
Synthesis of Gelatin Methacryloyl Analogs and Their Use in the Fabrication of pH-Responsive Microspheres


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
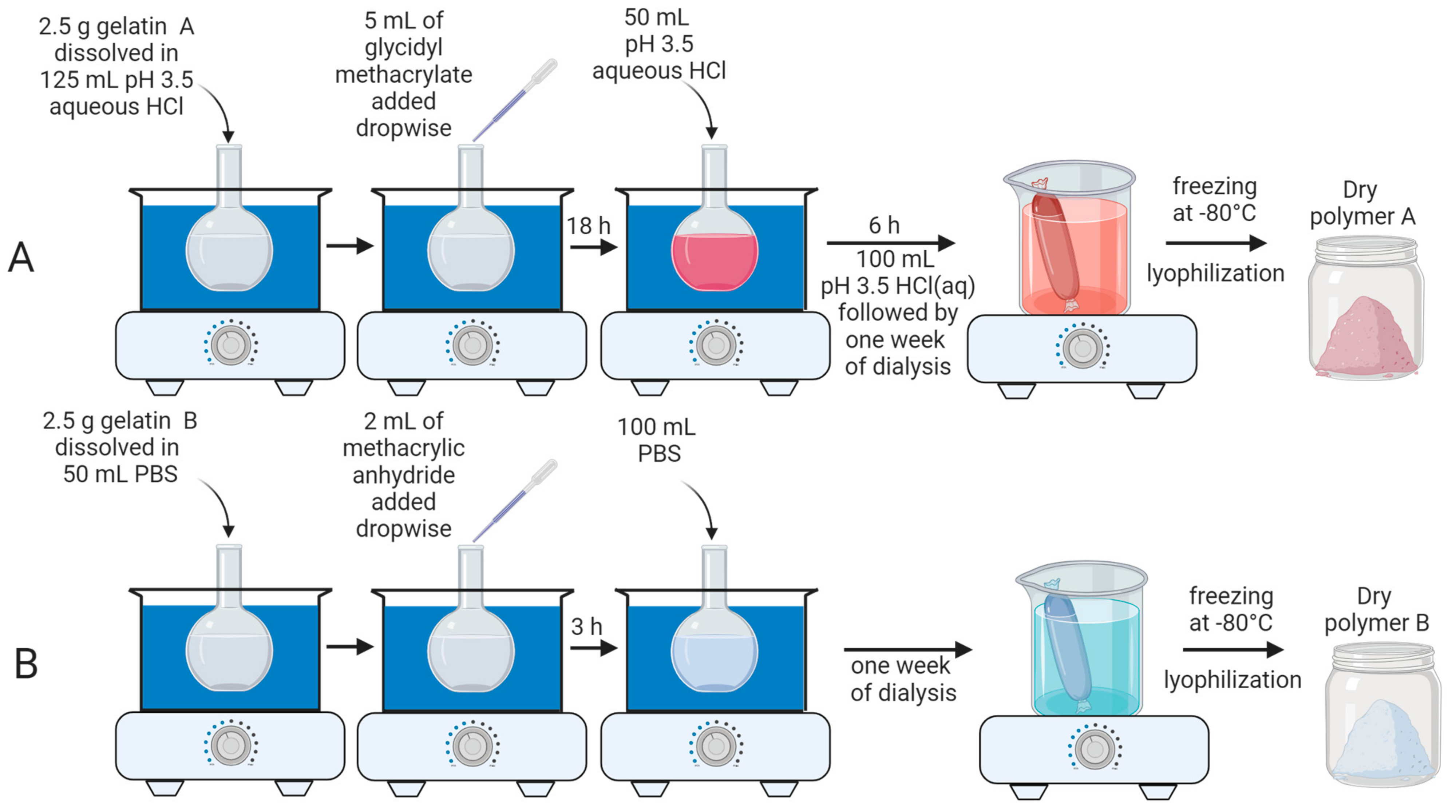
2.2. Synthesis of Polymers A and B
2.3. Characterization of Polymers A and B
2.4. Fabrication of Microfluidic Device
2.5. Synthesis and Characterization of Hydrogel Microspheres
3. Results and Discussion
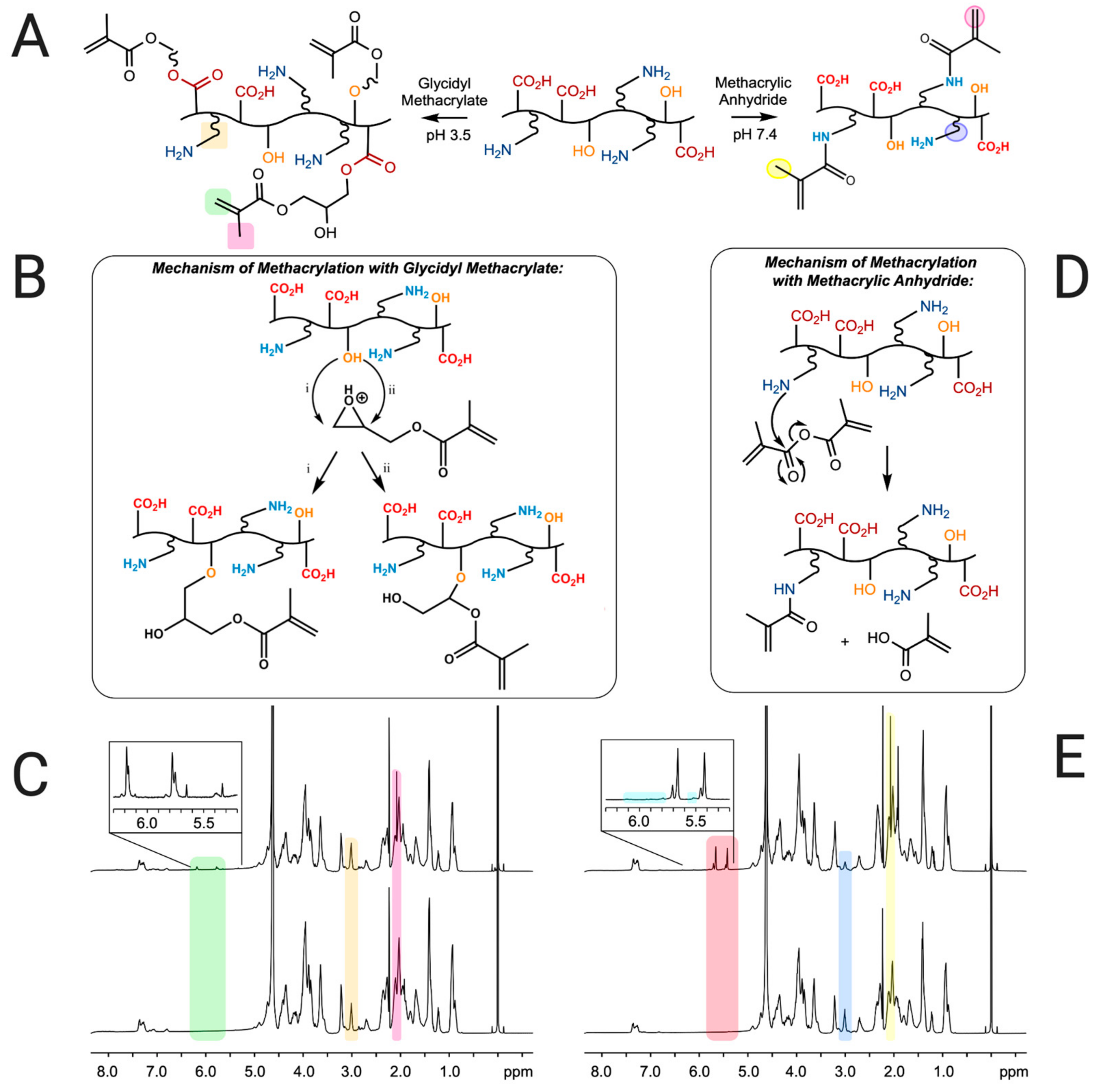
3.1. Determination of Degree of Modification of Polymers A and B
3.2. Fabrication of Hydrogel Microspheres
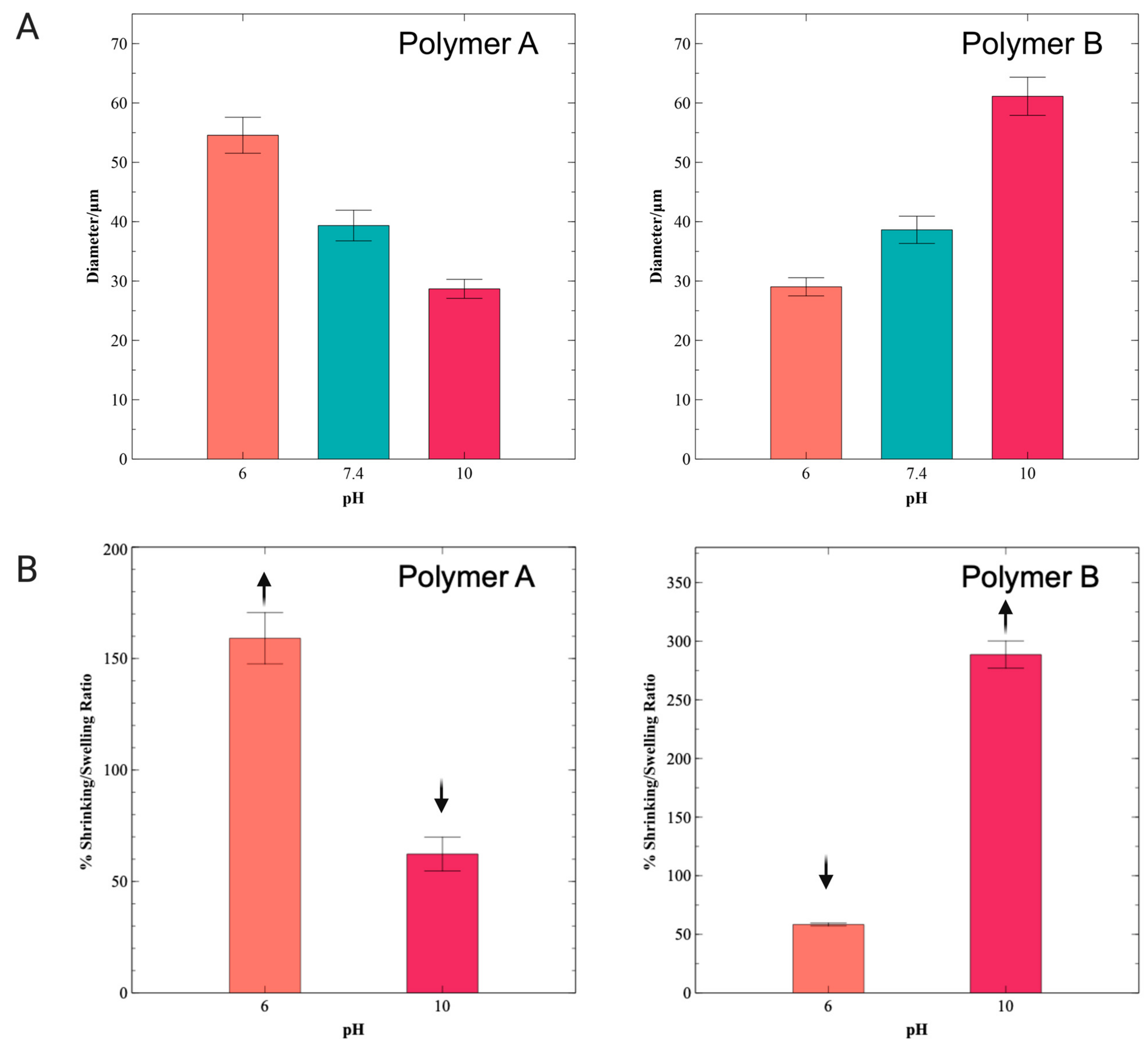
3.3. Swelling and Shrinking Behavior of Hydrogel Microspheres
4. Conclusions
5. Patent
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-Laden Microengineered Gelatin Methacrylate Hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, H.-W. Emerging Properties of Hydrogels in Tissue Engineering. J. Tissue Eng. 2018, 9, 2041731418768285. [Google Scholar] [CrossRef]
- Drury, J.L.; Mooney, D.J. Hydrogels for Tissue Engineering: Scaffold Design Variables and Applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing Hydrogels for Controlled Drug Delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Daly, A.C.; Riley, L.; Segura, T.; Burdick, J.A. Hydrogel Microparticles for Biomedical Applications. Nat. Rev. Mater. 2020, 5, 20–43. [Google Scholar] [CrossRef]
- Rizwan, M.; Yahya, R.; Hassan, A.; Yar, M.; Azzahari, A.; Selvanathan, V.; Sonsudin, F.; Abouloula, C.; Rizwan, M.; Yahya, R.; et al. pH Sensitive Hydrogels in Drug Delivery: Brief History, Properties, Swelling, and Release Mechanism, Material Selection and Applications. Polymers 2017, 9, 137. [Google Scholar] [CrossRef]
