.G.; et al. Phase Ib Study of Lumretuzumab Plus Cetuximab or Erlotinib in Solid Tumor Patients and Evaluation of HER3 and Heregulin as Potential Biomarkers of Clinical Activity. Clin. Cancer Res. 2017, 23, 5406–5415. [Google Scholar] [CrossRef]
- Cleary, J.M.; McRee, A.J.; Shapiro, G.I.; Tolaney, S.M.; O’Neil, B.H.; Kearns, J.D.; Mathews, S.; Nering, R.; MacBeath, G.; Czibere, A.; et al. A Phase 1 Study Combining the HER3 Antibody Seribantumab (MM-121) and Cetuximab with and without Irinotecan. Investig. New Drugs 2017, 35, 68–78. [Google Scholar] [CrossRef]
- Uliano, J.; Corvaja, C.; Curigliano, G.; Tarantino, P. Targeting HER3 for Cancer Treatment: A New Horizon for an Old Target. ESMO Open 2023, 8, 100790. [Google Scholar] [CrossRef]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody–Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.-Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential Mechanisms for Thrombocytopenia Development with Trastuzumab Emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of Resistance to Trastuzumab Emtansine (T-DM1) in HER2-Positive Breast Cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Z.; Wang, Y. Bispecific Antibody Drug Conjugates: Making 1+1>2. Acta Pharm. Sin. B 2024, 14, 1965–1986. [Google Scholar] [CrossRef]
- Spring, L.M.; Nakajima, E.; Hutchinson, J.; Viscosi, E.; Blouin, G.; Weekes, C.; Rugo, H.; Moy, B.; Bardia, A. Sacituzumab Govitecan for Metastatic Triple-Negative Breast Cancer: Clinical Overview and Management of Potential Toxicities. Oncologist 2021, 26, 827–834. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef]
- Bardia, A.; Jhaveri, K.; Im, S.-A.; Simon, S.P.; Laurentiis, M.D.; Wang, S.; Martinez, N.; Borges, G.S.; Cescon, D.W.; Hattori, M.; et al. LBA11 Datopotamab Deruxtecan (Dato-DXd) vs Chemotherapy in Previously-Treated Inoperable or Metastatic Hormone Receptor-Positive, HER2-Negative (HR+/HER2–) Breast Cancer (BC): Primary Results from the Randomised Phase III TROPION-Breast01 Trial. Ann. Oncol. 2023, 34, S1264–S1265. [Google Scholar] [CrossRef]
- Soares, L.R.; Vilbert, M.; Rosa, V.D.L.; Oliveira, J.L.; Deus, M.M.; Freitas-Junior, R. Incidence of Interstitial Lung Disease and Cardiotoxicity with Trastuzumab Deruxtecan in Breast Cancer Patients: A Systematic Review and Single-Arm Meta-Analysis. ESMO Open 2023, 8, 101613. [Google Scholar] [CrossRef]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient Generation of Bispecific IgG Antibodies by Split Intein Mediated Protein Trans-Splicing System. Sci. Rep. 2017, 7, 8360. [Google Scholar] [CrossRef]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-Dependent Solid Tumours–Molecular Diagnosis and Targeted Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef]
- Stern, Y.E.; Al-Ghabkari, A.; Monast, A.; Fiset, B.; Aboualizadeh, F.; Yao, Z.; Stagljar, I.; Walsh, L.A.; Duhamel, S.; Park, M. Met-HER3 Crosstalk Supports Proliferation via MPZL3 in MET-Amplified Cancer Cells. Cell. Mol. Life Sci. CMLS 2022, 79, 178. [Google Scholar] [CrossRef]
- Yamasaki, A.; Miyake, R.; Hara, Y.; Okuno, H.; Imaida, T.; Okita, K.; Okazaki, S.; Akiyama, Y.; Hirotani, K.; Endo, Y.; et al. Dual-Targeting Therapy against HER3/MET in Human Colorectal Cancers. Cancer Med. 2023, 12, 9684–9696. [Google Scholar] [CrossRef]
- Klempner, S.; Strickler, J.; Gourley, L.; Jacquemont, C.; Bhatia, V.; Hunder, N.; Odegard, V.; Piha-Paul, S. 393 A Phase 1/2 Study of SBT6050 Combined with Trastuzumab Deruxtecan (T-DXd) or Trastuzumab and Tucatinib with or without Capecitabine in Patients with HER2-Expressing or HER2-Amplified Cancers. J. Immunother. Cancer 2021, 9 (Suppl. S2), A426. [Google Scholar] [CrossRef]
- Silverback Discontinues Targeted Oncology Programs, Reduces Staff. Available online: https://www.precisionmedicineonline.com/cancer/silverback-discontinues-targeted-oncology-programs-reduces-staff (accessed on 26 May 2024).
