
Powder Self-Emulsifying Drug Delivery System for Mitotane: In Vitro and In Vivo Evaluation
 , , ,
, , ,
Abstract
:
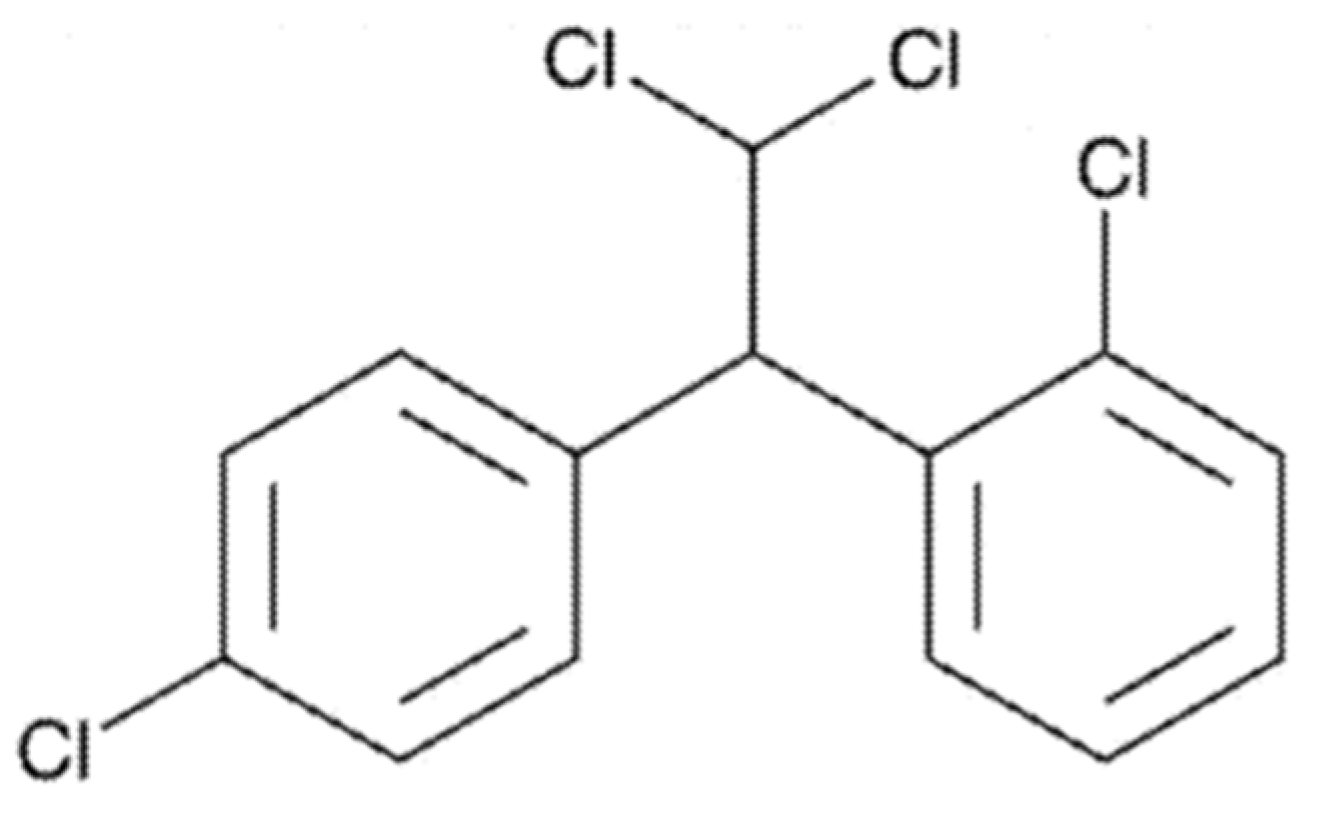
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of a Mitotane-Loaded P-SEDDS
- -
- Preparation of a mitotane-loaded P-SEDDS (Mito F1):
- -
- Preparation of a mitotane-loaded P-SEDDS with absorption promoter (Mito F2):
2.2.2. In Vitro Drug Release
Dissolution Studies
Drug Release Mathematical Modeling
UV-HPLC Assays
2.2.3. Pharmacokinetics Profile
In-Vivo Study and Pharmacokinetic Analysis
- GLP regulation requirements
- Animals
- Protocol
HPLC Analysis
- Chromatographic conditions
- Validation method
Statistical Analysis
3. Results and Discussion
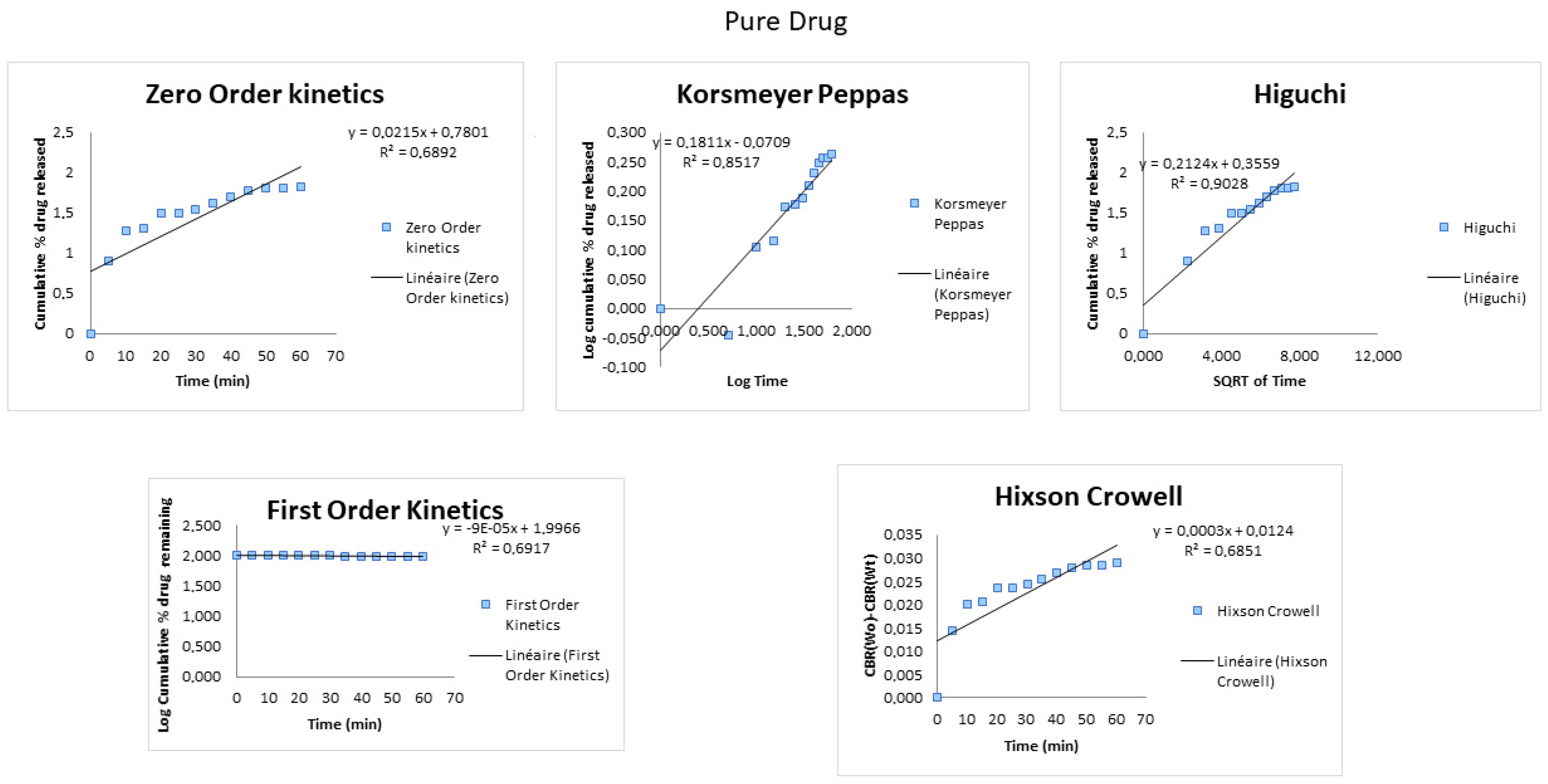
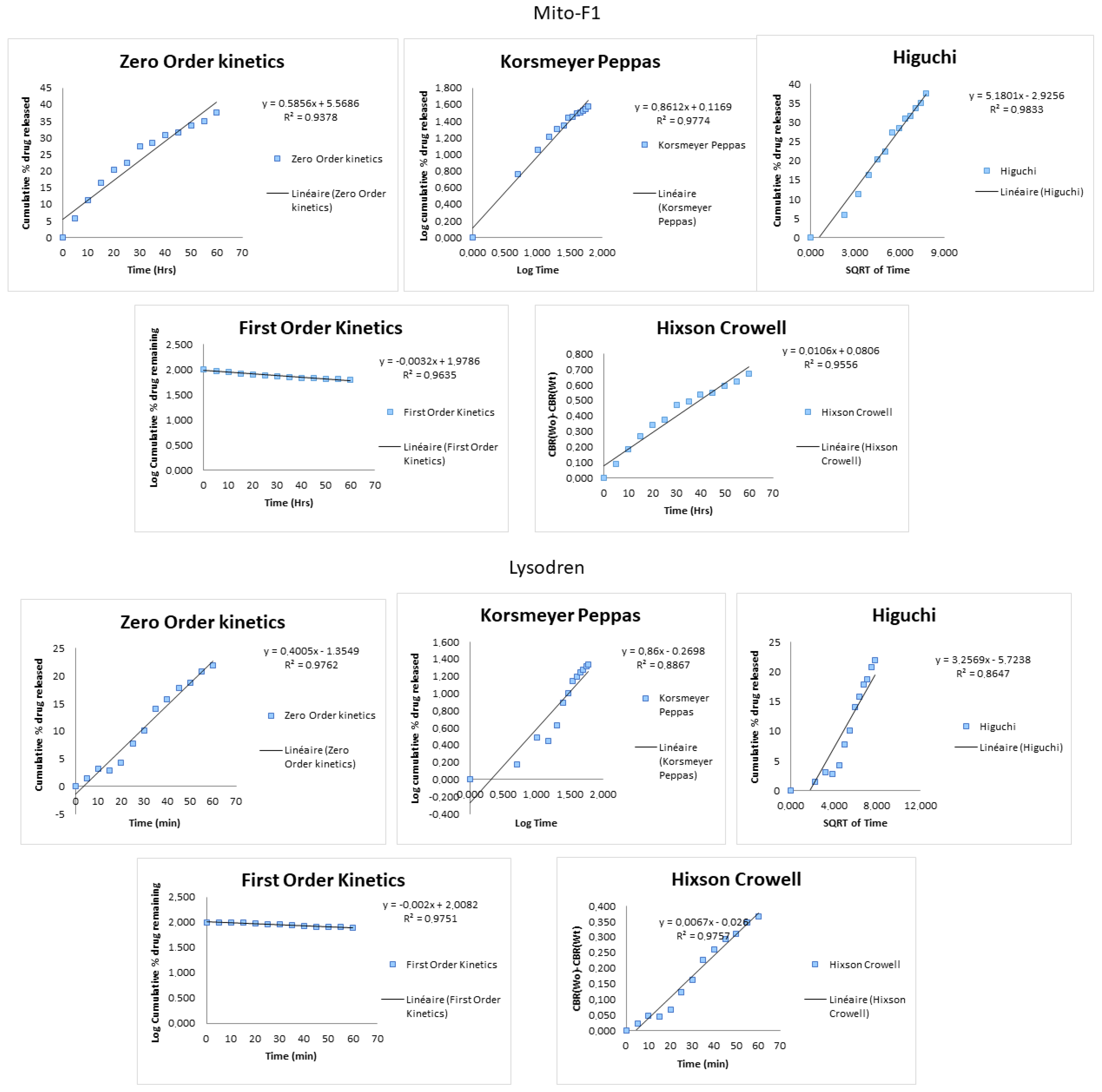
3.1. Dissolution
3.2. Pharmacokinetics Profile
3.2.1. Method Validation
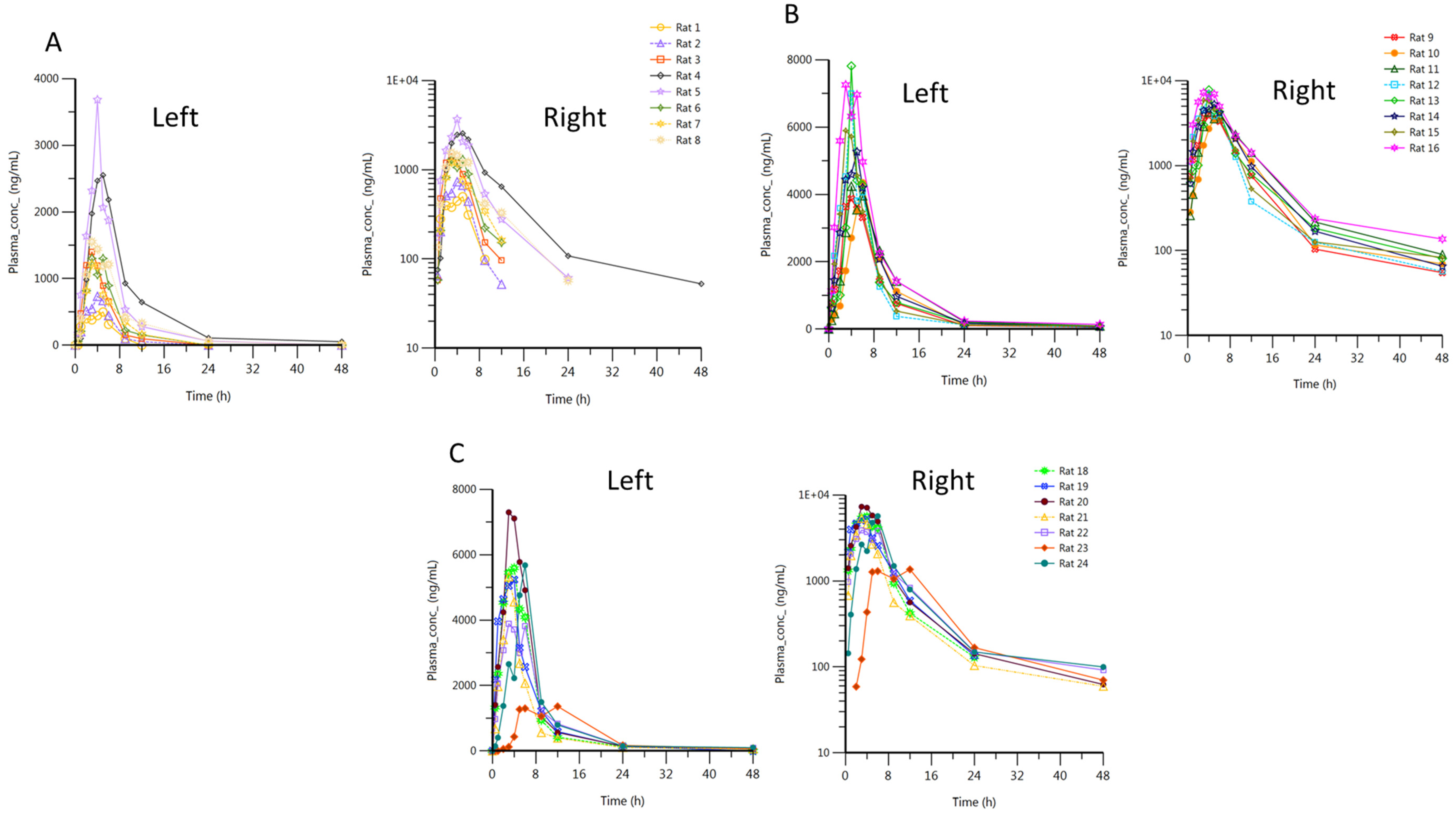
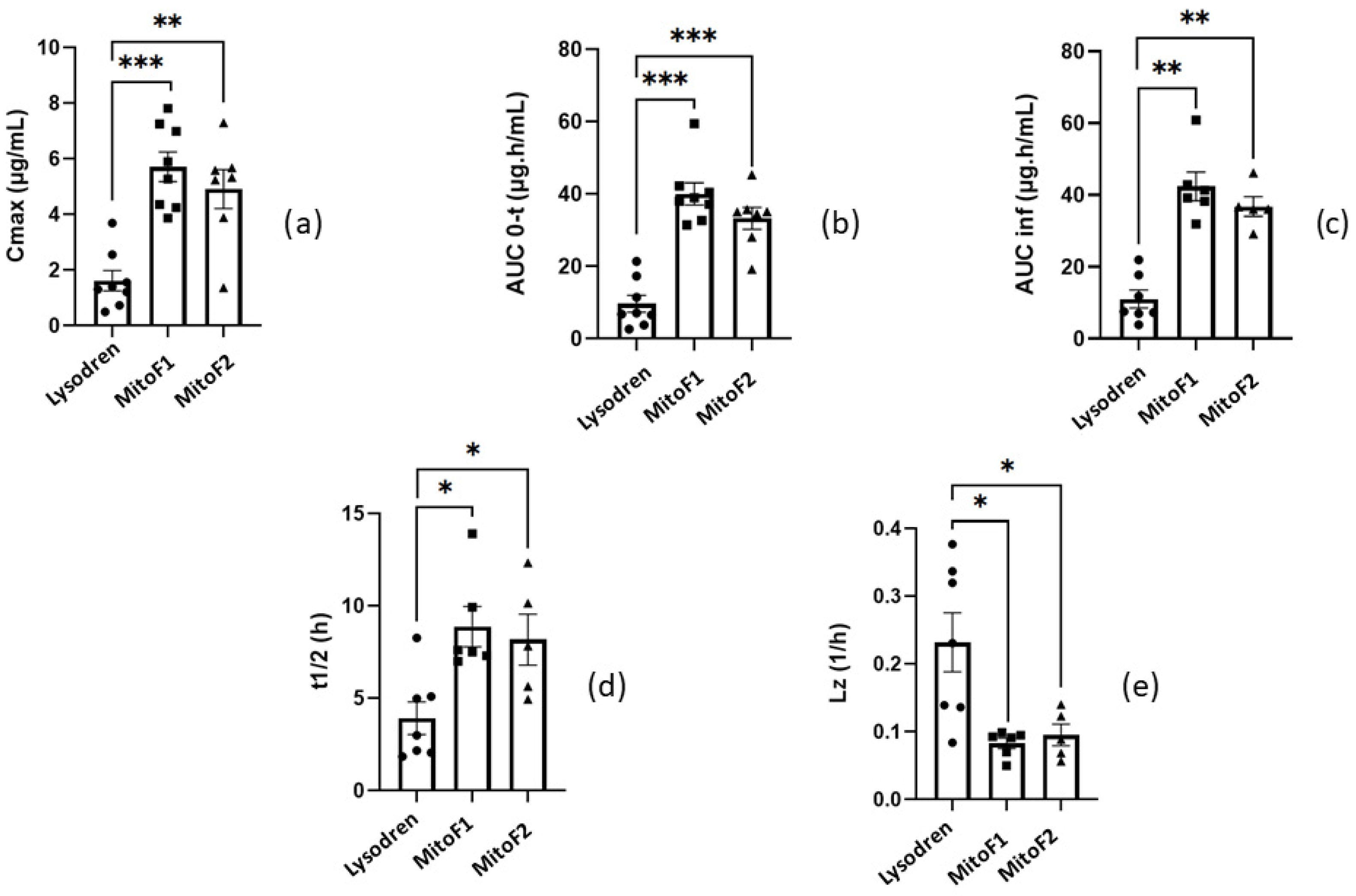
3.2.2. In Vivo Study and Pharmacokinetic Analysis
