Abstract
Small interfering RNA (siRNA) therapeutics, characterized by high specificity, potency, and durability, hold great promise in the treatment of cancer and other diseases. However, the clinic implementation of siRNA therapeutics critically depends on the safe and on–demand delivery of siRNA to the target cells. Here, we reported a family of ferrocenyl amphiphilic dendrimers (Fc-AmDs) for on–demand delivery of siRNA in response to the high ROS content in cancer cells. These dendrimers bear ROS–sensitive ferrocene moieties in the hydrophobic components and positively chargeable poly(amidoamine) dendrons as the hydrophilic entities, possessing favorable safety profiles and ROS responsive properties. One of these ferrocenyl amphiphilic dendrimers, Fc-C8-AmD 8A, outperforms in siRNA delivery, benefiting from its optimal balance of hydrophobicity and hydrophilicity. Its ROS feature facilitates specific and efficient disassembly of its complex with siRNA in ROS–rich cancer cells for effective siRNA delivery and gene silencing. Moreover, Fc-C8-AmD 8A also integrates the features and beneficial properties of both lipid and dendrimer vectors. Therefore, it represents a novel on–demand delivery system for cancer cell–specific siRNA delivery. This work opens new perspectives for designing self–assembly nanosystems for on–demand drug delivery.
1. Introduction
RNA interference (RNAi), triggered by small interfering RNA (siRNA), is a sequence-specific post–transcriptional gene silencing process that provides a powerful approach to interfere with the function of any disease–associated protein in a highly selective manner [1,2]. Therefore, siRNAs have been emerging as promising therapeutic reagents for the treatment of inherited and acquired diseases [2]. So far, six siRNA drugs have been approved for marketing worldwide [3]. A growing number of clinical trials of RNAi–based solid tumors treatment further demonstrate the potential of siRNA therapeutics in treating cancer patients by specifically turning off gene expression that promotes cancer cell survival, migration, and tumorigenicity [4]. However, the therapeutic outcomes extremely rely on the safe and efficient delivery of siRNA therapeutics in target cells, as they are susceptible to nuclease degradation and renal clearance [5]. Moreover, siRNA molecules are hydrophilic biomacromolecules with strongly negative charge, which prevents them from spontaneously crossing cell membranes [6,7]. During the past decades, tremendous efforts have been dedicated to exploit delivery vehicles for siRNA therapeutics [5,6,7]. However, there is still an unmet need for the specific delivery of siRNA therapeutics to disease–related cells [7,8]. Given this, the development of on–demand delivery vehicles capable of transporting siRNA specifically to cancer cells is highly desirable and also of paramount importance for RNAi–based cancer therapy.
Lipids and polymers are the two most advanced vectors for siRNA therapeutics in clinical and preclinical studies [9]. For instance, the first siRNA drug approved by FDA, Onpattro [10], is a lipid nanoparticle formulation, while the first clinic trial of siRNA therapeutics is a polymer–based formulation [11]. Dendrimers, a special type of polymers, are emerging as promising delivery vehicles for siRNA therapeutics by virtue of their well-defined molecular structure and unique multivalent cooperativity [12,13]. In particular, amphiphilic dendrimers, a kind of lipid/dendrimer hybrid with judiciously designed hydrophilic and hydrophobic components, exhibit excellent performance on siRNA delivery thanks to the combined advantageous of lipid and polymer vectors as well as unique characteristics of dendrimer vectors [14,15]. We and others have recently established several types of amphiphilic dendrimers, including amphiphilic poly(amidoamine) (PAMAM) dendrimers [16,17], amphiphilic peptide dendrimers [18], amphiphilic poly(aminoester) dendrimers [19], and amphiphilic polyglycerol dendrimers [20], all of which possess the excellent capability of delivering siRNA therapeutics for the treatment of different diseases. Therefore, amphiphilic dendrimers are able to be a promising platform for elaborating on–demand vectors for cancer cell–specific siRNA delivery.
The tumor microenvironment has a variety of atypical hallmarks, such as hypoxia, abnormal redox environment, low pH, upregulated secretion of specific enzymes, overexpression of inflammatory meditators, etc. [21]. Notably, the hypoxia environment within the tumor tissues leads to the continuous production of reactive oxygen species (ROS) within cancer cells [22,23]. Thus, high levels of ROS are one of the intrinsic characteristics specifically associated with cancer cells [23,24]. Harnessing this factor to exploit on–demand vectors that are capable of ROS–triggered siRNA release in cancer cells constitute a promising strategy to achieve cancer cell–specific siRNA delivery for RNAi–based cancer therapy.
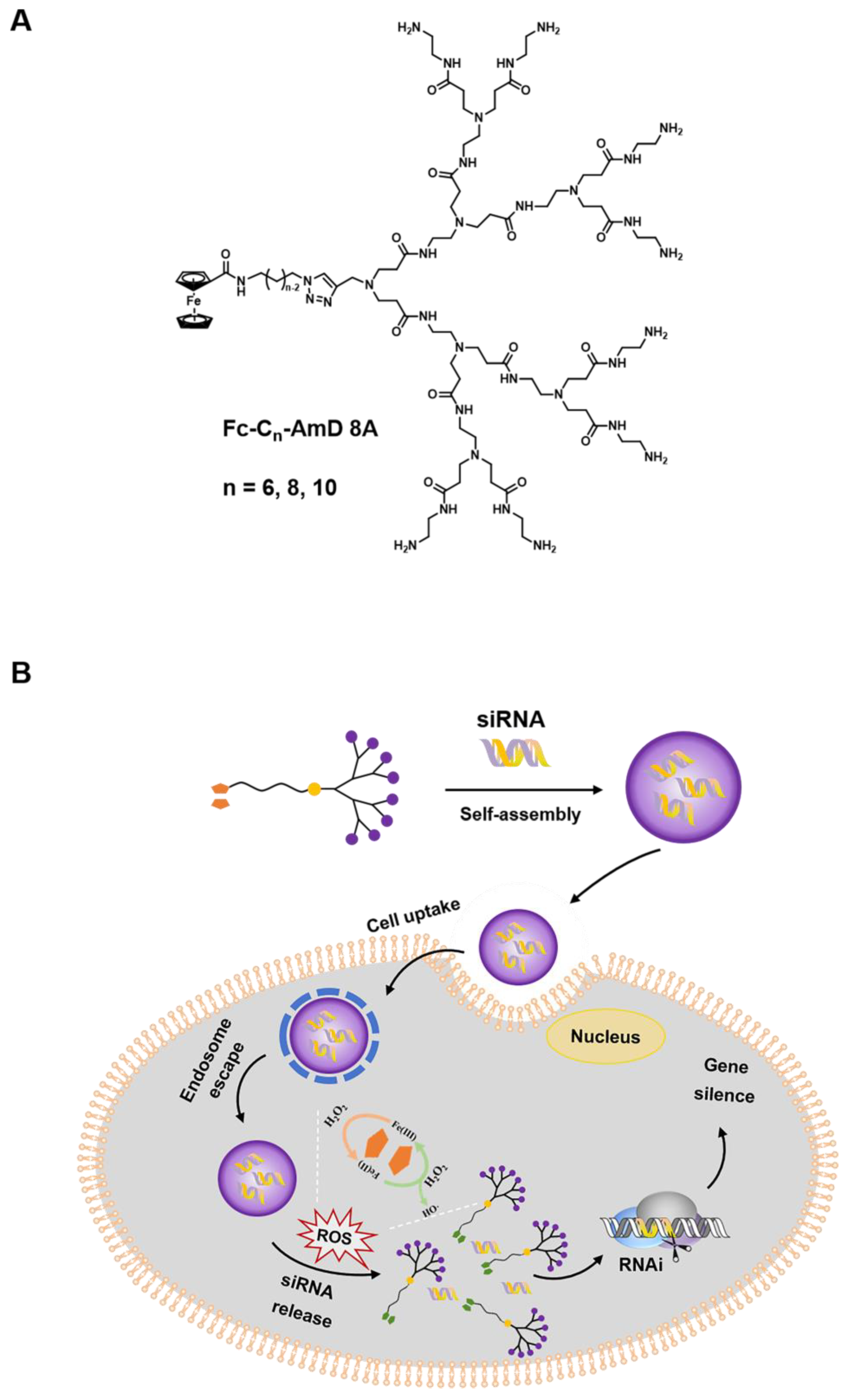
Herein, we report a class of ROS–responsive amphiphilic dendrimers (Fc-AmDs) for on–demand siRNA delivery upon the exposure to the elevated ROS level in cancer cells (Scheme 1). These dendrimers are composed of hydrophilic PAMAM dendron and hydrophobic alkyl chains with different chain lengths containing ROS–sensitive ferrocene groups (Scheme 1A). The hydrophilic PAMAM dendrons, which feature with amine groups and are positively charged at physiological pH, have been designed to interact with the negatively charged siRNA through electrostatic interactions. Meanwhile, the tertiary amine groups presented in PAMAM dendrons can be further protonated in acidic environments (e.g., pH 5.0 in endosome), thus promoting endosome escape via the proton sponge effect [15,25,26]. Moreover, the hydrophobic alkyl chains with ferrocene groups endow Fc-AmDs with good assembly properties. Particularly, the hydrophobic ferrocene is converted to the hydrophilic ferrocenium cation triggered by elevated ROS in cancer cells [27,28]. This hydrophobic–to–hydrophilic transition is anticipated to disrupt the hydrophobic/hydrophilic balance of Fc-AmD nanoassemblies and cause their complete disintegration, eventually initiating siRNA release for effective gene silencing within cancer cells (Scheme 1B).
