
Impact of Hot-Melt Extrusion on Glibenclamide’s Physical and Chemical States and Dissolution Behavior: Case Studies with Three Polymer Blend Matrices
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Hot-Melt Extrusion
2.3. Thermogravimetric Analysis (TGA)
2.4. Temperature Modulated Differential Scanning Calorimetry (MDSC)
2.5. X-ray Diffraction (XRD)
2.6. High-Performance Liquid Chromatography (HPLC) Analysis and Drug Content Determination
2.7. Mass Spectrometry (MS) Analysis
2.8. Determination of the Saturation Concentration and the Two-Stage Glibenclamide Dissolution Kinetics
2.9. Water Content and Dry Mass Loss Kinetics of the Extrudates
2.10. Optical Imaging of Extrudates
2.11. Scanning Electron Microscopy (SEM) Coupled with Energy Dispersive X-ray (EDX)
3. Results and Discussion
3.1. Hot-Melt Extrusion Process Parameters
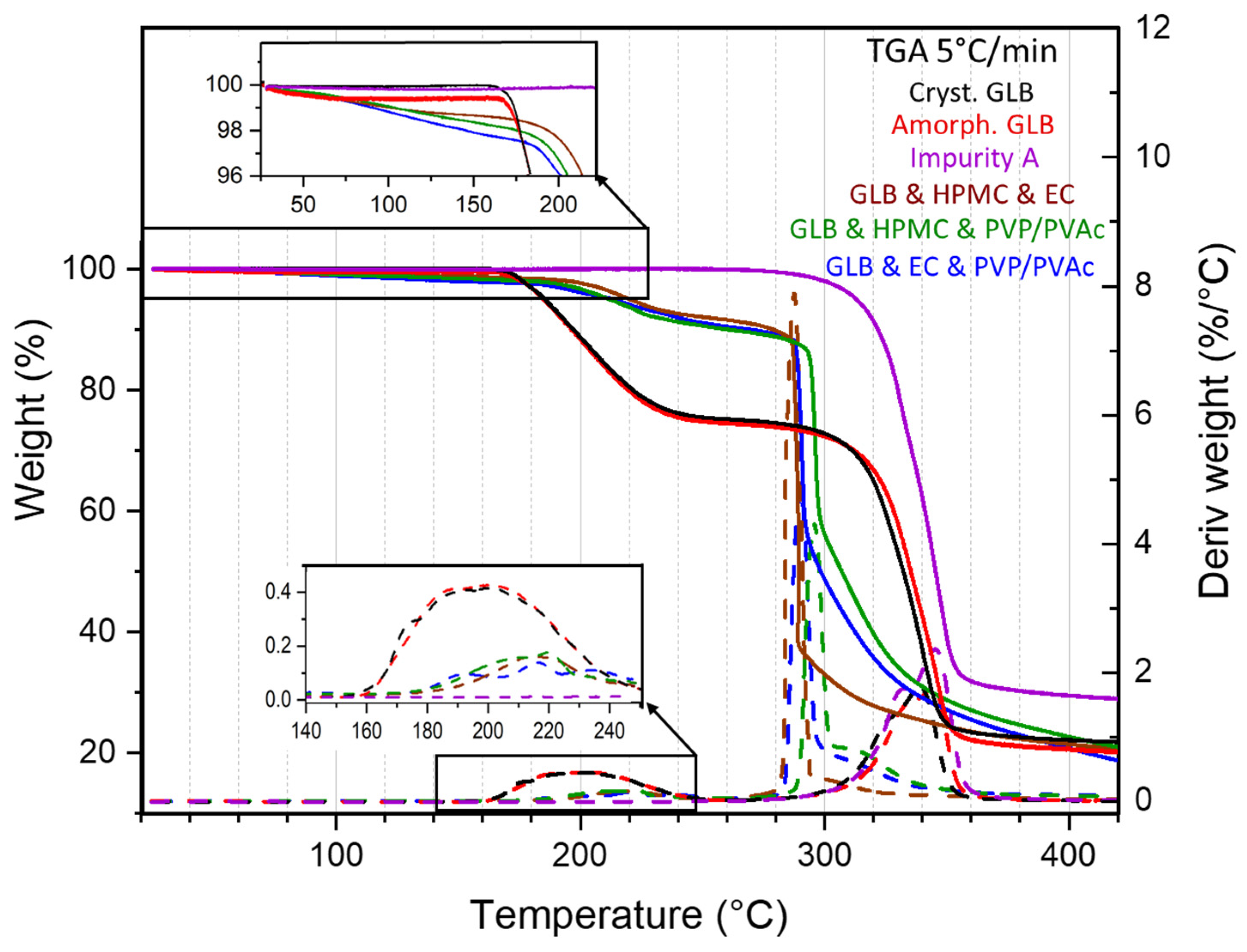
3.2. The Thermal Degradation of Glibenclamide
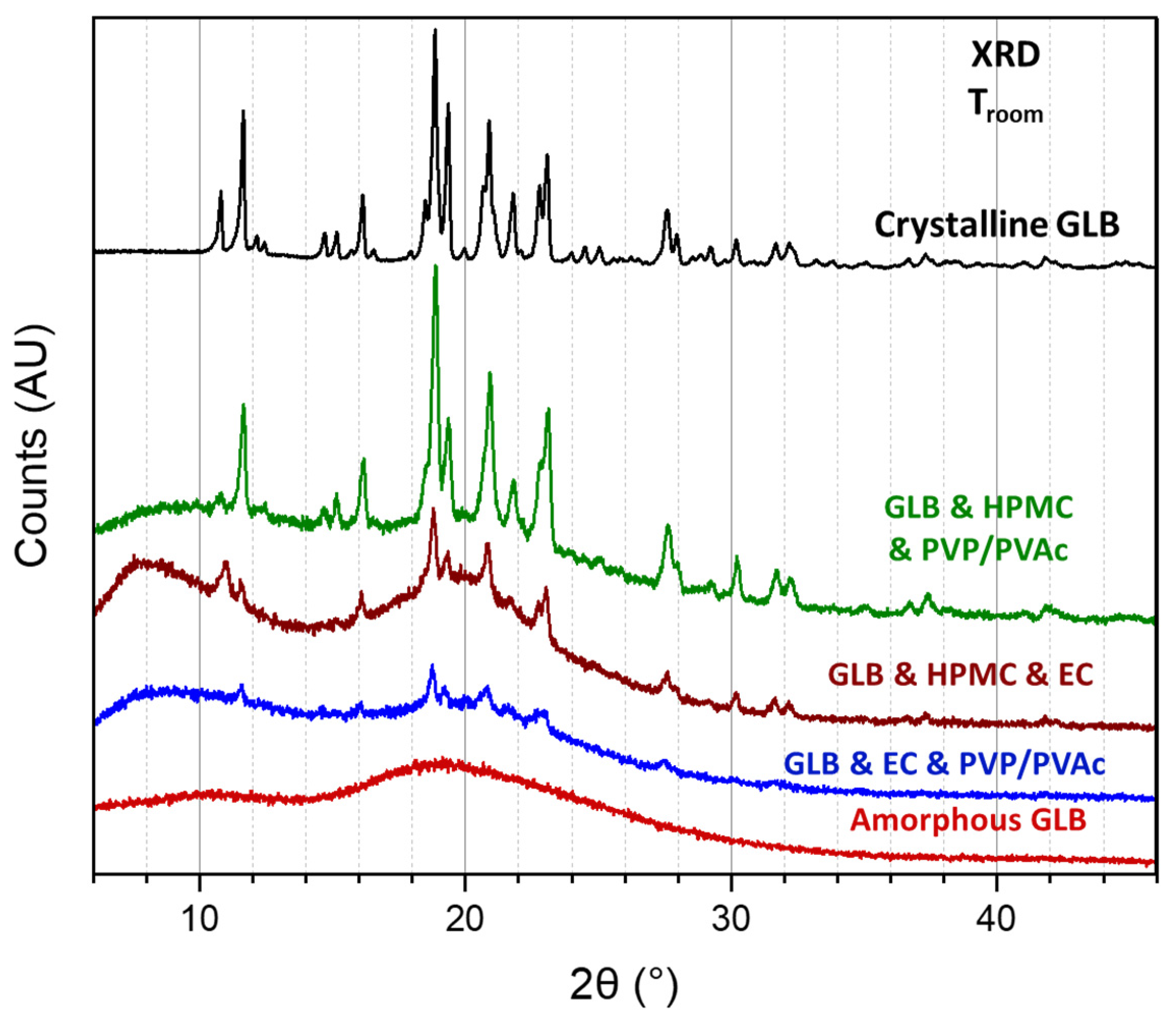
3.3. Physical State Analysis
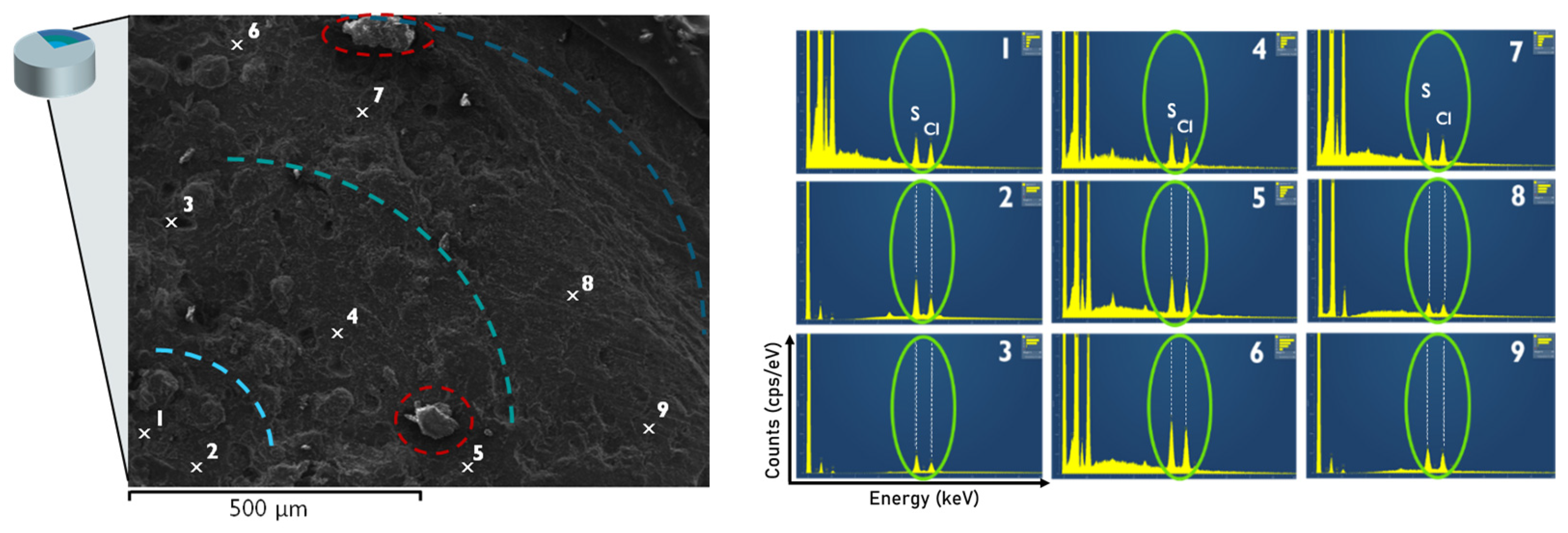
3.4. Optical Appearance of Extruded Matrices via SEM Coupled with EDX Analysis
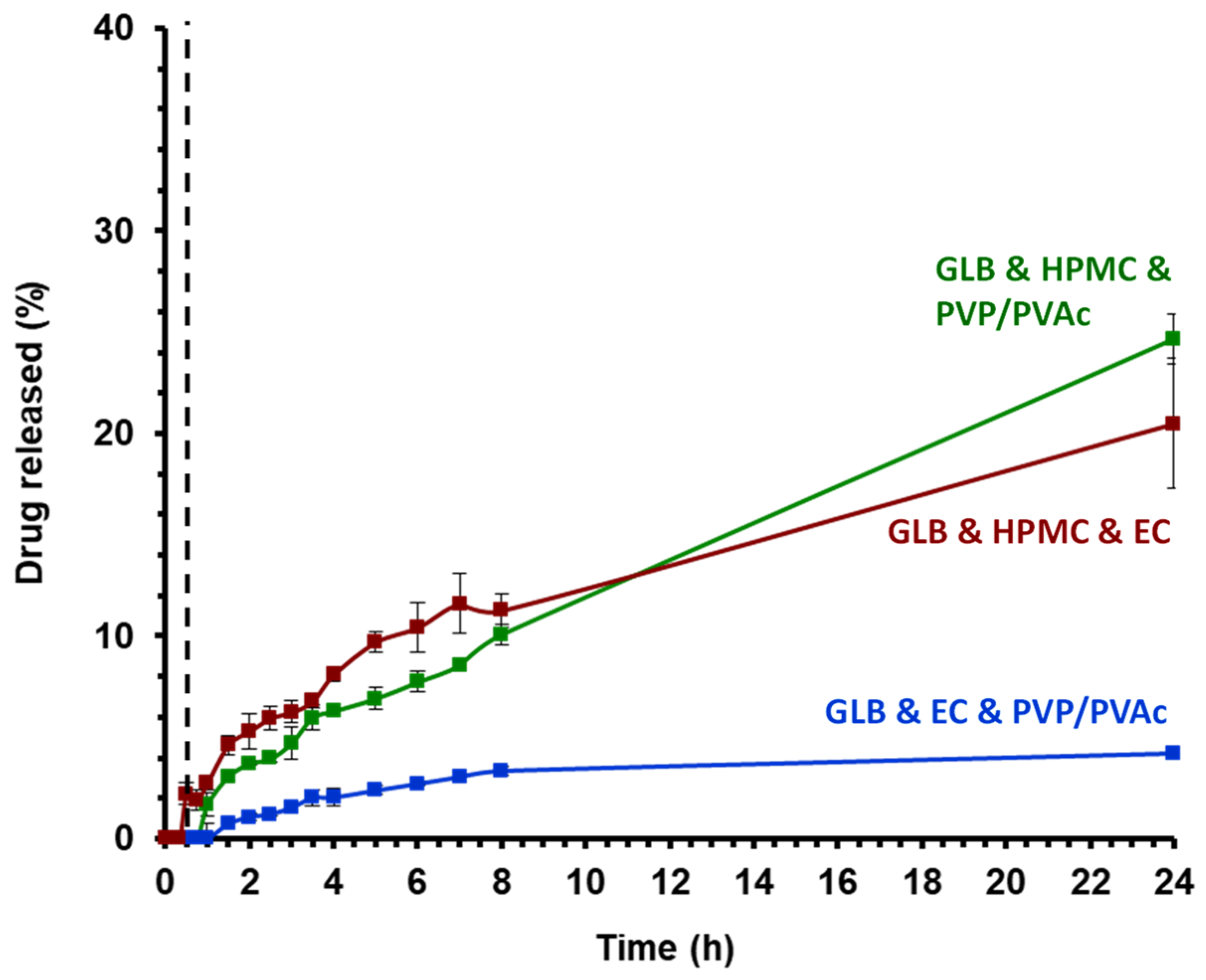
3.5. Dissolution Kinetics and Optical Appearance of Ternary Blends Obtained by HME
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choiri, S.; Sulaiman, T.N.S.; Rohman, A. Characterization of Eudragit types and Kollidon SR inter-polymer complexes and their effects on the drug release. J. Appl. Pharm. Sci. 2019, 9, 58–68. [Google Scholar] [CrossRef]
- Borde, S.; Paul, S.K.; Chauhan, H. Ternary solid dispersions: Classification and formulation considerations. Drug Dev. Ind. Pharm. 2021, 47, 1011–1028. [Google Scholar] [CrossRef]
- Crowley, M.M.; Zhang, F.; Repka, M.A.; Thumma, S.; Upadhye, S.B.; Battu, S.K.; McGinity, J.W.; Martin, C. Pharmaceutical Applications of Hot-Melt Extrusion: Part I. Drug Dev. Ind. Pharm. 2007, 33, 909–926. [Google Scholar] [CrossRef]
