
Pharmacokinetic Models of Tafenoquine: Insights for Optimal Malaria Treatment Strategies

Abstract
:1. Introduction
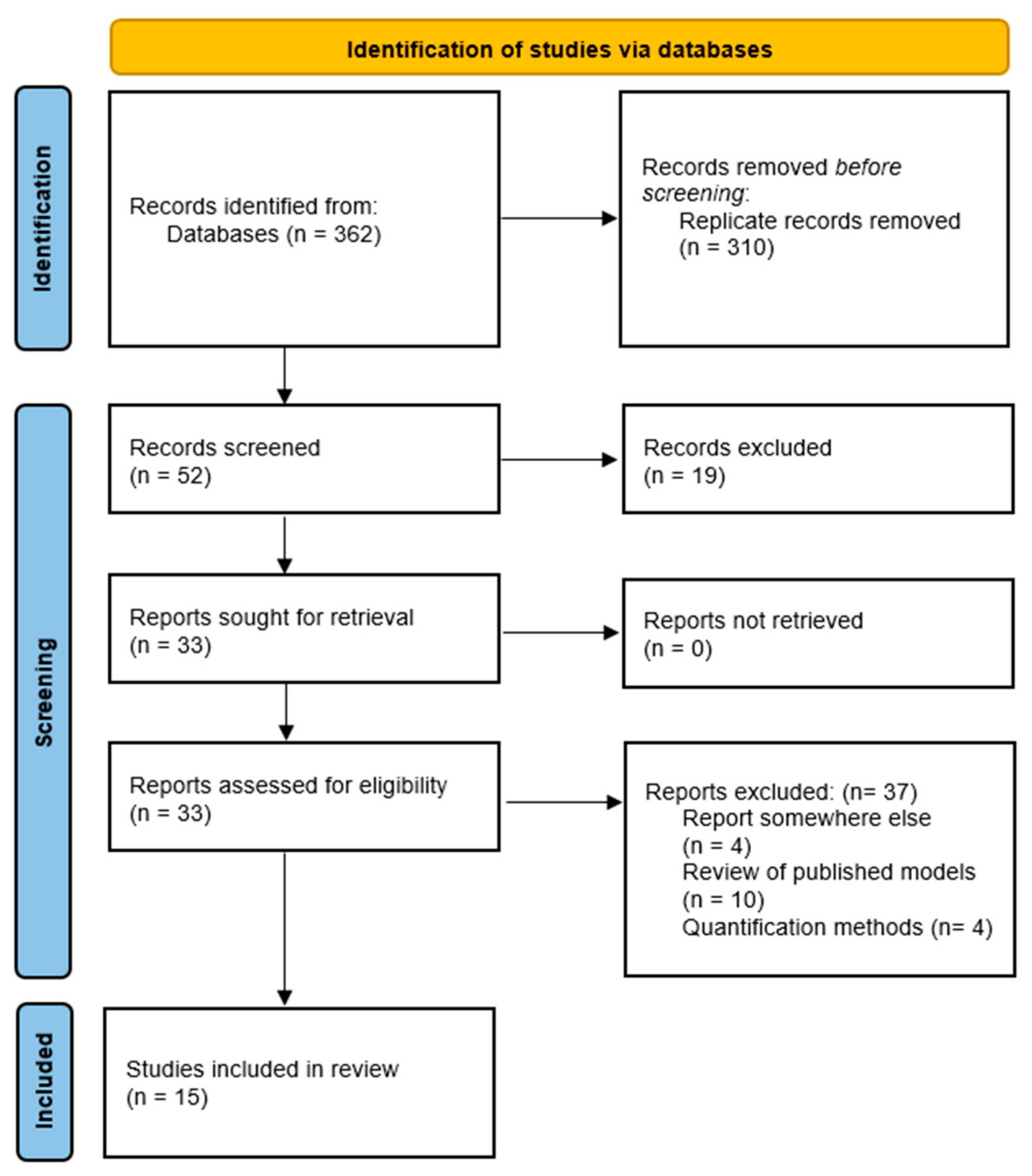
2. Methodology
2.1. Research Question
2.2. Search Strategy
2.3. Study Selection
2.4. Data Collection and Data Analysis
2.5. Data Presentation
3. Results and Discussion
3.1. Non-Compartmental Analysis
3.2. Population Pharmacokinetic Analysis
3.3. Pharmacokinetic/Pharmacodynamic Analysis
3.4. Results Summary
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). WHO Guidelines for Malaria; WHO: Geneva, Switzerland, 2023; Available online: https://iris.who.int/bitstream/handle/10665/373339/WHO-UCN-GMP-2023.01-Rev.1-eng.pdf (accessed on 24 June 2024).