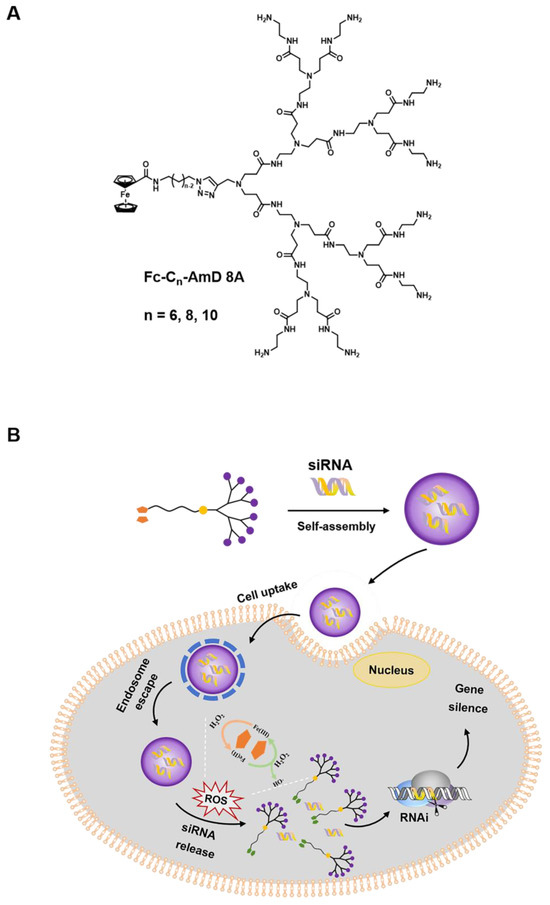
Scheme 1.
Schematic presentation of ferrocenyl amphiphilic dendrimer-mediated siRNA delivery. (A) Chemical structure of the Fc-AmDs. (B) Cartoon illustration of Fc-AmD–mediated siRNA delivery.
2. Materials and Methods
2.1. Materials
The human AKT2 siRNA (forward primer: 5′–GCUCCUUCAUUGGGUACAAdTdT–3′; reverse primer: 5′–UAAUGUGCCCGUCCUUGUCdTdT–3′) and GAPDH (forward primer: 5′–AATCCCATCACCATCTTCCA–3′; reverse primer: 5′–TGGACTCCACGACGTACTCA–3′) were purchased from GenScript Biotech Corp. (Nanjing, China). The scramble siRNA (forward primer: 5′–ACGUGACACGUUCGGAGAAdTdT–3′; reverse primer: 5′–UUCUCCGAACGUGUCACGUdTdT–3′) was purchased from Biosyntech Co., Ltd. (Suzhou, China). All the biochemical reagents are suitable for cell culture and were purchased from Sigma–Aldrich (Shanghai, China), Servicebio (Wuhan, China), Cell signaling Technology (Danvers, MA, USA), Vazyme Biotech Co., Ltd. (Nanjing, China) and Thermo Fisher Scientific Inc. (Carlsbad, CA, USA). All the reagents were used without any further purification from commercial sources.
All the chemical reagents are analytical grade and were purchased from Aladdin Ltd. (Shanghai, China), Energy Chemical Ltd. (Shanghai, China), J&K Scientific (Beijing, China) or Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Methyl acrylate and ethylenediamine were distilled before use. The compounds 8E and 8A were synthesized according to the reported literature [17]. All the other chemicals were purchased from commercial sources and used as received. All the solvents used were freshly distilled. Dialysis tubes were purchased from Yuanye Bio–Technology Co., Ltd. (Shanghai, China).
A Bruker AC (300 MHz) spectrometer was employed to determine the 1H NMR and 13C NMR spectra of compounds. All the chemical shifts were recorded in parts per million (δ, ppm) with reference to tetramethylsilane (TMS). Mass spectra were measured by using the Agilent 6230 time–of–flight LC/MS (LC/TOF) system. The infrared spectra were recorded by using a Bruker ALPHA FT–IR spectrophotometer (Bruker, Ettlingen, Germany) in the range of 400–4000 cm−1.
2.2. Synthesis and Characterization of Fc-AmDs
The synthetic protocols of Fc-AmDs are detailed in Supplementary Materials.
Fc-C6-AmD 8A: 1H NMR (300 MHz, CD3OD/CDCl3) δ 7.89 (s, 1H), 4.79 (br, 2H), 4.47–4.34 (m, 4H), 4.18 (s, 5H), 3.81 (s, 2H), 3.50–3.15 (m, 30H), 2.96–2.67 (m, 44H), 2.66–2.50 (m, 12H), 2.49–2.25 (m, 28H), 2.05–1.89 (m, 2H), 1.67–1.55 (m, 2H), 1.52–1.34 (m, 4H). 13C NMR (75 MHz, CD3OD/CDCl3) δ 174.04, 173.36, 173.18, 171.93, 143.24, 123.85, 75.58, 70.43, 69.54, 68.05, 52.12, 49.81, 49.25,47.87, 40.39, 39.26, 37.26, 33.47, 30.05, 29.48, 29.01, 28.79, 26.70, 26.23. ESI–HRMS (m/z): calcd for C90H167FeN33O15, [M+2H]2+ 1004.6424, found 1004.6393. HPLC (RT = 16.1 min). IR (cm−1): υ 1630.50 and 1542.97 (–NH(CO)–).
Fc-C8-AmD 8A: 1H NMR (300 MHz, CD3OD/CDCl3) δ 7.87 (s, 1H), 4.79 (br, 2H), 4.46–4.33 (m, 4H), 4.18 (s, 5H), 3.82 (s, 2H), 3.47–3.14 (m, 30H), 2.98–2.69 (m, 44H), 2.68–2.53 (m, 12H), 2.49–2.28 (m, 28H), 1.98–1.86 (m, 2H), 1.67–1.52 (m, 2H), 1.45–1.27 (m, 8H). 13C NMR (75 MHz, CD3OD/CDCl3) δ 173.86, 173.36, 173.17, 171.93, 143.35, 123.84, 75.56, 70.42, 69.52, 68.05, 52.12, 49.80, 49.05, 47.68, 41.26, 40.61, 39.10, 37.26, 33.46, 30.00, 29.31, 26.13, 25.97. ESI–HRMS (m/z): calcd for C92H171FeN33O15, [M+2H]2+ 1018.6581, found 1018.6541. HPLC (RT = 19.1 min). IR (cm−1): υ 1631.17 and 1541.60 (–NH(CO)–).
Fc-C10-AmD 8A: 1H NMR (300 MHz, CD3OD/CDCl3) δ 7.87 (s, 1H), 4.79(br, 2H), 4.44–4.32 (m, 4H), 4.19 (s, 5H), 3.82 (s, 2H), 3.45–3.16 (m, 30H), 2.93–2.68 (m, 44H), 2.66–2.53 (m, 12H), 2.50–2.16 (m, 28H), 1.97–1.85 (m, 2H), 1.66–1.53 (m, 2H), 1.46–1.23(m, 12H). 13C NMR (75 MHz, CD3OD/CDCl3): δ 175.52, 174.94, 145.36, 125.29, 72.07, 71.26, 69.78, 54.08, 51.77, 43.41, 42.54, 42.54, 40.90, 39.13, 35.48, 35.25, 31.68, 31.21, 31.04, 31.04, 30.65, 30.43, 28.40, 27.71. ESI-HRMS (m/z): calcd for C94H175FeN33O15, [M+2H]2+ 1032.6737, found 1032.6722. HPLC (RT = 20.2 min). IR (cm−1): υ 1630.66 and 1542.24 (–NH(CO)–).
2.3. HPLC of Fc-AmDs
A high–performance liquid chromatography (HPLC) analysis was conducted on a Waters Empower system (Waters 1525, binary HPLC pump, Waters Corp., The Capricorn, Singapore) equipped with a photodiode array detector (Waters 2998, Waters Corp., The Capricorn, Singapore) and a SinoChrom C8 column (5 μm, 4.6 mm × 250 mm). A gradient elution mode of 10–50% acetonitrile in water was employed over 40 min. The mobile phases consisted of acetonitrile and water, both of which contained 0.04% TFA. The flow rate was 0.8 mL/min, with an injection volume of 20 μL. Peaks were detected at 210 nm. The retention times (RTs) were recorded in minutes. All reagents were HPLC–gradient grade and purchased from Anhui Tedia High Purity Solvents Co., Ltd. (Anqing, China) and Aladdin Ltd. (Shanghai, China).
2.4. Critical Aggregation Concentration (CAC)
CAC was determined using pyrene as a fluorescent probe. The solutions of Fc-AmDs at different concentrations varying from 1.0 × 10−7 to 2.0 × 10−4 mol/L were prepared, and the final pyrene concentration was 3 × 10−7 mol/L in water. The solutions were sonicated for 30 min and kept at room temperature for 2 h to promote the micelle formation prior to fluorescence measurement. Fluorescence spectra were recorded at the emission wavelength of 334 nm on FL8500 fluorescence spectrophotometer at room temperature. The excitation bandwidth was 20 nm, and the emission bandwidth was 1 nm. The fluorescence intensity ratio of I373/I384 was analyzed as a function of dendrimer concentration.
2.5. Potentiometric pH Titration
The solution of Fc-AmDs (5 mg in 5 mL) was adjusted to a pH of around 2–3 with 1.0 M HCl. The pH titration was performed with 0.05 M NaOH using METTLER TOLEDO FE28–Bio pH meter (Mettler–Toledo International Inc., Shanghai, China).