- Thakkar, R.; Thakkar, R.; Pillai, A.; Ashour, E.A.; Repka, M.A. Systematic screening of pharmaceutical polymers for hot melt extrusion processing: A comprehensive review. Int. J. Pharm. 2020, 576, 118989. [Google Scholar] [CrossRef]
- Censi, R.; Gigliobianco, M.; Casadidio, C.; Di Martino, P. Hot Melt Extrusion: Highlighting Physicochemical Factors to Be Investigated While Designing and Optimizing a Hot Melt Extrusion Process. Pharmaceutics 2018, 10, 89. [Google Scholar] [CrossRef]
- Stanković, M.; Frijlink, H.W.; Hinrichs, W.L.J. Polymeric formulations for drug release prepared by hot melt extrusion: Application and characterization. Drug Discov. Today 2015, 20, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Van Den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, 79–85. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef] [PubMed]
- Kolter, K.; Karl, M.; Gryczke, A. Hot-Melt Extrusion with BASF Pharma Polymers, 2nd ed.; BASF SE: Ludwigshafen, Germany, 2012; pp. 19–34. [Google Scholar]
- Matić, J.; Alva, C.; Witschnigg, A.; Eder, S.; Reusch, K.; Paudel, A.; Khinast, J. Towards predicting the product quality in hot-melt extrusion: Small scale extrusion. Int. J. Pharm. 2020, 2, 100062. [Google Scholar] [CrossRef]
- Prasad, E.; Islam, M.T.; Goodwin, D.J.; Megarry, A.J.; Halbert, G.W.; Florence, A.J.; Robertson, J. Development of a hot-melt extrusion (HME) process to produce drug loaded AffinisolTM 15LV filaments for fused filament fabrication (FFF) 3D printing. Addit. Manuf. 2019, 29, 100776. [Google Scholar] [CrossRef]
- Kukkonen, J.; Ervasti, T.; Laitinen, R. Production and characterization of glibenclamide incorporated PLA filaments for 3D printing by fused deposition modeling. J. Drug Deliv. Sci. Technol. 2022, 77, 103843. [Google Scholar] [CrossRef]