- World Health Organization (WHO). World Malaria Report 2023; WHO: Geneva, Switzerland, 2023; Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023 (accessed on 24 June 2024).
- Elewa, H.; Wilby, K.J. A review of pharmacogenetics of antimalarials and associated clinical implications. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Matin, M.A.; Ferdous, N.E.N.; Hasan, A.; Sazed, S.A.; Neogi, A.K.; Chakma, S.; Islam, M.A.; Khan, A.A.; Haque, M.E.; et al. Adherence to anti-malarial treatment in malaria endemic areas of Bangladesh. Pathogens 2023, 12, 1392. [Google Scholar] [CrossRef]
- Almeida, E.D.; Vieira, J.L.F. Factors associated with non-adherence to the treatment of vivax malaria in a rural community from the Brazilian Amazon Basin. Rev. Soc. Bras. Med. Trop. 2016, 49, 248–251. [Google Scholar] [CrossRef]
- Brueckner, R.P.; Coster, T.; Wesche, D.L.; Shmuklarsky, M.; Schuster, B.G. Prophylaxis of Plasmodium falciparum infection in a human challenge model with WR 238605, a new 8-aminoquinoline antimalarial. Antimicrob. Agents Chemother. 1998, 42, 1293–1294. [Google Scholar] [CrossRef]
- Brueckner, R.P.; Lasseter, K.C.; Lin, E.T.; Schuster, B.G. First-time-in-humans safety and pharmacokinetics of WR 238605, a new antimalarial. Am. J. Trop. Med. Hyg. 1998, 58, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.K. 8-aminoquinoline therapy for latent malaria. Clin. Microbiol. Rev. 2019, 32, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ebstie, Y.A.; Abay, S.M.; Tadesse, W.T.; Ejigu, D.A. Tafenoquine and its potential in the treatment and relapse prevention of Plasmodium vivax malaria: The evidence to date. Drug Des. Devel Ther. 2016, 26, 2387–2399. [Google Scholar] [CrossRef]
- Usman, M.; Khadka, S.; Saleem, M.; Rasheed, H.; Kunwar, B.; Ali, M. Pharmacometrics: A new era of pharmacotherapy and drug development in low- and middle-income countries. Adv. Pharmacol. Pharm. Sci. 2023, 2023, 10. [Google Scholar] [CrossRef]
- Lim, H.S. Evolving role of modeling and simulation in drug development. Transl. Clin. Pharmacol. 2019, 27, 19–23. [Google Scholar] [CrossRef]
- Whittemore, R.; Knafl, K. The integrative review: Updated methodology. J. Adv. Nurs. 2005, 52, 546–553. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, 9. [Google Scholar]
- Voung, C.; Xie, L.H.; Potter, B.M.; Zhang, J.; Zhang, P.; Duan, D.; Nolan, C.K.; Sciotti, R.J.; Zottig, V.E.; Nanayakkara, N.D.; et al. Differential cytochrome P450 2D metabolism alters tafenoquine pharmacokinetics. Antimicrob. Agents Chemother. 2015, 59, 3864–3869. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; O’neil, M.; Xie, L.; Caridha, D.; Zeng, Q.; Zhang, J.; Pybus, B.; Hickman, M.; Melendez, V. Assessment of the prophylactic activity and pharmacokinetic profile of oral tafenoquine compared to primaquine for inhibition of liver stage malaria infections. Malar. J. 2014, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Green, J.A.; Mohamed, K.; Goyal, N.; Bouhired, S.; Hussaini, A.; Jones, S.W.; Koh, G.C.; Kostov, I.; Taylor, M.; Wolstenholm, A.; et al. Pharmacokinetic interactions between tafenoquine and dihydroartemisinin-piperaquine or artemether-lumefantrine in healthy adult subjects. Antimicrob. Agents Chemother. 2016, 60, 7321–7332. [Google Scholar] [CrossRef]
