
The Evolving Paradigm of Antibody–Drug Conjugates Targeting the ErbB/HER Family of Receptor Tyrosine Kinases

 , , , and
, , , and
Abstract
:1. Introduction
1.1. HER Activation and Signaling Mechanisms
1.2. The HER Family in Cancer
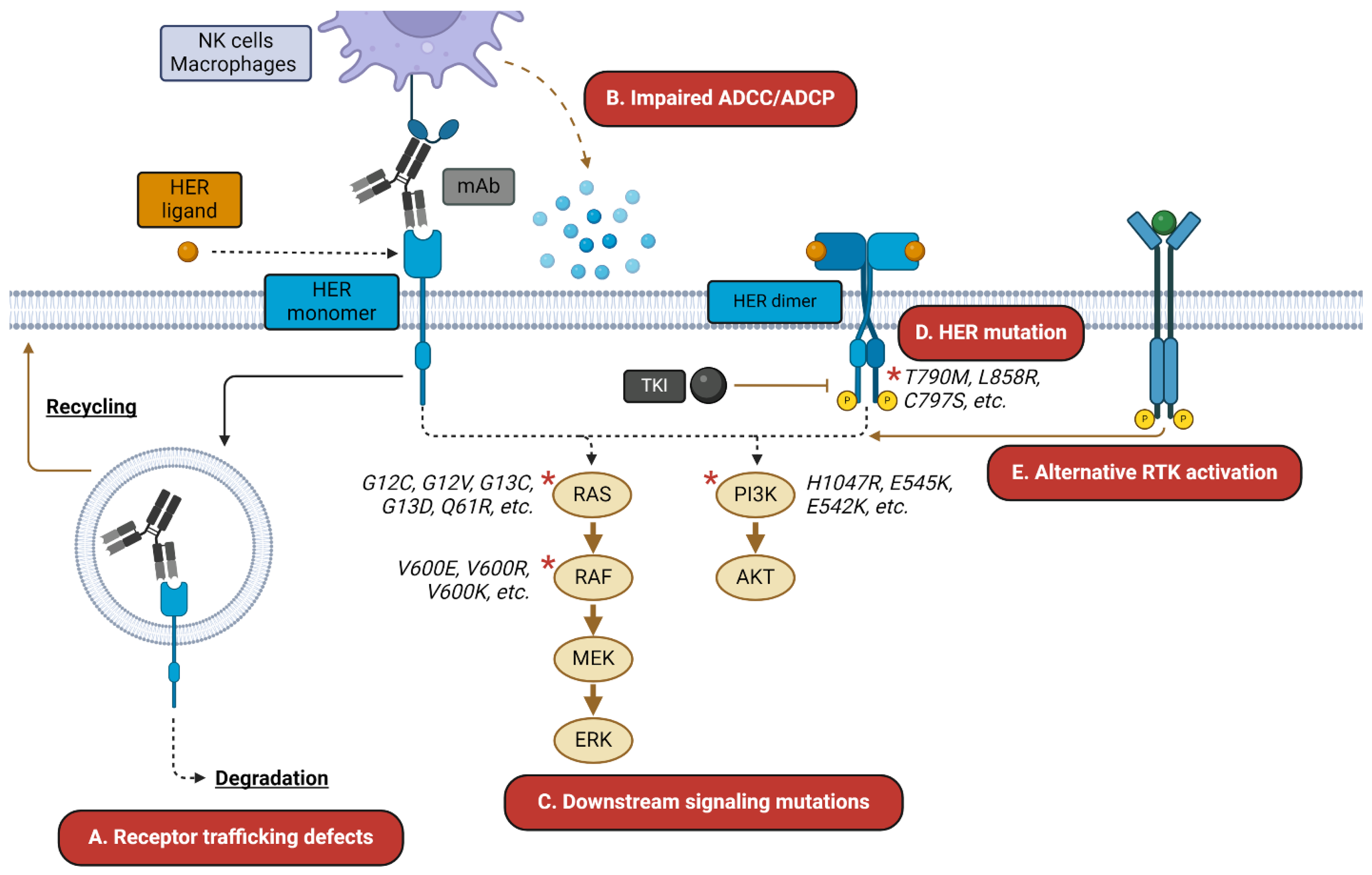
1.3. HER-Targeted TKIs and mAbs: Successes and Challenges
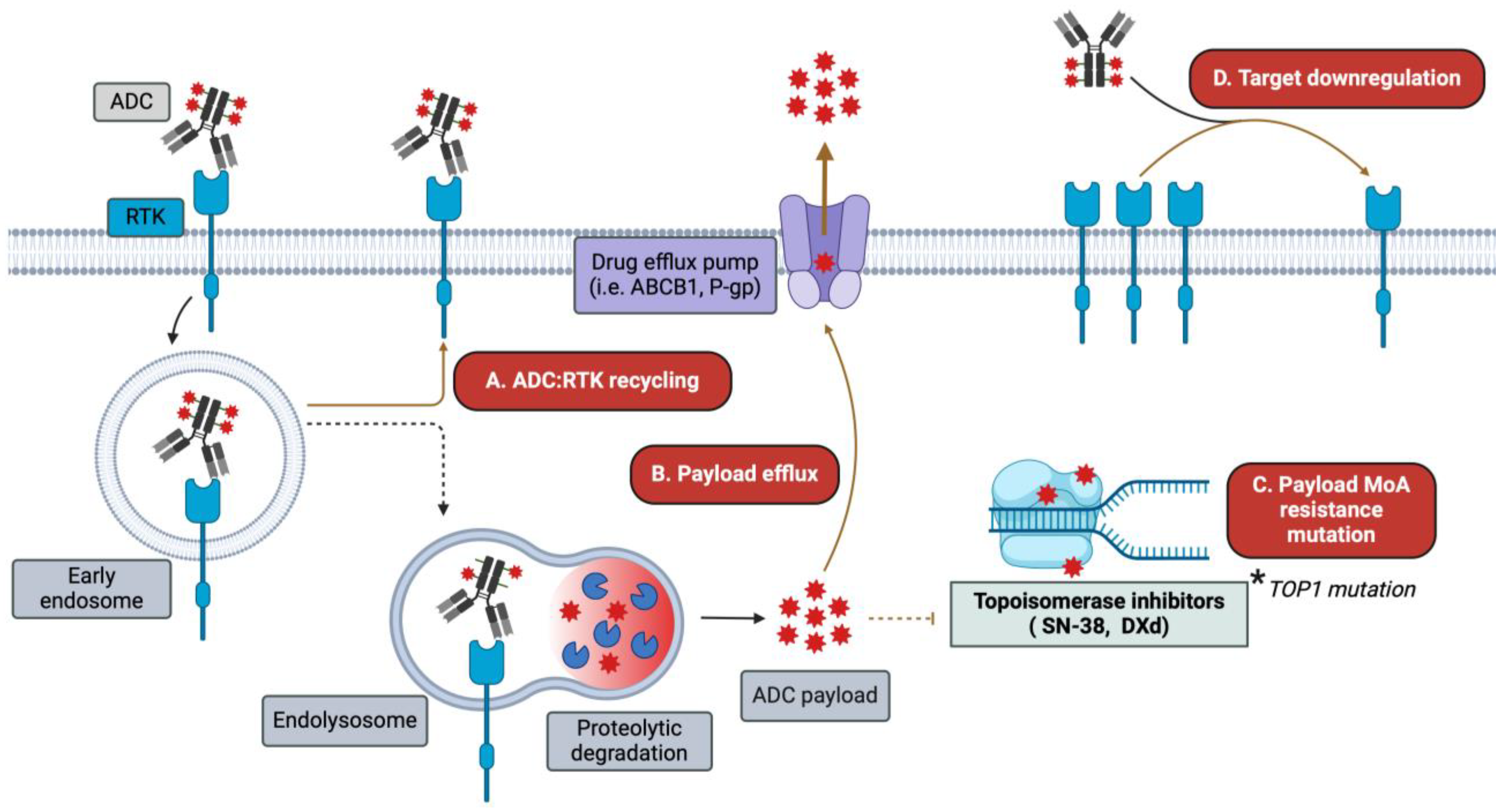
1.4. Antibody–Drug Conjugates: The Anti-Cancer Biological Missiles
2. Monospecific ADCs Targeting the ErbB Receptor Family
2.1. EGFR-Targeting ADCs
2.1.1. Depatuxizumab Mafodotin (ABT-414)
2.1.2. MRG003
2.1.3. HLX42
2.2. HER2-Targeting ADCs
2.2.1. Trastuzumab Emtansine (T-DM1)
2.2.2. Trastuzumab Deruxtecan (T-DXd)
2.2.3. Disitamab Vedotin (RC48)
2.2.4. MRG002
2.2.5. DP303c
2.2.6. FS-1502
2.2.7. Trastuzumab Duocarmazine (SYD985)
2.2.8. Emergent Clinical-Stage HER2-Targeting ADCs
2.3. HER3-Targeting ADCs
2.3.1. Patritumab Deruxtecan (U3-1402, HER3-DXd)
2.3.2. AMT-562
2.3.3. DB-1310
2.3.4. YL202/BNT326
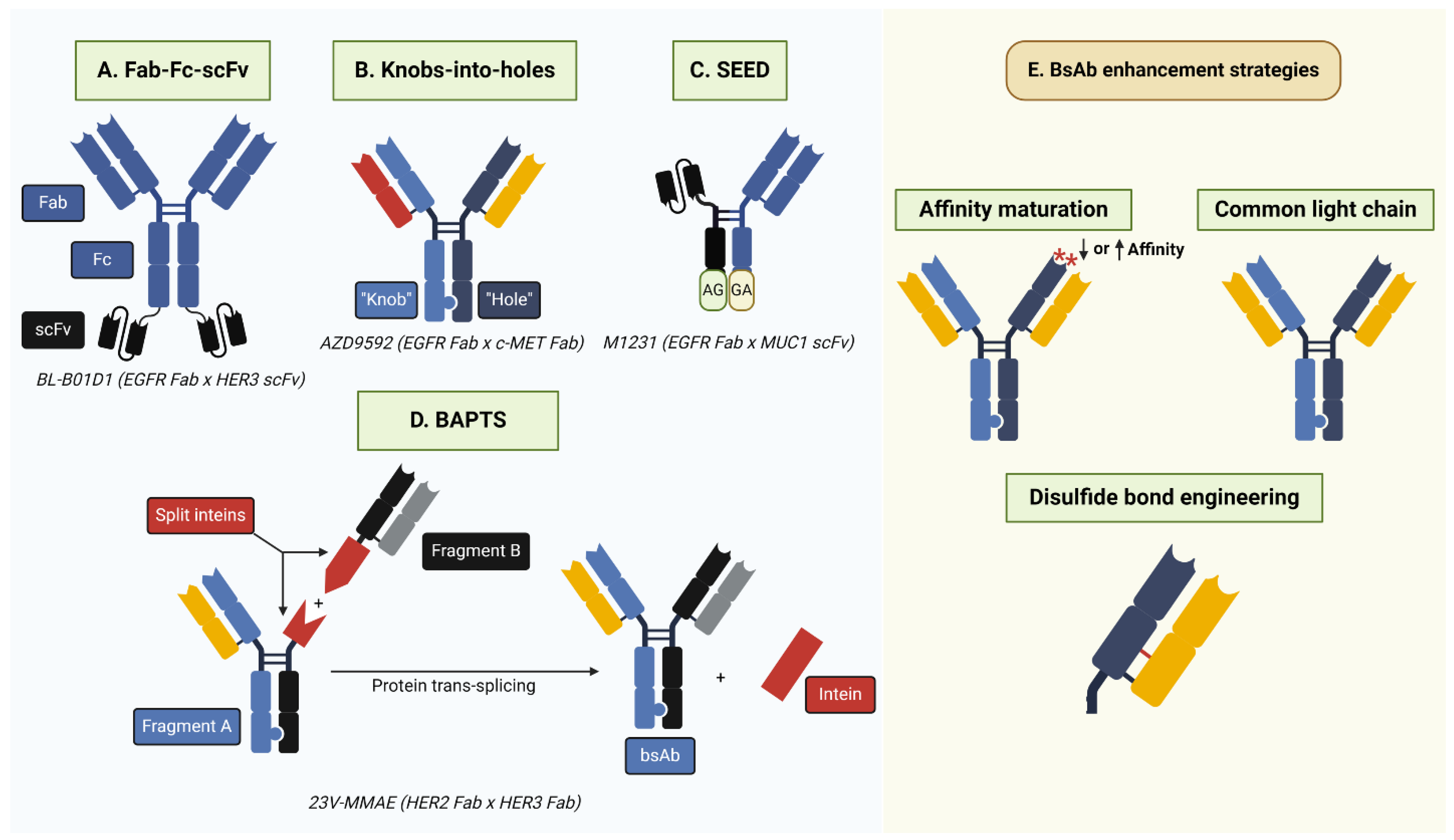
3. Bispecific ADCs Targeting the HER Family
3.1. EGFR-Targeting bsADCs
3.1.1. BL-B01D1 (EGFR x HER3)
3.1.2. M1231 (EGFR x MUC1)
3.1.3. AZD9592 (EGFR x c-MET)
3.1.4. VBC101-F11 (EGFR x c-MET)
3.1.5. DM001 (EGFR x TROP2)
3.2. HER2-Targeting bsADCs
3.2.1. Zanidatamab Zovodotin (ZW49; HER2 Biparatopic)
3.2.2. MEDI4276 (HER2 Biparatopic)
3.2.3. JSKN003 (HER2 Biparatopic)
3.2.4. 23V-MMAE (HER2 x HER3)
3.2.5. YH012 (HER2 x TROP2)
3.2.6. BIO-201 (HER2 x TROP2)
3.3. HER3-Targeting bsADCs
3.3.1. BCG022 (HER3 x c-MET)
3.3.2. DM002 (HER3 x MUC1)
4. Emergent Trends
4.1. ADCs Incorporating Novel Payloads
4.1.1. Immune-Stimulating Antibody Conjugates (ISACs)
4.1.2. Degrader-Antibody Conjugates (DACs)
4.1.3. ADCs Incorporating BCL-XL or CDK Inhibitors
4.2. Internalization-Enhancing bsADCs
4.3. HER Ligand-Targeted ADCs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wieduwilt, M.J.; Moasser, M.M. The Epidermal Growth Factor Receptor Family: Biology Driving Targeted Therapeutics. Cell. Mol. Life Sci. CMLS 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in Cancer Cells Is Mediated by Loss of IGF-Binding Proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef]
- Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Flynn, P.; Suryaprakash, S.; Grossman, D.; Panier, V.; Wu, J. The Antibody–Drug Conjugate Landscape. Nat. Rev. Drug Discov. 2024. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions That Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Leahy, D.J. Structure of the Extracellular Region of HER3 Reveals an Interdomain Tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the Preferred Heterodimerization Partner of All ErbB Receptors, Is a Mediator of Lateral Signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF Receptor Ligands: Recent Advances. F1000Research 2016, 5, 2270. [Google Scholar] [CrossRef]
- Gutierrez, C.; Schiff, R. HER2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef]
- Honegger, A.M.; Kris, R.M.; Ullrich, A.; Schlessinger, J. Evidence That Autophosphorylation of Solubilized Receptors for Epidermal Growth Factor Is Mediated by Intermolecular Cross-Phosphorylation. Proc. Natl. Acad. Sci. USA 1989, 86, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the Biology of HER3 Receptor as a Therapeutic Target in Human Cancer. Acta Pharm. Sin. B 2018, 8, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Stacey, M.M.; Liu, B.A.; Pawson, T. Molecular Mechanisms of SH2- and PTB-Domain-Containing Proteins in Receptor Tyrosine Kinase Signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a008987. [Google Scholar] [CrossRef]
- Beerli, R.R.; Hynes, N.E. Epidermal Growth Factor-Related Peptides Activate Distinct Subsets of ErbB Receptors and Differ in Their Biological Activities. J. Biol. Chem. 1996, 271, 6071–6076. [Google Scholar] [CrossRef]
- Riese, D.J.; Bermingham, Y.; van Raaij, T.M.; Buckley, S.; Plowman, G.D.; Stern, D.F. Betacellulin Activates the Epidermal Growth Factor Receptor and erbB-4, and Induces Cellular Response Patterns Distinct from Those Stimulated by Epidermal Growth Factor or Neuregulin-Beta. Oncogene 1996, 12, 345–353. [Google Scholar]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal Growth Factor Receptor (EGFR) in Lung Cancer: An Overview and Update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- O’Leary, C.; Gasper, H.; Sahin, K.B.; Tang, M.; Kulasinghe, A.; Adams, M.N.; Richard, D.J.; O’Byrne, K.J. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals 2020, 13, 273. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational Up-Regulation of the EGFR by Tumor Hypoxia Provides a Nonmutational Explanation for Its Overexpression in Human Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Pabla, B.; Bissonnette, M.; Konda, V.J. Colon Cancer and the Epidermal Growth Factor Receptor: Current Treatment Paradigms, the Importance of Diet, and the Role of Chemoprevention. World J. Clin. Oncol. 2015, 6, 133–141. [Google Scholar] [CrossRef]
- Hynes, N.E.; Stern, D.F. The Biology of erbB-2/Neu/HER-2 and Its Role in Cancer. Biochim. Biophys. Acta 1994, 1198, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Amisha, F.; Malik, P.; Saluja, P.; Gautam, N.; Patel, T.H.; Roy, A.M.; Singh, S.R.K.; Malapati, S.J. A Comprehensive Review on the Role of Human Epidermal Growth Factor Receptor 2 (HER2) as a Biomarker in Extra-Mammary and Extra-Gastric Cancers. Onco 2023, 3, 96–124. [Google Scholar] [CrossRef]
- Alimandi, M.; Romano, A.; Curia, M.C.; Muraro, R.; Fedi, P.; Aaronson, S.A.; Di Fiore, P.P.; Kraus, M.H. Cooperative Signaling of ErbB3 and ErbB2 in Neoplastic Transformation and Human Mammary Carcinomas. Oncogene 1995, 10, 1813–1821. [Google Scholar] [PubMed]
- Majumder, A. HER3: Toward the Prognostic Significance, Therapeutic Potential, Current Challenges, and Future Therapeutics in Different Types of Cancer. Cells 2023, 12, 2517. [Google Scholar] [CrossRef]
- Jia, X.; Wang, H.; Li, Z.; Yan, J.; Guo, Y.; Zhao, W.; Gao, L.; Wang, B.; Jia, Y. HER4 Promotes the Progression of Colorectal Cancer by Promoting Epithelial-Mesenchymal Transition. Mol. Med. Rep. 2020, 21, 1779–1788. [Google Scholar] [CrossRef]
- Canfield, K.; Li, J.; Wilkins, O.M.; Morrison, M.M.; Ung, M.; Wells, W.; Williams, C.R.; Liby, K.T.; Vullhorst, D.; Buonanno, A.; et al. Receptor Tyrosine Kinase ERBB4 Mediates Acquired Resistance to ERBB2 Inhibitors in Breast Cancer Cells. Cell Cycle Georget. Tex 2015, 14, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.R.; Edelblum, K.L.; Mullane, M.T.; Liang, D.; Polk, D.B. The ErbB4 Growth Factor Receptor Is Required for Colon Epithelial Cell Survival in the Presence of TNF. Gastroenterology 2009, 136, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gong, L.; Qian, Z.; Song, G.; Liu, J. ERBB4 Promotes the Proliferation of Gastric Cancer Cells via the PI3K/Akt Signaling Pathway. Oncol. Rep. 2018, 39, 2892–2898. [Google Scholar] [CrossRef] [PubMed]