2.6. ROS Response Measurement of Fc-AmDs
The oxidation process of Fc-AmDs was detected using UV–Vis spectrophotometry. Fc-AmDs were added to H2O2–containing acetate buffer solution and incubated at 37 °C with gentle shaking. Then, the UV absorption of the solution was measured by UV–1900i UV–Vis spectrophotometer (Shimadzu, Kyoto, Japan).
The hydroxyl radical (·OH) produced by Fenton–like reaction was detected using TMB. Briefly, Fc-AmDs and TMB were added to H2O2–containing acetate buffer solution and incubated at 37 °C with gentle shaking. Then, the UV absorption of the mixed solution was measured by UV–Vis spectrophotometry.
2.7. Dynamic Light Scattering (DLS)
2.7.1. DLS Analysis of Fc-C8-AmD 8A
A 200 μM solution of Fc-C8-AmD 8A was prepared by dissolving the compound in ultrapure water. The sizes of Fc-C8-AmD 8A solutions were measured using a NanoBrookOmni (Brookhaven, Holtsville, NY, USA) equipped with a standard 633 nm laser at 25 °C.
2.7.2. DLS Analysis of siRNA/Fc-C8-AmD 8A Nanoparticles
The solution of siRNA/Fc-C8-AmD 8A complexes was prepared via mixing the siRNA solution and Fc-C8-AmD 8A solution at N/P ratio of 10, resulting in a final concentration of 1.0 μM for the siRNA. Ultrapure water was used for the solvent. The sizes of the siRNA/Fc-C8-AmD 8A complexes solutions were measured by the same DLS method as described above.
2.7.3. DLS Analysis of the ROS–Triggered Disassembly of siRNA/Fc-C8-AmD 8A Nanoparticles
The solutions of siRNA/Fc-C8-AmD 8A complexes were prepared as described above. PB solution (pH 5.0) was used for the solvent. The sizes of the siRNA/Fc-C8-AmD 8A complexes in the absence and presence of H2O2 were measured at different time points (0, 1, 2, 4, 6, 8, 12, and 24 h) using the same DLS method as described above.
2.8. Gel Retardation Analysis
The solutions of Fc-AmDs and siRNA were mixed at different N/P ratios ranging from 0.2 to 10 and incubated at 37 °C for 30 min. The final concentration of siRNA in each sample was 200 ng/well. The siRNA/Fc-AmDs complexes were analyzed via electrophoretic mobility–shift assays conducted using 2% agarose gels in standard TAE buffer for 15 min. Following staining with GoodView nucleic acid dyes, the siRNA bands were imaged using an automatic chemiluminescence imaging system (5200 Multi) (Tanon, Shanghai, China).
2.9. Stability of siRNA/Fc-C8-AmD 8A Complex against RNase
An aliquot containing 200 ng of siRNA and the indicated amounts of Fc-C8-AmD 8A solution at N/P ratio of 10 was incubated in the presence of RNase A (1.0 μg/mL) at 37 °C for different times (0, 10, 30, 60, 90, and 120 min). Afterwards, aliquots (10.0 μL) of the corresponding solution were added 1.0 μL of 2% SDS solution on ice. The mixtures were analyzed by gel retardation as described above. Naked siRNA served as a control.
2.10. RNA Dissociation Assay
A solution containing 0.1 μg ethidium bromide (EB), 1.32 μg siRNA, and an appropriate amount of Fc-AmDs at a N/P ratio of 10 in PB buffer at pH 5.0 was added to the black 96–well plate and incubated at 25 °C for 30 min. Then, to 60 μL of the above siRNA/Fc-AmDs complexes was added the 40 μL of heparin solution in PB at different concentrations varying from 0 to 50 U/mL to achieve a total volume of 100 μL. After a further incubation of 30 min at 25 °C, the mixture was measured at 590 nm using a Cytation5 Microplate Reader (BioTek, Winooski, VT, USA) with 360 nm fluorescence excitation. The fluorescence values were normalized to wells containing EB siRNA solution only. All samples were assayed in triplicate.
2.11. Cell Culture
Mouse fibroblast L929 cells, madin-darby canine kidney MDCK cells, human normal liver L02 cells, and human ovarian cancer SKOV–3 cells were purchased from Tongpai Biotechnology Co., Ltd. (Shanghai, China). L929 cells were maintained in DMEM (Hyclone, Logan, UT, USA), supplemented with 10% FBS. MDCK cells were maintained in MEM (Hyclone, Logan, UT, USA) with 10% FBS. Human ovarian cancer SKOV–3 cells were cultured in McCOY’S 5A (Hyclone, Logan, UT, USA) with 10% FBS. L02 cells were maintained in RMPI–1640 (Hyclone, Logan, UT, USA) with 10% FBS. All cells were cultured in an incubator with a humidified environment of 5% CO2 and a constant temperature of 37 °C.
2.12. MTT(3–(4,5–Dimethylthiazol–2–yl)–2,5–Diphenyltetrazolium Bromide) Assay
Cancer cells (SKOV–3 cells) and normal cells (MDCK cells, L929 cells, and L02 cells) (8 × 103) were seeded in 96–well plates and cultured for 24 h. Cells were then treated with Fc-AmDs at different concentrations, ranging from 0.25 to 50 μM, for 8 h. After transfection, the medium was replaced with fresh medium. Then, 48 h later, the cells were treated with MTT solution and incubated for a further 4 h. After removing the solution, the cells were re–suspended in DMSO. Cytation5 (BioTek, Winooski, VT, USA) was used to measure the optical density (OD) of DMSO solutions, which was read at 570 nm. The viability of cells was assessed by the difference between the OD values of treated and untreated cells. All samples were assayed in triplicate.
2.13. Flow Cytometry
One day prior to the transfection, SKOV–3 cells were seeded at 8.0 × 104 cells/well in a 24–well plate. The cells were incubated with Cy5–labeled siRNA/Fc-C8-AmD 8A complex (50 nM Cy5–siRNA, N/P ratio 10) for 1, 2, 4, 6, and 8 h. After three washes with cold PBS, the cells were analyzed by flow cytometry (Attune NxT, Thermo Fisher Scientific). All samples were assayed in triplicate.
2.14. Confocal Microscopy
One day prior to the transfection, SKOV–3 cells were seeded in 2.5 dishes (8.0 × 104 cells/dish). The cells were incubated with Cy5–labeled siRNA/Fc-C8-AmD 8A complex (50 nM Cy5–siRNA, N/P ratio of 10) for 1, 2, 4, 6, and 8 h at 37 °C. After three washes with cold PBS, the cells were stained by PBS mixed with Hoechst33342 (10 μg/mL) and Lyso-Tracker Red (0.10 μM) for 10 min at 37 °C. Images were acquired with a Zeiss LSM880 Meta laser scanning confocal microscope (Carl Zeiss, Jena, Germany) using ZEN2.3 pro software (Carl Zeiss GmbH).
2.15. ROS Level Measurement
SKOV–3 and NAC–treated SKOV–3 cells (1.5 × 105 cells/group) were treated with the CellROX® orange Reagent (Thermo Fisher Scientific, Carlsbad, CA, USA) at a final concentration of 1.0 μM and incubated at 37 °C for 30 min. Following three washes with PBS, the mean fluorescence intensity of the cells was quantified by flow cytometry (Attune NxT, Thermo Fisher Scientific). Each assay was performed in triplicate.
2.16. In Vitro Transfection
SKOV–3 cells and NAC–pretreated SKOV–3 cells were seeded in 6–well plates and grown for 24 h. The solutions of siAKT2/Fc-AmDs complexes with 50 nM siRNA at an N/P ratio of 10 were prepared before transfection. After incubation with siAKT2/Fc-AmDs complexes for 8 h, the transfection mixture was replaced with the complete medium. And then, the cells were incubated for an additional 48 h for qRT–PCR assay and 72 h for Western blot assay.
2.16.1. Effect of Dioleoylphosphatidylethanolamine (DOPE)
The effect of DOPE was assessed by the transfection experiments as described above. Before transfection, the solutions of siAKT2/Fc-C8-AmD 8A complexes were prepared with DOPE at mole ratio of 1.0 (DOPE/dendrimer).
2.16.2. Effect of Bafilomycin A1
After preincubation of SKOV–3 cells with 200 nM bafilomycin A1 at 37 °C for 1 h, the effect of bafilomycin A1 was assessed by the transfection experiments as described above.
2.17. Quantitative Real-Time (qRT)–PCR Analysis
The transfection experiments were performed in SKOV–3 cells as described above. After isolation with the Trizol method (Vazyme Biotech Co., Ltd., Nanjing, China), the total RNAs of SKOV–3 cells were reverse–transcribed by a Reverse Transcription Kit (Vazyme Biotech Co., Ltd., Nanjing, China). The expression of AKT2 was analyzed by quantitative real–time PCR performed on the QuantStudio3™ real–time PCR System (Applied Bisystems, Thermo Fisher Scientific) using 2 × SYBR Green (Vazyme Biotech Co., Ltd., Nanjing, China). The expression of GAPDH was used for normalization of the qPCR data.