- Vanachayangkul, P.; Kodchakorn, C.; Ta-Aksorn, W.; Im-Erbsin, R.; Tungtaeng, A.; Tipthara, P.; Tarning, J.; Lugo-Roman, L.A.; Wojnarski, M.; Vesely, B.A.; et al. Safety, pharmacokinetics, and potential neurological interactions of ivermectin, tafenoquine, and chloroquine in Rhesus macaques. Antimicrob. Agents Chemother. 2024, 68, 19. [Google Scholar] [CrossRef]
- Miller, A.K.; Harrell, E.; Ye, L.; Baptiste-Brown, S.; Kleim, J.; Ohrt, C.; Duparc, S.; Möhrle, J.J.; Webster, A.; Stinnett, S.; et al. Pharmacokinetic interactions and safety evaluations of coadministered tafenoquine and chloroquine in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 858–867. [Google Scholar] [CrossRef]
- Green, J.A.; Patel, A.K.; Patel, B.R.; Hussaini, A.; Harrell, E.J.; McDonald, M.J.; Carter, N.; Mohamed, K.; Duparc, S.; Miller, A.K. Tafenoquine at therapeutic concentrations does not prolong Fridericia-corrected QT interval in healthy subjects. J. Clin. Pharmacol. 2014, 54, 995–1005. [Google Scholar] [CrossRef]
- Barber, B.E.; Abd-Rahman, A.N.; Webster, R.; Potter, A.J.; Llewellyn, S.; Marquart, L.; Sahai, N.; Leelasena, I.; Birrell, G.W.; Edstein, M.D.; et al. Characterizing the blood-stage antimalarial activity of tafenoquine in healthy volunteers experimentally infected with Plasmodium falciparum. Clin. Infect. Dis. 2023, 76, 1919–1927. [Google Scholar] [CrossRef]
- Brueckner, R.P.; Fleckenstein, L. Simultaneous modeling of the pharmacokinetics and methemoglobin pharmacodynamics of an 8-aminoquinoline candidate antimalarial (WR 238605). Pharm. Res. 1991, 8, 1505–1510. [Google Scholar] [CrossRef]
- Tenero, D.; Green, J.A.; Goyal, N. Exposure-response analyses for tafenoquine after administration to patients with Plasmodium vivax malaria. Antimicrob. Agents Chemother. 2015, 59, 6188–6194. [Google Scholar] [CrossRef]
- Thakkar, N.; Green, J.A.; Koh, G.C.K.W.; Duparc, S.; Tenero, D.; Goyal, N. Population pharmacokinetics of tafenoquine, a novel antimalarial. Antimicrob. Agents Chemother. 2018, 62, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Edstein, M.D.; Kocisko, D.A.; Brewer, T.G.; Walsh, D.S.; Eamsila, C.; Charles, B.G. Population pharmacokinetics of the new antimalarial agent tafenoquine in Thai soldiers. Br. J. Clin. Pharmacol. 2001, 52, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Charles, B.G.; Miller, A.K.; Nasveld, P.E.; Reid, M.G.; Harris, I.E.; Edstein, M.D. Population pharmacokinetics of tafenoquine during malaria prophylaxis in healthy subjects. Antimicrob. Agents Chemother. 2007, 51, 2709–2715. [Google Scholar] [CrossRef] [PubMed]
- Bachhav, S.S.; Taylor, M.; Martin, A.; Green, J.A.; Duparc, S.; Rolfe, K.; Sharma, H.; Tan, L.K.; Goyal, N. A Pharmacometrics approach to assess the feasibility of capillary microsampling to replace venous sampling in clinical studies: Tafenoquine study. Br. J. Clin. Pharmacol. 2023, 89, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Vélez, I.D.; Hien, T.T.; A Green, J.; Martin, A.; Sharma, H.; Rousell, V.M.; Breton, J.J.; Ernest, T.B.; Rolfe, K.; Taylor, M.; et al. Tafenoquine exposure assessment, safety, and relapse prevention efficacy in children with Plasmodium vivax malaria: Open-label, single-arm, non-comparative, multicentre, pharmacokinetic bridging, phase 2 trial. Lancet Child. Adolesc. Health 2022, 6, 86–95. [Google Scholar] [CrossRef]