3.3. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical Carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Libé, R.; Kroiss, M.; Allolio, B. Adrenocortical carcinoma: A clinician’s update. Nat. Rev. Endocrinol. 2011, 7, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Kroiss, M.; Allolio, B. Update in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, 4551–4564. [Google Scholar] [CrossRef] [PubMed]
- Assie, G.; Antoni, G.; Tissier, F.; Caillou, B.; Abiven, G.; Gicquel, C.; Leboulleux, S.; Travagli, J.-P.; Dromain, C.; Bertagna, X.; et al. Prognostic Parameters of Metastatic Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2007, 92, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Bergenstal, D.; Hertz, R.; Lipsett, M.B.; Moy, R.H. Chemotherapy of adrenocortical cancer with o,p′DDD. Ann. Int. Med. 1960, 53, 672–682. [Google Scholar] [CrossRef]
- Lacroix, A.; Feelders, R.A.; Stratakis, C.A.; Nieman, L.K. Cushing’s syndrome. Lancet 2015, 386, 913–927. [Google Scholar] [CrossRef]
- Biller, B.M.K.; Grossman, A.B.; Stewart, P.M.; Melmed, S.; Bertagna, X.; Bertherat, J.; Buchfelder, M.; Colao, A.; Hermus, A.R.; Hofland, L.J.; et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: A consensus statement. J. Clin. Endocrinol. Metab. 2008, 93, 2454–2462. [Google Scholar] [CrossRef]
- Pivonello, R.; De Leo, M.; Cozzolino, A.; Colao, A. The Treatment of Cushing’s Disease. Endocr. Rev. 2015, 36, 385–486. [Google Scholar] [CrossRef]
- Pivonello, R.; De Martino, M.C.; De Leo, M.; Lombardi, G.; Colao, A. Cushing’s Syndrome. Endocrinol. Metab. Clin. N. Am. 2008, 37, 135–149. [Google Scholar] [CrossRef]
- Tritos, N.A.; Biller, B.M.K.; Swearingen, B. Management of Cushing disease. Nat. Rev. Endocrinol. 2011, 7, 279–289. [Google Scholar] [CrossRef]
- Dimopoulou, C.; Schopohl, J.; Rachinger, W.; Buchfelder, M.; Honegger, J.; Reincke, M.; Stalla, G.K. Long-term remission and recurrence rates after first and second transsphenoidal surgery for Cushing’s disease: Care reality in the Munich Metropolitan Region. Eur. J. Endocrinol. 2013, 170, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Hahner, S.; Fassnacht, M. Mitotane for adrenocortical carcinoma treatment. Curr. Opin. Investig. Drugs 2005, 6, 386–394. [Google Scholar]