- Janku, F.; Han, S.-W.; Doi, T.; Amatu, A.; Ajani, J.A.; Kuboki, Y.; Cortez, A.; Cellitti, S.E.; Mahling, P.C.; Subramanian, K.; et al. Preclinical Characterization and Phase I Study of an Anti-HER2-TLR7 Immune-Stimulator Antibody Conjugate in Patients with HER2+ Malignancies. Cancer Immunol. Res. 2022, 10, 1441–1461. [Google Scholar] [CrossRef]
- Ackerman, S.E.; Gonzalez, J.C.; Gregorio, J.D.; Paik, J.C.; Hartmann, F.J.; Kenkel, J.A.; Lee, A.; Luo, A.; Pearson, C.I.; Nguyen, M.L.; et al. Abstract 1559: TLR7/8 Immune-Stimulating Antibody Conjugates Elicit Robust Myeloid Activation Leading to Enhanced Effector Function and Anti-Tumor Immunity in Pre-Clinical Models. Cancer Res. 2019, 79 (Suppl. S13), 1559. [Google Scholar] [CrossRef]
- Li, B.T.; Pegram, M.D.; Lee, K.-W.; Sharma, M.; Lee, J.; Spira, A.I.; Hanna, G.J.; Kang, Y.-K.; Rasco, D.W.; Moore, K.N.; et al. A Phase 1/2 Study of a First-in-Human Immune-Stimulating Antibody Conjugate (ISAC) BDC-1001 in Patients with Advanced HER2-Expressing Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), 2538. [Google Scholar] [CrossRef]