2.18. Western Blot Analysis
Samples containing equal amounts of protein (15 μg) from SKOV–3 cells were separated by SDS–PAGE gradient gel and transferred to the PVDF membrane after electrophoresis. After being blocked in 5% skimmed milk, the PVDF membranes were incubated with anti–human vinculin rabbit polyclonal antibody (Cell signaling Technology, Danvers, MA, USA, 1:5000) or anti–human AKT2 rabbit polyclonal antibody (Cell signaling Technology, Danvers, MA, USA, 1:1000) at 4 °C for 12 h. Then, the membranes were washed and incubated with anti–rabbit or anti-mouse monoclonal secondary antibodies (Invitrogen, Boston, MA, USA, 1:5000) for 2 h at 25 °C. Specific proteins were recorded by an enhanced-chemiluminescence Western blotting analysis system (Tanon, Shanghai, China).
2.19. Statistical Tests
All data are presented as mean ± SD unless otherwise indicated. One–way analysis of variance (ANOVA), two–way ANOVA, unpaired Student’s t-test or Mann–Whitney test (GraphPad Prism 8.01) were used for statistical analysis. A p value < 0.05 was considered statistically significant, whereby all significant values in various figures are indicated as follows: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001.
3. Results and Discussions
3.1. Ferrocenyl Amphiphilic Dendrimer Synthesis
Ferrocenyl amphiphilic dendrimers, denoted as Fc-AmDs, were prepared according to the synthetic routes shown in Scheme 2 and Scheme S1. Briefly, haloalkanols or dihaloalkanes were used as the starting materials and followed by Gabriel amine synthesis, azidation, and amidation with ferrocenecarboxylic acids to build the ferrocene and azido–bearing hydrophobic alkyl chains Fc-Cn-N3 with different chain lengths (Scheme S1). Then, the obtained Fc-Cn-N3 were coupled with the alkynyl–bearing PAMAM dendron 8E by the copper(I)–catalyzed azide–alkyne cycloaddition (CuAAC) click reaction to generate the ester–terminated dendrimer conjugates Fc-Cn-AmD 8E, respectively. These conjugates were further subjected to amidation with ethylenediamine to yield the desired ferrocenyl amphiphilic dendrimers Fc-Cn-AmD 8A (n = 6, 8, 10, Scheme 2). The structural integrities and purities of all the obtained dendrimers were characterized and confirmed by 1H–NMR, 13C–NMR, electrospray high–resolution mass spectrometry (ESI–HRMS), infrared spectrum (IR), and high–performance liquid chromatography (HPLC) (Figures S1–S3 and Table S1). Moreover, all established dendrimers Fc-Cn-AmD 8A harbor tertiary amines in the interior and primary amines at the surface, with pKa of about 5 and 9, respectively, as revealed by pH titration (Figure S4). Therefore, the primary amines in these dendrimers are able to protonate at physiological pH 7.4, while the tertiary amines can be further protonated under acidic conditions (pH 5.0), implying that they have a buffering effect [29].
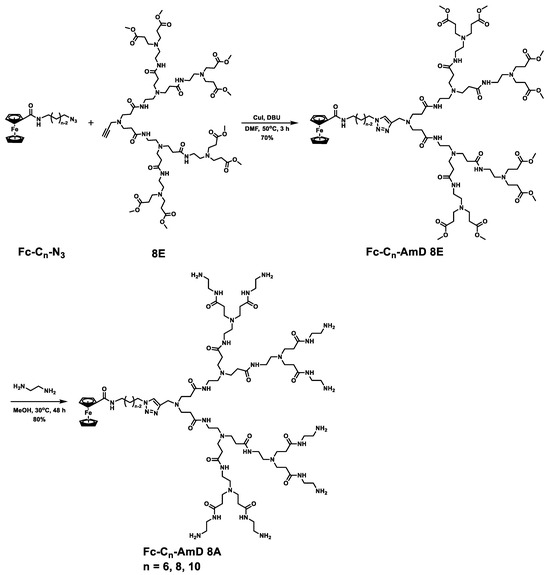
Scheme 2.
Synthesis routes of Fc-AmDs.
3.2. ROS Response of Fc-AmDs
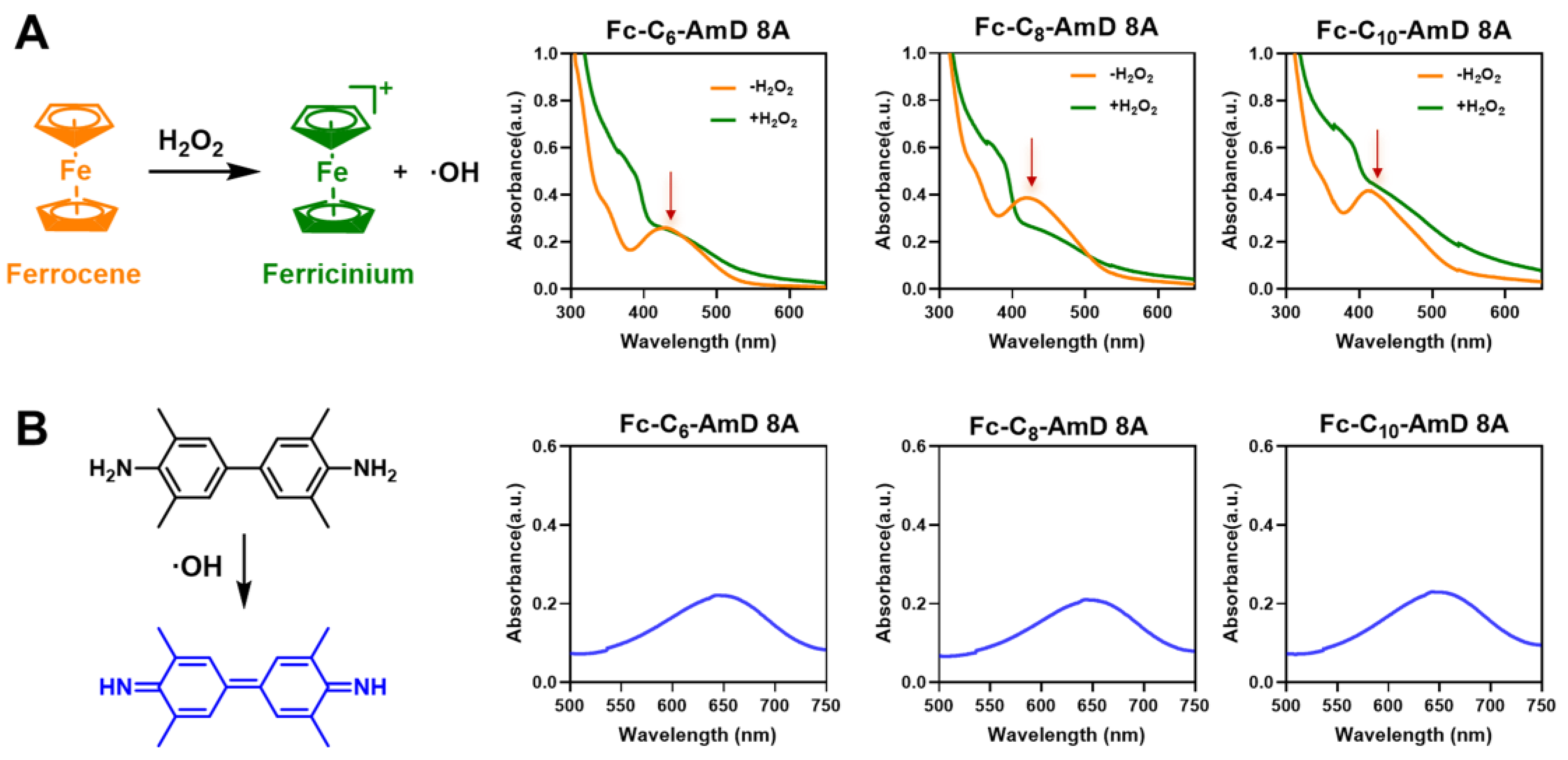
After obtaining ferrocenyl amphiphilic dendrimers (Fc-AmDs), we wished to study their ROS–responsive characteristics under simulated ROS conditions (in the presence of H2O2). As a typical Fenton reagent, the ferrocenyl group is prone to a Fenton–like reaction upon treatment with H2O2, which generates oxidized ferrocenium and hydroxyl radicals (·OH) [27]. Thus, we incubated Fc-AmDs with H2O2 and found that the characteristic UV absorptions (430–450 nm) originating from the ferrocenyl groups rapidly disappeared (Figure 1A). This result indicated that the ferrocene moieties in Fc-AmDs indeed undergo a Fenton–like reaction and change into their oxidative forms following exposure to H2O2. Moreover, hydroxyl radicals (·OH) produced by the Fenton reaction can be detected by 3,3′,5,5′–tetramethylbenzidine (TMB), as it can be oxidized by hydroxyl radicals (·OH) to oxTMB, which has a characteristic UV absorption at around 650 nm [30]. Therefore, we added TMB after exposing Fc-AmDs to H2O2. The appearance of the characteristic UV absorptions from oxTMB at around 650 nm further confirmed the production of ·OH during the oxidation of ferrocenyl groups in Fc-AmDs (Figure 1B and Figure S5). These findings suggest that the ferrocene–bearing Fc-AmDs indeed can undergo Fenton–like reactions under ROS conditions and, hence, possess the potential for on–demand drug delivery in response to ROS.

Figure 1.
Characterization of the ROS–responsive properties of Fc-AmDs. (A) UV–vis absorption spectra of Fc-AmDs before and after hydrogen peroxide (H2O2) oxidation; (B) the hydroxyl radicals produced by the Fenton reaction of Fc-AmDs with H2O2 using TMB by UV–vis spectrophotometer.