- Igaz, P.; Tombol, Z.; Szabo, P.M.; Liko, I.; Racz, K. Steroid biosynthesis inhibitors in the therapy of hypercortisolism: Theory and practice. Med. Chem. 2008, 15, 2734–2747. [Google Scholar] [CrossRef] [PubMed]
- Terzolo, M.; Pia, A.; Berruti, A.; Osella, G.; Alì, A.; Carbone, V.; Testa, E.; Dogliotti, L.; Angeli, A. Low-dose monitored mitotane treatment achieves the therapeutic range with manageable side effects in patients with adrenocortical cancer. J. Clin. Endocrinol. Metab. 2000, 85, 2234–2238. [Google Scholar] [CrossRef]
- Terzolo, M.; Zaggia, B.; Allasino, B.; De Francia, S. Practical treatment using mitotane for adrenocortical carcinoma. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.S.; Ahmad, T.; Groll, J.; Scherf-Clavel, O.; Kroiss, M.; Luxenhofer, R. The Challenging Pharmacokinetics of Mitotane: An Old Drug in Need of New Packaging. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 575–593. [Google Scholar] [CrossRef]
- Schmouchkovitch, A.; Herry, H.; Thuillier, P.; Kerlan, V.; Fleuret, C.; Le Toux, G.; Boisramé, S. Oral and vulvo-vaginal lichenoid reactions due to mitotane (Lysodren®): A case report. Medicine 2017, 96, e5057. [Google Scholar] [CrossRef]
- Lung, B.Y.; Valentino, E.; Gerst, S.R.; Untch, B.R.; Katz, S.; Strong, V.E.; Raj, N.P.; Olino, K.; Saltz, L.; Reidy, D.L. Low objective response and high toxicity to single-agent mitotane in patients with metastatic adrenocortical carcinoma (ACC): A 25 year experience at MSKCC. J. Clin. Oncol. 2015, 33, 4105. [Google Scholar] [CrossRef]
- Grove, M.; Müllertz, A. Chapter 5: Liquid Self-Microemulsifying Drug Delivery Systems. In Oral Lipid-Based Formulations—Enhancing the Bioavailability of Poorly Water-Soluble Drugs; Hauss, D.J., Ed.; Informa Healthcare: New York, NY, USA, 2007; pp. 107–128. ISBN 9780429133695. [Google Scholar]
- Skiba, M.; Lahiani-Skiba, M.; Bounoure, F.; Thomas, M.; Lefebvre, H. Pharmaceutical Composition Comprising Mitotane Administered Orally for Treatment of Adrenocortical Carcinoma and Cushing’s Syndrome. Application PCT/IB2020/059218, 1 October 2020. [Google Scholar]