- Yue, C.S.; Ducharme, M.P. Empirical models, mechanistic models, statistical moments, and noncompartmental analysis. In Applied Biopharmaceutics and Pharmacokinetics, 7th ed.; Shargel, L., Yu, A.B.C., Eds.; Mc Graw Hill Education: New York, NY, USA, 2016; pp. 817–849. [Google Scholar]
- Gabrielsson, J.; Weiner, D. Non-compartmental analysis. Methods Mol. Biol. 2012, 929, 377–389. [Google Scholar]
- Gibaldi, M.; Perrier, D. Noncompartmental analysis based on statistical moment theory. In Pharmacokinetics, 2nd ed.; Gibaldi, M., Perrier, M., Eds.; Informa Healthcare: New York, NY, USA, 2007; pp. 409–417. [Google Scholar]
- Ojara, F.W.; Kawuma, A.N.; Waitt, C. A systematic review on maternal-to-infant transfer of drugs through breast milk during the treatment of malaria, tuberculosis, and neglected tropical diseases. PLoS Negl. Trop. Dis. 2023, 17, 26. [Google Scholar] [CrossRef]
- Andes, D.; Craig, W.A. Animal model pharmacokinetics and pharmacodynamics: A critical review. Int. J. Antimicrob. Agents 2002, 4, 261–268. [Google Scholar] [CrossRef]
- Pasquiers, B.; Benamara, S.; Felices, M.; Nguyen, L.; Declèves, X. Review of the Existing Translational Pharmacokinetics Modeling Approaches Specific to Monoclonal Antibodies (mAbs) to Support the First-In-Human (FIH) Dose Selection. Int. J. Mol. Sci. 2022, 23, 12754. [Google Scholar] [CrossRef]
- MacIntyre, A.C.; Cutler, D.J. The potential role of lysosomes in tissue distribution of weak bases. Biopharm. Drug Dispos. 1988, 9, 513–526. [Google Scholar] [CrossRef]
- Guidi, M.; Csajka, C.; Buclin, T. Parametric approaches in population pharmacokinetics. J. Clin. Pharmacol. 2022, 62, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Samara, E.; Granneman, R. Role of population pharmacokinetics in drug development: A pharmaceutical industry perspective. Clin. Pharmacokinet. 1997, 32, 294–312. [Google Scholar] [CrossRef]
- Byon, W.; Smith, M.; Chan, P.; Tortorici, M.; Riley, S.; Dai, H.; Dong, J.; Ruiz-Garcia, A.; Sweeney, K.; Cronenberger, C. Establishing best practices and guidance in population modeling: An experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Melariri, P.; Kalombo, L.; Nkuna, P.; Dube, A.; Hayeshi, R.; Ogutu, B.; Gibhard, L.; Dekock, C.; Smith, P.; Weisner, L.; et al. Oral lipid-based nanoformulation of tafenoquine enhanced bioavailability and blood stage antimalarial efficacy and led to a reduction in human red blood cell loss in mice. Int. J. Nanomed. 2015, 10, 1493–1503. [Google Scholar] [CrossRef]
- Wu, B.; Sy, S.K.B.; Derendorf, H. Principles of applied pharmacokinetic-pharmacodynamic modeling. In Fundamentals of Antimicrobial Pharmacokinetics and Pharmacodynamics, 1st ed.; Vinks, A.A., Mounton, J.W., Derendorf, H., Eds.; Springer: New York, NY, USA, 2014; pp. 63–79. [Google Scholar]
- Mi, K.; Zhou, K.; Sun, L.; Hou, Y.; Ma, W.; Xu, X.; Huo, M.; Liu, Z.; Huang, L. Application of semi-mechanistic pharmacokinetic and pharmacodynamic model in antimicrobial resistance. Pharmaceutics 2022, 14, 246. [Google Scholar] [CrossRef]
- Bueters, T.; Ploeger, B.; Visser, S.A.G. The virtue of translational PKPD modeling in drug discovery: Selecting the right clinical candidate while sparing animal lives. Drug Discov. Today 2013, 18, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.; Bohnert, T.; Damian-Iordache, V.; Gibson, C.; Hsu, C.-P.; Krishnatry, A.S.; Liederer, B.M.; Lin, J.; Lu, Q.; Mettetal, J.T.; et al. Translational pharmacokinetic-pharmacodynamic analysis in the pharmaceutical industry: An IQ Consortium PK-PD Discussion Group perspective. Drug Discov. Today 2017, 22, 1447–1459. [Google Scholar] [CrossRef]