- Kocak, G.; Tuncer, C.; Bütün, V. pH-Responsive Polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Deligkaris, K.; Tadele, T.S.; Olthuis, W.; van den Berg, A. Hydrogel-Based Devices for Biomedical Applications. Sens. Actuators B Chem. 2010, 147, 765–774. [Google Scholar] [CrossRef]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging Applications of Stimuli-Responsive Polymer Materials. Nature Materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Vegad, U.; Patel, M.; Khunt, D.; Zupancic, O.; Chauhan, S.; Paudel, A. pH stimuli-responsive hydrogels from non-cellulosic biopolymers for drug delivery. Front. Bioeng. Biotechnol. 2023, 11, 1270364. [Google Scholar] [CrossRef]
- Valente, K.P.; Khetani, S.; Kolahchi, A.R.; Sanati-Nezhad, A.; Suleman, A.; Akbari, M. Microfluidic Technologies for Anticancer Drug Studies. Drug Discov. Today 2017, 22, 1654–1670. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Du, J.Z.; Sun, T.M.; Song, W.J.; Wu, J.; Wang, J. A Tumor-Acidity-Activated Charge-Conversional Nanogel as an Intelligent Vehicle for Promoted Tumoral-Cell Uptake and Drug Delivery. Angew. Chem.—Int. Ed. 2010, 49, 3621–3626. [Google Scholar] [CrossRef]
- Johnson, R.P.; Jeong, Y.-I.; Choi, E.; Chung, C.-W.; Kang, D.H.; Oh, S.-O.; Suh, H.; Kim, I. Biocompatible Poly(2-Hydroxyethyl Methacrylate)-b-Poly(L-Histidine) Hybrid Materials for pH-Sensitive Intracellular Anticancer Drug Delivery. Adv. Funct. Mater. 2012, 22, 1058–1068. [Google Scholar] [CrossRef]
- Du, H.; Liu, M.; Yang, X.; Zhai, G. The Design of pH-Sensitive Chitosan-Based Formulations for Gastrointestinal Delivery. Drug Discov. Today 2015, 20, 1004–1011. [Google Scholar] [CrossRef]
- Gao, X.; He, C.; Xiao, C.; Zhuang, X.; Chen, X. Biodegradable pH-Responsive Polyacrylic Acid Derivative Hydrogels with Tunable Swelling Behavior for Oral Delivery of Insulin. Polymer 2013, 54, 1786–1793. [Google Scholar] [CrossRef]
