
Pharmacokinetic Simulation and Area under the Curve Estimation of Drugs Subject to Enterohepatic Circulation
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
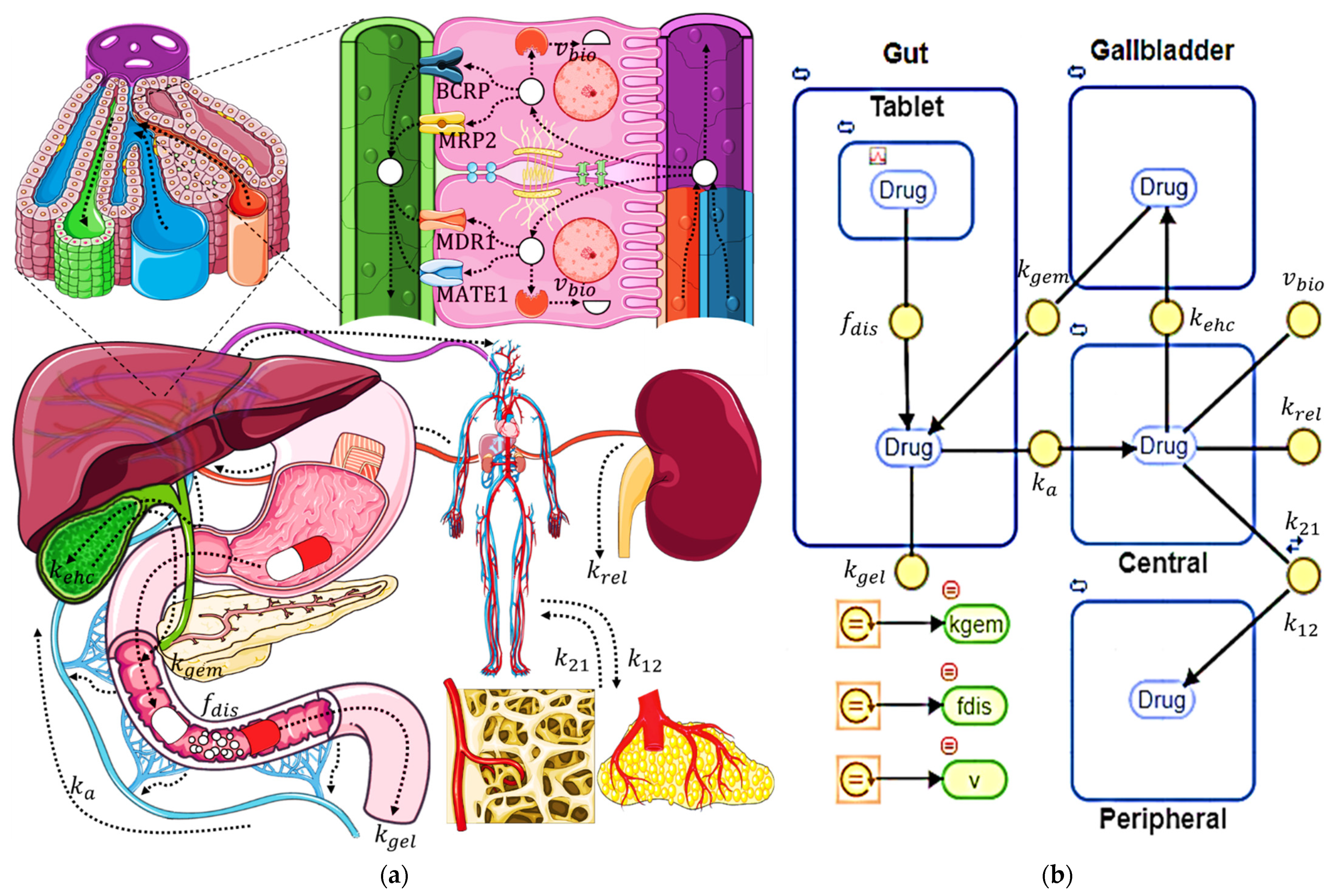
2.1. Compartmental Pharmacokinetic Model
2.2. Compartmental Model Assumptions
- Compartments are homogeneous, so drug concentration within each of the compartments reaches an instantaneous kinetic equilibrium;
- Liver is located within the central compartment, so there is an instantaneous kinetic equilibrium between plasma and intrahepatic drug concentrations;
- Elimination by routes other than renal, fecal, or biotransformation is insignificant;
- Fraction of drug that is biotransformed does not need to be reconverted to the original drug to undergo EHC, as this usually happens under the presence of microbiota glucuronidases;
- Elimination by biotransformation occurs through a single pathway involving a single enzyme;
- Drug dissolved in the bile is excreted to the intestinal compartment only from the bile compartment, so direct excretion from the central compartment is non-existent;
- Fraction of drug dissolved, transfer of drug from the gallbladder to the intestine, and rate of biotransformation can be realistically represented by equations, while the rest of the parameters follow first-order kinetics that do not consider changes in transport rates between compartments due to physiological phenomena.