- White, N.J. Cardiotoxicity of antimalarial drugs. Lancet Infect. Dis. 2007, 7, 549–558. [Google Scholar] [CrossRef]
- Mzayek, F.; Deng, H.; Mather, F.J.; Wasilevich, E.C.; Liu, H.; Hadi, C.M.; Chansolme, D.H.; A Murphy, H.; Melek, B.H.; Tenaglia, A.N.; et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin. Trials 2007, 2, 15. [Google Scholar] [CrossRef]
- Crockett, M.; Kain, K.C. Tafenoquine: A promising new antimalarial agent. Expert. Opin. Investig. Drugs 2007, 16, 705–715. [Google Scholar] [CrossRef]
- Idowu, O.R.; Peggins, J.; Brewer, T.G.; Kelley, C. Metabolism of a candidate 8-aminoquinoline antimalarial agent, WR 238605, by rat liver microsomes. Drug Metab. Dispos. 1995, 23, 1–17. [Google Scholar] [PubMed]
- Rueangweerayut, R.; Bancone, G.; Harrell, E.J.; Beelen, A.P.; Kongpatanakul, S.; Möhrle, J.J.; Rousell, V.; Mohamed, K.; Qureshi, A.; Narayan, S.; et al. Hemolytic potential of tafenoquine in female volunteers heterozygous for glucose-6-phosphate dehydrogenase (G6PD) deficiency (G6PD Mahidol variant) versus G6PD-normal volunteers. Am. J. Trop. Med. Hyg. 2017, 97, 702–711. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Authors | Type of Model | Patients/Animal Study Characteristics | Dosage Regimen | Outcomes |
|---|---|---|---|---|
| Voung, et al., 2015 [14] | NCA | 7 healthy male mice KO and WT | 20 mg/kg (IG) | The study demonstrated that decreased CYP2D metabolism affected the PK of TQ in mice, and a human study with poor metabolizers of the CYP2D6 phenotype is required. |
| Li, et al., 2014 [15] | NCA | 3 healthy male mice | 5 mg/kg (IV) and 20 mg/kg (IG) TQ | They found that TQ was 5-fold more potent than PQ as a liver schizonticide, with prolonged elimination, longer half-life, and increased drug exposure in plasma and the liver. |
| Green, et al., 2016 [16] | NCA | 118 healthy adult volunteers | 300 mg TQ p.o.; 120 mg/960 mg DHA-PQP p.o.; 80 mg/480 mg AL p.o.; TQ+DHA-PQP; TQ+AL | The study found no clinically significant drug interactions between TQ and the first-line ACTs, and there is no need for dose adjustment for co-administration. |
| Vanachayangkul, et al., 2024 [17] | NCA | 4 healthy male macaques | 1 mL/kg (NG) TQ, IVM and CQ | TQ exposure was elevated by co-administration of CQ but not IVM, which suggests that IVM and TQ combinations for MDAs or prophylaxis are a viable option. |
| Miller, et al., 2013 [18] | NCA and PK/PD | 58 healthy adults volunteers | 300 mg CQ p.o.; 900 mg TQ p.o.; CQ plus TQ | This study demonstrated no clinically significant pharmacokinetic or pharmacodynamic interactions between CQ and TQ in healthy subjects, with no changes in PK parameters or QT intervals. |
| Green, et al., 2014 [19] | NCA and PK/PD | 260 healthy adult volunteers | 300 and 600 TQ p.o.; 400 mg TQ p.o. (3 days); 400 mg MXF p.o. | This study found no effect of clinical doses of TQ (300 and 600 mg) and the supratherapeutic dose (1200 mg) on QTcF interval prolongation compared to placebo. |