- Alshafiee, M.; Aljammal, M.K.; Markl, D.; Ward, A.; Walton, K.; Blunt, L.; Korde, S.; Pagire, S.K.; Kelly, A.; Paradkar, A.; et al. Hot-melt extrusion process impact on polymer choice of glyburide solid dispersions: The effect of wettability and dissolution. Int. J. Pharm. 2019, 559, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, F.; Zhang, C.; Ping, Q. Use of polymer combinations in the preparation of solid dispersions of a thermally unstable drug by hot-melt extrusion. Acta Pharm. Sin. B 2013, 3, 263–272. [Google Scholar] [CrossRef]
- Huang, S.; O’Donnell, K.P.; Delpon De Vaux, S.M.; O’Brien, J.; Stutzman, J.; Williams, R.O. Processing thermally labile drugs by hot-melt extrusion: The lesson with gliclazide. Eur. J. Pharm. Biopharm. 2017, 119, 56–67. [Google Scholar] [CrossRef]
- Baronsky-Probst, J.; Möltgen, C.-V.; Kessler, W.; Kessler, R.W. Process design and control of a twin screw hot melt extrusion for continuous pharmaceutical tamper-resistant tablet production. Eur. J. Pharm. Sci. 2016, 87, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Bansal, G.; Singh, M.; Jindal, K.C.; Singh, S. Ultraviolet-Photodiode Array and High-Performance Liquid Chromatographic/Mass Spectrometric Studies on Forced Degradation Behavior of Glibenclamide and Development of a Validated Stability-Indicating Method. J. AOAC Int. 2008, 91, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.; Brunsteiner, M.; Modhave, D.; Paudel, A. Role of Crystal Disorder and Mechanoactivation in Solid-State Stability of Pharmaceuticals. J. Pharm. Sci. 2023, 112, 1539–1565. [Google Scholar] [CrossRef] [PubMed]
- DiNunzio, J.C.; Brough, C.; Hughey, J.R.; Miller, D.A.; Williams, R.O., III; McGinity, J.W. Fusion production of solid dispersions containing a heat-sensitive active ingredient by hot melt extrusion and Kinetisol® dispersing. Eur. J. Pharm. Biopharm. 