2.3. Pharmacokinetic Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Units | Value | Description |
|---|---|---|---|
| h−1 | 0.9 | Intestinal absorption rate | |
| h−1 | 0.1 | Renal elimination rate | |
| h−1 | 0.1 | Fecal elimination rate | |
| - | See Equation (2) | Dissolved drug fraction | |
| h−1 | 0.5 | Scale parameter of the Weibull function | |
| h | 0 | Weibull function delay time | |
| - | 1 | Shape parameter of the Weibull function | |
| h−1 | See Equation (3) | Rate of transfer from gallbladder to intestine | |
| h−1 | 3.0 | Maximum transfer rate from gallbladder to intestine | |
| - | 300 | Biliary emptying cycle duration parameter | |
| - | 5, 11, 17 | Times of maximum gallbladder emptying rate | |
| h−1 | 0 | Transfer rate from central to peripheral compartment | |
| h−1 | 0 | Transfer rate from peripheral to central compartment | |
| mg/h | See Equation (4) | Instantaneous biotransformation rate | |
| mg/h | 0.1 | Maximum biotransformation rate | |
| mg/L | See Table 3 | Michaelis–Menten constant for biotransformation | |
| h−1 | See Table 3 | Transfer rate from the central compartment to the gallbladder |
| Scenarios | Parameter | Value | a | a | Description |
|---|---|---|---|---|---|
| 1.5 | I | A | Rapid absorption in the intestinal compartment | ||
| 0.3 | I | A | Slow absorption in the intestinal compartment | ||
| 0.5 | II | B | Rapid renal elimination from central compartment | ||
| 0.02 | III | C | Slow renal elimination from central compartment | ||
| 0.5 | I | A | Rapid elimination from intestinal compartment | ||
| 0.02 | I | A | Slow elimination from intestinal compartment | ||
| 0.0 | I | A | Drug release without delay time | ||
| 2.0 | I | A | Time-delayed drug release | ||
| 0.5 | I | A | Parabolic drug release | ||
| 2.0 | I | A | Sigmoidal drug release | ||
| 6.0 | I | A | Rapid gallbladder emptying | ||
| 1.5 | I | A | Slow gallbladder emptying | ||
| B | 600 | I | A | Short gallbladder emptying duration | |
| 100 | I | A | Long gallbladder emptying duration | ||
| 1.0, 0.1 | II | D | Fast peripheral distribution with slow central return | ||
| 0.1, 1.0 | I | E | Slow peripheral distribution with fast central return | ||
| 1.0 | IV | A | Broad first pass elimination | ||
| 0.02 | I | A | Reduced first pass removal |
| km | Effect on EHC% a | kehc | EHCmax% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | A | B | C | D | E | ||
| 10 | 1 | 100 | 100 | + | 0.0666 | 0.3333 | 0.0133 | 0.7333 | 0.1333 | 20% |
| 0.1 | 0.01 | 1 | 10 | ++ | 0.15 | 0.75 | 0.03 | 1.65 | 0.3 | 60% |
| 0.001 | 0.0001 | 0.01 | 1 | +++ | 0.4 | 2.0 | 0.08 | 4.4 | 0.8 | 80% |
2.4. Empirical Pharmacokinetic Models
2.5. Compilation of Pharmacokinetic Profiles from the Literature
2.6. Data Fitting, Statistical Analysis, and Evaluation of Empirical Models
3. Results
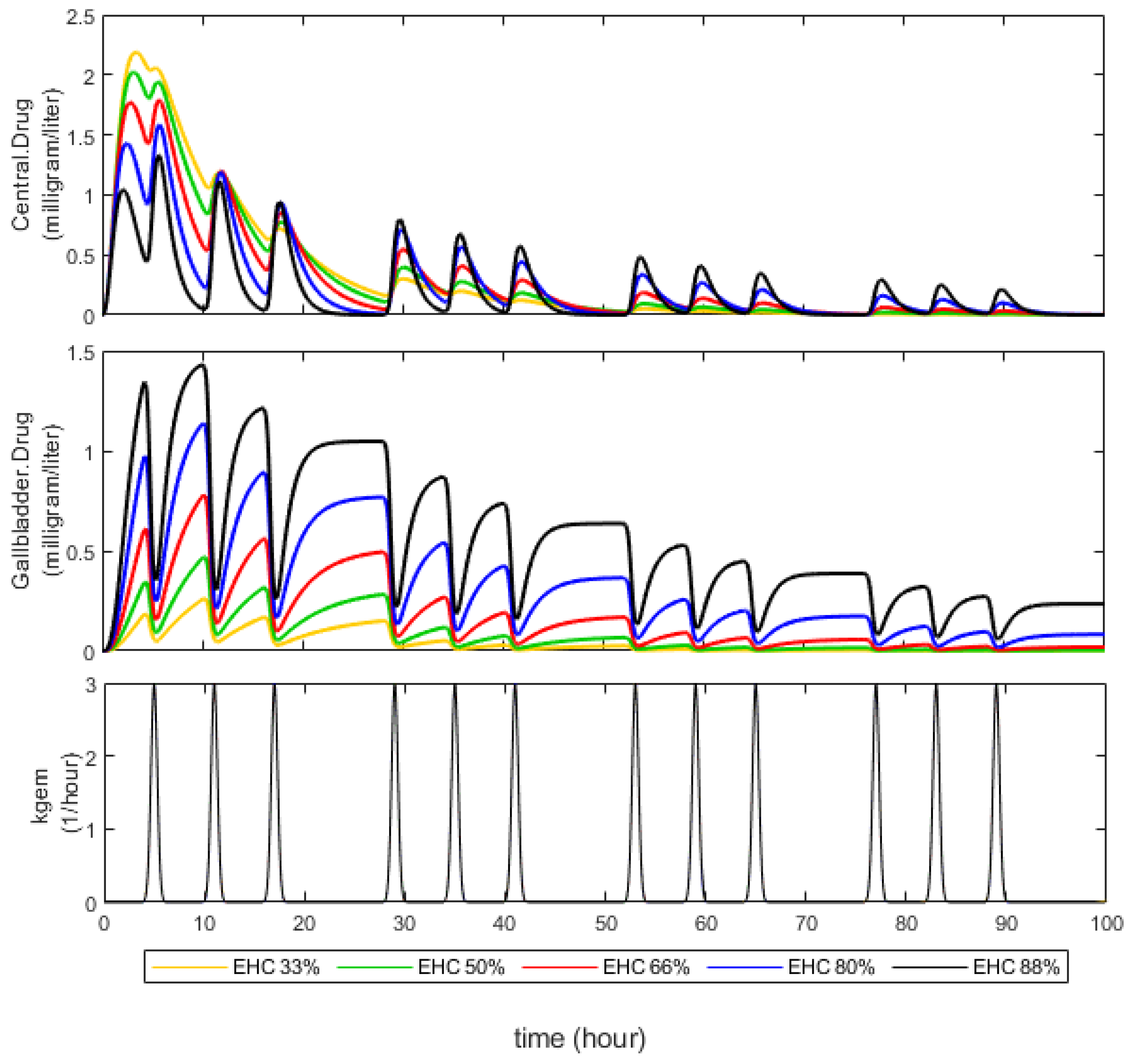
3.1. Effect of EHC Degree on AUC
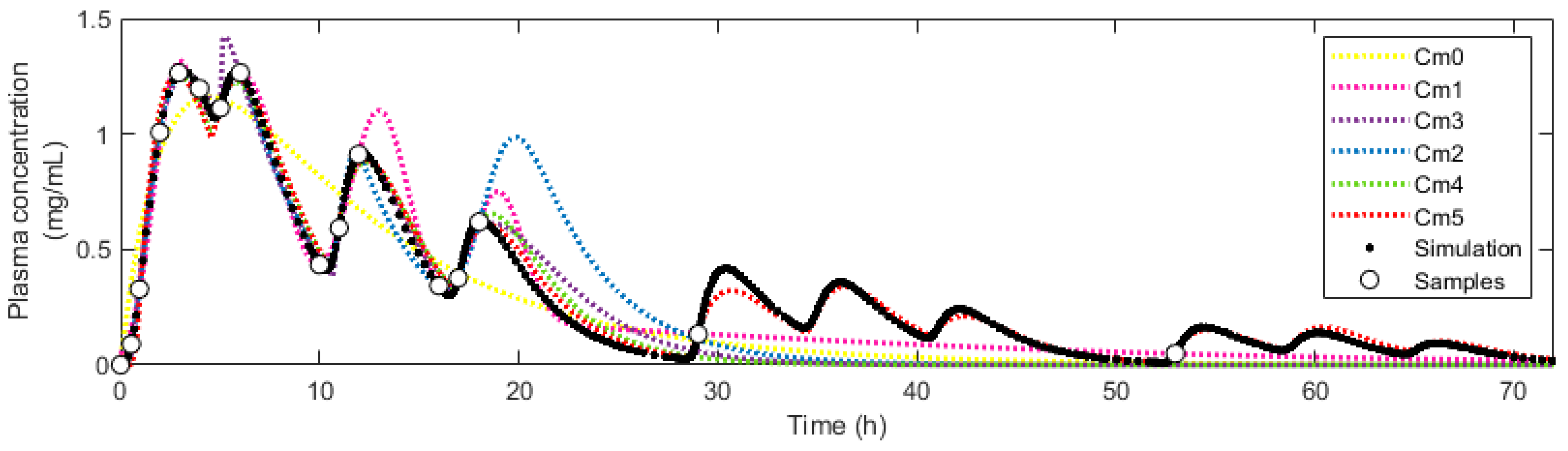
3.2. Simulation of Pharmacokinetic Scenarios and Fitting to Empirical Models
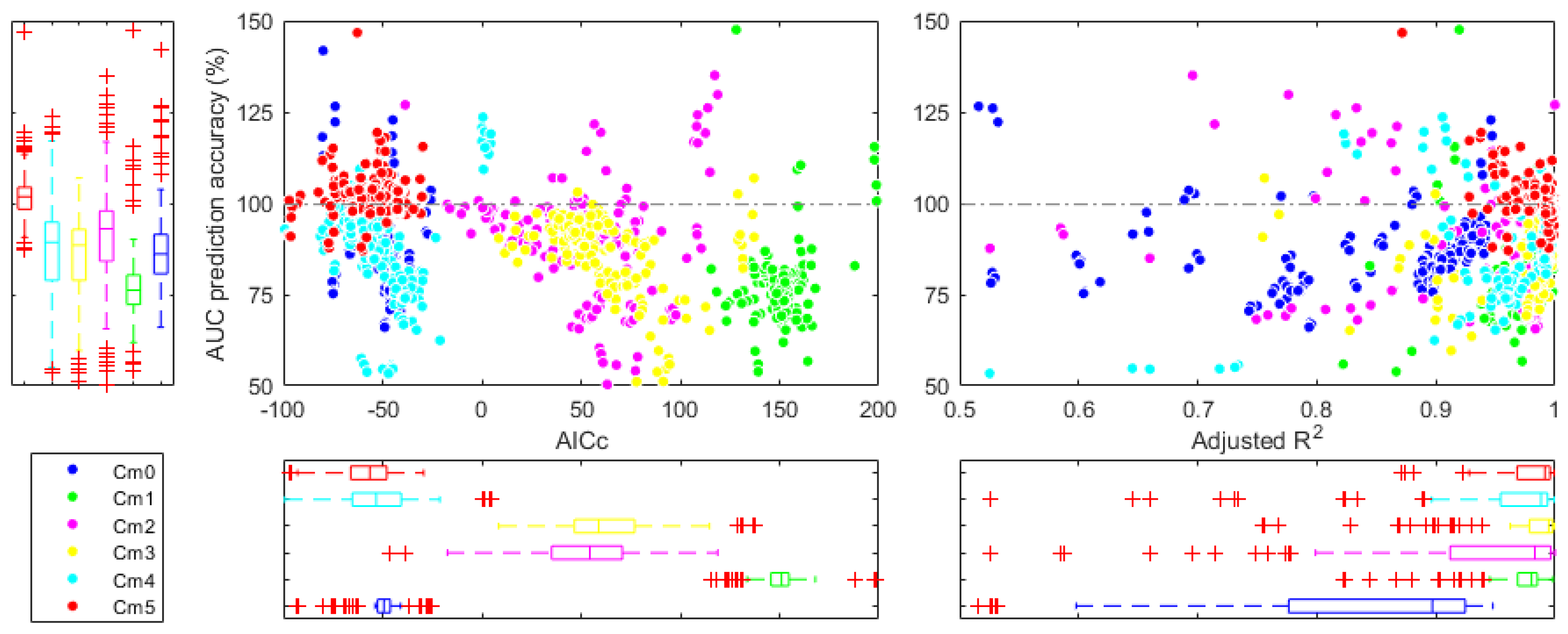
3.3. Estimation of AUC in Different Pharmacokinetic Scenarios
3.4. Estimation of AUC under Different Sampling Schemes
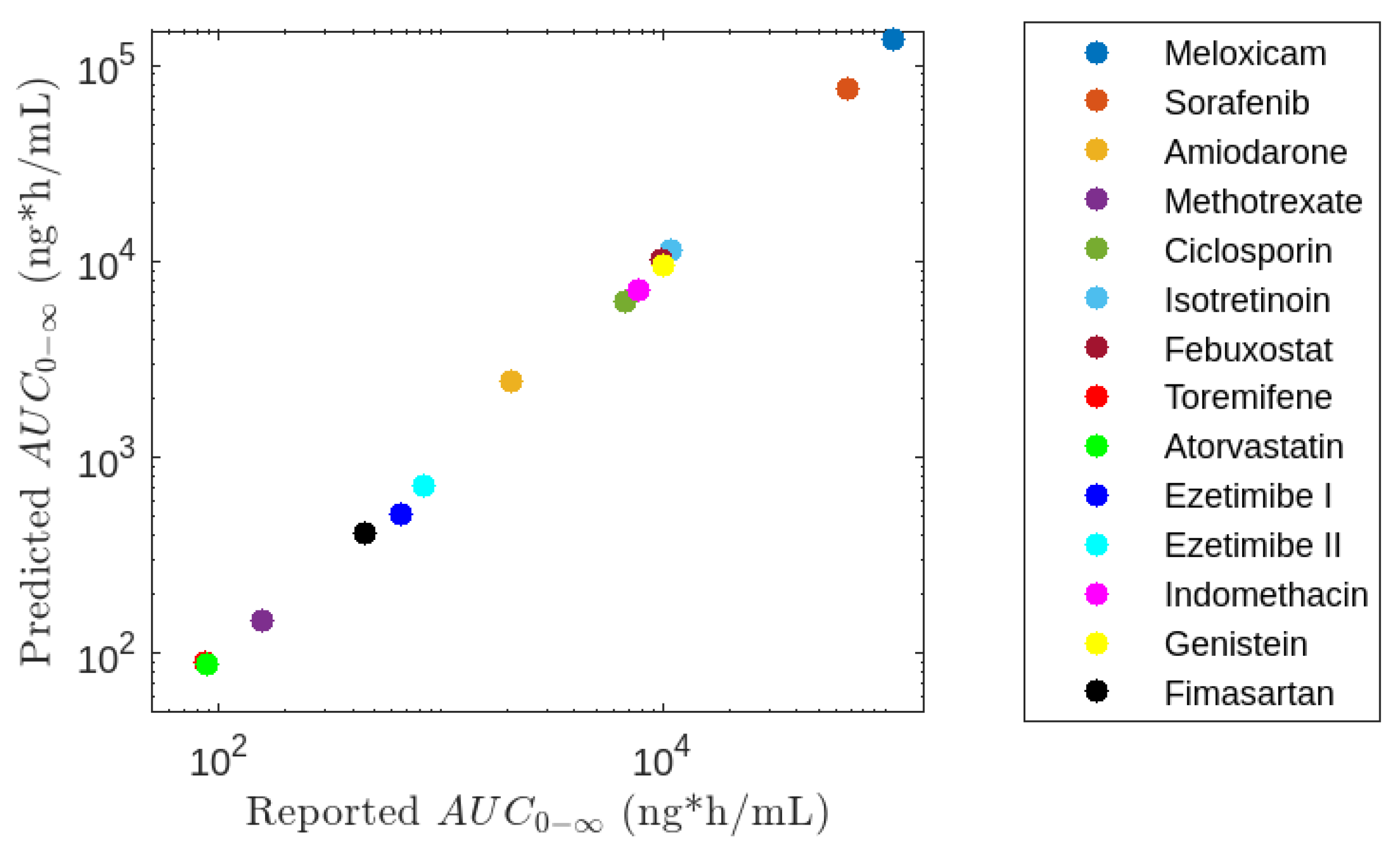
3.5. Application of Model M5
4. Discussion
4.1. Effect of EHC Degree on AUC
4.2. Simulation of Pharmacokinetic Scenarios and Fit to Empirical Models
4.3. Estimation of AUC in Different Pharmacokinetic Scenarios
4.4. Estimation of AUC under Different Sampling Schemes
4.5. Application of Model M5
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, Y.; Shao, J.; Jiang, Z.; Chen, J.; Gu, S.; Yu, S.; Zheng, K.; Jia, L. Drug Enterohepatic Circulation and Disposition: Constituents of Systems Pharmacokinetics. Drug Discov. Today 2014, 19, 326–340. [Google Scholar] [CrossRef]