- Portier, B.P.; Minca, E.C.; Wang, Z.; Lanigan, C.; Gruver, A.M.; Downs-Kelly, E.; Budd, G.T.; Tubbs, R.R. HER4 Expression Status Correlates with Improved Outcome in Both Neoadjuvant and Adjuvant Trastuzumab Treated Invasive Breast Carcinoma. Oncotarget 2013, 4, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Gullick, W.J. C-erbB-4/HER4: Friend or Foe? J. Pathol. 2003, 200, 279–281. [Google Scholar] [CrossRef]
- Lucas, L.M.; Dwivedi, V.; Senfeld, J.I.; Cullum, R.L.; Mill, C.P.; Piazza, J.T.; Bryant, I.N.; Cook, L.J.; Miller, S.T.; Lott, J.H.; et al. The Yin and Yang of ERBB4: Tumor Suppressor and Oncoprotein. Pharmacol. Rev. 2022, 74, 18–47. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Haap, M.; Kopp, H.-G.; Lipp, H.-P. Tyrosine Kinase Inhibitors—A Review on Pharmacology, Metabolism and Side Effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa) Tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Erlotinib (Tarceva) Tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef]
- Starling, N.; Neoptolemos, J.; Cunningham, D. Role of Erlotinib in the Management of Pancreatic Cancer. Ther. Clin. Risk Manag. 2006, 2, 435–445. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448 Pt 3, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Soulières, D.; Melezínek, I.; Moecks, J.; Keil, L.; Mok, T.; Rosell, R.; Klughammer, B. Clinical Outcomes in Non-Small-Cell Lung Cancer Patients with EGFR Mutations: Pooled Analysis. J. Cell. Mol. Med. 2010, 14, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, E.; De Carlo, E.; Del Conte, A.; Stanzione, B.; Revelant, A.; Fassetta, K.; Spina, M.; Bearz, A. Acquired Resistance to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer: How Do We Overcome It? Int. J. Mol. Sci. 2022, 23, 6936. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Yang, J.C.-H.; Lee, C.K.; Kurata, T.; Kim, D.-W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation–Positive Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nakashima, C.; Komiya, K.; Kitera, K.; Hirai, M.; Kimura, S.; Aragane, N. Mechanisms of Acquired Resistance to Afatinib Clarified with Liquid Biopsy. PLoS ONE 2018, 13, e0209384. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018, 13, 727–731. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, V.; Raimondo, L.; Formisano, L.; Giuliano, M.; De Placido, S.; Rosa, R.; Bianco, R. Mechanisms of Lapatinib Resistance in HER2-Driven Breast Cancer. Cancer Treat. Rev. 2015, 41, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Eli, L.D.; Kavuri, S.M. Mechanisms of Neratinib Resistance in HER2-Mutant Metastatic Breast Cancer. Cancer Drug Resist. 2022, 5, 873–881. [Google Scholar] [CrossRef]
- Freeman, D.; Sun, J.; Bass, R.; Jung, K.; Ogbagabriel, S.; Elliott, G.; Radinsky, R. Panitumumab and Cetuximab Epitope Mapping and in Vitro Activity. J. Clin. Oncol. 2008, 26 (Suppl. S15), 14536. [Google Scholar] [CrossRef]
- Hao, Y.; Yu, X.; Bai, Y.; McBride, H.J.; Huang, X. Cryo-EM Structure of HER2-Trastuzumab-Pertuzumab Complex. PLoS ONE 2019, 14, e0216095. [Google Scholar] [CrossRef] [PubMed]
- Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.E.; Lyu, H.; Liu, B. HER3-Targeted Therapeutic Antibodies and Antibody–Drug Conjugates in Non-Small Cell Lung Cancer Refractory to EGFR-Tyrosine Kinase Inhibitors. Chin. Med. J. Pulm. Crit. Care Med. 2023, 1, 11–17. [Google Scholar] [CrossRef]
- Thakkar, D.; Paliwal, S.; Kar, S.; Gandhi, N.; Paszkiewicz, K.; Ingram, P.; Boyd-Kirkup, J. Abstract P197: An Anti-HER3 Antibody, HMBD-001, That Uniquely Binds to and Blocks the HER3 Heterodimerization Interface, Shows Superior Tumor Growth Inhibition in Biomarker-Defined Preclinical Cancer Models Including NRG1-Fusion Driven Cancers. Mol. Cancer Ther. 2021, 20 (Suppl. S12), P197. [Google Scholar] [CrossRef]
- Li, W.; Xie, C.; Zhu, X.; Tang, J.; Wang, L.; Lou, L. SIBP-03, a Novel Anti-HER3 Antibody, Exerts Antitumor Effects and Synergizes with EGFR- and HER2-Targeted Drugs. Acta Pharmacol. Sin. 2024, 45, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Thavaneswaran, S.; Chan, W.Y.; Asghari, R.; Grady, J.P.; Deegan, M.; Jansen, V.M.; Thomas, D.M. Clinical Response to Seribantumab, an Anti–Human Epidermal Growth Factor Receptor-3 Immunoglobulin 2 Monoclonal Antibody, in a Patient With Metastatic Pancreatic Ductal Adenocarcinoma Harboring an NRG1 Fusion. JCO Precis. Oncol. 2022, 6, e2200263. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-Q.; Zeng, L.-S.; Wang, L.-F.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Han, Z.-W.; Zhou, Y.; Huang, S.-L.; Wang, X.-W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-Positive Breast Cancer: Advances and Future Directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Torka, P.; Barth, M.; Ferdman, R.; Hernandez-Ilizaliturri, F.J. Mechanisms of Resistance to Monoclonal Antibodies (mAbs) in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 426–438. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular Mechanisms of Resistance to the EGFR Monoclonal Antibody Cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef]
- Wang, K.; Du, R.; Myall, N.J.; Lewis, W.E.; Uy, N.; Hong, L.; Skoulidis, F.; Byers, L.A.; Tsao, A.; Cascone, T.; et al. Real-World Efficacy and Safety of Amivantamab for EGFR-Mutant NSCLC. J. Thorac. Oncol. 2024, 19, 500–506. [Google Scholar] [CrossRef]
- Tsuchikama, K.; Anami, Y.; Ha, S.Y.Y.; Yamazaki, C.M. Exploring the next Generation of Antibody-Drug Conjugates. Nat. Rev. Clin. Oncol. 2024, 21, 203–223. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload Diversification: A Key Step in the Development of Antibody–Drug Conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Ricciuti, B.; Pradhan, S.M.; Tolaney, S.M. Optimizing the Safety of Antibody–Drug Conjugates for Patients with Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 558–576. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Matsuda, Y. Novel Formats of Antibody Conjugates: Recent Advances in Payload Diversity, Conjugation, and Linker Chemistry. Expert Opin. Biol. Ther. 2023, 23, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-Drug Conjugates: Recent Advances in Conjugation and Linker Chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- High, P.; Carmon, K.S. G Protein-Coupled Receptor-Targeting Antibody-Drug Conjugates: Current Status and Future Directions. Cancer Lett. 2023, 564, 216191. [Google Scholar] [CrossRef]
- Smith, R.A.; Zammit, D.J.; Damle, N.K.; Usansky, H.; Reddy, S.P.; Lin, J.-H.; Mistry, M.; Rao, N.S.; Denis, L.J.; Gupta, S. ASN004, A 5T4-Targeting scFv-Fc Antibody-Drug Conjugate with High Drug-to-Antibody Ratio, Induces Complete and Durable Tumor Regressions in Preclinical Models. Mol. Cancer Ther. 2021, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Francisco, L.E.; Chatterjee, T.; Liang, Z.; Subramanian, S.; Liu, Q.J.; Rowe, J.H.; Carmon, K.S. An Antibody-Drug Conjugate Targeting GPR56 Demonstrates Efficacy in Preclinical Models of Colorectal Cancer. Br. J. Cancer 2023, 128, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, Z.; Hu, C.; Zhang, S.; Zi, M.; Yuan, L.; Cheng, X. Targeting CLDN18.2 in Cancers of the Gastrointestinal Tract: New Drugs and New Indications. Front. Oncol. 2023, 13, 1132319. [Google Scholar] [CrossRef]