2010, 74, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Hughey, J.R.; DiNunzio, J.C.; Bennett, R.C.; Brough, C.; Miller, D.A.; Ma, H.; Williams, R.O., III; McGinity, J.W. Dissolution Enhancement of a Drug Exhibiting Thermal and Acidic Decomposition Characteristics by Fusion Processing: A Comparative Study of Hot Melt Extrusion and KinetiSol® Dispersing. AAPS PharmSciTech 2010, 11, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Mah, P.T.; Laaksonen, T.; Rades, T.; Aaltonen, J.; Peltonen, L.; Strachan, C.J. Unravelling the Relationship between Degree of Disorder and the Dissolution Behavior of Milled Glibenclamide. Mol. Pharm. 2014, 11, 234–242. [Google Scholar] [CrossRef]
- Wojnarowska, Z.; Grzybowska, K.; Adrjanowicz, K.; Kaminski, K.; Paluch, M.; Hawelek, L.; Wrzalik, R.; Dulski, M.; Sawicki, W.; Mazgalski, J.; et al. Study of the Amorphous Glibenclamide Drug: Analysis of the Molecular Dynamics of Quenched and Cryomilled Material. Mol. Pharm. 2010, 7, 1692–1707. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Grohganz, H.; Strachan, C.; Rades, T. Amino Acids as Co-amorphous Excipients for Simvastatin and Glibenclamide: Physical Properties and Stability. Mol. Pharm. 2014, 11, 2381–2389. [Google Scholar] [CrossRef]
- Hassan, M.A.; Najib, N.M.; Suleiman, M.S. Characterization of glibenclamide glassy state. Int. J. Pharm. 1991, 67, 131–137. [Google Scholar] [CrossRef]
- Patterson, J.E.; James, M.B.; Forster, A.H.; Lancaster, R.W.; Butler, J.M.; Rades, T. The Influence of Thermal and Mechanical Preparative Techniques on the Amorphous State of Four Poorly Soluble Compounds. J. Pharm. Sci. 2005, 94, 1998–2012. [Google Scholar] [CrossRef]
- Rehder, S.; Sakmann, A.; Rades, T.; Leopold, C.S. Thermal degradation of amorphous glibenclamide. Eur. J. Pharm. Biopharm. 2012, 80, 203–208. [Google Scholar] [CrossRef]
- Yuce, C. Twin-Screw Extrusion Applications in the Pharmaceutical Industry. In Proceedings of the Master 2 Pharmacie Galénique Industrielle Program Formation, Lille, France, 11 January 2024. [Google Scholar]