- Jetter, A.; Kullak-Ublick, G.A. Drugs and Hepatic Transporters: A Review. Pharmacol. Res. 2020, 154, 104234. [Google Scholar] [CrossRef]
- Durník, R.; Šindlerová, L.; Babica, P.; Jurček, O. Bile Acids Transporters of Enterohepatic Circulation for Targeted Drug Delivery. Molecules 2022, 27, 2961. [Google Scholar] [CrossRef]
- Guiastrennec, B.; Sonne, D.; Hansen, M.; Bagger, J.; Lund, A.; Rehfeld, J.; Alskär, O.; Karlsson, M.; Vilsbøll, T.; Knop, F.; et al. Mechanism-Based Modeling of Gastric Emptying Rate and Gallbladder Emptying in Response to Caloric Intake. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 692–700. [Google Scholar] [CrossRef]
- Ivy, A.C. A Hormone Mechanism for Gall-Bladder Contraction & Evacuation. Am. J. Surg. 1929, 7, 455–459. [Google Scholar] [CrossRef]
- Shaffer, E.A.; McOrmond, P.; Duggan, H. Quantitative Cholescintigraphy: Assessment of Gallbladder Filling and Emptying and Duodenogastric Reflux. Gastroenterology 1980, 79, 899–906. [Google Scholar] [CrossRef]
- Portincasa, P.; Moschetta, A.; Giampaolo, M.; Palasciano, G. Diffuse Gastrointestinal Dysmotility by Ultrasonography, Manometry and Breath Tests in Colonic Inertia. Eur. Rev. Med. Pharmacol. Sci. 2000, 4, 81–87. [Google Scholar]
- Howard, P.J.; Murphy, G.M.; Dowling, R.H. Gall Bladder Emptying Patterns in Response to a Normal Meal in Healthy Subjects and Patients with Gall Stones: Ultrasound Study. Gut 1991, 32, 1406–1411. [Google Scholar] [CrossRef]
- Okour, M.; Brundage, R.C. Modeling Enterohepatic Circulation. Curr. Pharmacol. Rep. 2017, 3, 301–313. [Google Scholar] [CrossRef]
- Harrison, L.I.; Gibaldi, M. Influence of Cholestasis on Drug Elimination: Pharmacokinetics. J. Pharm. Sci. 1976, 65, 1346–1348. [Google Scholar] [CrossRef]
- Steimer, J.-L.; Plusquellec, Y.; Guillaume, A.; Boisvieux, J.-F. A Time-Lag Model for Pharmacokinetics of Drugs Subject to Enterohepatic Circulation. J. Pharm. Sci. 1982, 71, 297–302. [Google Scholar] [CrossRef]
- Colburn, W.A.; Hirom, P.C.; Parker, R.J.; Milburn, P. A Pharmacokinetic Model for Enterohepatic Recirculation in the Rat: Phenolphthalein, a Model Drug. Drug Metab. Dispos. 1979, 7, 100–102. [Google Scholar]
- Colburn, W.A. Pharmacokinetic and Biopharmaceutic Parameters During Enterohepatic Circulation of Drugs. J. Pharm. Sci. 1982, 71, 131–133. [Google Scholar] [CrossRef]
- Pedersen, P.V.; Miller, R. Pharmacokinetics and Bioavailability of Cimetidine in Humans. J. Pharm. Sci. 1980, 69, 394–398. [Google Scholar] [CrossRef]
- Pedersen, P.V.; Miller, R. Pharmacokinetics of Doxycycline Reabsorption. J. Pharm. Sci. 1980, 69, 204–207. [Google Scholar] [CrossRef]
- Miller, R. Pharmacokinetics and Bioavailability of Ranitidine in Humans. J. Pharm. Sci. 1984, 73, 1376–1379. [Google Scholar] [CrossRef]
- Ide, T.; Sasaki, T.; Maeda, K.; Higuchi, S.; Sugiyama, Y.; Ieiri, I. Quantitative Population Pharmacokinetic Analysis of Pravastatin Using an Enterohepatic Circulation Model Combined with Pharmacogenomic Information on SLCO1B1 and ABCC2 Polymorphisms. J. Clin. Pharmacol. 2009, 49, 1309–1317. [Google Scholar] [CrossRef]
- Funaki, T. Enterohepatic Circulation Model for Population Pharmacokinetic Analysis. J. Pharm. Pharmacol. 2010, 51, 1143–1148. [Google Scholar] [CrossRef]
- Strandgården, K.; Höglund, P.; Grönquist, L.; Svensson, L.; Gunnarsson, P.O. Absorption and Disposition Including Enterohepatic Circulation of (14C) Roquinimex after Oral Administration to Healthy Volunteers. Biopharm. Drug Dispos. 2000, 21, 53–67. [Google Scholar] [CrossRef]
- Rosner, G.; Panetta, J.; Innocenti, F.; Ratain, M. Pharmacogenetic Pathway Analysis of Irinotecan. Clin. Pharmacol. Ther. 2008, 84, 393–402. [Google Scholar] [CrossRef]