3.3. Best Performance of Fc-AmDs in siRNA Delivery and the Underlying Rationale
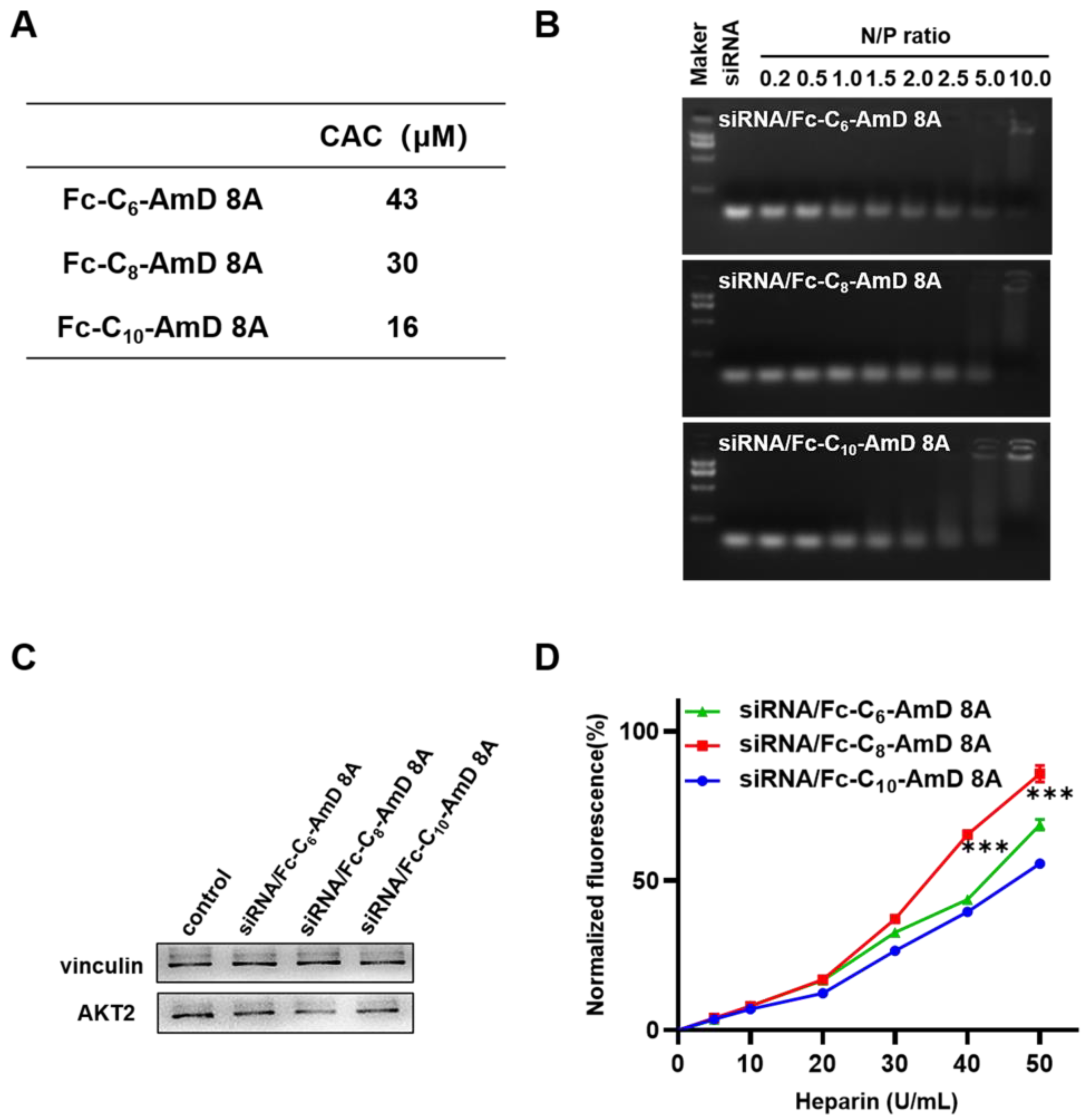
The obtained ferrocenyl amphiphilic dendrimers, Fc-AmDs, are speculated to be able to self–assemble in aqueous solution by virtue of their amphiphilic nature. Therefore, we investigated the self–assembly behavior of Fc-AmDs by measuring the value of their critical aggregation concentration (CAC) using hydrophobic fluorescent probe pyrene. The detected CAC values of Fc-C6-AmD 8A, Fc-C8-AmD 8A, and Fc-C10-AmD 8A were 43, 30, and 16 μmol/L, respectively (Figure 2A and Figure S6), confirming the inverse relationship between the CAC values of Fc-AmDs 8A and the length of their hydrophobic portions, i.e., Fc-C10-AmD 8A bearing the longest hydrophobic chain self-assembles and packs most efficiently among these three dendrimers.
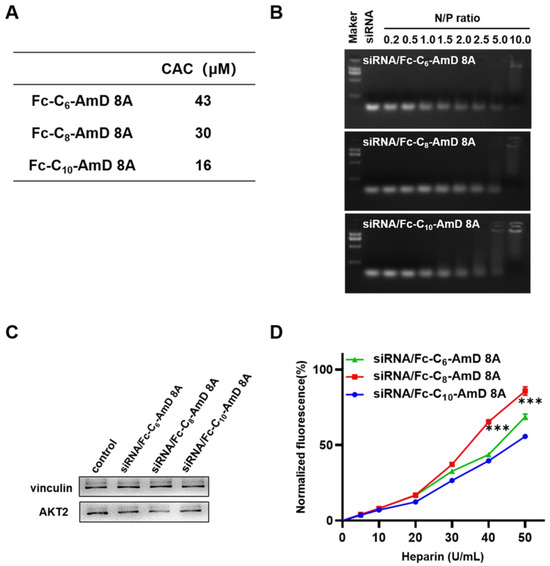
Figure 2.
(A) The CAC values of the Fc-AmDs were determined using fluorescent probe pyrene. (B) Gel retardation assay of siRNA with Fc-AmDs at N/P ratios ranging from 0.2 to 10 (200 ng siRNA per well). (C) AKT2 protein expression on SKOV–3 cells determined by Western blotting after treating with siAKT2/Fc-AmDs complexes (50 nM siRNA, N/P ratio of 10). (D) siRNA release from the siRNA/Fc-AmDs complexes was determined by heparin replacement assay (1.32 μg siRNA per well, N/P ratio of 10). *** p ≤ 0.001 (mean ± SD, n = 3).
We next examined their ability to bind siRNA at varying N/P ratios using agarose gel electrophoresis migration assays. As shown in Figure 2B, all three Fc-AmDs can efficiently bind siRNA and completely retard the migration of siRNA at an N/P ratio of 10, while Fc-C10-AmD 8A exhibited relatively better binding ability than other two dendrimers and started to retard the migration of siRNA from an N/P ratio of 2.0. These results denoted that Fc-C10-AmD 8A, with the best self–assembly capability, achieved siRNA binding most efficiently.
Prior to assessing the siRNA delivery capacity of Fc-AmDs, we first examined their safety, which is an important parameter for developing siRNA delivery vectors. The 3– (4,5–dimethylthiazol–2–yl)–2,5–diphenyltetrazolium bromide (MTT) assay was used to detect metabolic toxicity by measuring cell viability. As shown in Figure S7 and Table S2, all Fc-AmDs exhibited no significant influence on the proliferation of all the tested cell lines, including mouse fibroblast L929 cells, human–derived normal liver L02 cells, Madin–Darby canine kidney cells, and ovarian cancer SKOV–3 cells. These results indeed confirmed that all the three Fc-AmDs are devoid of any notable toxicity.
Encouraged by the favorable safety profile of Fc-AmDs, we then evaluated their ability to deliver siRNA targeting AKT2 in SKOV–3 cells. AKT2 is a proto–oncogene involved in the PI3K/AKT/mTOR signaling pathway, and its amplification contributes to tumorigenesis, cancer cell proliferation, invasion, metastasis and angiogenesis, even to protecting cancer cells from drug–induced apoptosis [31,32]. Therefore, AKT2 is recognized as an important therapeutic target in ovarian cancer. Among the ferrocenyl amphiphilic dendrimers, dendrimer Fc-C8-AmD 8A exhibited effective gene silencing with AKT2 siRNA (siAKT2), whereas no significant gene silencing was observed after siAKT2 delivered by the other two ferrocenyl amphiphilic dendrimers carrying the shortest or longest alkyl chains (Fc-C6-AmD 8A or Fc-C10-AmD 8A) (Figure 2C and Figure S8).
In order to understand why Fc-C8-AmD 8A performed better in siRNA delivery than the other two Fc-AmDs, we further studied the release of siRNA from the corresponding siRNA/dendrimer complexes using the heparin replacement assay. Heparin is a kind of highly negatively charged polysaccharide that can compete with siRNA for binding to dendrimers. As the concentration of heparin increased, the siRNA molecules dissociated more efficiently from their complex with Fc-C8-AmD 8A than from their complexes formed with the two other dendrimers (Fc-C6-AmD 8A and Fc-C10-AmD 8A) (Figure 2D). This finding confirmed that the siRNA release process is indeed more efficient for siRNA/Fc-C8-AmD 8A than for siRNA/Fc-C6-AmD 8A and siRNA/Fc-C10-AmD 8A.
Further investigations showed that the average sizes of empty Fc-C8-AmD 8A and siRNA/Fc-C8-AmD 8A complexes measured by dynamic light scattering were approximately 100 and 70 nm, respectively. The smaller size distribution of the siRNA/Fc-C8-AmD 8A complexes (Figure S9) indicated that the Fc-C8-AmD 8A was able to compact siRNA into stable nanoparticles. Such a nanoscale complex indeed can effectively protect siRNA from enzymatic degradation (Figure S10), facilitate efficient uptake of the siRNA cargos by cells (Figure S11), and release the loaded siRNA at its final destination.