- Sau, S.; Petrovici, A.; Alsaab, H.O.; Bhise, K.; Iyer, A.K. PDL-1 Antibody Drug Conjugate for Selective Chemo-Guided Immune Modulation of Cancer. Cancers 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Reilly, E.B.; Phillips, A.C.; Buchanan, F.G.; Kingsbury, G.; Zhang, Y.; Meulbroek, J.A.; Cole, T.B.; DeVries, P.J.; Falls, H.D.; Beam, C.; et al. Characterization of ABT-806, a Humanized Tumor-Specific Anti-EGFR Monoclonal Antibody. Mol. Cancer Ther. 2015, 14, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; DeVries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol. Cancer Ther. 2016, 15, 661–669. [Google Scholar] [CrossRef]
- Gan, H.K.; Burge, M.; Solomon, B.; Lee, S.T.; Holen, K.D.; Zhang, Y.; Ciprotti, M.; Lee, F.T.; Munasinghe, W.; Fischer, J.; et al. A Phase 1 and Biodistribution Study of ABT-806i, an 111In-Radiolabeled Conjugate of the Tumor-Specific Anti-EGFR Antibody ABT-806. J. Nucl. Med. 2021, 62, 787–794. [Google Scholar] [CrossRef]
- van den Bent, M.; Gan, H.K.; Lassman, A.B.; Kumthekar, P.; Merrell, R.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Efficacy of Depatuxizumab Mafodotin (ABT-414) Monotherapy in Patients with EGFR-Amplified, Recurrent Glioblastoma: Results from a Multi-Center, International Study. Cancer Chemother. Pharmacol. 2017, 80, 1209–1217. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; Vogelbaum, M.A.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab Mafodotin in EGFR-Amplified Newly Diagnosed Glioblastoma: A Phase III Randomized Clinical Trial. Neuro-Oncology 2023, 25, 339–350. [Google Scholar] [CrossRef]
- Gan, H.K.; Parakh, S.; Lassman, A.B.; Seow, A.; Lau, E.; Lee, S.T.; Ameratunga, M.; Perchyonok, Y.; Cao, D.; Burvenich, I.J.G.; et al. Tumor Volumes as a Predictor of Response to the Anti-EGFR Antibody Drug Conjugate Depatuxizumab Mafadotin. Neuro-Oncol. Adv. 2021, 3, vdab102. [Google Scholar] [CrossRef]
- Marin, B.-M.; Porath, K.A.; Jain, S.; Kim, M.; Conage-Pough, J.E.; Oh, J.-H.; Miller, C.L.; Talele, S.; Kitange, G.J.; Tian, S.; et al. Heterogeneous Delivery across the Blood-Brain Barrier Limits the Efficacy of an EGFR-Targeting Antibody Drug Conjugate in Glioblastoma. Neuro-Oncology 2021, 23, 2042–2053. [Google Scholar] [CrossRef]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Falls, H.D.; Mitten, M.J.; DeVries, P.J.; Benatuil, L.; Hsieh, C.-M.; Meulbroek, J.A.; Panchal, S.C.; et al. Characterization of ABBV-221, a Tumor-Selective EGFR-Targeting Antibody Drug Conjugate. Mol. Cancer Ther. 2018, 17, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Falls, H.D.; Mitten, M.J.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Gao, W.; Palma, J.P.; Cao, D.; Chia, P.-L.; et al. Targeting Multiple EGFR-Expressing Tumors with a Highly Potent Tumor-Selective Antibody-Drug Conjugate. Mol. Cancer Ther. 2020, 19, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Calvo, E.; Moreno, V.; Juric, D.; Shapiro, G.I.; Vanderwal, C.A.; Hu, B.; Gifford, M.; Barch, D.; Roberts-Rapp, L.; et al. A Phase 1 Study Evaluating Safety and Pharmacokinetics of Losatuxizumab Vedotin (ABBV-221), an Anti-EGFR Antibody-Drug Conjugate Carrying Monomethyl Auristatin E, in Patients with Solid Tumors Likely to Overexpress EGFR. Investig. New Drugs 2020, 38, 1483–1494. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Papadopoulos, K.P.; Strickler, J.H.; Lassman, A.B.; Waqar, S.N.; Chae, Y.K.; Patel, J.D.; Shacham-Shmueli, E.; Kelly, K.; Khasraw, M.; et al. Phase I Study of Anti-Epidermal Growth Factor Receptor Antibody-Drug Conjugate Serclutamab Talirine: Safety, Pharmacokinetics, and Antitumor Activity in Advanced Glioblastoma. Neuro-Oncol. Adv. 2023, 5, vdac183. [Google Scholar] [CrossRef]
- Qiu, M.-Z.; Zhang, Y.; Guo, Y.; Guo, W.; Nian, W.; Liao, W.; Xu, Z.; Zhang, W.; Zhao, H.-Y.; Wei, X.; et al. Evaluation of Safety of Treatment With Anti–Epidermal Growth Factor Receptor Antibody Drug Conjugate MRG003 in Patients With Advanced Solid Tumors. JAMA Oncol. 2022, 8, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Liu, R.; Song, G.; Song, H.; Jiang, J.; Jia, C.; Huang, X.; Yuan, X.; Yang, W.-J.; Wang, X.; et al. 683P Preclinical Evaluation of HLX42, a Novel EGFR-Targeting ADC, for Cetuximab or TKI Resistant Cancer. Ann. Oncol. 2023, 34, S477–S478. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V.J. Ado-Trastuzumab Emtansine (T-DM1): An Antibody-Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab Emtansine: Mechanisms of Action and Drug Resistance. Breast Cancer Res. BCR 2014, 16, 209. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Wedam, S.; Fashoyin-Aje, L.; Gao, X.; Bloomquist, E.; Tang, S.; Sridhara, R.; Goldberg, K.B.; King-Kallimanis, B.L.; Theoret, M.R.; Ibrahim, A.; et al. FDA Approval Summary: Ado-Trastuzumab Emtansine for the Adjuvant Treatment of HER2-Positive Early Breast Cancer. Clin. Cancer Res. 2020, 26, 4180–4185. [Google Scholar] [CrossRef]
- Kowalczyk, L.; Bartsch, R.; Singer, C.F.; Farr, A. Adverse Events of Trastuzumab Emtansine (T-DM1) in the Treatment of HER2-Positive Breast Cancer Patients. Breast Care 2017, 12, 401–408. [Google Scholar] [CrossRef]
- Azar, I.; Alkassis, S.; Fukui, J.; Alsawah, F.; Fedak, K.; Al Hallak, M.N.; Sukari, A.; Nagasaka, M. Spotlight on Trastuzumab Deruxtecan (DS-8201,T-DXd) for HER2 Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer Targets Ther. 2021, 12, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Baba, E.; Fujitani, K.; Oki, E.; Fujii, S.; Yamaguchi, K. Discovery and Development of Trastuzumab Deruxtecan and Safety Management for Patients with HER2-Positive Gastric Cancer. Gastric Cancer 2021, 24, 780–789. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Kumagai, K.; Aida, T.; Tsuchiya, Y.; Kishino, Y.; Kai, K.; Mori, K. Interstitial Pneumonitis Related to Trastuzumab Deruxtecan, a Human Epidermal Growth Factor Receptor 2-Targeting Ab-Drug Conjugate, in Monkeys. Cancer Sci. 2020, 111, 4636–4645. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Hegg, R.; Chung, W.-P.; Im, S.-A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine in Patients with HER2-Positive Metastatic Breast Cancer: Updated Results from DESTINY-Breast03, a Randomised, Open-Label, Phase 3 Trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Curigliano, G.; Hu, X.; Dent, R.A.; Yonemori, K.; Barrios, C.H.; O’Shaughnessy, J.; Wildiers, H.; Zhang, Q.; Im, S.-A.; Saura, C.; et al. Trastuzumab Deruxtecan (T-DXd) vs Physician’s Choice of Chemotherapy (TPC) in Patients (pts) with Hormone Receptor-positive (HR+), Human Epidermal Growth Factor Receptor 2 (HER2)-Low or HER2-Ultralow Metastatic Breast Cancer (mBC) with Prior Endocrine Therapy (ET): Primary Results from DESTINY-Breast06 (DB-06). Available online: https://meetings.asco.org/abstracts-presentations/238015 (accessed on 3 June 2024).