- Jiao, Z.; Ding, J.; Shen, J.; Liang, H.; Zhong, L.; Wang, Y.; Zhong, M.; Lu, W. Population Pharmacokinetic Modelling for Enterohepatic Circulation of Mycophenolic Acid in Healthy Chinese and the Influence of Polymorphisms in UGT1A9. Br. J. Clin. Pharmacol. 2008, 65, 893–907. [Google Scholar] [CrossRef]
- Sam, W.-J.; Akhlaghi, F.; Rosenbaum, S.E. Population Pharmacokinetics of Mycophenolic Acid and Its 2 Glucuronidated Metabolites in Kidney Transplant Recipients. J. Clin. Pharmacol. 2009, 49, 185–195. [Google Scholar] [CrossRef]
- de Winter, B.C.M.; van Gelder, T.; Sombogaard, F.; Shaw, L.M.; van Hest, R.M.; Mathot, R.A.A. Pharmacokinetic Role of Protein Binding of Mycophenolic Acid and Its Glucuronide Metabolite in Renal Transplant Recipients. J. Pharmacokinet. Pharmacodyn. 2009, 36, 541–564. [Google Scholar] [CrossRef]
- Yau, W.-P.; Vathsala, A.; Lou, H.-X.; Zhou, S.; Chan, E. Mechanism-Based Enterohepatic Circulation Model of Mycophenolic Acid and Its Glucuronide Metabolite: Assessment of Impact of Cyclosporine Dose in Asian Renal Transplant Patients. J. Clin. Pharmacol. 2009, 49, 684–699. [Google Scholar] [CrossRef]
- Berg, A.K.; Mandrekar, S.J.; Ziegler, K.L.A.; Carlson, E.C.; Szabo, E.; Ames, M.M.; Boring, D.; Limburg, P.J.; Reid, J.M. Population Pharmacokinetic Model for Cancer Chemoprevention with Sulindac in Healthy Subjects. J. Clin. Pharmacol. 2013, 53, 403–412. [Google Scholar] [CrossRef]
- Shepard, T.A.; Reuning, R.H.; Aarons, L.J. Estimation of Area under the Curve for Drugs Subject to Enterohepatic Cycling. J. Pharmacokinet. Biopharm. 1985, 13, 589–608. [Google Scholar] [CrossRef]
- Jain, L.; Woo, S.; Gardner, E.R.; Dahut, W.L.; Kohn, E.C.; Kummar, S.; Mould, D.R.; Giaccone, G.; Yarchoan, R.; Venitz, J.; et al. Population Pharmacokinetic Analysis of Sorafenib in Patients with Solid Tumours. Br. J. Clin. Pharmacol. 2011, 72, 294–305. [Google Scholar] [CrossRef]
- Huntjens, D.R.H.; Strougo, A.; Chain, A.; Metcalf, A.; Summerfield, S.; Spalding, D.J.M.; Danhof, M.; Della Pasqua, O. Population Pharmacokinetic Modelling of the Enterohepatic Recirculation of Diclofenac and Rofecoxib in Rats. Br. J. Pharmacol. 2008, 153, 1072–1084. [Google Scholar] [CrossRef]
- Wajima, T.; Yano, Y.; Oguma, T. A Pharmacokinetic Model for Analysis of Drug Disposition Profiles Undergoing Enterohepatic Circulation. J. Pharm. Pharmacol. 2010, 54, 929–934. [Google Scholar] [CrossRef]
- Edginton, A.N.; Zimmerman, E.I.; Vasilyeva, A.; Baker, S.D.; Panetta, J.C. Sorafenib Metabolism, Transport, and Enterohepatic Recycling: Physiologically Based Modeling and Simulation in Mice. Cancer Chemother. Pharmacol. 2016, 77, 1039–1052. [Google Scholar] [CrossRef]
- Abbiati, R.A.; Manca, D. Enterohepatic Circulation Effect in Physiologically Based Pharmacokinetic Models: The Sorafenib Case. Ind. Eng. Chem. Res. 2017, 56, 3156–3166. [Google Scholar] [CrossRef]
- Voronova, V.; Sokolov, V.; Al-Khaifi, A.; Straniero, S.; Kumar, C.; Peskov, K.; Helmlinger, G.; Rudling, M.; Angelin, B. A Physiology-Based Model of Bile Acid Distribution and Metabolism Under Healthy and Pathologic Conditions in Human Beings. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 149–170. [Google Scholar] [CrossRef]
- Gerner, B.; Scherf-Clavel, O. Physiologically Based Pharmacokinetic Modelling of Cabozantinib to Simulate Enterohepatic Recirculation, Drug–Drug Interaction with Rifampin and Liver Impairment. Pharmaceutics 2021, 13, 778. [Google Scholar] [CrossRef]
- Perry, C.; Davis, G.; Conner, T.M.; Zhang, T. Utilization of Physiologically Based Pharmacokinetic Modeling in Clinical Pharmacology and Therapeutics: An Overview. Curr. Pharmacol. Rep. 2020, 6, 71–84. [Google Scholar] [CrossRef]
- Tan, Y.-M.; Worley, R.R.; Leonard, J.A.; Fisher, J.W. Challenges Associated with Applying Physiologically Based Pharmacokinetic Modeling for Public Health Decision-Making. Toxicol. Sci. 2018, 162, 341–348. [Google Scholar] [CrossRef]