Collectively, these findings demonstrate that the proper balance of the hydrophobic chain length and the hydrophilic dendritic component of Fc-AmDs is crucial for their self-assembly behavior, which in turn plays an important role in their performance in siRNA delivery. One of these amphiphilic dendrimers, Fc-C8-AmD 8A, possesses an optimal balance of hydrophobicity and hydrophilicity and is, therefore, able to generate stable nanosized assemblies for optimal siRNA binding and the most efficient siRNA release. These properties make Fc-C8-AmD 8A outperform in siRNA delivery and gene silencing compared to the two other dendrimers (Fc-C6-AmD 8A and Fc-C10-AmD 8A).
3.4. Specific ROS–Responsive siRNA Delivery Mediated by Fc-C8-AmD 8A
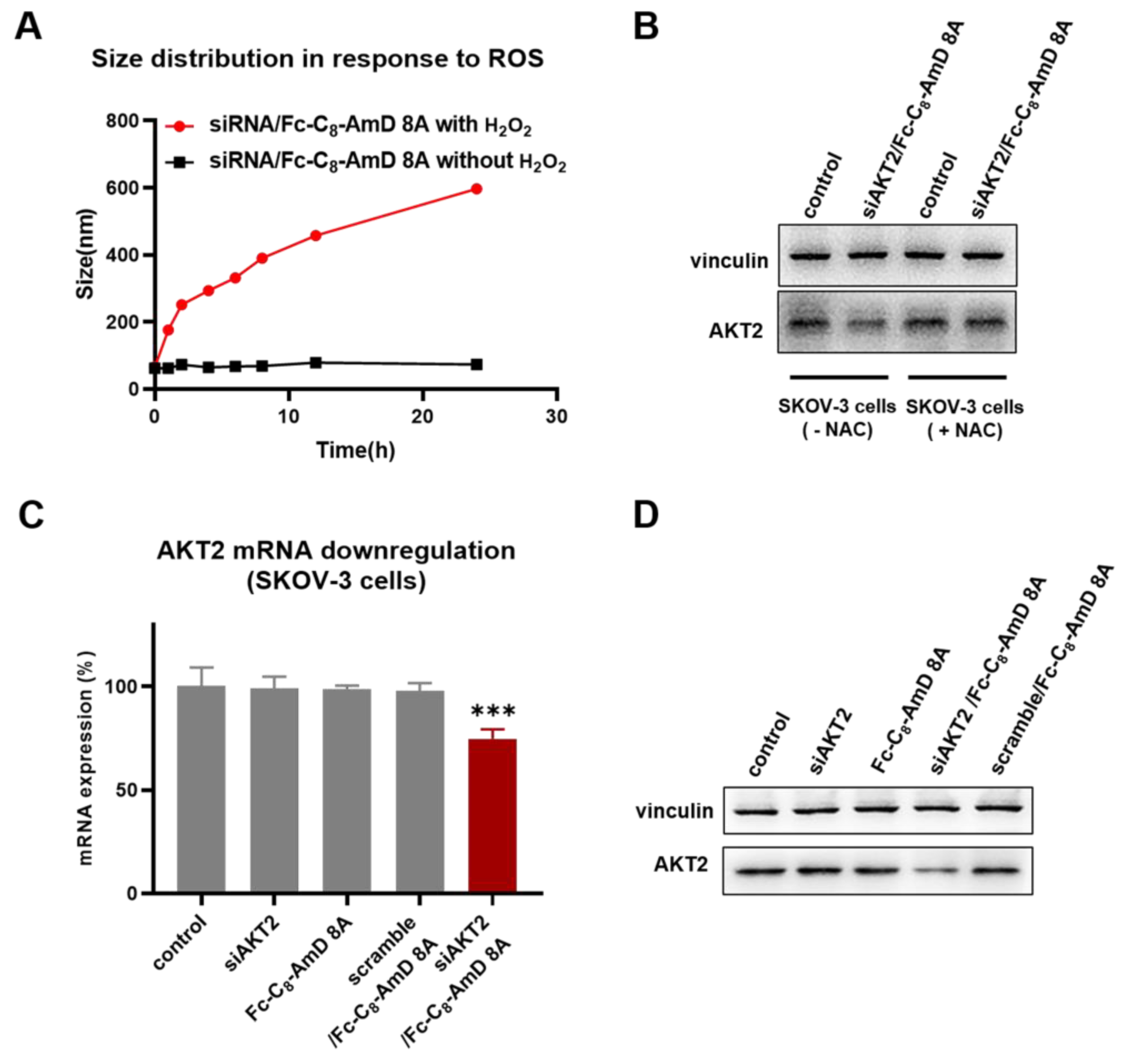
Benefiting from the optimal siRNA delivery performance of Fc-C8-AmD 8A alongside its ROS–responsive characteristics, it is expected to be able to achieve on–demand delivery of siRNA therapeutics to cancer cells that typically have high levels of ROS. First, the specific disassembly of siRNA/Fc-C8-AmD 8A nanoparticles under high ROS conditions in cancer cells is a prerequisite for on–demand delivery of siRNA. We, thus, studied the siRNA/Fc-C8-AmD 8A nanoparticles in the presence of H2O2, simulating the high ROS conditions in cancer cells. As revealed by DLS analysis, the size of the siRNA/Fc-C8-AmD 8A complex was increased from the original value of supposedly 70 nm to over 600 nm following the exposure to H2O2. This result confirms that the siRNA/Fc-C8-AmD 8A complex becomes loose due to the good ROS–responsive characteristics of Fc-C8-AmD 8A, which will facilitate the release of loaded siRNA within the complex. (Figure 3A). Furthermore, Fc-C8-AmD 8A-mediated delivery of siAKT2 was indeed specific to ROS–rich ovarian cancer SKOV-3 cells, as a significantly decreased AKT2 gene silencing was observed in N–Acetyl–L–cysteine (NAC) pretreated SKOV–3 cells (Figure 3B and Figure S12). NAC, an antioxidant, has been demonstrated to significantly reduce the ROS levels in SKOV–3 cells (Figure S13).
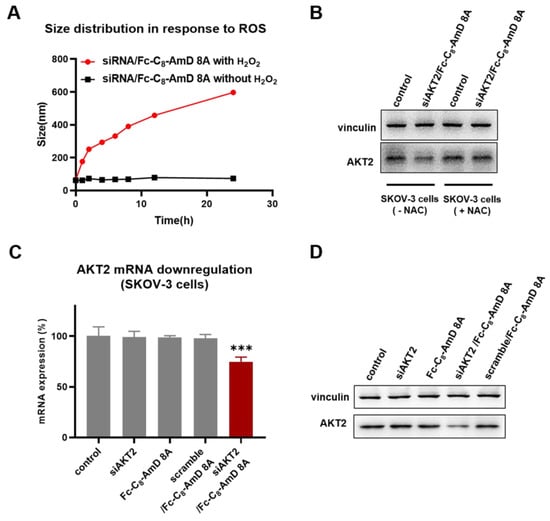
Figure 3.
(A) DLS analysis of the ROS–responsive disassembly of siRNA/Fc-C8-AmD 8A nanoparticles (1.0 μM siRNA, N/P ratio of 10). (B) ROS–responsive siRNA delivery mediated by Fc-C8-AmD 8A in ROS–rich SKOV–3 cells and ROS–poor SKOV–3 cells (pretreated with antioxidant NAC) (50 nM siRNA, N/P ratio of 10). (C) mRNA and (D) protein expression of AKT2 determined by qRT–PCR and Western blot after treating with Fc-C8-AmD 8A with siAKT2 in comparison with control, siAKT2 alone, Fc-C8-AmD 8A alone, Fc-C8-AmD 8A with scrambled siRNA (50 nM siRNA, N/P ratio of 10). *** p ≤ 0.001 (mean ± SD, n = 3).
Moreover, following Fc-C8-AmD 8A–mediated delivery of siAKT2, the expression of AKT2 was significantly and specifically suppressed at both mRNA and protein levels in ROS–rich SKOV3 cells, whereas no detectable gene silencing was observed with the treatment of either Fc-C8-AmD 8A or siRNA alone or with a scrambled siRNA/Fc-C8-AmD 8A complex. Altogether, these results suggest that Fc-C8-AmD 8A enables efficient delivery of siRNA therapeutics to cancer cells and achieves potent gene silencing in response to high ROS content, which is in line with our design concept for Fc-C8-AmD 8A as a ROS–responsive delivery vehicle.