- Smit, E.F.; Felip, E.; Uprety, D.; Nagasaka, M.; Nakagawa, K.; Rodríguez, L.P.-A.; Pacheco, J.M.; Li, B.T.; Planchard, D.; Baik, C.; et al. Trastuzumab Deruxtecan in Patients with Metastatic Non-Small-Cell Lung Cancer (DESTINY-Lung01): Primary Results of the HER2-Overexpressing Cohorts from a Single-Arm, Phase 2 Trial. Lancet Oncol. 2024, 25, 439–454. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Fam-Trastuzumab Deruxtecan-Nxki for Unresectable or Metastatic HER2-Positive Solid Tumors. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-unresectable-or-metastatic-her2 (accessed on 30 June 2024).
- Xu, Y.; Wang, Y.; Gong, J.; Zhang, X.; Peng, Z.; Sheng, X.; Mao, C.; Fan, Q.; Bai, Y.; Ba, Y.; et al. Phase I Study of the Recombinant Humanized Anti-HER2 Monoclonal Antibody-MMAE Conjugate RC48-ADC in Patients with HER2-Positive Advanced Solid Tumors. Gastric Cancer 2021, 24, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, X.; Xu, Z.; Li, L.; Liu, W.; Dai, Z.; Zhao, Z.; Xiao, L.; Li, H.; Hu, C. Preclinical Evaluation of MRG002, a Novel HER2-Targeting Antibody-Drug Conjugate with Potent Antitumor Activity against HER2-Positive Solid Tumors. Antib. Ther. 2021, 4, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Yuan, C.; Cao, W.; Ge, W.; Zhang, D.; Dan, M.; Zhao, Q.; Liu, B.; Yao, B. An Innovative Site-Specific Anti-HER2 Antibody-Drug Conjugate with High Homogeneity and Improved Therapeutic Index. OncoTargets Ther. 2022, 15, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Y.; Tong, Z.; Liu, Y.; Wang, X.; Yan, M.; Chang, J.; Wang, S.; Du, C.; Li, L.; et al. FS-1502 in HER2-positive advanced breast cancer: Results from an open-label, phase 1 study. J. Clin. Oncol. 2023, 41, 3044. [Google Scholar] [CrossRef]
- Aftimos, P.G.; Turner, N.; O’Shaughnessy, J.; van den Tweel, E.; Oesterholt, M.; Escrivá-de-Romaní, S.; Tueux, N.Q.; Tan, T.J.Y.; Lim, J.; Ladoire, S.; et al. 386MO Trastuzumab Duocarmazine versus Physician’s Choice Therapy in Pre-Treated HER2-Positive Metastatic Breast Cancer: Final Results of the Phase III TULIP Trial. Ann. Oncol. 2023, 34, S340–S341. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Issues Complete Response Letter for Byondis’ [Vic-]Trastuzumab Duocarmazine. Available online: https://www.byondis.com/media/press-releases/us-food-and-drug-administration-issues-complete-response-letter-for-vic-trastuzumab-duocarmazine (accessed on 26 May 2024).
- Hu, X.; Zhang, J.; Liu, R.; Gao, S.; Qing, Y.; Yi, S.; Yuan, J.; Chen, H.; Fan, B.; Zheng, H.; et al. Phase I Study of A166 in Patients with HER2-Expressing Locally Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2021, 39 (Suppl. S15), 1024. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; Liu, R.; Gao, S.; Wu, J.; Wang, Y.; Hao, Y.; Ge, J.; Qing, Y.; Yi, S.; et al. Updated Results and Biomarker Analyses from the Phase I Trial of A166 in Patients with HER2-Expressing Locally Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2022, 40 (Suppl. S16), 1037. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, R.; Gao, S.; Li, W.; Chen, Y.; Meng, Y.; Liu, C.; Jin, W.; Wu, J.; Wang, Y.; et al. Phase I Study of A166, an Antibody–drug Conjugate in Advanced HER2-Expressing Solid Tumours. Npj Breast Cancer 2023, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Huang, Y.; Lu, Y.; Rao, X.; Wang, F.; Huang, Y.; Huang, X. 438P Phase I Study of ZV0203, a First in Class Pertuzumab ADC, in Patients with HER2+ Advanced Solid Tumors. Ann. Oncol. 2023, 34, S365. [Google Scholar] [CrossRef]
- Miao, Z.; Lin, F.; Rao, X.; Huang, Y.; Wang, F.; Lu, Y.; Huang, X.; Huang, Y. Phase 1 study of ZV0203, a first in class pertuzumab ADC, in patients with HER2 positive advanced solid tumors. J. Clin. Oncol. 2024, 42, e15004. [Google Scholar] [CrossRef]
- Barok, M.; Le Joncour, V.; Martins, A.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H. ARX788, a Novel Anti-HER2 Antibody-Drug Conjugate, Shows Anti-Tumor Effects in Preclinical Models of Trastuzumab Emtansine-Resistant HER2-Positive Breast Cancer and Gastric Cancer. Cancer Lett. 2020, 473, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-Specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-Low and T-DM1-Resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, D.; Shen, W.; Xiao, Q.; Gu, Y.; O’Shaughnessy, J.; Hu, X. Phase I Trial of a Novel Anti-HER2 Antibody-Drug Conjugate, ARX788, for the Treatment of HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2022, 28, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Yonesaka, K.; Takegawa, N.; Watanabe, S.; Haratani, K.; Kawakami, H.; Sakai, K.; Chiba, Y.; Maeda, N.; Kagari, T.; Hirotani, K.; et al. An HER3-Targeting Antibody-Drug Conjugate Incorporating a DNA Topoisomerase I Inhibitor U3-1402 Conquers EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC. Oncogene 2019, 38, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Jänne, P.A.; Oliveira, M.; Rizvi, N.; Malburg, L.; Keedy, V.; Yee, L.; Copigneaux, C.; Hettmann, T.; Wu, C.-Y.; et al. Phase I Study of U3-1287, a Fully Human Anti-HER3 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 3078–3087. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.J.; Ogbagabriel, S.; Bready, J.; Sun, J.-R.; Radinsky, R.; Hettmann, T. Abstract A182: U3–1287 (AMG 888), a Fully Human Anti-HER3 mAb, Demonstrates in Vitro and in Vivo Efficacy in the FaDu Model of Human Squamous Cell Carcinoma of the Head and Neck (SCCHN). Mol. Cancer Ther. 2011, 10 (Suppl. S11), A182. [Google Scholar] [CrossRef]
- Koganemaru, S.; Kuboki, Y.; Koga, Y.; Kojima, T.; Yamauchi, M.; Maeda, N.; Kagari, T.; Hirotani, K.; Yasunaga, M.; Matsumura, Y.; et al. U3-1402, a Novel HER3-Targeting Antibody-Drug Conjugate, for the Treatment of Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody-Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Masuda, N.; Mukohara, T.; Takahashi, S.; Nakayama, T.; Inoue, K.; Iwata, H.; Toyama, T.; Yamamoto, Y.; Hansra, D.M.; et al. Results from the Phase 1/2 Study of Patritumab Deruxtecan, a HER3-Directed Antibody-Drug Conjugate (ADC), in Patients with HER3-Expressing Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 1002. [Google Scholar] [CrossRef]
- Krop, I.E.; Masuda, N.; Mukohara, T.; Takahashi, S.; Nakayama, T.; Inoue, K.; Iwata, H.; Yamamoto, Y.; Alvarez, R.H.; Toyama, T.; et al. Patritumab Deruxtecan (HER3-DXd), a Human Epidermal Growth Factor Receptor 3–Directed Antibody-Drug Conjugate, in Patients With Previously Treated Human Epidermal Growth Factor Receptor 3–Expressing Metastatic Breast Cancer: A Multicenter, Phase I/II Trial. J. Clin. Oncol. 2023, 41, 5550–5560. [Google Scholar] [CrossRef]
- Pistilli, B.; Ibrahimi, N.; Lacroix-Triki, M.; D’Hondt, V.; Vicier, C.; Frenel, J.-S.; Dalenc, F.; Bachelot, T.; Benderra, M.-A.; Loirat, D.; et al. 189O A Phase II Study of Patritumab Deruxtecan (HER3-DXd), in Patients (Pts) with Advanced Breast Cancer (ABC), with Biomarker Analysis to Characterize Response to Therapy (ICARUS-BREAST01). ESMO Open 2023, 8, 101378. [Google Scholar] [CrossRef]
- Oliveira, M.; Falato, C.; Cejalvo, J.M.; Vila, M.M.; Tolosa, P.; Salvador-Bofill, F.J.; Cruz, J.; Arumi, M.; Luna, A.M.; Guerra, J.A.; et al. Patritumab Deruxtecan in Untreated Hormone Receptor-Positive/HER2-Negative Early Breast Cancer: Final Results from Part A of the Window-of-Opportunity SOLTI TOT-HER3 Pre-Operative Study. Ann. Oncol. 2023, 34, 670–680. [Google Scholar] [CrossRef]
- Pascual, T.; Oliveira, M.; Ciruelos, E.; Bellet Ezquerra, M.; Saura, C.; Gavilá, J.; Pernas, S.; Muñoz, M.; Vidal, M.J.; Margelí Vila, M.; et al. SOLTI-1805 TOT-HER3 Study Concept: A Window-of-Opportunity Trial of Patritumab Deruxtecan, a HER3 Directed Antibody Drug Conjugate, in Patients With Early Breast Cancer. Front. Oncol. 2021, 11, 638482. [Google Scholar] [CrossRef]
- Steuer, C.E.; Hayashi, H.; Su, W.-C.; Nishio, M.; Johnson, M.L.; Kim, D.-W.; Koczywas, M.; Felip, E.; Gold, K.A.; Murakami, H.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in Advanced/Metastatic Non-Small Cell Lung Cancer (NSCLC) without EGFR-Activating Mutations. J. Clin. Oncol. 2022, 40 (Suppl. S16), 9017. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Dosunmu, O.; Shastry, M.; Finney, L.; Sellami, D.B.; Sternberg, D.W.; Wright-Browne, V.; Toppmeyer, D.; Gwin, W.R.; Thaddeus, J.T.; et al. A Phase 2 Study of HER3-DXd in Patients (Pts) with Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2023, 41 (Suppl. S16), 1004. [Google Scholar] [CrossRef]