- Paiva, S.-L.; Crews, C.M. Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review. Explor. Target. Anti-Tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Kihlberg, J. PROTAC Cell Permeability and Oral Bioavailability: A Journey into Uncharted Territory. Future Med. Chem. 2022, 14, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.; Bai, C.; Yi, Y.; Skaletskaya, A.; Takrouri, K.; Wong, W.; Kim, M.-S.; Choi, D.-K.; Kim, D.-Y.; Yang, Y.; et al. Abstract 3933: ORM-5029: A First-in-Class Targeted Protein Degradation Therapy Using Antibody Neodegrader Conjugate (AnDC) for HER2-Expressing Breast Cancer. Cancer Res. 2022, 82 (Suppl. S12), 3933. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Hamilton, E.P.; Spira, A.I.; Pohlmann, P.R.; Giordano, A.; Clifton, K.; Anderson, B.D.; Dutta, S.; Mangipudi, U.; Saini, S.; et al. A Phase 1, First-in-Human, Open Label, Escalation and Expansion Study of ORM-5029, a Highly Potent GSPT1 Degrader Targeting HER2, in Patients with HER2-Expressing Advanced Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS1114. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 Regulated Apoptosis in Cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.K.; Waqar, S.N.; Papadopoulos, K.P.; Hung, J.-Y.; Spira, A.I.; Gan, H.; Yoshida, T.; Kuo, C.