- Bassand, C.; Benabed, L.; Freitag, J.; Verin, J.; Siepmann, F.; Siepmann, J. How bulk fluid renewal can affect in vitro drug release from PLGA implants: Importance of the experimental set-up. Int. J. Pharm. X 2022, 4, 100131. [Google Scholar] [CrossRef]
- Gupta, S.S.; Solanki, N.; Serajuddin, A.T.M. Investigation of Thermal and Viscoelastic Properties of Polymers Relevant to Hot Melt Extrusion, IV: AffinisolTM HPMC HME Polymers. AAPS PharmSciTech 2016, 17, 148–157. [Google Scholar] [CrossRef]
- Huang, S.; O’Donnell, K.P.; Keen, J.M.; Rickard, M.A.; McGinity, J.W.; Williams, R.O. A New Extrudable Form of Hypromellose: AFFINISOLTM HPMC HME. AAPS PharmSciTech 2016, 17, 106–119. [Google Scholar] [CrossRef]
- Prodduturi, S.; Urman, K.L.; Otaigbe, J.U.; Repka, M.A. Stabilization of hot-melt extrusion formulations containing solid solutions using polymer blends. AAPS PharmSciTech 2007, 8, 152–161. [Google Scholar] [CrossRef]
- Martin, C. Twin Screw Extrusion for Pharmaceutical Processes. In Melt Extrusion; Repka, M.A., Langley, N., DiNunzio, J., Eds.; Springer: New York, NY, USA, 2013; Volume 9, pp. 47–79. [Google Scholar] [CrossRef]
- Hanada, M.; Jermain, S.V.; Thompson, S.A.; Furuta, H.; Fukuda, M.; Williams, R.O. Ternary Amorphous Solid Dispersions Containing a High-Viscosity Polymer and Mesoporous Silica Enhance Dissolution Performance. Mol. Pharm. 2021, 18, 198–213. [Google Scholar] [CrossRef]
- Thompson, S.A.; Williams, R.O. Specific mechanical energy—An essential parameter in the processing of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2021, 173, 374–393. [Google Scholar] [CrossRef]
- Radjenović, J.; Pérez, S.; Petrović, M.; Barceló, D. Identification and structural characterization of biodegradation products of atenolol and glibenclamide by liquid chromatography coupled to hybrid quadrupole time-of-flight and quadrupole ion trap mass spectrometry. J. Chromatogr. A 2008, 1210, 142–153. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016; p. 2670. [Google Scholar] [CrossRef]