- Yu, H.A.; Goto, Y.; Hayashi, H.; Felip, E.; Yang, J.C.-H.; Reck, M.; Yoh, K.; Lee, S.-H.; Paz-Ares, L.; Besse, B.; et al. HERTHENA-Lung01, a Phase II Trial of Patritumab Deruxtecan (HER3-DXd) in Epidermal Growth Factor Receptor-Mutated Non-Small-Cell Lung Cancer After Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy and Platinum-Based Chemotherapy. J. Clin. Oncol. 2023, 41, 5363–5375. [Google Scholar] [CrossRef]
- Mok, T.; Jänne, P.A.; Nishio, M.; Novello, S.; Reck, M.; Steuer, C.; Wu, Y.-L.; Fougeray, R.; Fan, P.-D.; Meng, J.; et al. HERTHENA-Lung02: Phase III Study of Patritumab Deruxtecan in Advanced EGFR-Mutated NSCLC after a Third-Generation EGFR TKI. Future Oncol. 2024, 20, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Baik, C.S.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated (EGFRm) Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2021, 39 (Suppl. S15), 9007. [Google Scholar] [CrossRef]
- Weng, W.; Meng, T.; Pu, J.; Ma, L.; Shen, Y.; Wang, Z.; Pan, R.; Wang, M.; Chen, C.; Wang, L.; et al. AMT-562, a Novel HER3-Targeting Antibody-Drug Conjugate, Demonstrates a Potential to Broaden Therapeutic Opportunities for HER3-Expressing Tumors. Mol. Cancer Ther. 2023, 22, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, J.; Qu, C.; Luo, L.; Li, B.; Zhang, Y.; Zhu, Z.; Qiu, Y.; Hua, H. DB-1310, an ADC Comprised of a Novel Anti-HER3 Antibody Conjugated to a DNA Topoisomerase I Inhibitor, Is Highly Effective for the Treatment of HER3-Positive Solid Tumors. J. Transl. Med. 2024, 22, 362. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, X.; Liu, J.; Huang, Z.; Lv, D.; Zhang, Y.; Wang, X.; Chaudhry, A.; Amin, H.; Damodaran, S.; et al. YL202/BNT326, a HER3-Targeted ADC, in Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer and Breast Cancer: Preliminary Results from a First-in-Human Phase I Trial. J. Clin. Oncol. 2024, 42 (Suppl. S16), 3034. [Google Scholar] [CrossRef]
- Xu, J.; Zong, Q.; Zhu, L.; Liu, Q.; Stann, S.; Cai, J. Abstract 563: Preclinical Development of YL202, a Novel HER3-Targeting Antibody-Drug Conjugate (ADC) with Novel DNA Topoisomerase I Inhibitor for Treatment of Solid Tumors. Cancer Res. 2023, 83 (Suppl. S7), 563. [Google Scholar] [CrossRef]
- Wan, W.; Zhao, S.; Zhuo, S.; Zhang, Y.; Chen, L.; Li, G.; Renshaw, B.; Khalili, J.S.; Xiao, S.; Zhu, Y. Abstract 2642: BL-B01D1, a Novel EGFR×HER3-Targeting ADC, Demonstrates Robust Anti-Tumor Efficacy in Preclinical Evaluation. Cancer Res. 2023, 83 (Suppl. S7), 2642. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, Y.; Zhao, Y.; Fang, W.; Zhao, H.; Huang, Y.; Yang, Y.; Chen, L.; Hou, X.; Zou, W.; et al. BL-B01D1, a First-in-Class EGFRxHER3 Bispecific Antibody-Drug Conjugate (ADC), in Patients with Locally Advanced or Metastatic Solid Tumor: Results from a First-in-Human Phase 1 Study. J. Clin. Oncol. 2023, 41 (Suppl. S16), 3001. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, Y.; Zhao, Y.; Zhao, S.; Xue, J.; Yang, Y.; Fang, W.; Guo, Y.; Han, Y.; Yang, K.; et al. BL-B01D1, a First-in-Class EGFR–HER3 Bispecific Antibody–Drug Conjugate, in Patients with Locally Advanced or Metastatic Solid Tumours: A First-in-Human, Open-Label, Multicentre, Phase 1 Study. Lancet Oncol. 2024, 25, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shang, C.; An, G.; Guo, C.; An, W.; Yang, Y. A Novel EGFR×HER3-Targeting Bispecific Antibody Drug-Conjugate, BCG019, Demonstrates Robust Anti-Tumor Efficacy in Preclinical Evaluation. Available online: https://www.abstractsonline.com/pp8/#!/20272/presentation/6184 (accessed on 26 May 2024).
- Knuehl, C.; Toleikis, L.; Dotterweich, J.; Ma, J.; Kumar, S.; Ross, E.; Wilm, C.; Schmitt, M.; Grote, H.J.; Amendt, C. Abstract 5284: M1231 Is a Bispecific Anti-MUC1xEGFR Antibody-Drug Conjugate Designed to Treat Solid Tumors with MUC1 and EGFR Co-Expression. Cancer Res. 2022, 82 (Suppl. S12), 5284. [Google Scholar] [CrossRef]
- Zutshi, A.; Neuteboom, B.; Kumar, S.; Sloot, W.; Knuehl, C.; Dotterweich, J.; Ma, J.; Amendt, C.; Venkatakrishnan, K.; Park, T.; et al. Abstract 5423: Translational PK/PD/Efficacy Modeling and Efficacious Human Dose Prediction for a First-in-Class MUC1-EGFR (M1231) Bispecific Antibody Drug Conjugate. Cancer Res. 2022, 82 (Suppl. S12), 5423. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, A.; Zhang, J.; Liu, Y.; Li, H.; Li, X.; An, G.; An, W.F.; Guo, C.; et al. 1164 BSA01, a Bispecific Antibody-Drug Conjugate Targeting EGFR and Membrane-Bound MUC-1-C, Exhibits Anti-Tumor Efficacy In Vivo. J. Immunother. Cancer 2023, 11 (Suppl. S1). [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, A.; Zhang, J.; Liu, Y.; Li, H.; Li, X.; An, G.; Hui, L.; An, F.; et al. Abstract 6325: A Novel EGFR x MUC1 Bispecific Antibody-Drug Conjugate, BSA01, Targets MUC1 Transmembrane Cleavage Products and Improves Tumor Selectivity. Cancer Res. 2023, 83 (Suppl. S7), 6325. [Google Scholar] [CrossRef]
- Comer, F.; Mazor, Y.; Hurt, E.; Yang, C.; Fleming, R.; Shandilya, H.; Vijayakrishnan, B.; Sterba, M.; Chen, R.; Rosfjord, E.; et al. Abstract 5736: AZD9592: An EGFR-cMET Bispecific Antibody-Drug Conjugate (ADC) Targeting Key Oncogenic Drivers in Non-Small-Cell Lung Cancer (NSCLC) and Beyond. Cancer Res. 2023, 83 (Suppl. S7), 5736. [Google Scholar] [CrossRef]
- McGrath, L.; Zheng, Y.; Christ, S.; Sachs, C.C.; Khelifa, S.; Windmüller, C.; Sweet, S.; Kim, Y.J.; Sutton, D.; Sulikowski, M.; et al. Abstract 5737: Evaluation of the Relationship between Target Expression and in Vivo Anti-Tumor Efficacy of AZD9592, an EGFR/c-MET Targeted Bispecific Antibody Drug Conjugate. Cancer Res. 2023, 83 (Suppl. S7), 5737. [Google Scholar] [CrossRef]
- McGrath, L.; Rosfjord, E.; Christ, S.; Zheng, Y.; Floch, N.; Sachs, C.C.; Cotoi, C.; Batelli, S.; Cantu, E.; Makaretz, S.; et al. P1.12-04 In Vivo Efficacy of AZD9592, an EGFR-cMET Bispecific ADC, in a Broad Panel of NSCLC Patient-Derived Xenograft Models. J. Thorac. Oncol. 2023, 18, S208. [Google Scholar] [CrossRef]
- Aggarwal, C.; Azzoli, C.G.; Spira, A.I.; Solomon, B.J.; Le, X.; Rolfo, C.; Planchard, D.; Felip, E.; Wu, Y.-L.; Ahn, M.-J.; et al. EGRET: A First-in-Human Study of the Novel Antibody-Drug Conjugate (ADC) AZD9592 as Monotherapy or Combined with Other Anticancer Agents in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS3156. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Ma, L.; Xu, M.; Yin, Q. Abstract LB043: VBC101-F11: An Innovative EGFR/cMet Bispecific Antibody Drug Conjugate (ADC) Targeting Key Oncogenic Drivers in Solid Tumors. Cancer Res. 2024, 84 (Suppl. S7), LB043. [Google Scholar] [CrossRef]
- Li, Z.; Shang, C.; Guan, X.; An, G.; Guo, Y.; Zhang, E.; Lin, Q.; Yang, Y. Abstract LB215: A First-in-Class Anti-TROP2/EGFR Bispecific Antibody-Drug Conjugate, DM001, Exhibits Potent Anti-Tumor Efficacy. Cancer Res. 2023, 83 (Suppl. S8), LB215. [Google Scholar] [CrossRef]
- Weisser, N.E.; Sanches, M.; Escobar-Cabrera, E.; O’Toole, J.; Whalen, E.; Chan, P.W.Y.; Wickman, G.; Abraham, L.; Choi, K.; Harbourne, B.; et al. An Anti-HER2 Biparatopic Antibody That Induces Unique HER2 Clustering and Complement-Dependent Cytotoxicity. Nat. Commun. 2023, 14, 1394. [Google Scholar] [CrossRef]
- Hamblett, K.; Barnscher, S.; Davies, R.; Hammond, P.; Hernandez, A.; Wickman, G.; Fung, V.; Ding, T.; Garnett, G.; Galey, A.; et al. Abstract P6-17-13: ZW49, a HER2 Targeted Biparatopic Antibody Drug Conjugate for the Treatment of HER2 Expressing Cancers. Cancer Res. 2019, 79 (Suppl. S4), P6-17-13. [Google Scholar] [CrossRef]
- Moody, P.R.; Sayers, E.J.; Magnusson, J.P.; Alexander, C.; Borri, P.; Watson, P.; Jones, A.T. Receptor Crosslinking: A General Method to Trigger Internalization and Lysosomal Targeting of Therapeutic Receptor:Ligand Complexes. Mol. Ther. 2015, 23, 1888–1898. [Google Scholar] [CrossRef]
- Barnscher, S.D.; Rojas, A.H.; Hamblett, K.J.; Escalante, N. Abstract 2633: Zanidatamab Zovodotin (ZW49) Induces Hallmarks of Immunogenic Cell Death and Is Active in Patient-Derived Xenograft Models of Gastric Cancer. Cancer Res. 2023, 83 (Suppl. S7), 2633. [Google Scholar] [CrossRef]
- Jhaveri, K.; Han, H.; Dotan, E.; Oh, D.-Y.; Ferrario, C.; Tolcher, A.; Lee, K.-W.; Liao, C.-Y.; Kang, Y.-K.; Kim, Y.H.; et al. 460MO Preliminary Results from a Phase I Study Using the Bispecific, Human Epidermal Growth Factor 2 (HER2)-Targeting Antibody-Drug Conjugate (ADC) Zanidatamab Zovodotin (ZW49) in Solid Cancers. Ann. Oncol. 2022, 33, S749–S750. [Google Scholar] [CrossRef]
- Waldron, J. Zymeworks Halts Plans for Phase 2 ADC Trial to Mull ‘Evolving Clinical Landscape’. Available online: https://www.fiercebiotech.com/biotech/zymeworks-halts-plans-phase-2-adc-trial-mull-evolving-clinical-landscape (accessed on 26 June 2024).