-H.S.; de Spéville, B.D.; Felip, E.; et al. 1318MO First-in-Human Study of ABBV-637, an EGFR-Targeting BCL-XL–Inhibiting Antibody-Drug Conjugate Combined with Osimertinib (OSI) in Relapsed/Refractory, EGFR-Mutated Non-Small Cell Lung Cancer (NSCLC). Ann. Oncol. 2023, 34, S759. [Google Scholar] [CrossRef]
- Thibault, S.; Hu, W.; Hirakawa, B.; Kalabat, D.; Franks, T.; Sung, T.; Khoh-Reiter, S.; Lu, S.; Finkelstein, M.; Jessen, B.; et al. Intestinal Toxicity in Rats Following Administration of CDK4/6 Inhibitors Is Independent of Primary Pharmacology. Mol. Cancer Ther. 2019, 18, 257–266. [Google Scholar] [CrossRef]
- van Aken, E.S.M.; Beeker, A.; Houtenbos, I.; Pos, F.J.; Linn, S.C.; Elkhuizen, P.H.M.; de Jong, M.C. Unexpected Toxicity of CDK4/6 Inhibitor Palbociclib and Radiotherapy. Cancer Rep. 2022, 5, e1470. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, M.E.; Ciaccio, A.; Danesi, R.; Duhoux, F.P.; Girmenia, C.; Zaman, K.; Lindman, H.; Luppi, F.; Mavroudis, D.; Paris, I.; et al. Late Onset Toxicities Associated with the Use of CDK 4/6 Inhibitors in Hormone Receptor Positive (HR+), Human Epidermal Growth Factor Receptor-2 Negative (HER2−) Metastatic Breast Cancer Patients: A Multidisciplinary, Pan-EU Position Paper Regarding Their Optimal Management. The GIOCONDA Project. Front. Oncol. 2023, 13, 1247270. [Google Scholar] [CrossRef]