- Budavari, S.; O’Neil, M.J.; Smith, A.; Heckelman, P.E.; Obenchain, J.R., Jr.; Gallipeau, J.A.R.; D’Arecea, M.A. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; Merck & Co. Inc.: Whitehouse Station, NJ, USA, 1996; Volume 450, p. 1674. [Google Scholar]
- Suresh, K.; Khandavilli, U.B.R.; Gunnam, A.; Nangia, A. Polymorphism, isostructurality and physicochemical properties of glibenclamide salts. CrystEngComm 2017, 19, 918–929. [Google Scholar] [CrossRef]
- Klumpp, L.; Dressman, J. Physiologically based pharmacokinetic model outputs depend on dissolution data and their input: Case examples glibenclamide and dipyridamole. Eur. J. Pharm. Sci. 2020, 151, 105380. [Google Scholar] [CrossRef]
- Mann, J.; Dressman, J.; Rosenblatt, K.; Ashworth, L.; Muenster, U.; Frank, F.; Hutchins, P.; Williams, J.; Klumpp, L.; Wielockx, K.; et al. Validation of Dissolution Testing with Biorelevant Media: An OrBiTo Study. Mol. Pharm. 2017, 14, 4192–4201. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Zhang, G.G.Z.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef]
- Langer, R.; Peppas, N. Chemical and Physical Structure of Polymers as Carriers for Controlled Release of Bioactive Agents: A Review. J. Macromol. Sci. 1983, 23 Pt C, 61–126. [Google Scholar] [CrossRef]
- Siepmann, F.; Eckart, K.; Maschke, A.; Kolter, K.; Siepmann, J. Modeling drug release from PVAc/PVP matrix tablets. J. Control. Release 2010, 141, 216–222. [Google Scholar] [CrossRef]
- Aita, I.E.; Breitkreutz, J.; Quodbach, J. Investigation of semi-solid formulations for 3D printing of drugs after prolonged storage to mimic real-life applications. Eur. J. Pharm. Sci. 2020, 146, 105266. [Google Scholar] [CrossRef]
- De Brabander, C.; Vervaet, C.; Remon, J.P. Development and evaluation of sustained release mini-matrices prepared via hot melt extrusion. J. Control. Release 2003, 89, 235–247. [Google Scholar] [CrossRef]