3.5. Fc-C8-AmD 8A Capitalizes on the Delivery Advantage of Both Lipid and Dendrimer Vectors
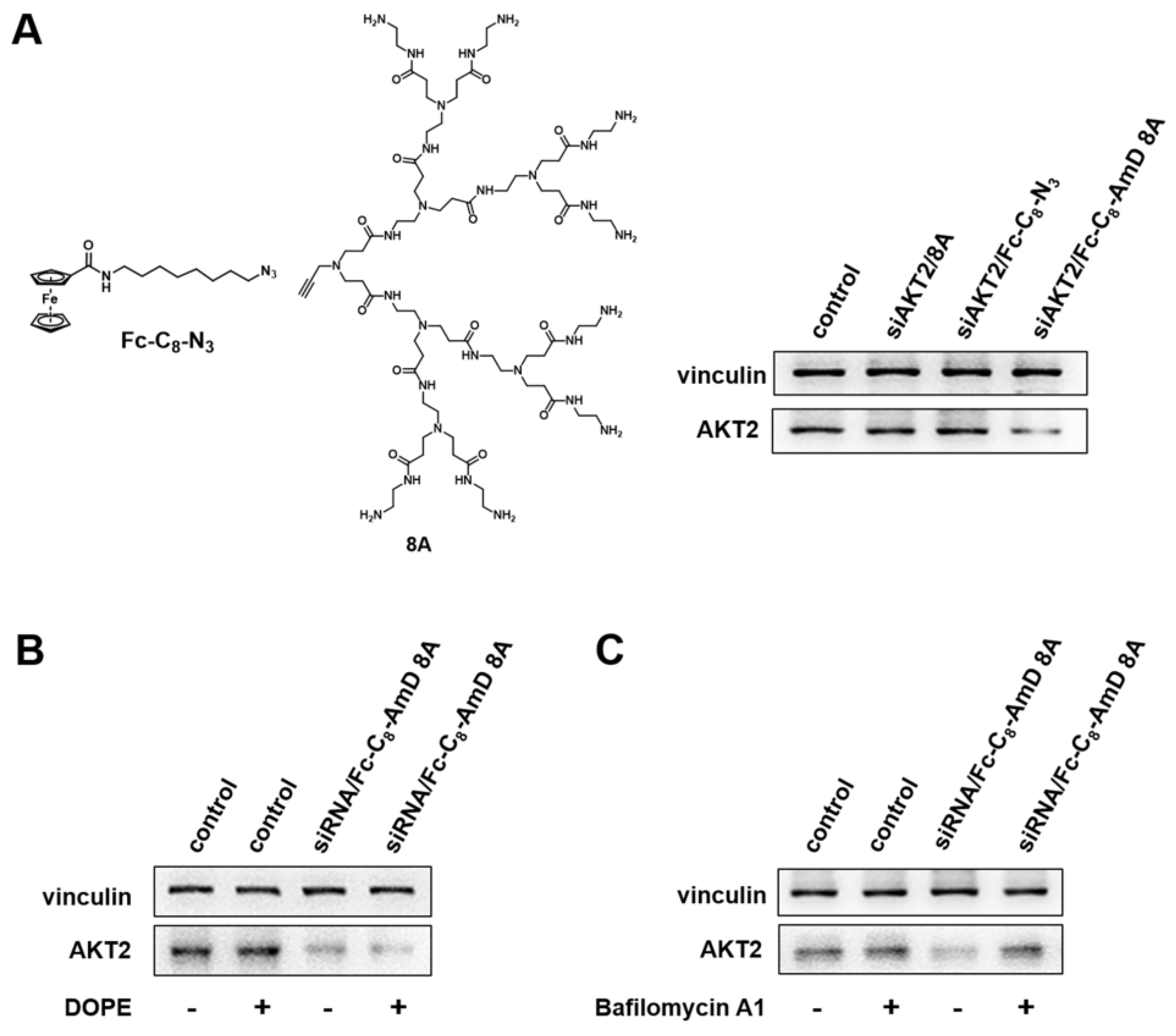
Ferrocenyl amphiphilic dendrimer Fc-C8-AmD 8A is a lipid/dendrimer hybrid. Therefore, it is conceived to take the delivery advantages of both lipid and dendrimer vectors. To confirm this, we evaluated the siRNA delivery efficacy of the alkyl chain containing the ferrocenyl moiety (Fc-C8-N3) and the PAMAM dendron (8A) in comparison with Fc-C8-AmD 8A. As shown in Figure 4A and Figure S14, except for Fc-C8-AmD 8A, the alkyl chain bearing the Fc moiety Fc-C8-N3 and the PAMAM dendron 8A was unable to produce notable gene silencing, indicating the amphiphilic structure of Fc-C8-AmD 8A is of great importance for its effective siRNA delivery.
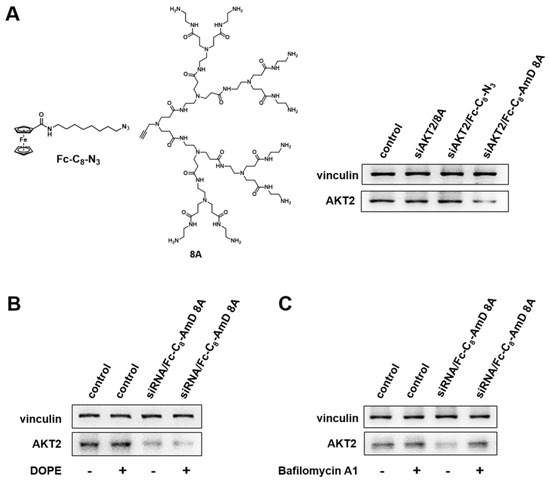
Figure 4.
Fc-C8-AmD 8A–mediated siRNA delivery combines the beneficial properties of both lipid and dendrimer vectors. (A) Compared to Fc-C8-AmD 8A, neither the Fc alkyl chain entity Fc-C8-N3 nor the dendron 8A led to any gene silencing with 50 nM siRNA at N/P ratio of 10. (B) Dioleoylphosphatidylethanolamine (DOPE) enhanced the gene silencing of AKT2 after treatment of siRNA/Fc-C8-AmD 8A complexes with 50 nM siRNA at N/P ratio of 10 on SKOV–3 cells. (C) The presence of bafilomycin A1 decreased the Fc-C8-AmD 8A–mediated gene silencing of AKT2 on SKOV–3 on cells (50 nM siRNA, N/P ratio of 10).
We further assessed the efficacy of siRNA delivered by Fc-C8-AmD 8A in the presence of dioleoylphosphatidyl–ethanolamine (DOPE). DOPE is a fusogenic lipid that promotes membrane fusion and is often used as a helper lipid to improve lipid–mediated delivery. The addition of DOPE significantly improved the AKT2 silencing achieved by Fc-C8-AmD 8A–mediated siRNA delivery (Figure 4B and Figure S15), confirming that Fc-C8-AmD 8A possesses the delivery characteristics of lipid vectors.
We next examined whether Fc-C8-AmD 8A benefits from the “proton sponge effect” inherited from PAMAM dendrimer vectors, resulting in efficient siRNA delivery. PAMAM dendrimers frequently utilize the “proton sponge effect” to promote endosome escape and subsequent cargo release in the cytoplasm. We therefore verified Fc-C8-AmD 8A–mediated siRNA delivery in the presence of bafilomycin A1, a proton pump inhibitor that impedes the acidification of endosomes. As presented in Figure 4C and Figure S16, the siRNA delivery and AKT2 silencing efficacy mediated by Fc-C8-AmD 8A was significantly reduced in the presence of bafilomycin A1, implying Fc-C8-AmD 8A–mediated siRNA delivery is also critically dependent on the “proton sponge effect”. In addition, the pH titration profile of Fc-C8-AmD 8A showed that it is able to absorb protons in endosomes (at pH 5.0) via protonation of its interior tertiary amines (Figure S4), further supporting the involvement of the proton sponge effect.
Altogether, these findings demonstrated that Fc-C8-AmD 8A indeed harnesses the beneficial properties of lipid and dendrimer vectors for effective delivery of siRNA therapeutics.