- Cheung, A.; Chenoweth, A.M.; Johansson, A.; Laddach, R.; Guppy, N.; Trendell, J.; Esapa, B.; Mavousian, A.; Navarro-Llinas, B.; Haider, S.; et al. Anti-EGFR Antibody-Drug Conjugate Carrying an Inhibitor Targeting CDK Restricts Triple-Negative Breast Cancer Growth. Clin. Cancer Res. 2024. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [PubMed]
- DeVay, R.M.; Delaria, K.; Zhu, G.; Holz, C.; Foletti, D.; Sutton, J.; Bolton, G.; Dushin, R.; Bee, C.; Pons, J.; et al. Improved Lysosomal Trafficking Can Modulate the Potency of Antibody Drug Conjugates. Bioconjugate Chem. 2017, 28, 1102–1114. [Google Scholar] [CrossRef]
- de Goeij, B.E.C.G.; Vink, T.; ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- Zhuang, W.; Zhang, W.; Wang, L.; Xie, L.; Feng, J.; Zhang, B.; Hu, Y. Generation of a Novel SORT1×HER2 Bispecific Antibody–Drug Conjugate Targeting HER2-Low-Expression Tumor. Int. J. Mol. Sci. 2023, 24, 16056. [Google Scholar] [CrossRef]