- Weiss, M. Empirical Models for Fitting of Oral Concentration Time Curves with and without an Intravenous Reference. J. Pharmacokinet. Pharmacodyn. 2017, 44, 193–201. [Google Scholar] [CrossRef]
- Prémaud, A.; Debord, J.; Rousseau, A.; Le Meur, Y.; Toupance, O.; Lebranchu, Y.; Hoizey, G.; Le Guellec, C.; Marquet, P. A Double Absorption-Phase Model Adequately Describes Mycophenolic Acid Plasma Profiles in de Novo Renal Transplant Recipients given Oral Mycophenolate Mofetil. Clin. Pharmacokinet. 2005, 44, 837–847. [Google Scholar] [CrossRef]
- Alpízar-Salazar, M.; Alejandro Trejo-Rangel, M.; de Jesús Reséndiz-Rojas, J.; Frydman, T.D.; Ramos-Mundo, C. A Modified Compartmental Pharmacokinetic Model of Enterohepatic Circulation for Simvastatin in Healthy Mexican Subjects. A Pilot Study. J. Pharm. Pharmacol. Res. 2020, 4. [Google Scholar] [CrossRef]
- Zahr, N.; Amoura, Z.; Debord, J.; Hulot, J.-S.; Saint-Marcoux, F.; Marquet, P.; Piette, J.C.; Lechat, P. Pharmacokinetic Study of Mycophenolate Mofetil in Patients with Systemic Lupus Erythematosus and Design of Bayesian Estimator Using Limited Sampling Strategies. Clin. Pharmacokinet. 2008, 47, 277–284. [Google Scholar] [CrossRef]
- Ibarra, M.; Trocóniz, I.F.; Fagiolino, P. Enteric Reabsorption Processes and Their Impact on Drug Pharmacokinetics. Sci. Rep. 2021, 11, 5794. [Google Scholar] [CrossRef]
- van Gelder, T.; Le Meur, Y.; Shaw, L.M.; Oellerich, M.; DeNofrio, D.; Holt, C.; Holt, D.W.; Kaplan, B.; Kuypers, D.; Meiser, B.; et al. Therapeutic Drug Monitoring of Mycophenolate Mofetil in Transplantation. Ther. Drug Monit. 2006, 28, 145–154. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and Comparison of Dissolution Profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Drevon, D.; Fursa, S.R.; Malcolm, A.L. Intercoder Reliability and Validity of WebPlotDigitizer in Extracting Graphed Data. Behav. Modif. 2017, 41, 323–339. [Google Scholar] [CrossRef]
- Bae, J.-W.; Choi, C.-I.; Jang, C.-G.; Lee, S.-Y. Effects of CYP2C9*1/*13 on the Pharmacokinetics and Pharmacodynamics of Meloxicam. Br. J. Clin. Pharmacol. 2011, 71, 550–555. [Google Scholar] [CrossRef]
- Huang, F.; Ajavon, A.; Huang, E.; Lettieri, J.; Liu, R.; Peña, C.; Berse, M. No Effect of Levothyroxine and Levothyroxine-Induced Subclinical Thyrotoxicosis on the Pharmacokinetics of Sorafenib in Healthy Male Subjects. Thyroid 2017, 27, 1118–1127. [Google Scholar] [CrossRef]
- Filho, H.; Ilha, J.; Silva, L.; Borges, A.; Mendes, G.; Nucci, G. Comparative Bioavailability Study with Two Amiodarone Tablet Formulations Administered with and without Food in Healthy Subjects. Arzneimittelforschung 2011, 57, 582–590. [Google Scholar] [CrossRef]
- Danafar, H.; Hamidi, M. Pharmacokinetics and Bioequivalence of Methotrexate in Human Plasma Studied by Liquid Chromatography-Mass Spectrometry (LC-MS). Jundishapur J. Nat. Pharm. Prod. 2016, 11. [Google Scholar] [CrossRef]
- Ducharme, M.P.; Warbasse, L.H.; Edwards, D.J. Disposition of Intravenous and Oral Cyclosporine after Administration with Grapefruit Juice*. Clin. Pharmacol. Ther. 1995, 57, 485–491. [Google Scholar] [CrossRef]
- Madan, S.; Kumar, S.; Segal, J. Comparative Pharmacokinetic Profiles of a Novel Low-Dose Micronized-Isotretinoin 32 Mg Formulation and Lidose-Isotretinoin 40 Mg in Fed and Fasted Conditions: Two Open-Label, Randomized, Crossover Studies in Healthy Adult Participants. Acta Derm. Venereol. 2019, 100, adv00049. [Google Scholar] [CrossRef]
- Patel, D.S.; Sharma, N.; Patel, M.C.; Patel, B.N.; Shrivastav, P.S.; Sanyal, M. Liquid Chromatography Tandem Mass Spectrometry Method for Determination of Febuxostat in Human Plasma to Support a Bioequivalence Study. J. Adv. Pharm. Sci. Technol. 2013, 1, 37–50. [Google Scholar] [CrossRef]