- Oganesyan, V.; Peng, L.; Bee, J.S.; Li, J.; Perry, S.R.; Comer, F.; Xu, L.; Cook, K.; Senthil, K.; Clarke, L.; et al. Structural Insights into the Mechanism of Action of a Biparatopic Anti-HER2 Antibody. J. Biol. Chem. 2018, 293, 8439–8448. [Google Scholar] [CrossRef]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef]
- Pegram, M.D.; Hamilton, E.P.; Tan, A.R.; Storniolo, A.M.; Balic, K.; Rosenbaum, A.I.; Liang, M.; He, P.; Marshall, S.; Scheuber, A.; et al. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody–Drug Conjugate MEDI4276 in Patients with HER2-Positive Advanced Breast or Gastric Cancer. Mol. Cancer Ther. 2021, 20, 1442–1453. [Google Scholar] [CrossRef]
- Wang, P.; Guo, K.; Peng, J.; Sun, J.; Xu, T. JSKN003, A NOVEL BIPARATOPIC ANTI-HER2 ANTIBODY-DRUG CONJUGATE, EXHIBITS POTENT ANTITUMOR EFFICACY. Antib. Ther. 2023, 6 (Suppl. S1), tbad014.009. [Google Scholar] [CrossRef]
- Park, J.J.; Gao, B.; Beecroft, C.; Wilkinson, K.J.; Parsonson, A.; Zhang, K.; Yan, X.; Wang, N. Safety and efficacy of JSKN003 in patients with advanced/metastatic solid tumors: A first-in-human, dose-escalation, multicenter, open-label, phase I study. J. Clin. Oncol. 2024, 42, 3038. [Google Scholar] [CrossRef]
- Beecroft, C.; Gao, B.; Park, J.; Wilkinson, K.; Zhang, K.; Yan, X.; Lv, Y. Abstract CT179: Safety and Efficacy of JSKN003 in Patients with Advanced/Metastatic Solid Tumors: A First-in-Human, Dose-Escalation, Multicenter, Open-Label, Phase I Study. Cancer Res. 2024, 84 (Suppl. S7), CT179. [Google Scholar] [CrossRef]
- Schaefer, G.; Haber, L.; Crocker, L.M.; Shia, S.; Shao, L.; Dowbenko, D.; Totpal, K.; Wong, A.; Lee, C.V.; Stawicki, S.; et al. A Two-in-One Antibody against HER3 and EGFR Has Superior Inhibitory Activity Compared with Monospecific Antibodies. Cancer Cell 2011, 20, 472–486. [Google Scholar] [CrossRef]
- Shang, C.; An, G.; Guo, Y.; Zhang, E.; Lin, Q.; Yang, Y. Abstract 2977: A First-in-Class Anti-HER2/TROP2 Bispecific Antibody-Drug Conjugate (YH012) Exhibits Potent Anti-Tumor Efficacy. Cancer Res. 2023, 83 (Suppl. S7), 2977. [Google Scholar] [CrossRef]
- Shang, C.; Yang, L.; Zhang, J.; Han, Y.; Li, Z.; Han, Z.; Li, J.; Meng, Y.; An, G.; Yang, H.; et al. Abstract 4256: YH012, a Novel Bispecific Anti-HER2 and TROP2 Antibody-Drug Conjugate, Exhibits Potent Antitumor Efficacy. Cancer Res. 2022, 82 (Suppl. S12), 4256. [Google Scholar] [CrossRef]
- Zhong, H.; Xiong, Y.; Huang, H.; Pan, Y.; Cao, L.; Liu, S.; Wang, N.; Sun, B.; Liu, D.; Yuan, W.; et al. Preclinical Antitumor Activity of an Anti-HER2/Trop-2 Bispecific Antibody-Drug Conjugate with a New DNA Topoisomerase I Inhibitor. J. Clin. Oncol. 2023, 41 (Suppl. S16), e15013. [Google Scholar] [CrossRef]
- Li, Z.; Shang, C.; Han, Z.; An, G.; Guo, C.; An, W.F.; Yang, Y. Abstract 2617: BCG022, a Novel HER3 × MET Bispecific Antibody-Drug Conjugate (bsADC), Demonstrates Promising Anti-Tumor Efficacy. Cancer Res. 2024, 84 (Suppl. S6), 2617. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, N.; An, G.; Zhang, E.; Lin, Q.; Yang, Y. Abstract LB214: A First-in-Class Bispecific Antibody-Drug Conjugate (DM002) Targeting HER3 and the Juxtamembrane Domain of MUC1. Cancer Res. 2023, 83 (Suppl. S8), LB214. [Google Scholar] [CrossRef]
- Wahab, A.; Rafae, A.; Mushtaq, K.; Masood, A.; Ehsan, H.; Khakwani, M.; Khan, A. Ocular Toxicity of Belantamab Mafodotin, an Oncological Perspective of Management in Relapsed and Refractory Multiple Myeloma. Front. Oncol. 2021, 11, 678634. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W. Antibody Drug Conjugates: Lessons from 20 Years of Clinical Experience. Ann. Oncol. 2016, 27, 2168–2172. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Makker, V.; Oaknin, A.; Oh, D.-Y.; Banerjee, S.; González-Martín, A.; Jung, K.H.; Ługowska, I.; Manso, L.; Manzano, A.; et al. Efficacy and Safety of Trastuzumab Deruxtecan in Patients With HER2-Expressing Solid Tumors: Primary Results From the DESTINY-PanTumor02 Phase II Trial. J. Clin. Oncol. 2024, 42, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.K.; Manore, S.G.; Regua, A.T.; Lo, H.-W. Antibody-Drug Conjugates for the Treatment of HER2-Positive Breast Cancer. Genes 2022, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Chic, N.; Brasó-Maristany, F.; Paré, L.; Pascual, T.; Conte, B.; Martínez-Sáez, O.; Adamo, B.; Vidal, M.; Barnadas, E.; et al. Clinical, Pathological, and PAM50 Gene Expression Features of HER2-Low Breast Cancer. NPJ Breast Cancer 2021, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Gandini, S.; Nicolò, E.; Trillo, P.; Giugliano, F.; Zagami, P.; Vivanet, G.; Bellerba, F.; Trapani, D.; Marra, A.; et al. Evolution of Low HER2 Expression between Early and Advanced-Stage Breast Cancer. Eur. J. Cancer 2022, 163, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Inokuchi, M.; Takagi, Y.; Yamada, H.; Kojima, K.; Kumagai, J.; Kawano, T.; Sugihara, K. High Expression of HER3 Is Associated with a Decreased Survival in Gastric Cancer. Clin. Cancer Res. 2008, 14, 7843–7849. [Google Scholar] [CrossRef]
- Reschke, M.; Mihic-Probst, D.; van der Horst, E.H.; Knyazev, P.; Wild, P.J.; Hutterer, M.; Meyer, S.; Dummer, R.; Moch, H.; Ullrich, A. HER3 Is a Determinant for Poor Prognosis in Melanoma. Clin. Cancer Res. 2008, 14, 5188–5197. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, R.; Yan, H.; Zhao, P.; Wu, L.; Wang, H.; Li, T.; Cao, B. Prognostic Significance of HER3 in Patients with Malignant Solid Tumors. Oncotarget 2017, 8, 67140–67151. [Google Scholar] [CrossRef]
- Mendell, J.; Freeman, D.J.; Feng, W.; Hettmann, T.; Schneider, M.; Blum, S.; Ruhe, J.; Bange, J.; Nakamaru, K.; Chen, S.; et al. Clinical Translation and Validation of a Predictive Biomarker for Patritumab, an Anti-Human Epidermal Growth Factor Receptor 3 (HER3) Monoclonal Antibody, in Patients With Advanced Non-Small Cell Lung Cancer. eBioMedicine 2015, 2, 264–271. [Google Scholar] [CrossRef]
- Meulendijks, D.; Jacob, W.; Voest, E.E.; Mau-Sorensen, M.; Martinez-Garcia, M.; Taus, A.; Fleitas, T.; Cervantes, A.; Lolkema, M.P.; Langenberg, M.H.G.; et al. Phase Ib Study of Lumretuzumab Plus Cetuximab or Erlotinib in Solid Tumor Patients and Evaluation of HER3 and Heregulin as Potential Biomarkers of Clinical Activity. Clin. Cancer Res. 2017, 23, 5406–5415. [Google Scholar] [CrossRef]
- Cleary, J.M.; McRee, A.J.; Shapiro, G.I.; Tolaney, S.M.; O’Neil, B.H.; Kearns, J.D.; Mathews, S.; Nering, R.; MacBeath, G.; Czibere, A.; et al. A Phase 1 Study Combining the HER3 Antibody Seribantumab (MM-121) and Cetuximab with and without Irinotecan. Investig. New Drugs 2017, 35, 68–78. [Google Scholar] [CrossRef]
- Uliano, J.; Corvaja, C.; Curigliano, G.; Tarantino, P. Targeting HER3 for Cancer Treatment: A New Horizon for an Old Target. ESMO Open 2023, 8, 100790. [Google Scholar] [CrossRef]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody–Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.-Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential Mechanisms for Thrombocytopenia Development with Trastuzumab Emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of Resistance to Trastuzumab Emtansine (T-DM1) in HER2-Positive Breast Cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Z.; Wang, Y. Bispecific Antibody Drug Conjugates: Making 1+1>2. Acta Pharm. Sin. B 2024, 14, 1965–1986. [Google Scholar] [CrossRef]
- Spring, L.M.; Nakajima, E.; Hutchinson, J.; Viscosi, E.; Blouin, G.; Weekes, C.; Rugo, H.; Moy, B.; Bardia, A. Sacituzumab Govitecan for Metastatic Triple-Negative Breast Cancer: Clinical Overview and Management of Potential Toxicities. Oncologist 2021, 26, 827–834. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef]
- Bardia, A.; Jhaveri, K.; Im, S.-A.; Simon, S.P.; Laurentiis, M.D.; Wang, S.; Martinez, N.; Borges, G.S.; Cescon, D.W.; Hattori, M.; et al. LBA11 Datopotamab Deruxtecan (Dato-DXd) vs Chemotherapy in Previously-Treated Inoperable or Metastatic Hormone Receptor-Positive, HER2-Negative (HR+/HER2–) Breast Cancer (BC): Primary Results from the Randomised Phase III TROPION-Breast01 Trial. Ann. Oncol. 2023, 34, S1264–S1265. [Google Scholar] [CrossRef]