| Barber, et al., 2023 [20] | NCA and PK/PD | 12 healthy adult volunteers infected with P. falciparum | 200, 300, 400 and 600 mg TQ p.o. | Single low dose (200–300 mg) of TQ was not effective at clearing asexual parasitemia. However, higher doses (400–600 mg) were effective, demonstrating that single-dose TQ exhibits potent P. falciparum blood-stage activity. |
| Brueckner and Fleckenstein, 1991 [21] | NCA and PK/PD | 6 healthy male Beagles | Four daily doses of 6 mg/kg TQ p.o. | The PK/PD model provided a good estimate of plasma TQ concentrations and MHb levels as well as the times of their occurrences, and the model can be useful to guide the design of further animal studies and initial phase 1 human studies. |
| Brueckner, et al., 1998 [7] | NCA and popPK | 80 healthy adults volunteers | 4, 16, 36, 72, 100, 144, 192, 240, 250, 288, 300, 350, 400, 500 and 600 mg TQ p.o. | They observed gastrointestinal side effects at higher doses (300–600 mg), as well as linear PK. A one-compartment model with no impact of covariates best described the data. |
| Tenero, et al., 2015 [22] | popPK and PK/PD | 180 patients with uncomplicated P. vivax mono-infection | 600 mg CQ (day 1 and 2); 300 mg CQ (day 3); 50, 100, 300 or 600 mg TQ (day 1 or 2); 15 mg PQ (day 2–14) | The results of this phase 2b dose–response study showed that doses of 300 and 600 mg of TQ plus CQ had significantly improved relapse-free efficacy at 6 months compared with CQ alone. They also observed that the efficacy of PQ was lower than that of TQ at 300 and 600 mg. The 300 mg dose was proposed for further clinical studies. |
| Thakkar, et al., 2018 [23] | popPK | 841 subjects collected from 5 studies (193 healthy adult volunteers and 648 patients) | 50, 100, 300 and 600 mg TQ p.o.; 400 mg TQ p.o. (3 days) | The study found the final model to be a two-compartment model with allometric scaling for clearance and volume of distribution. No dose adjustment is needed based on other demographic characteristics, and this model found no difference in exposure across different formulations. |
| Edstein, et al., 2001 [24] | popPK | 135 healthy adult volunteers | LD of 400 mg TQ p.o. (3 days); MD of 400 mg TQ p.o. monthly (5 months) | They observed a one-compartment model with first-order absorption and elimination. Covariates as age and weight did not have a significant impact on clearance and volume of distribution. |
| Charles, et al., 2007 [25] | popPK | 490 healthy adult volunteers | LD of 200 mg TQ p.o. (3 days); MD of 200 mg TQ p.o. weekly (6 months) | The results showed a one-compartment model with first-order absorption and elimination, with no impact of covariates such as age and CLCR. The study supports a weekly dosing regimen for prophylaxis prolonged periods. |
| Bachhav, et al., 2022 [26] | popPK | 59 pediatric patients with P. vivax malaria | 100, 150, 200 and 300 mg TQ p.o. | The study demonstrated with a popPK model that microsampling may be an alternative sampling technique for PK studies because key exposure parameters, such as AUCtotal and Cmax, are comparable across the venous sample model and the capillary microsampling model |
| Vélez, et al., 2022 [27] | popPK | 60 pediatric patients with P. vivax malaria | 100, 150, 200 and 300 mg TQ p.o. | A pediatric population pharmacokinetic model was developed, and they found a similar relative bioavailability between 50 mg dispersible tablets and 150 mg tablets. The 300 mg dose approved for adults demonstrated good exposure across all the weight and age bands. |