- Crowley, M.M.; Schroeder, B.; Fredersdorf, A.; Obara, S.; Talarico, M.; Kucera, S.; McGinity, J.W. Physicochemical properties and mechanism of drug release from ethyl cellulose matrix tablets prepared by direct compression and hot-melt extrusion. Int. J. Pharm. 2004, 269, 509–522. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ternary Blend Composition | Feeding Rate [g/min] | Screw Speed [rpm] | Torque [%] | Extrudate Output [g/min] | T Zone 2 [°C] | T Zone 3 [°C] | T Zone 4–6 [°C] | T Zone 7 [°C] | T Zone 8 [°C] | T Die [°C] | Drug Recovery [%] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GLB and HPMC and PVP/PVAc | 1.71 | 160 | 60 | 1.67 | 60 | 100 | 110 | 115 | 120 | 130 | 75.0 ± 6.7 |
| GLB and EC and PVP/PVAc | 1.92 | 200 | 40 | 1.85 | 60 | 100 | 110 | 110 | 125 | 130 | 74.9 ± 8.2 |
| GLB and HPMC and EC | 1.06 | 45 | 75 | 0.81 | 110 | 130 | 150 | 150 | 150 | 155 | 72.4 ± 10.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zupan, N.; Yous, I.; Danede, F.; Verin, J.; Kouach, M.; Foulon, C.; Dudognon, E.; Florin Muschert, S. Impact of Hot-Melt Extrusion on Glibenclamide’s Physical and Chemical States and Dissolution Behavior: Case Studies with Three Polymer Blend Matrices. Pharmaceutics 2024, 16, 1071. https://doi.org/10.3390/pharmaceutics16081071
Zupan N, Yous I, Danede F, Verin J, Kouach M, Foulon C, Dudognon E, Florin Muschert S. Impact of Hot-Melt Extrusion on Glibenclamide’s Physical and Chemical States and Dissolution Behavior: Case Studies with Three Polymer Blend Matrices. Pharmaceutics. 2024; 16(8):1071. https://doi.org/10.3390/pharmaceutics16081071
Chicago/Turabian StyleZupan, Nina, Ines Yous, Florence Danede, Jeremy Verin, Mostafa Kouach, Catherine Foulon, Emeline Dudognon, and Susanne Florin Muschert. 2024. "Impact of Hot-Melt Extrusion on Glibenclamide’s Physical and Chemical States and Dissolution Behavior: Case Studies with Three Polymer Blend Matrices" Pharmaceutics 16, no. 8: 1071. https://doi.org/10.3390/pharmaceutics16081071