- Samal, S.K.; Dash, M.; Van Vlierberghe, S.; Kaplan, D.L.; Chiellini, E.; Van Blitterswijk, C.; Moroni, L.; Dubruel, P. Cationic Polymers and Their Therapeutic Potential. Chem. Soc. Rev. 2012, 41, 7147–7194. [Google Scholar] [CrossRef]
- Loessner, D.; Meinert, C.; Kaemmerer, E.; Martine, L.C.; Yue, K.; Levett, P.A.; Klein, T.J.; Melchels, F.P.W.; Khademhosseini, A.; Hutmacher, D.W. Functionalization, Preparation and Use of Cell-Laden Gelatin Methacryloyl–Based Hydrogels as Modular Tissue Culture Platforms. Nat. Protoc. 2016, 11, 727–746. [Google Scholar] [CrossRef]
- Claaßen, C.; Claaßen, M.H.; Truffault, V.; Sewald, L.; Tovar, G.E.M.; Borchers, K.; Southan, A. Quantification of Substitution of Gelatin Methacryloyl: Best Practice and Current Pitfalls. Biomacromolecules 2018, 19, 42–52. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, Z.; Menard, F.; Kim, K. Comparative Study of Gelatin Methacrylate Hydrogels from Different Sources for Biofabrication Applications. Biofabrication 2017, 9, 044101. [Google Scholar] [CrossRef]
- Jaipan, P.; Nguyen, A.; Narayan, R.J. Gelatin-Based Hydrogels for Biomedical Applications. MRS Commun. 2017, 7, 416–426. [Google Scholar] [CrossRef]
- Yue, K.; Trujillo-de Santiago, G.; Alvarez, M.M.; Tamayol, A.; Annabi, N.; Khademhosseini, A. Synthesis, Properties, and Biomedical Applications of Gelatin Methacryloyl (GelMA) Hydrogels. Biomaterials 2015, 73, 254–271. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Kawazoe, N.; Chen, G. Fabrication of Highly Crosslinked Gelatin Hydrogel and Its Influence on Chondrocyte Proliferation and Phenotype. Polymers 2017, 9, 309. [Google Scholar] [CrossRef]