- DeGregorio, M.W.; Wurz, G.T.; Taras, T.L.; Erkkola, R.U.; Halonen, K.H.; Huupponen, R.K. Pharmacokinetics of (Deaminohydroxy)Toremifene in Humans: A New, Selective Estrogen-Receptor Modulator. Eur. J. Clin. Pharmacol. 2000, 56, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Rodríguez, O.; Torres-Roque, I.; Martínez-Delgado, M.; Escobedo-Moratilla, A.; Pérez-Urizar, J. Pharmacokinetic Non-Interaction Analysis in a Fixed-Dose Formulation in Combination of Atorvastatin and Ezetimibe. Front. Pharmacol. 2014, 5, 261. [Google Scholar] [CrossRef]
- Palmer, J.L.; Kunhihitlu, A.; Costantini, A.; Esquivel, F.; Roush, J.; Edwards, K.; Hill, T.W.K. Pharmacokinetic Bioequivalence Crossover Study of Branded Generic and Innovator Formulations of the Cholesterol Lowering Agent Ezetimibe. Clin. Pharmacol. Drug Dev. 2014, 3, 242–248. [Google Scholar] [CrossRef]
- Olugemo, K.; Solorio, D.; Sheridan, C.; Young, C.L. Pharmacokinetics and Safety of Low-Dose Submicron Indomethacin 20 and 40 Mg Compared with Indomethacin 50 Mg. Postgrad. Med. 2015, 127, 223–231. [Google Scholar] [CrossRef]
- Limopasmanee, W.; Chansakaow, S.; Rojanasthien, N.; Manorot, M.; Sangdee, C.; Teekachunhatean, S. Effects of the Chinese Herbal Formulation (Liu Wei Di Huang Wan) on the Pharmacokinetics of Isoflavones in Postmenopausal Women. Biomed. Res. Int. 2015, 2015, 902702. [Google Scholar] [CrossRef]
- Yang, E.; Lee, S.; Lee, H.; Hwang, I.; Jang, I.-J.; Yu, K.-S.; Lee, S. Pharmacokinetic Comparison between Fixed-Dose Combination of Fimasartan/Amlodipine 60/10 Mg and the Corresponding Loose Combination through Partial Replicated Crossover Study in Healthy Subjects. Transl. Clin. Pharmacol. 2019, 27, 134. [Google Scholar] [CrossRef]
- Okour, M.; Brundage, R.C. A Gallbladder-Based Enterohepatic Circulation Model for Pharmacokinetic Studies. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 493–504. [Google Scholar] [CrossRef]
- Gandelman, K.; Malhotra, B.; LaBadie, R.R.; Crownover, P.; Bergstrom, T. Analytes of Interest and Choice of Dose: Two Important Considerations in the Design of Bioequivalence Studies with Atorvastatin. J. Bioequivalence Bioavailab. 2011, 3, 4. [Google Scholar] [CrossRef]
- Granger, D.K. Enteric-Coated Mycophenolate Sodium: Results of Two Pivotal Global Multicenter Trials. Transplant. Proc. 2001, 33, 3241–3244. [Google Scholar] [CrossRef]
- Ichikawa, T.; Ishida, S.; Sakiya, Y.; Sawada, Y.; Hanano, M. Biliary Excretion and Enterohepatic Cycling of Glycyrrhizin in Rats. J. Pharm. Sci. 1986, 75, 672–675. [Google Scholar] [CrossRef]
- Shou, M.; Lu, W.; Kari, P.H.; Xiang, C.; Liang, Y.; Lu, P.; Cui, D.; Emary, W.B.; Michel, K.B.; Adelsberger, J.K.; et al. Population Pharmacokinetic Modeling for Enterohepatic Recirculation in Rhesus Monkey. Eur. J. Pharm. Sci. 2005, 26, 151–161. [Google Scholar] [CrossRef]
- Zhong, Z.-Y.; Sun, B.-B.; Shu, N.; Xie, Q.-S.; Tang, X.-G.; Ling, Z.-L.; Wang, F.; Zhao, K.-J.; Xu, P.; Zhang, M.; et al. Ciprofloxacin Blocked Enterohepatic Circulation of Diclofenac and Alleviated NSAID-Induced Enteropathy in Rats Partly by Inhibiting Intestinal β-Glucuronidase Activity. Acta Pharmacol. Sin. 2016, 37, 1002–1012. [Google Scholar] [CrossRef]
- Meaney, C.J.; Sudchada, P.; Consiglio, J.D.; Wilding, G.E.; Cooper, L.M.; Venuto, R.C.; Tornatore, K.M. Influence of Calcineurin Inhibitor and Sex on Mycophenolic Acid Pharmacokinetics and Adverse Effects Post-Renal Transplant. J. Clin. Pharmacol. 2019, 59, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Shepard, T.A.; Reuning, R.H.; Aarons, L.J. Interpretation of Area Under the Curve Measurements for Drugs Subject to Enterohepatic Cycling. J. Pharm. Sci. 1985, 74, 227–228. [Google Scholar] [CrossRef]