| Reference | Parameters | |||||
|---|---|---|---|---|---|---|
| AUCtotal | Cmax | Tmax | t1/2 | CL | Vd | |
| Li et al., 2014 [15] | 1.31 μg*h/mL (PQ) | 0.53 μg/mL (PQ) | 0.50 h (PQ) | 1.84 h (PQ) | 57.94 L/h/kg (PQ) | 154.66 L/kg (PQ) |
| 139.18 μg*h/mL (TQ) | 2.04 μg/mL (TQ) | 11.33 h (TQ) | 50.87 h (TQ) | 0.15 L/h/kg (TQ) | 10.60 L/kg (TQ) | |
| Voung et al., 2015 [14] | 85.6 μg*h/mL (WT) | 1.2 μg/mL (WT) | 5 h (WT) | 53.8 h (WT) | 6.0 mL/h/kg (WT) | 18.5 L/kg (WT) |
| 123.2 μg*h/mL (KO) | 1.4 μg/mL (KO) | 10 h (KO) | 72.4 h (KO) | 2.0 mL/h/kg (KO) | 19.9 L/kg (KO) | |
| Vanachayangkul et al., 2024 [17] | 3597 ng*h/mL (TQ) | 67.4 ng/mL (TQ) | 12 h (TQ, TQ+IVM, TQ+IVM+CQ) | 45.8 h (TQ) | 0.64 L/h/kg (TQ) | 40.2 L/kg (TQ) |
| 3248 ng*h/mL (TQ+IVM) | 56 ng/mL (TQ+IVM) | 49.4 h (TQ+IVM) | 0.75 L/h/kg (TQ+IVM) | 49 L/kg (TQ+IVM) | ||
| 6508 ng*h/mL (TQ+IVM+CQ) | 84.4 ng/mL (TQ+IVM+CQ) | 63.2 (TQ+IVM+CQ) | 0.33 L/h/kg (TQ+IVM+CQ) | 29.7 L/kg (TQ+IVM+CQ) | ||
| Brueckner et al., 1998 [7] | n.a | n.a | 13.8 h | 325 h | 6.1 L/h | 2534 L |
| Miller et al., 2013 [18] | 0.84–1.4 (TQ) | 1.17–1.64 (TQ) | n.a | 0.94–1.20 (TQ) | n.a | n.a |
| 0.84–1.18 (TQ+CQ) | 0.74–1.08 (TQ+CQ) | n.a | 0.78–1.12 (TQ+CQ) | n.a | n.a | |
| Green et al., 2014 [19] | 10,611 ng*h/mL (300 mg) # | 186 ng/mL (300 mg) | 15 h (300 mg) | n.a | n.a. | n.a |
| 22,986 ng*h/mL (600 mg) # | 422 ng/mL (600 mg) | 12 h (600 mg) | n.a | |||
| 41,896 ng*h/mL (1200 mg) # | 724 ng/mL (1200 mg) | 12 h (1200 mg) | n.a | |||
| Green et al., 2016 [16] | 97,195.5 ng*h/mL (TQ) | 199.6 ng/mL (TQ) | 12.1 h (TQ) | 375.2 h (TQ) | 3 L/h (TQ) | n.a |
| 109,333.7 ng*h/mL (TQ+DHA-PQP) | 274.7 ng/mL (TQ+DHA-PQP) | 6 h (TQ+DHA-PQP) | 483.9 h (TQ+DHA-PQP) | 2.7 L/h (TQ+DHA-PQP) | ||
| 102,328.4 ng*h/mL (TQ+AL) | 208.4 ng/mL (TQ+AL) | 12.1 h (TQ+AL) | 396.5 h (TQ+AL) | 2.9 L/h (TQ+AL) | ||
| Barber et al., 2023 [20] | 47,091 ng*h/mL (200 mg) | 138.2 ng/mL (200 mg) | 12.2 h (200 mg) | 309.4 h (200 mg) | 3.8 L/h (200 mg) | 1708 L (200 mg) |
| 67,303 ng*h/mL (300 mg) | 205.8 ng/mL (300 mg) | 12 h (300 mg) | 357.3 h (300 mg) | 3.9 L/h (300 mg) | 2003 L (300 mg) | |
| 117,107 ng*h/mL (400 mg) | 324.8 ng/mL (400 mg) | 10 h (400 mg) | 313.9 h (400 mg) | 3.1 L/h (400 mg) | 1379 L (400 mg) | |
| 128,140 ng*h/mL (600 mg) | 340.2 ng/mL (600 mg) | 12.1 h (600 mg) | 314.4 h (600 mg) | 4.1 L/h (600 mg) | 1863 L (600 mg) | |
| Reference | Model | Covariates | Parameters | ||||
|---|---|---|---|---|---|---|---|
| Brueckner et al., 1998 [7] | One-compartment model | n.a | Vd = 2550 L; CL = 4.71 L/h; ka = 0.391 h−1 | ||||
| Edstein et al., 2001 [24] | One-compartment model | n.a | Vd = 1820 L; CL = 3.20 L/h; Tmax = 8.6 h; ka = 0.694 h−1; t1/2 = 16.4 dias | ||||
| Charles et al., 2007 [25] | One-compartment model | n.a | Vd = 1110 L; CL = 3.02 L/h; Tmax = 21.4 h; ka = 0.243 h−1; t1/2 = 12.7 dias | ||||
| Tenero et al., 2015 [22] | Two-compartment model with absorption lag time | n.a | n.a | n.a | n.a | n.a | n.a |
| Thakkar et al., 2018 [23] | Two-compartment model with absorption lag time | Weight was included in CL and Vd; formulation status was associated with ka and bioavailability; health status was included as a covariate on Vd | V2/F = 915 L; V3/F = 664 L CL/F = 2.96 L/h; Q/F = 5.09 L/h Ka = 0.252 h−1 | ||||