- Lee, B.H.; Shirahama, H.; Cho, N.-J.; Tan, L.P. Efficient and Controllable Synthesis of Highly Substituted Gelatin Methacrylamide for Mechanically Stiff Hydrogels. RSC Adv. 2015, 5, 106094–106097. [Google Scholar] [CrossRef]
- Wu, T.; Xu, Z.; Zhang, Y.; Wang, H.; Cui, C.; Chang, B.; Feng, X.; Liu, W. A pH-Responsive Biodegradable High-Strength Hydrogel as a Potential Gastric Resident Filler. Macro-Mol. Mater. Eng. 2018, 303, 1800290. [Google Scholar] [CrossRef]
- Du, M.; Jin, J.; Zhou, F.; Chen, J.; Jiang, W. Dual drug-loaded hydrogels with pH-responsive and antibacterial activity for skin wound dressing. Colloids Surf. B Biointerfaces 2023, 222, 113063. [Google Scholar] [CrossRef]
- Oh, J.; Kim, B. Mucoadhesive and pH-responsive behavior of gelatin containing hydrogels for protein drug delivery applications. Korea-Aust. Rheol. J. 2020, 32, 41–46. [Google Scholar] [CrossRef]
- Hoch, E.; Schuh, C.; Hirth, T.; Tovar, G.E.M.; Borchers, K. Stiff Gelatin Hydrogels Can Be Photo-Chemically Synthesized from Low Viscous Gelatin Solutions Using Molecularly Functionalized Gelatin with a High Degree of Methacrylation. J. Mater. Sci. Mater. Med. 2012, 23, 2607–2617. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Y.; Ferracci, G.; Zheng, J.; Cho, N.; Lee, B. Gelatin Methacryloyl and its hydrogels with an exceptional degree of controllability and batch-to-batch consistency. Sci Rep. 2019, 9, 6863. [Google Scholar] [CrossRef]
- Lai, T.C.; Yu, J.; Tsai, W.B. Gelatin Methacrylate/Carboxybetaine Methacrylate Hydrogels with Tunable Crosslinking for Controlled Drug Release. J. Mater. Chem. B 2016, 4, 2304–2313. [Google Scholar] [CrossRef]
- Jung, J.; Oh, J. Swelling Characterization of Photo-Cross-Linked Gelatin Methacrylate Spherical Microgels for Bioencapsulation. e-Polymers 2014, 14, 161–168. [Google Scholar] [CrossRef]