- Scarfia, R.V.; Clementi, A.; Granata, A. Rhabdomyolysis and Acute Kidney Injury Secondary to Interaction between Simvastatin and Cyclosporine. Ren. Fail. 2013, 35, 1056–1057. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Nies, A.T.; Brom, M.; Hummel-Eisenbeiss, J.; Spring, H.; Keppler, D. A Common Dubin-Johnson Syndrome Mutation Impairs Protein Maturation and Transport Activity of MRP2 (ABCC2). Am. J. Physiol. -Gastrointest. Liver Physiol. 2003, 284, G165–G174. [Google Scholar] [CrossRef]
- Dong, J.; Yu, X.; Wang, L.; Sun, Y.; Chen, X.; Wang, G. Effects of Cyclosporin A and Itraconazole on the Pharmacokinetics of Atorvastatin in Rats. Acta Pharmacol. Sin. 2008, 29, 1247–1252. [Google Scholar] [CrossRef]
- Le Meur, Y.; Büchler, M.; Thierry, A.; Caillard, S.; Villemain, F.; Lavaud, S.; Etienne, I.; Westeel, P.-F.; Hurault de Ligny, B.; Rostaing, L.; et al. Individualized Mycophenolate Mofetil Dosing Based on Drug Exposure Significantly Improves Patient Outcomes after Renal Transplantation. Am. J. Transpl. 2007, 7, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Bullingham, R.E.; Nicholls, A.J.; Kamm, B.R. Clinical Pharmacokinetics of Mycophenolate Mofetil. Clin. Pharmacokinet. 1998, 34, 429–455. [Google Scholar] [CrossRef]
- Qian, L.; Jiao, Z.; Zhong, M. Effect of Meal Timings and Meal Content on the AUC0-12h of Mycophenolic Acid: A Simulation Study. Clin. Pharmacol. Drug Dev. 2022, 11, 1331–1340. [Google Scholar] [CrossRef]
- Li, J.; Chai, H.; Li, Y.; Chai, X.; Zhao, Y.; Zhao, Y.; Tao, T.; Xiang, X. A Three-Pulse Release Tablet for Amoxicillin: Preparation, Pharmacokinetic Study and Physiologically Based Pharmacokinetic Modeling. PLoS ONE 2016, 11, e0160260. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.L.; Chaudhary, I.; Kollins, S.H.; Padilla, A. A Pharmacokinetic Study of Methylphenidate Hydrochloride Multilayer Extended-Release Capsules (Aptensio XR®) in Preschool-Aged Children with Attention-Deficit/Hyperactivity Disorder. Paediatr. Drugs 2020, 22, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.-B.; Debord, J.; Marquet, P. Comment on “Population Pharmacokinetics of Mycophenolic Acid: An Update”. Clin. Pharmacokinet. 2018, 57, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Stoeckel, H.; Hengstmann, J.H.; Schüttler, J. Pharmacokinetics of Fentanyl as a Possible Explanation for Recurrence of Respiratory Depression. Br. J. Anaesth. 1979, 51, 741–745. [Google Scholar] [CrossRef]
- Estebe, J.P.; Le Corre, P.; Levron, J.C.; Le Moing, J.P.; Le Naoures, A.; Ecoffey, C. Pilot Study on the Effect of Tourniquet Use on Sufentanil Pharmacokinetics. J. Clin. Anesth. 2002, 14, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Cockshott, I.D.; Briggs, L.P.; Douglas, E.J.; White, M. Pharmacokinetics of Propofol in Female Patients. Studies Using Single Bolus Injections. Br. J. Anaesth. 1987, 59, 1103–1110. [Google Scholar] [CrossRef]
- Fischler, M.; Levron, J.C.; Trang, H.; Vaxelaire, J.F.; Flaisler, B.; Vourc’h, G. Pharmacokinetics of Phenoperidine in Anaesthetized Patients Undergoing General Surgery. Br. J. Anaesth. 1985, 57, 872–876. [Google Scholar] [CrossRef]
- Sherwin, C.M.T.; Fukuda, T.; Brunner, H.I.; Goebel, J.; Vinks, A.A. The Evolution of Population Pharmacokinetic Models to Describe the Enterohepatic Recycling of Mycophenolic Acid in Solid Organ Transplantation and Autoimmune Disease. Clin. Pharmacokinet. 2011, 50, 1–24. [Google Scholar] [CrossRef]
- Kim, T.H.; Shin, S.; Landersdorfer, C.B.; Chi, Y.H.; Paik, S.H.; Myung, J.; Yadav, R.; Horkovics-Kovats, S.; Bulitta, J.B.; Shin, B.S. Population Pharmacokinetic Modeling of the Enterohepatic Recirculation of Fimasartan in Rats, Dogs, and Humans. AAPS J. 2015, 17, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Koloskoff, K.; Benito, S.; Chambon, L.; Dayan, F.; Marquet, P.; Jacqz-Aigrain, E.; Woillard, J.-B. Limited Sampling Strategy and Population Pharmacokinetic Model of Mycophenolic Acid in Pediatric Patients with Systemic Lupus Erythematosus: Application of a Double Gamma Absorption Model with SAEM Algorithm. Eur. J. Clin. Pharmacol. 2024, 80, 83–92. [Google Scholar] [CrossRef]