- Krytska, K.; Casey, C.E.; Pogoriler, J.; Martinez, D.; Rathi, K.S.; Farrel, A.; Berko, E.R.; Tsang, M.; Sano, R.R.; Kendsersky, N.; et al. Evaluation of the DLL3-Targeting Antibody–Drug Conjugate Rovalpituzumab Tesirine in Preclinical Models of Neuroblastoma. Cancer Res. Commun. 2022, 2, 616–623. [Google Scholar] [CrossRef]
- Capone, E.; Lamolinara, A.; Pastorino, F.; Gentile, R.; Ponziani, S.; Di Vittorio, G.; D’Agostino, D.; Bibbò, S.; Rossi, C.; Piccolo, E.; et al. Targeting Vesicular LGALS3BP by an Antibody-Drug Conjugate as Novel Therapeutic Strategy for Neuroblastoma. Cancers 2020, 12, 2989. [Google Scholar] [CrossRef]
- Montero, J.C.; Rodríguez-Barrueco, R.; Ocaña, A.; Díaz-Rodríguez, E.; Esparís-Ogando, A.; Pandiella, A. Neuregulins and Cancer. Clin. Cancer Res. 2008, 14, 3237–3241. [Google Scholar] [CrossRef]
- Su, X.; Lai, T.; Tao, Y.; Zhang, Y.; Zhao, C.; Zhou, J.; Chen, E.; Zhu, M.; Zhang, S.; Wang, B.; et al. miR-33a-3p Regulates METTL3-Mediated AREG Stability and Alters EMT to Inhibit Pancreatic Cancer Invasion and Metastasis. Sci. Rep. 2023, 13, 13587. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, H.; Lu, J.; Zhang, Z.; Wu, H.; Liang, Z. AREG Mediates the Epithelial-Mesenchymal Transition in Pancreatic Cancer Cells via the EGFR/ERK/NF-κB Signalling Pathway. Oncol. Rep. 2020, 43, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-L.; Feng, P.