- Reis, A.V.; Fajardo, A.R.; Schuquel, I.T.A.; Guilherme, M.R.; Vidotti, G.J.; Rubira, A.F.; Muniz, E.C. Reaction of Glycidyl Methacrylate at the Hydroxyl and Carboxylic Groups of Poly(Vinyl Alcohol) and Poly(Acrylic Acid): Is This Reaction Mechanism Still Unclear? J. Org. Chem. 2009, 74, 3750–3757. [Google Scholar] [CrossRef]
- Suvarnapathaki, S.; Ramos, R.; Sawyer, S.W.; McLoughlin, S.; Ramos, A.; Venn, S.; Soman, P. Generation of Cell-Laden Hydrogel Microspheres Using 3D Printing-Enabled Microfluidics. J. Mater. Res. 2018, 33, 2012–2018. [Google Scholar] [CrossRef]
- Strutt, J.W., VI. On the capillary phenomena of jets. Proc. R. Soc. Lond. 1879, 29, 71–97. [Google Scholar]
- Samanipour, R.; Wang, Z.; Ahmadi, A.; Kim, K. Experimental and Computational Study of Microfluidic Flow-Focusing Generation of Gelatin Methacrylate Hydrogel Droplets. J. Appl. Polym. Sci 2016, 133, 43701. [Google Scholar] [CrossRef]
- Hafidz, M.; Yaakob, C.M.; Amin, I.; Noorfaizan, A. Chemical and Functional Properties of Bovine and Porcine Skin Gelatin. Int. Food Res. J. 2011, 18, 813–817. [Google Scholar]
- Neves, N.M.; Reis, R.L. (Eds.) Biomaterials from Nature for Advanced Devices and Therapies; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Gu, L.; Li, T.; Song, X.; Yang, X.; Li, S.; Chen, L.; Liu, P.; Gong, X.; Chen, C.; Sun, L. Preparation and Characterization of Methacrylated Gelatin/Bacterial Cellulose Composite Hydrogels for Cartilage Tissue Engineering. Regen. Biomater. 2020, 7, 195–202. [Google Scholar] [CrossRef]
- Xiao, S.; Zhao, T.; Wang, J.; Wang, C.; Du, J.; Ying, L.; Lin, J.; Zhang, C.; Hu, W.; Wang, L.; et al. Gelatin Methacrylate (GelMA)-Based Hydrogels for Cell Transplantation: An Effective Strategy for Tissue Engineering. Stem Cell Rev. Rep. 2019, 15, 664–679. [Google Scholar] [CrossRef]