| Bachhlav et al., 2022 [26] | Two-compartment model with absorption lag time | Weight was included in CL and Vd; formulation status was associated with ka and bioavailability; health status was included as a covariate on Vd | V2/F = 798 L; V3/F = 732 L CL/F = 3.4 L/h; Q/F = 6.33 L/h Ka = 0.253 h−1 | ||||
| Vélez et al., 2022 [27] | Two-compartment model with absorption lag time | Weight was included in CL and Vd; formulation status was associated with ka and bioavailability; health status was included as a covariate on Vd | V2/F = 917 L; V3/F = 724 L CL/F = 3.67 L/h; Q/F = 5.94 L/h Ka = 0.253 h−1 | ||||
| Reference | Model | Parameters |
|---|---|---|
| Miller, et al., 2013 [18] | PK analysis was conducted using NCA, PD assessment used electrocardiographs, and PK/PD assessment was performed with graphical analyses | For TQ: AUCtotal = 0.84–1.14 ng*h/mL; Cmax = 1.17–1.64 ng/mL; t1/2 = 0.94–1.20 h For TQ+CQ: AUCtotal = 0.84–1.18 ng*h/mL; Cmax = 0.74–1.08 ng/mL; t1/2 = 0.78–1.12 h |
| Green et al., 2014 [19] | Population analysis was used to establish the PK/PD relationship between TQ concentrations and the QTcF interval. It included the covariates sex and race describing the baseline, 2 cosine functions (8-h cycle and 24-h cycle), and 2 slope terms | For 300 mg: AUCtotal = 10,611 ng*h/mL; Cmax = 186 ng/mL; Tmax = 15 h For 600 mg: AUCtotal = 22,986 ng*h/mL; Cmax = 422 ng/mL; Tmax = 12 h For 1200 mg: AUCtotal = 41,896 ng*h/mL; Cmax = 724 ng/mL; Tmax = 12 h |
| Tenero et al., 2015 [22] | The exposure–response model used nonlinear mixed-effects and the recurrence of P. vivax infection was modeled using a time-to-event approach where drug exposure affects the time to a recurrent episode | n.a |
| Barber et al., 2023 [20] | PK/PD model used a nonlinear mixed-effects and individual parameters from a population model as regression parameters | For 200 mg: AUCtotal = 47,091 ng*h/mL; Cmax = 138.2 ng/mL; Tmax = 12.2 h; t1/2 = 309.4 h; CL = 3.8 L/h; Vd = 1708 L For 300 mg: AUCtotal = 67,303 ng*h/mL; Cmax = 205.8 ng/mL; Tmax = 12 h; t1/2 = 357.3 h; CL = 3.9 L/h; Vd = 2003 L For 400 mg: AUCtotal = 117,107 ng*h/mL; Cmax = 324.8 ng/mL; Tmax = 10 h; t1/2 = 313.9 h; CL = 3.1 L/h; Vd = 1379 L For 600 mg: AUCtotal = 128,140 ng*h/mL; Cmax = 340.2 ng/mL; Tmax = 12.1 h; t1/2 = 314.4 h; CL = 4.1 L/h; Vd = 1863 L |
| Brueckner and Fleckenstein, 1991 [21] | Nonlinear least-squares regression was used for the PK/PD model with two-compartments, first-order absorption and elimination, linked to an effect compartment; the PD model used a sigmoid Emax to describe the %MHb | AUCtotal = 311,097 ng*h/mL; Vd = 18.5 L/kg; CL = 83 mL/h/kg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, L.O.; Alves, I.A.; Azeredo, F.J. Pharmacokinetic Models of Tafenoquine: Insights for Optimal Malaria Treatment Strategies. Pharmaceutics 2024, 16, 1124. https://doi.org/10.3390/pharmaceutics16091124
Santos LO, Alves IA, Azeredo FJ. Pharmacokinetic Models of Tafenoquine: Insights for Optimal Malaria Treatment Strategies. Pharmaceutics. 2024; 16(9):1124. https://doi.org/10.3390/pharmaceutics16091124
Chicago/Turabian StyleSantos, Luisa Oliveira, Izabel Almeida Alves, and Francine Johansson Azeredo. 2024. "Pharmacokinetic Models of Tafenoquine: Insights for Optimal Malaria Treatment Strategies" Pharmaceutics 16, no. 9: 1124. https://doi.org/10.3390/pharmaceutics16091124