4. Conclusions
In summary, we have successfully established ROS–responsive ferrocenyl amphiphilic dendrimers (Fc-AmDs) as on–demand delivery platforms for cancer cell specific siRNA delivery. These Fc-AmDs are composed of hydrophobic alkyl chains with ferrocene moieties and hydrophilic PAMAM dendrons, possessing good self–assembly behavior in aqueous solutions, ROS-–responsive properties, and favorable safety profiles. One of these dendrimers, Fc-C8-AmD 8A, was endowed with the optimal balance between the hydrophobic chain length and the hydrophilic dendron, exhibiting the optimal siRNA binding strength and best siRNA release ability. It was capable of packing siRNA into nanoparticles to protect siRNA and promoting cellular uptake, resulting in the best performance in siRNA delivery. This ferrocenyl amphiphilic dendrimer is also able to harness the features and the delivery advantages of both lipid and dendrimer vectors. Notably, the ROS properties of Fc-C8-AmD 8A allows efficient disassembly of its complex with siRNA in ROS–rich environments, thereby inducing specific and significant silencing of the AKT2 gene in ovarian cancer SKOV–3 cells. All in all, our results demonstrate that this ferrocenyl amphiphilic dendrimer Fc-C8-AmD 8A does provide a novel on–demand delivery approach for cancer cell–specific siRNA delivery by exploiting the high levels of ROS in cancer cells. This work opens a new perspective on the design of on-demand drug delivery nanosystems based on self–assembling dendrimers.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/pharmaceutics16070936/s1, Scheme S1: Synthetic route of the hydrophobic part; Figures S1–S3: 1H and 13C NMR, HPLC, ESI–HRMS, and IR spectra of Fc-C6-AmD 8A, Fc-C8-AmD 8A, and Fc-C10-AmD 8A; Figure S4: Potentiometric pH titration profile of Fc-AmDs; Figure S5: The ROS–responsive performance of Fc-C8-AmD 8A; Figure S6: The CAC measurements of Fc-AmDs; Figure S7: The MTT assay of the metabolic toxicity of Fc-AmDs in MDCK cells, L02 cells, L929 cells, and SKOV–3 cells; Figure S8: AKT2 protein expression on SKOV–3 cells after treatment with siRNA/Fc-AmDs complexes quantified by Western blotting; Figure S9: Size distribution of Fc-C8-AmD 8A and siRNA/Fc-C8-AmD 8A complexes by intensity determined using dynamic light scattering analysis; Figure S10: Agarose gel retardation of siRNA/Fc-C8-AmD 8A complexes at different times after incubation with RNase A and SDS (200 ng siRNA/well, N/P ratio of 10); Figure S11: (A) Flow cytometry analysis and (B) confocal imaging of the cell uptake of the siRNA/Fc-C8-AmD 8A complexes in SKOV–3 cells (50 nM Cy5–labeled siRNA, N/P ratio of 10). Red: Cy5–labeled siRNA; Blue: Hoechst33342 labeling nuclei, Scale 20 μm; Figure S12: ROS–responsive siRNA delivery mediated by Fc-C8-AmD 8A in ROS-rich SKOV–3 cells and ROS–poor SKOV–3 cells (pretreated with antioxidant NAC) (50 nM siRNA, N/P ratio of 10). *** p ≤ 0.001 (mean ± SD, n = 3); Figure S13: ROS levels in human ovarian cancer SKOV–3 cells and SKOV–3 cells pretreated with the antioxidant N–acetyl–cysteine (NAC) (10 mM) quantified using CellROX orange reagent by flow cytometry. *** p ≤ 0.001 (mean ± SD, n = 3); Figure S14: Gene silencing following siAKT2 delivery mediated by 8A, Fc-C8-N3, and Fc-C8-AmD 8A (50 nM siRNA, N/P ratio of 10). *** p ≤ 0.001 (mean ± SD, n = 3); Figure S15: Dioleoylphosphatidylethanolamine (DOPE) enhanced the gene silencing of AKT2 after treatment of siRNA/Fc-C8-AmD 8A complexes with 50 nM siRNA at N/P ratio 10 on SKOV–3 cells. *** p ≤ 0.001 (mean ± SD, n = 3); Figure S16: AKT2 protein downregulation following treatment with the siAKT2/Fc-C8-AmD 8A complex (50 nM siRNA, N/P ratio of 10) on SKOV–3 cells in the presence and absence of the proton pump inhibitor, bafilomycin A1. *** p ≤ 0.001 (mean ± SD, n = 3); Table S1: The information on Fc-AmDs from HPLC detection; Table S2: The IC50 of Fc-AmDs was assessed by MTT assay on normal and cancer cells.
Author Contributions
Conceptualization, P.C., Z.W., X.W., D.Z. and X.L.; methodology, P.C., Z.W., J.G., X.W., J.S., Y.P. and D.Z.; formal analysis, J.G., J.S. and Y.P.; investigation, Z.W., J.G., X.W., J.S. and Y.P.; resources, X.L.; data curation, P.C. and Z.W.; writing—original draft preparation, P.C., Z.W., J.G. and J.S.; writing—review and editing, P.C., D.Z. and X.L.; supervision and funding acquisition, X.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Cell Therapy Research Project from the National Center of Technology Innovation for Biopharmaceuticals (NCTIB2023XB02003), the National Natural Science Foundation of China (No. 81701815), the Youth Thousand-Talents Program of China, and the National Key Research & Development Program of China for International S&T Cooperation Projects (2018YFE0117800).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All study data are included within the article and/or Supplementary Materials.
Acknowledgments
We thank Haijuan Dong from the Public Laboratory Platform of China Pharmaceutical University for her help in MS analysis.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, V.; Vaishnaw, A.; Fitzgerald, K.; Maier, M.A. RNA interference in the era of nucleic acid therapeutics. Nat. Biotechnol. 2024, 42, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Khvorova, A. RNAi-based drug design: Considerations and future directions. Nat. Rev. Drug Discov. 2024, 23, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Kara, G.; Calin, G.A.; Ozpolat, B. RNAi-based therapeutics and tumor targeted delivery in cancer. Adv. Drug Deliv. Rev. 2022, 182, 114113. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Moazzam, M.; Zhang, M.; Hussain, A.; Yu, X.; Huang, J.; Huang, Y. The landscape of nanoparticle-based siRNA delivery and therapeutic development. Mol. Ther. 2024, 32, 284–312. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhen, X.; Zhang, Y.; Li, Y.; Koo, S.; Saiding, Q.; Kong, N.; Liu, G.; Chen, W.; Tao, W. Chemically modified platforms for better RNA therapeutics. Chem. Rev. 2024, 124, 929–1033. [Google Scholar] [CrossRef] [PubMed]
- Charbe, N.B.; Amnerkar, N.D.; Ramesh, B.; Tambuwala, M.M.; Bakshi, H.A.; Aljabali, A.A.A.; Khadse, S.C.; Satheeshkumar, R.; Satija, S.; Metha, M.; et al. Small interfering RNA for cancer treatment: Overcoming hurdles in delivery. Acta Pharm. Sin. B 2020, 10, 2075–2109. [Google Scholar] [CrossRef]
- Mendes, B.B.; Conniot, J.; Avital, A.; Yao, D.; Jiang, X.; Zhou, X.; Sharf-Pauker, N.; Xiao, Y.; Adir, O.; Liang, H.; et al. Nano delivery of nucleic acids. Nat. Rev. Methods Primers 2022, 2, 24. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First global approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Peng, L. Potent drugless dendrimers. Nat. Biomed. Eng. 2017, 1, 686–688. [Google Scholar] [CrossRef]
- Lyu, Z.; Ding, L.; Tintaru, A.; Peng, L. Self-assembling supramolecular dendrimers for biomedical applications: Lessons learned from poly(amidoamine) sendrimers. Acc. Chem. Res. 2020, 53, 2936–2949. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, D.; Liu, X.; Peng, L. Amphiphilic dendrimer vectors for RNA delivery: State-of-the-art and future perspective. Acc. Mater. Res. 2022, 3, 484–497. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Xiao, C.; Li, M.; Tian, H.; Chen, X. Cationic dendron-bearing lipids: Investigating structure–activity relationships for small interfering RNA delivery. Biomacromolecules 2013, 14, 4289–4300. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, D.; Lian, B.; Shi, K.; Chen, P.; Li, Y.; Lin, W.; Ding, L.; Long, Q.; Wang, Y.; et al. Cargo-selective and adaptive delivery of nucleic acid therapeutics by bola-amphiphilic dendrimers. Proc. Natl. Acad. Sci. USA 2023, 120, e2220787120. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Chen, Y.; Zhu, D.; Shi, K.; Ma, C.; Zhang, W.; Rocchi, P.; Jiang, L.; Liu, X. Self-assembly of amphiphilic phospholipid peptide dendrimer-based nanovectors for effective delivery of siRNA therapeutics in prostate cancer therapy. J. Control. Release 2020, 322, 416–425. [Google Scholar] [CrossRef]
- Ma, C.; Zhu, D.; Lin, W.; Li, Y.; Huang, Y.; Zhu, H.; Ye, M.; Wang, Y.; Peng, L.; Liu, X. A biodegradable amphiphilic poly(aminoester) dendrimer for safe and effective siRNA delivery. Chem. Commun. 2022, 58, 4168–4171. [Google Scholar] [CrossRef]
- Malhotra, S.; Bauer, H.; Tschiche, A.; Staedtler, A.M.; Mohr, A.; Calderón, M.; Parmar, V.S.; Hoeke, L.; Sharbati, S.; Einspanier, R.; et al. Glycine-terminated dendritic amphiphiles for nonviral gene delivery. Biomacromolecules 2012, 13, 3087–3098. [Google Scholar] [CrossRef]
- Lu, Y.; Aimetti, A.A.; Langer, R.; Gu, Z. Bioresponsive materials. Nat. Rev. Mater. 2016, 2, 16075. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, Y.; Shi, J. Reactive oxygen species (ROS)-based nanomedicine. Chem. Rev. 2019, 119, 4881–4985. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Bus, T.; Traeger, A.; Schubert, U.S. The great escape: How cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [Google Scholar] [CrossRef] [PubMed]
- Staab, H.A.; Saupe, T. “Proton sponges” and the geometry of hydrogen bonds: Aromatic nitrogen bases with exceptional basicities. Angew. Chem. Int. Ed. 1988, 27, 865–879. [Google Scholar] [CrossRef]
- Astruc, D. The numerous paths of ferrocene. Nat. Chem. 2023, 15, 1650. [Google Scholar] [CrossRef] [PubMed]
- Guchhait, C.; Suriyaa, V.; Sahu, N.; Sarkar, S.D.; Adhikari, B. Ferrocene: An exotic building block for supramolecular assemblies. Chem. Commun. 2023, 59, 14482–14496. [Google Scholar] [CrossRef] [PubMed]
- Mengerink, Y.; Mure, M.; de Brabander, E.M.M.; van der Wal, S. Exclusion chromatography of polypropylenamine dendrimers. J. Chromatogr. A 1996, 730, 75–81. [Google Scholar] [CrossRef]
- Tan, J.; Li, H.; Hu, X.; Abdullah, R.; Xie, S.; Zhang, L.; Zhao, M.; Luo, Q.; Li, Y.; Sun, Z.; et al. Size-tunable assemblies based on ferrocene-containing DNA polymers for spatially uniform penetration. Chem 2019, 5, 1775–1792. [Google Scholar] [CrossRef]
- Honardoost, M.; Rad, S.M.A.H. Triangle of AKT2, miRNA, and tumorigenesis in different cancers. Appl. Biochem. Biotechnol. 2018, 185, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Chen, J.; Wang, J.; Liu, J.; Jiang, Y. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021, 14, 128. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).