- Soares, L.R.; Vilbert, M.; Rosa, V.D.L.; Oliveira, J.L.; Deus, M.M.; Freitas-Junior, R. Incidence of Interstitial Lung Disease and Cardiotoxicity with Trastuzumab Deruxtecan in Breast Cancer Patients: A Systematic Review and Single-Arm Meta-Analysis. ESMO Open 2023, 8, 101613. [Google Scholar] [CrossRef]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient Generation of Bispecific IgG Antibodies by Split Intein Mediated Protein Trans-Splicing System. Sci. Rep. 2017, 7, 8360. [Google Scholar] [CrossRef]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-Dependent Solid Tumours–Molecular Diagnosis and Targeted Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef]
- Stern, Y.E.; Al-Ghabkari, A.; Monast, A.; Fiset, B.; Aboualizadeh, F.; Yao, Z.; Stagljar, I.; Walsh, L.A.; Duhamel, S.; Park, M. Met-HER3 Crosstalk Supports Proliferation via MPZL3 in MET-Amplified Cancer Cells. Cell. Mol. Life Sci. CMLS 2022, 79, 178. [Google Scholar] [CrossRef]
- Yamasaki, A.; Miyake, R.; Hara, Y.; Okuno, H.; Imaida, T.; Okita, K.; Okazaki, S.; Akiyama, Y.; Hirotani, K.; Endo, Y.; et al. Dual-Targeting Therapy against HER3/MET in Human Colorectal Cancers. Cancer Med. 2023, 12, 9684–9696. [Google Scholar] [CrossRef]
- Klempner, S.; Strickler, J.; Gourley, L.; Jacquemont, C.; Bhatia, V.; Hunder, N.; Odegard, V.; Piha-Paul, S. 393 A Phase 1/2 Study of SBT6050 Combined with Trastuzumab Deruxtecan (T-DXd) or Trastuzumab and Tucatinib with or without Capecitabine in Patients with HER2-Expressing or HER2-Amplified Cancers. J. Immunother. Cancer 2021, 9 (Suppl. S2), A426. [Google Scholar] [CrossRef]
- Silverback Discontinues Targeted Oncology Programs, Reduces Staff. Available online: https://www.precisionmedicineonline.com/cancer/silverback-discontinues-targeted-oncology-programs-reduces-staff (accessed on 26 May 2024).
- Janku, F.; Han, S.-W.; Doi, T.; Amatu, A.; Ajani, J.A.; Kuboki, Y.; Cortez, A.; Cellitti, S.E.; Mahling, P.C.; Subramanian, K.; et al. Preclinical Characterization and Phase I Study of an Anti-HER2-TLR7 Immune-Stimulator Antibody Conjugate in Patients with HER2+ Malignancies. Cancer Immunol. Res. 2022, 10, 1441–1461. [Google Scholar] [CrossRef]
- Ackerman, S.E.; Gonzalez, J.C.; Gregorio, J.D.; Paik, J.C.; Hartmann, F.J.; Kenkel, J.A.; Lee, A.; Luo, A.; Pearson, C.I.; Nguyen, M.L.; et al. Abstract 1559: TLR7/8 Immune-Stimulating Antibody Conjugates Elicit Robust Myeloid Activation Leading to Enhanced Effector Function and Anti-Tumor Immunity in Pre-Clinical Models. Cancer Res. 2019, 79 (Suppl. S13), 1559. [Google Scholar] [CrossRef]
- Li, B.T.; Pegram, M.D.; Lee, K.-W.; Sharma, M.; Lee, J.; Spira, A.I.; Hanna, G.J.; Kang, Y.-K.; Rasco, D.W.; Moore, K.N.; et al. A Phase 1/2 Study of a First-in-Human Immune-Stimulating Antibody Conjugate (ISAC) BDC-1001 in Patients with Advanced HER2-Expressing Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), 2538. [Google Scholar] [CrossRef]
- Paiva, S.-L.; Crews, C.M. Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review. Explor. Target. Anti-Tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Kihlberg, J. PROTAC Cell Permeability and Oral Bioavailability: A Journey into Uncharted Territory. Future Med. Chem. 2022, 14, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.; Bai, C.; Yi, Y.; Skaletskaya, A.; Takrouri, K.; Wong, W.; Kim, M.-S.; Choi, D.-K.; Kim, D.-Y.; Yang, Y.; et al. Abstract 3933: ORM-5029: A First-in-Class Targeted Protein Degradation Therapy Using Antibody Neodegrader Conjugate (AnDC) for HER2-Expressing Breast Cancer. Cancer Res. 2022, 82 (Suppl. S12), 3933. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Hamilton, E.P.; Spira, A.I.; Pohlmann, P.R.; Giordano, A.; Clifton, K.; Anderson, B.D.; Dutta, S.; Mangipudi, U.; Saini, S.; et al. A Phase 1, First-in-Human, Open Label, Escalation and Expansion Study of ORM-5029, a Highly Potent GSPT1 Degrader Targeting HER2, in Patients with HER2-Expressing Advanced Solid Tumors. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS1114. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 Regulated Apoptosis in Cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.K.; Waqar, S.N.; Papadopoulos, K.P.; Hung, J.-Y.; Spira, A.I.; Gan, H.; Yoshida, T.; Kuo, C.-H.S.; de Spéville, B.D.; Felip, E.; et al. 1318MO First-in-Human Study of ABBV-637, an EGFR-Targeting BCL-XL–Inhibiting Antibody-Drug Conjugate Combined with Osimertinib (OSI) in Relapsed/Refractory, EGFR-Mutated Non-Small Cell Lung Cancer (NSCLC). Ann. Oncol. 2023, 34, S759. [Google Scholar] [CrossRef]
- Thibault, S.; Hu, W.; Hirakawa, B.; Kalabat, D.; Franks, T.; Sung, T.; Khoh-Reiter, S.; Lu, S.; Finkelstein, M.; Jessen, B.; et al. Intestinal Toxicity in Rats Following Administration of CDK4/6 Inhibitors Is Independent of Primary Pharmacology. Mol. Cancer Ther. 2019, 18, 257–266. [Google Scholar] [CrossRef]
- van Aken, E.S.M.; Beeker, A.; Houtenbos, I.; Pos, F.J.; Linn, S.C.; Elkhuizen, P.H.M.; de Jong, M.C. Unexpected Toxicity of CDK4/6 Inhibitor Palbociclib and Radiotherapy. Cancer Rep. 2022, 5, e1470. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, M.E.; Ciaccio, A.; Danesi, R.; Duhoux, F.P.; Girmenia, C.; Zaman, K.; Lindman, H.; Luppi, F.; Mavroudis, D.; Paris, I.; et al. Late Onset Toxicities Associated with the Use of CDK 4/6 Inhibitors in Hormone Receptor Positive (HR+), Human Epidermal Growth Factor Receptor-2 Negative (HER2−) Metastatic Breast Cancer Patients: A Multidisciplinary, Pan-EU Position Paper Regarding Their Optimal Management. The GIOCONDA Project. Front. Oncol. 2023, 13, 1247270. [Google Scholar] [CrossRef]
- Cheung, A.; Chenoweth, A.M.; Johansson, A.; Laddach, R.; Guppy, N.; Trendell, J.; Esapa, B.; Mavousian, A.; Navarro-Llinas, B.; Haider, S.; et al. Anti-EGFR Antibody-Drug Conjugate Carrying an Inhibitor Targeting CDK Restricts Triple-Negative Breast Cancer Growth. Clin. Cancer Res. 2024. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [PubMed]
- DeVay, R.M.; Delaria, K.; Zhu, G.; Holz, C.; Foletti, D.; Sutton, J.; Bolton, G.; Dushin, R.; Bee, C.; Pons, J.; et al. Improved Lysosomal Trafficking Can Modulate the Potency of Antibody Drug Conjugates. Bioconjugate Chem. 2017, 28, 1102–1114. [Google Scholar] [CrossRef]
- de Goeij, B.E.C.G.; Vink, T.; ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- Zhuang, W.; Zhang, W.; Wang, L.; Xie, L.; Feng, J.; Zhang, B.; Hu, Y. Generation of a Novel SORT1×HER2 Bispecific Antibody–Drug Conjugate Targeting HER2-Low-Expression Tumor. Int. J. Mol. Sci. 2023, 24, 16056. [Google Scholar] [CrossRef]
- Krytska, K.; Casey, C.E.; Pogoriler, J.; Martinez, D.; Rathi, K.S.; Farrel, A.; Berko, E.R.; Tsang, M.; Sano, R.R.; Kendsersky, N.; et al. Evaluation of the DLL3-Targeting Antibody–Drug Conjugate Rovalpituzumab Tesirine in Preclinical Models of Neuroblastoma. Cancer Res. Commun. 2022, 2, 616–623. [Google Scholar] [CrossRef]
- Capone, E.; Lamolinara, A.; Pastorino, F.; Gentile, R.; Ponziani, S.; Di Vittorio, G.; D’Agostino, D.; Bibbò, S.; Rossi, C.; Piccolo, E.; et al. Targeting Vesicular LGALS3BP by an Antibody-Drug Conjugate as Novel Therapeutic Strategy for Neuroblastoma. Cancers 2020, 12, 2989. [Google Scholar] [CrossRef]
- Montero, J.C.; Rodríguez-Barrueco, R.; Ocaña, A.; Díaz-Rodríguez, E.; Esparís-Ogando, A.; Pandiella, A. Neuregulins and Cancer. Clin. Cancer Res. 2008, 14, 3237–3241. [Google Scholar] [CrossRef]
- Su, X.; Lai, T.; Tao, Y.; Zhang, Y.; Zhao, C.; Zhou, J.; Chen, E.; Zhu, M.; Zhang, S.; Wang, B.; et al. miR-33a-3p Regulates METTL3-Mediated AREG Stability and Alters EMT to Inhibit Pancreatic Cancer Invasion and Metastasis. Sci. Rep. 2023, 13, 13587. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, H.; Lu, J.; Zhang, Z.; Wu, H.; Liang, Z. AREG Mediates the Epithelial-Mesenchymal Transition in Pancreatic Cancer Cells via the EGFR/ERK/NF-κB Signalling Pathway. Oncol. Rep. 2020, 43, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-L.; Feng, P.-H.; Lee, K.-Y.; Chen, K.-Y.; Sun, W.-L.; Van Hiep, N.; Luo, C.-S.; Wu, S.-M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, K.A.; Sreekumar, S.; Jenkins, E.C.; Ernzen, K.J.; Kenny, P.A. Anti-Tumor Efficacy of an MMAE-Conjugated Antibody Targeting Cell Surface TACE/ADAM17-Cleaved Amphiregulin in Breast Cancer. Antib. Ther. 2021, 4, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, K.A.; Reker, N.C.; Sreekumar, S.; Kenny, P.A. Pan-Cancer Distribution of Cleaved Cell-Surface Amphiregulin, the Target of the GMF-1A3 Antibody Drug Conjugate. Antib. Ther. 2022, 5, 226–231. [Google Scholar] [CrossRef]