| Sampling Type | Sampling Times (h) | Number of Samples | Description | |
|---|---|---|---|---|
| Meal-based sampling | 0, 0.5, 1, 2, 3, 4, 5, 6, 10, 11, 12, 16, 17, 18, 29, 53, 77 | 17 | Focuses on sampling before and after food intake | |
| Conventional sampling | 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 7, 9, 12, 24, 48, 72 | 17 | Prioritize sampling at the beginning to characterize | |
| Sampling for TDM | TDM1 | 0, 1, 3, 4, 6 | 5 | Reduce the number of samples and restrict their taking to the first hours after administration |
| TDM2 | 0, 1, 3, 4, 6, 10 | 6 | ||
| TDM3 | 0, 1, 3, 4, 6, 10, 12 | 7 | ||
| TDM4 | 0, 1, 3, 4, 6, 10, 12, 16 | 8 | ||
| TDM5 | 0, 1, 3, 4, 6, 10, 12, 16, 18 | 9 | ||
| Scenario | Scenario | Scenario | |||
|---|---|---|---|---|---|
| b | 26.45 ± 2.73 | 23.91 ± 3.24 | 23.77 ± 3.31 | ||
| b | 16.51 ± 3.86 | 23.86 ± 3.25 | 24.10 ± 3.13 | ||
| b | 4.89 ± 0.64 | 23.89 ± 3.24 | a,b | 9.74 ± 4.34 | |
| a,b | 87.52 ± 9.84 | 23.89 ± 3.23 | 20.54 ± 4.54 | ||
| b | 12.86 ± 3.56 | 24.07 ± 3.15 | 23.49 ± 3.38 | ||
| b | 29.06 ± 2.09 | 23.38 ± 3.51 | 24.64 ± 3.25 |
| Model | Accuracy (%) | ||
|---|---|---|---|
| M0 | 0.827 ± 0.14 | −51.9 ± 12.3 | 88.4 ± 18.7 |
| M1 | 0.940 ± 0.17 | 150.4 ± 12.6 | 75.9 ± 13.0 |
| M2 | 0.888 ± 0.26 | 52.1 ± 31.8 | 89.6 ± 17.3 |
| M3 | 0.931 ± 0.20 | 63.3 ± 25.8 | 84.2 ± 14.0 |
| M4 | 0.943 ± 0.12 | −51.1 ± 19.1 | 86.4 ± 16.2 |
| M5 | 0.979 ± 0.02 | −58.1 ± 14.7 | 105.9 ± 21.4 |
| Drug | Number of Samples | Number of Parameters | Accuracy (%) | (h) | Reference | |
|---|---|---|---|---|---|---|
| Meloxicam | 11 | 9 | 124.1 | 0.8013 | 39.5 | [44] |
| Sorafenib | 14 | 9 | 110.6 | 0.8791 | 25.3 | [45] |
| Amiodarone | 15 | 9 | 117.6 | 0.9689 | 5.1 a | [46] |
| Methotrexate | 12 | 9 | 95.4 | 0.9684 | 5.3 | [47] |
| Cyclosporin | 11 | 9 | 93.6 | 0.9494 | 8.1 | [48] |
| Isotretinoin | 22 | 9 | 104.6 | 0.9591 | 23.5 | [49] |
| Febuxostat | 19 | 9 | 102.1 | 0.9652 | 2.78 | [50] |
| Toremifene | 10 | 9 | 104.1 | 0.9912 | 29.7 | [51] |
| Atorvastatin | 17 | 9 | 98.5 | 0.9400 | 3.9 | [52] |
| Ezetimibe | 25 | 9 | 78.4 | 0.8524 | 15.1 | [53] |
| Ezetimibe | 25 | 9 | 84.1 | 0.8586 | 13.8 | [53] |
| Indomethacin | 23 | 9 | 92.2 | 0.9736 | 6.8 | [54] |
| Genistein | 11 | 9 | 95.1 | 0.9527 | 4.1 | [55] |
| Fimasartan | 17 | 9 | 90.5 | 0.7552 | 5.6 | [56] |
| Model | Minimum Number of Parameters and Samples per Peak a | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| M0 | 3 | 3 | 3 | 3 |
| M1 | 5 | 8 | 11 | 14 |
| M2 | 4 | 7 | 10 | 13 |
| M3 | 4 | 7 | 10 | 13 |
| M4 | 3 | 5 | 7 | 9 |
| M5 | 3 | 5 | 7 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpízar, M.; de Jesús Reséndiz, J.; García Martínez, E.; Dwivedi, S.; Trejo, M.A. Pharmacokinetic Simulation and Area under the Curve Estimation of Drugs Subject to Enterohepatic Circulation. Pharmaceutics 2024, 16, 1044. https://doi.org/10.3390/pharmaceutics16081044
Alpízar M, de Jesús Reséndiz J, García Martínez E, Dwivedi S, Trejo MA. Pharmacokinetic Simulation and Area under the Curve Estimation of Drugs Subject to Enterohepatic Circulation. Pharmaceutics. 2024; 16(8):1044. https://doi.org/10.3390/pharmaceutics16081044
Chicago/Turabian StyleAlpízar, Melchor, José de Jesús Reséndiz, Elisa García Martínez, Sanyog Dwivedi, and Miguel Alejandro Trejo. 2024. "Pharmacokinetic Simulation and Area under the Curve Estimation of Drugs Subject to Enterohepatic Circulation" Pharmaceutics 16, no. 8: 1044. https://doi.org/10.3390/pharmaceutics16081044