- Gao, Q.; Niu, X.; Shao, L.; Zhou, L.; Lin, Z.; Sun, A.; Fu, J.; Chen, Z.; Hu, J.; Liu, Y.; et al. 3D Printing of Complex GelMA-Based Scaffolds with Nanoclay. Biofabrication 2019, 11, 035006. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Yan, M.; Wang, Y.; Fu, J.; Suo, H. 3D Bioprinting of Low-Concentration Cell-Laden Gelatin Methacrylate (GelMA) Bioinks with a Two-Step Cross-Linking Strategy. ACS Appl. Mater. Interfaces 2018, 10, 6849–6857. [Google Scholar] [CrossRef]
- Modaresifar, K.; Hadjizadeh, A.; Niknejad, H. Design and Fabrication of GelMA/Chitosan Nanoparticles Composite Hydrogel for Angiogenic Growth Factor Delivery. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1–10. [Google Scholar] [CrossRef]
- Vigata, M.; Meinert, C.; Pahoff, S.; Bock, N.; Hutmacher, D.W. Gelatin Methacryloyl Hydrogels Control the Localized Delivery of Albumin-Bound Paclitaxel. Polymers 2020, 12, 501. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valente, K.; Boice, G.N.; Polglase, C.; Belli, R.G.; Bourque, E.; Suleman, A.; Brolo, A. Synthesis of Gelatin Methacryloyl Analogs and Their Use in the Fabrication of pH-Responsive Microspheres. Pharmaceutics 2024, 16, 1016. https://doi.org/10.3390/pharmaceutics16081016
Valente K, Boice GN, Polglase C, Belli RG, Bourque E, Suleman A, Brolo A. Synthesis of Gelatin Methacryloyl Analogs and Their Use in the Fabrication of pH-Responsive Microspheres. Pharmaceutics. 2024; 16(8):1016. https://doi.org/10.3390/pharmaceutics16081016
Chicago/Turabian StyleValente, Karolina, Geneviève N. Boice, Cameron Polglase, Roman G. Belli, Elaina Bourque, Afzal Suleman, and Alexandre Brolo. 2024. "Synthesis of Gelatin Methacryloyl Analogs and Their Use in the Fabrication of pH-Responsive Microspheres" Pharmaceutics 16, no. 8: 1016. https://doi.org/10.3390/pharmaceutics16081016