- Jacob, J.; Anami, Y.; High, P.; Liang, Z.; Subramanian, S.; Ghosh, S.C.; AghaAmiri, S.; Guernsey, C.; Tran, H.; Liu, Q.J.; et al. Antibody-Drug Conjugates Targeting EGFR Ligand Epiregulin Inhibit Colorectal Tumor Growth Irrespective of RAS Mutational Status. bioRxiv 2024. [Google Scholar] [CrossRef]
- Jacob, J.; Liang, Z.; Carmon, K. Abstract 4251: Development and Characterization of an Epiregulin Antibody-Drug Conjugate for Targeting Colorectal Cancer Cell Plasticity. Cancer Res. 2022, 82 (Suppl. S12), 4251. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.; El-Kenawi, A.; Shonagh, R.; Weber, A.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.; Mulé, J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res 2016, 76, 1381–1390. [Google Scholar] [CrossRef]
- Cobb, P.W.; Zhou, H.; Nahar, A.; Marinello, P. Open-Label, Active-Control, Phase 2/3 Study of Zilovertamab Vedotin plus Standard of Care in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS7592. [Google Scholar] [CrossRef]
- Lugini, A.; Goldman, J.W.; Tanizaki, J.; Akamatsu, H.; Xia, S.; Ratajczak, C.; Li, M.; Bolotin, E.; Seraj, J.; Lu, S. 1501TiP A Phase III Global Study of Telisotuzumab Vedotin versus Docetaxel in Previously Treated Patients with C-Met Overexpressing, EGFR Wildtype, Locally Advanced/Metastatic Nonsquamous NSCLC (TeliMET NSCLC-01). Ann. Oncol. 2023, 34, S845. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EGFR-Targeted ADCs | ||||||||
| Name | Developer | Payload | Linker | Cleavable? | Conjugation | DAR | Clinical Stage | Ref. |
| ABT-414 | Abbvie | MMAF | mc | N | Interchain cysteines | 3.8 | Completed Phase III | [77,78,79,80,81,82,83] |
| ABBV-221 | Abbvie | MMAE | mc-vc-PABC | Y | Interchain cysteines | 3 | Discontinued | [84,85,86] |
| ABBV-321 | Abbvie | PBD | mc-va | Y | Site-specific (S238C) | 2 | Discontinued | [87] |
| MRG003 | Shanghai Miracogen | MMAE | mc-vc-PABC | Y | UD | UD | Phase II | [88] |
| HLX42 | Henlius | Topo I inhibitor | UD | Y | UD | 8 | Phase I | [89] |
| HER2-Targeted ADCs | ||||||||
| Name | Developer | Payload | Linker | Cleavable? | Conjugation | DAR | Clinical Stage | Ref. |
| T-DM1 | Genentech ImmunoGen | DM1 | Thioether | N | Lysine | 3.5 | Approved | [90,91,92,93,94] |
| T-DXd | Daichii Sankyo AstraZeneca | DXd | GGFG | Y | Interchain cysteines | 8 | Approved | [95,96,97,98,99,100,101,102] |
| RC48 | Remegen Biosciences Yantai Rongchang Biological Engineering | MMAE | mc-vc-PABC | Y | Interchain cysteines | 4 | Phase III | [103] |
| MRG002 | Shanghai Miracogen | MMAE | mc-vc-PABC | Y | Interchain cysteines | 3.8 | Phase III | [104] |
| DP303c | CSPC ZhongQi Pharmaceutical Technology | MMAE | NH2-PEG3-vc | Y | mTG site-specific | 2 | Phase III | [105] |
| FS-1502 | Iksuda Biotherapeutics | MMAF | β-glucuronide | Y | Prenyl transferase site-specific | 2 | Phase III | [106] |
| SYD985 | Byondis | seco-DUBA | vc | Y | Interchain cysteines | 2.8 | Completed Phase III | [107,108] |
| A166 | Klus Pharma | Duostatin-5 | vc | Y | Site-specific | 2 | Phase II | [109,110,111] |
| ZV0203 | Hangzhou Adcoris Biopharma | Duostatin-5 | vc | Y | UD | 2 | Phase I | [112,113] |
| ARX788 | Zheijang Medicine Ambrx | MMAF | Hydroxylamine-PEG4 | N | Unnatural amino acid | 1.9 | Phase II | [114,115,116] |
| HER3-Targeted ADCs | ||||||||
| Name | Developer | Payload | Linker | Cleavable? | Conjugation | DAR | Clinical Stage | Ref. |
| HER3-DXd | Daichii Sankyo Amgen | DXd | GGFG | Y | Interchain cysteines | 8 | Phase III | [117,118,119,120,121,122,123,124,125,126,127,128,129,130,131] |
| AMT-562 | Multitude Therapeutics | Exatecan | mc-va-T800 | Y | Site-specific | 8 | Phase I | [132] |
| DB-1310 | Duality Biologics | P1021 | Maleimide tetrapeptide | Y | UD | 8 | Phase I/IIa | [133] |
| YL202/BNT326 | MediLink Therapeutics | YL0010014 | Tripeptide | Y | UD | 8 | Phase II | [134,135] |
| Name | Developer | Targets | Structure | Strategy | Payload | Linker | Cleavable? | Conjugation | DAR | Clinical Stage | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BL-B01D1 | SystImmune Sichuan Baili Pharmaceutical | EGFR x HER3 | EGFR Fab x HER3 scFv | Fab-scFv | ED-04 | Tetrapeptide | Y | UD | 8 | Phase III | [136,137,138] |
| BCG019 | Biocytogen | EGFR x HER3 | EGFR Fab x HER3 Fab | Knobs-into-holes (RenLite) | MMAE BCPT02 | vc UD | Y Y | UD | 4 UD | Preclinical | [139] |
| M1231 | Merck Sutro Biopharma | EGFR x MUC1 | EGFR Fab x MUC1 scFv | SEED-antibody scaffold | SC209 | vc-PABA | Y | Site-specific, unnatural amino acid | 4 | Phase II | [140,141] |
| BSA01 | Biocytogen | EGFR x MUC1 | EGFR Fab x MUC1 Fab | Knobs-into-holes (RenLite) | MMAE | vc | Y | UD | 4 | Preclinical | [142,143] |
| AZD9592 | AstraZeneca | EGFR x c-MET | EGFR Fab x c-MET Fab | Knobs-into-holes (DuetMab) | AZ14170132 | Peptide-based | Y | UD | UD | Phase I | [144,145,146,147] |
| VBC101-F11 | VelaVigo | EGFR x c-MET | Nanobody-based | UD | MMAE | UD | UD | UD | 4 | Preclinical | [148] |
| DM001 | Biocytogen | EGFR x TROP2 | EGFR Fab x TROP2 Fab | Knobs-into-holes (RenLite) | MMAE | vc | Y | UD | 4 | IND submitted | [149] |
| ZW49 | Zymeworks | HER2 biparatopic | HER2 ECDII Fab x HER2 ECDIV scFv | UD | ZD02044 | Dipeptide | Y | Interchain cysteine | UD | Halted | [150,151,152,153,154,155] |
| MEDI4276 | AstraZeneca | HER2 biparatopic | HER2 ECDII Fab x HER2 ECDIV scFv | Fab-scFv | AZ13599185 | mc | Y | Site-specific cysteine | 4 | Phase I/II | [156,157,158] |
| JSKN-003 | Alphamab Oncology | HER2 biparatopic | HER ECDII Fab x HER2 ECDIV Fab | UD | Topo I inhibitor | Tetrapeptide | Y | Site-specific glycan | 4 | Phase III | [159,160,161] |
| 23V-MMAE | Shanghai Jiao Tong University Jecho Institute Co., Ltd. | HER2 x HER3 | HER2 Fab x HER3 Fab | MMAE | mc-vc-PAB | Dipeptide | Y | Interchain cysteine | 2.89 | Preclinical | [162] |
| YH012 | Biocytogen | HER2 x TROP2 | HER2 Fab x TROP2 Fab | Knobs-into-holes (RenLite) | MMAE | vc | Y | UD | 4 | Preclinical | [163,164] |
| BIO-201 | BiOneCure Therapeutics | HER2 x TROP2 | UD | UD | Topo I inhibitor | UD | Y | UD | UD | Preclinical | [165] |
| BCG022 | Biocytogen | HER3 x c-MET | HER3 Fab x c-MET Fab | Knobs-into-holes (RenLite) | MMAE BCPT02 | vc UD | Y Y | UD | 4 UD | Preclinical | [166] |
| DM002 | Biocytogen | HER3 x MUC1 | HER3 Fab x MUC1 Fab | Knobs-into-holes (RenLite) | MMAE | vc | Y | UD | 4 | Preclinical | [167] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
High, P.; Guernsey, C.; Subramanian, S.; Jacob, J.; Carmon, K.S. The Evolving Paradigm of Antibody–Drug Conjugates Targeting the ErbB/HER Family of Receptor Tyrosine Kinases. Pharmaceutics 2024, 16, 890. https://doi.org/10.3390/pharmaceutics16070890
High P, Guernsey C, Subramanian S, Jacob J, Carmon KS. The Evolving Paradigm of Antibody–Drug Conjugates Targeting the ErbB/HER Family of Receptor Tyrosine Kinases. Pharmaceutics. 2024; 16(7):890. https://doi.org/10.3390/pharmaceutics16070890
Chicago/Turabian StyleHigh, Peyton, Cara Guernsey, Shraddha Subramanian, Joan Jacob, and Kendra S. Carmon. 2024. "The Evolving Paradigm of Antibody–Drug Conjugates Targeting the ErbB/HER Family of Receptor Tyrosine Kinases" Pharmaceutics 16, no. 7: 890. https://doi.org/10.3390/pharmaceutics16070890
APA StyleHigh, P., Guernsey, C., Subramanian, S., Jacob, J., & Carmon, K. S. (2024). The Evolving Paradigm of Antibody–Drug Conjugates Targeting the ErbB/HER Family of Receptor Tyrosine Kinases. Pharmaceutics, 16(7), 890. https://doi.org/10.3390/pharmaceutics16070890