-H.; Lee, K.-Y.; Chen, K.-Y.; Sun, W.-L.; Van Hiep, N.; Luo, C.-S.; Wu, S.-M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, K.A.; Sreekumar, S.; Jenkins, E.C.; Ernzen, K.J.; Kenny, P.A. Anti-Tumor Efficacy of an MMAE-Conjugated Antibody Targeting Cell Surface TACE/ADAM17-Cleaved Amphiregulin in Breast Cancer. Antib. Ther. 2021, 4, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, K.A.; Reker, N.C.; Sreekumar, S.; Kenny, P.A. Pan-Cancer Distribution of Cleaved Cell-Surface Amphiregulin, the Target of the GMF-1A3 Antibody Drug Conjugate. Antib. Ther. 2022, 5, 226–231. [Google Scholar] [CrossRef]
- Jacob, J.; Anami, Y.; High, P.; Liang, Z.; Subramanian, S.; Ghosh, S.C.; AghaAmiri, S.; Guernsey, C.; Tran, H.; Liu, Q.J.; et al. Antibody-Drug Conjugates Targeting EGFR Ligand Epiregulin Inhibit Colorectal Tumor Growth Irrespective of RAS Mutational Status. bioRxiv 2024. [Google Scholar] [CrossRef]
- Jacob, J.; Liang, Z.; Carmon, K. Abstract 4251: Development and Characterization of an Epiregulin Antibody-Drug Conjugate for Targeting Colorectal Cancer Cell Plasticity. Cancer Res. 2022, 82 (Suppl. S12), 4251. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.; El-Kenawi, A.; Shonagh, R.; Weber, A.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.; Mulé, J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res 2016, 76, 1381–1390. [Google Scholar] [CrossRef]
- Cobb, P.W.; Zhou, H.; Nahar, A.; Marinello, P. Open-Label, Active-Control, Phase 2/3 Study of Zilovertamab Vedotin plus Standard of Care in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS7592. [Google Scholar] [CrossRef]
- Lugini, A.; Goldman, J.W.; Tanizaki, J.; Akamatsu, H.; Xia, S.; Ratajczak, C.; Li, M.; Bolotin, E.; Seraj, J.; Lu, S. 1501TiP A Phase III Global Study of Telisotuzumab Vedotin versus Docetaxel in Previously Treated Patients with C-Met Overexpressing, EGFR Wildtype, Locally Advanced/Metastatic Nonsquamous NSCLC (TeliMET NSCLC-01). Ann. Oncol. 2023, 34, S845. [Google Scholar] [CrossRef]
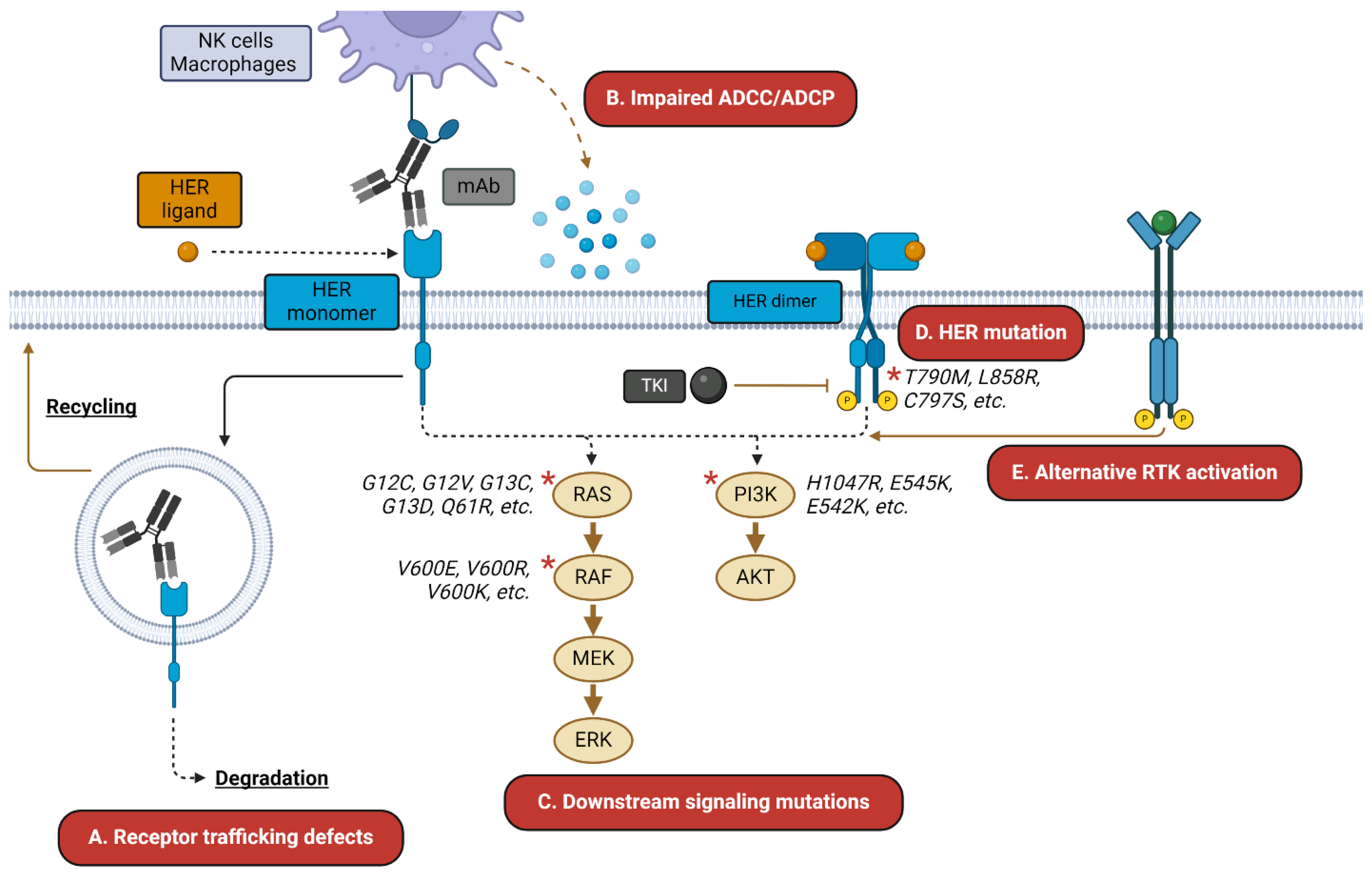
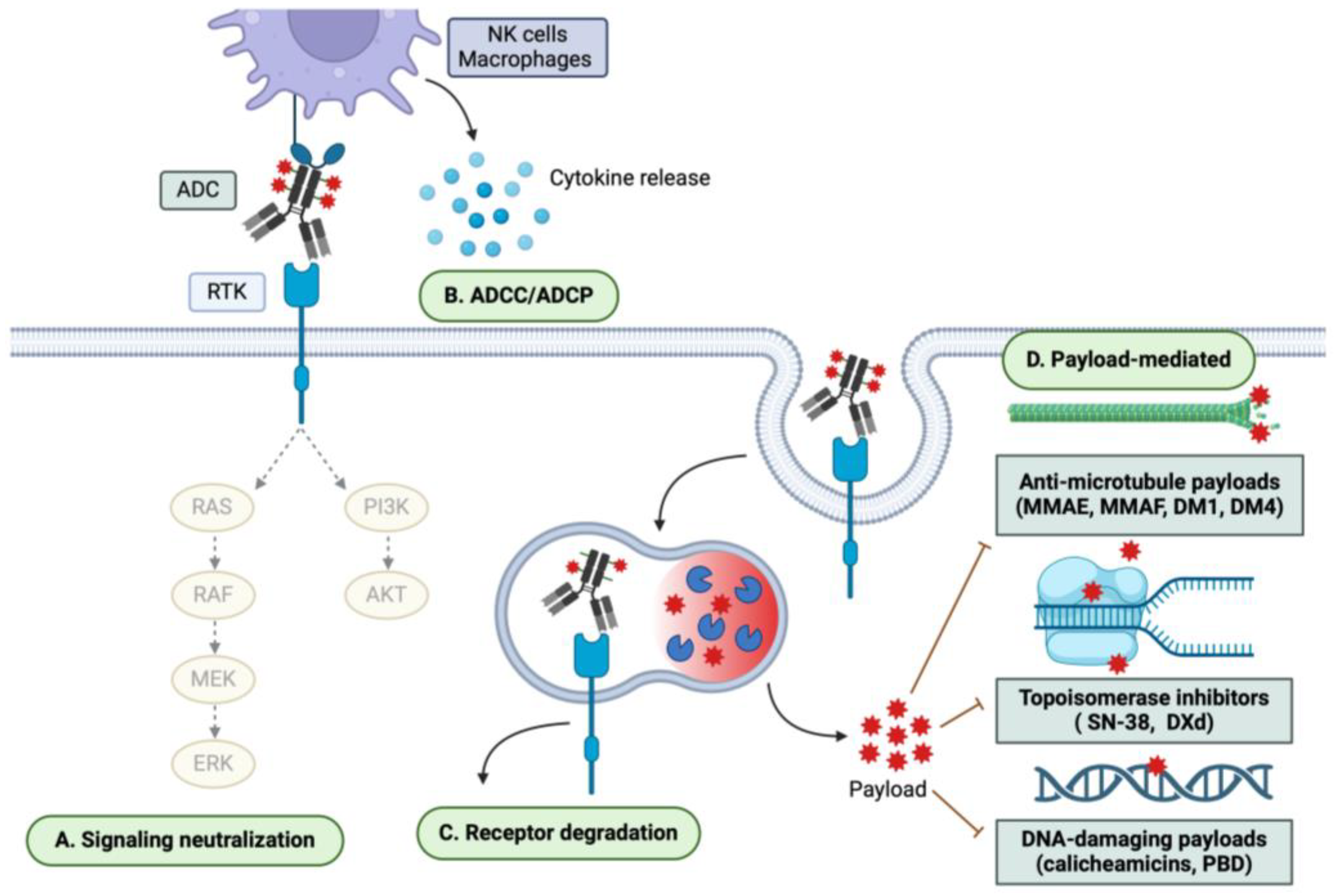
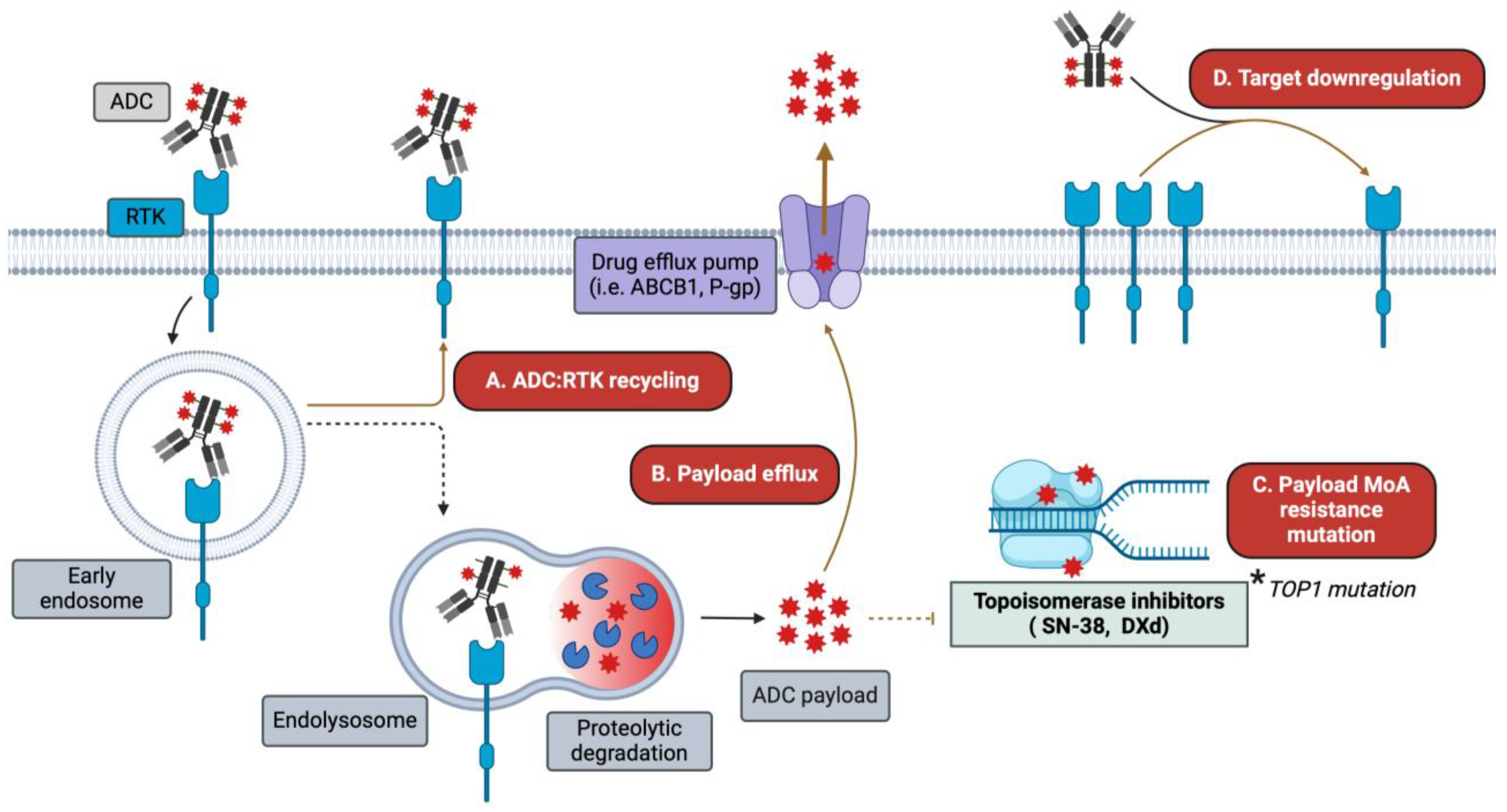
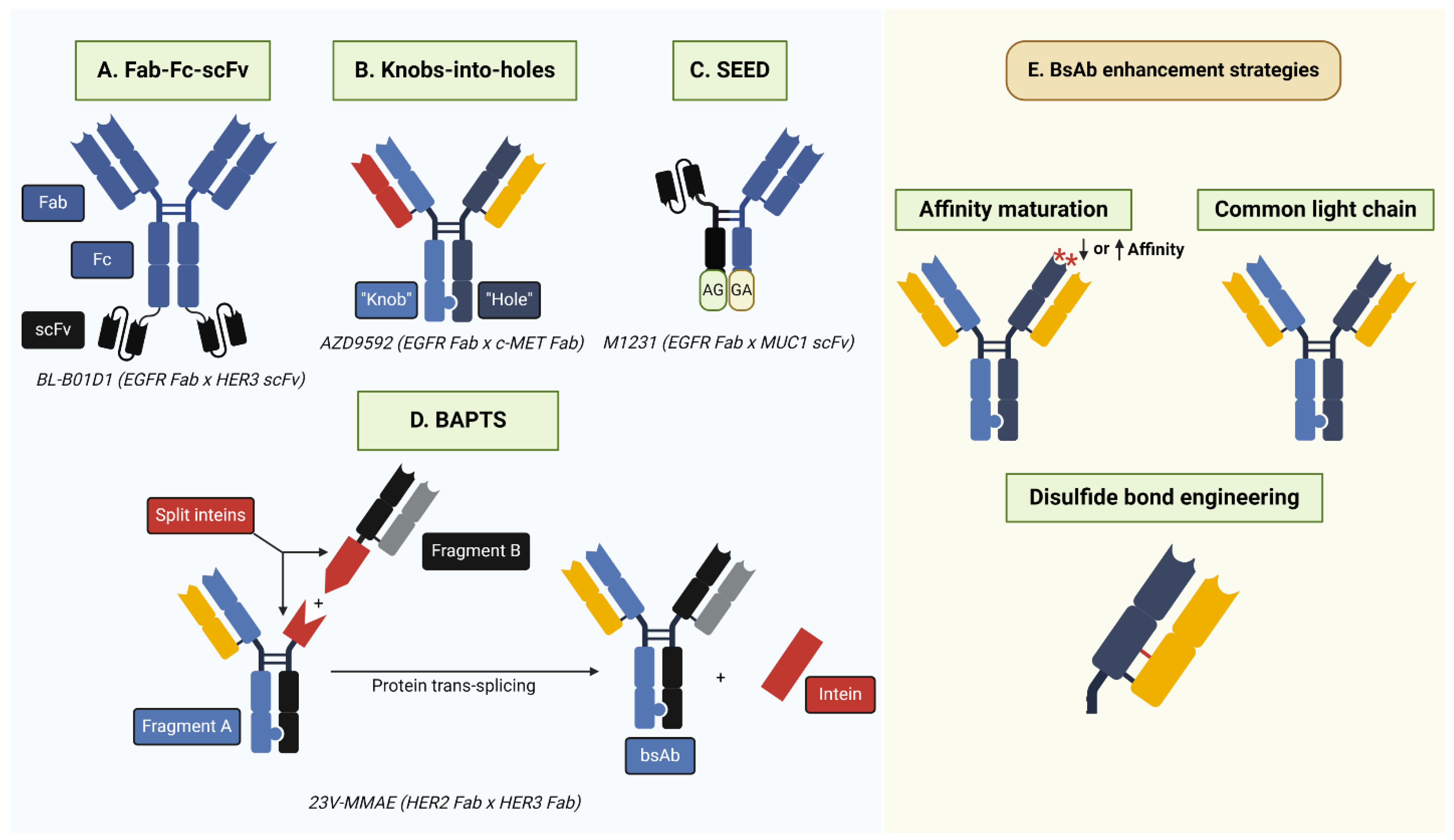
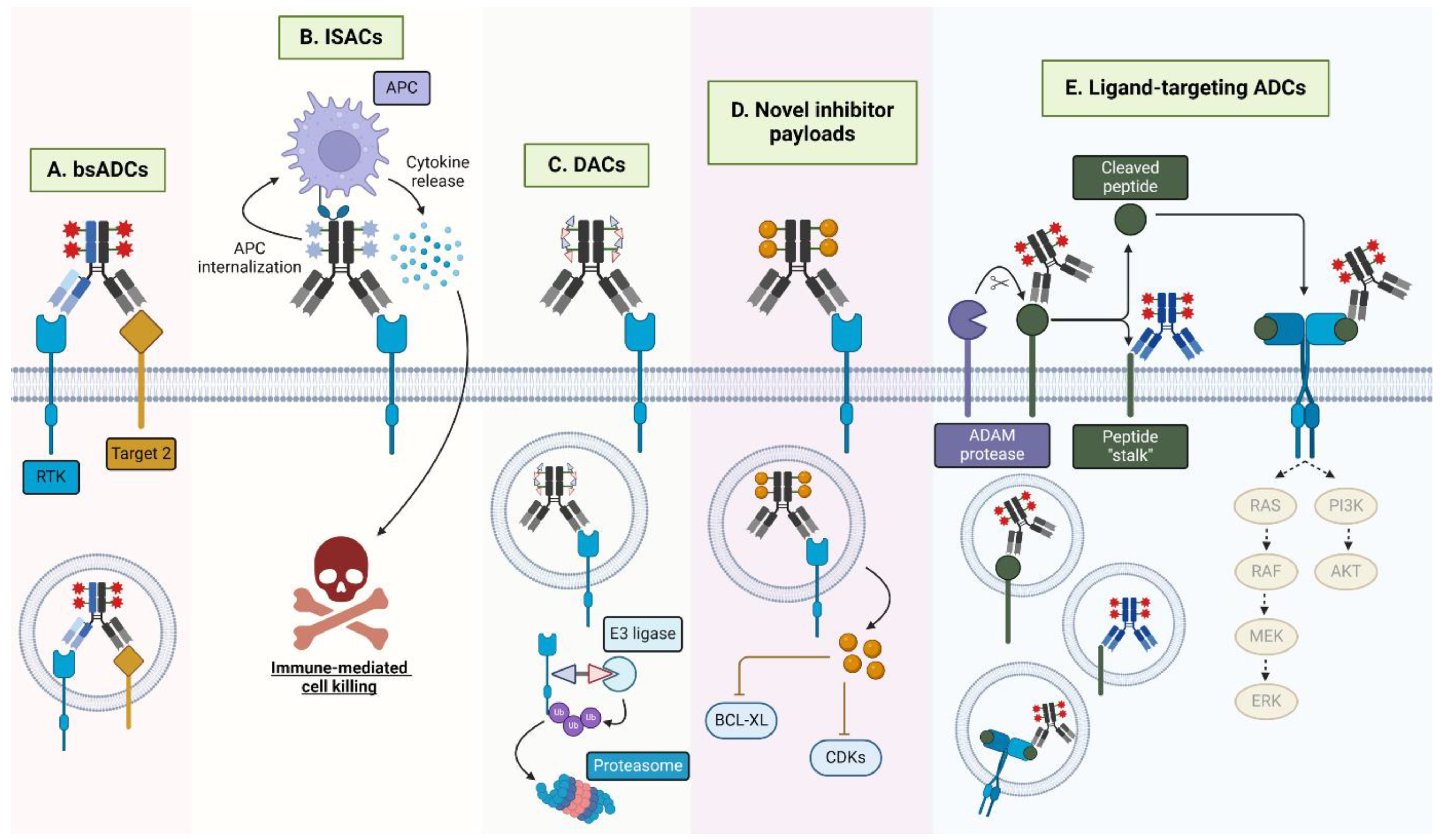
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).