- Stein, S.; Auel, T.; Kempin, W.; Bogdahn, M.; Weitschies, W.; Seidlitz, A. Influence of the test method on in vitro drug release from intravitreal model implants containing dexamethasone or fluorescein sodium in poly(d,l-lactide-co-glycolide) or polycaprolactone. Eur. J. Pharm. Biopharm. 2018, 127, 270–278. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar] [PubMed]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Hixon, A.W.; Crowell, J.H. Dependence of Reaction Velocity upon Surface and Agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Trotta, M.; Gallarate, M.; Pattarino, F.; Morel, S. Emulsions containing partially water-miscible solvents for the preparation of drug nanosuspensions. J. Control. Release 2001, 76, 119–128. [Google Scholar] [CrossRef]
- Attivi, D.; Ajana, I.; Astier, A.; Demoré, B.; Gibaud, S. Development of microemulsion of mitotane for improvement of oral bioavailability. Drug Dev. Ind. Pharm. 2010, 36, 421–427. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Triplicate Guideline on Validation of Analytical Procedures: Text and Methodology Q2 (R1). Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 1 January 2024).
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Corso, C.R.; Acco, A.; Bach, C.; Bonatto, S.J.R.; de Figueiredo, B.C.; de Souza, L.M. Pharmacological profile and effects of mitotane in adrenocortical carcinoma. Br. J. Clin. Pharmacol. 2020, 87, 2698–2710. [Google Scholar] [CrossRef]
- Lehmann, T.P.; Wrzesinski, T.; Jagodzinski, P.P. The effect of mitotane on viability, steroidogenesis and gene expression in NCI-H295R adrenocortical cells. Mol. Med. Rep. 2013, 7, 893–900. [Google Scholar] [CrossRef]
- Lund, B.O.; Bergman, A.; Brandt, I. In vitro macromolecular binding of 2-(2-chlorophenyl)-2-(4-chlorophenyl)-1,1-dichloroethane (o,p’-DDD) in the mouse lung and liver. Chem. Biol. Interact. 1989, 70, 63–72. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autosampler | Acquity UPLC Sample Manager FTN with Sample Organizer (Waters, Saint-Quentin-en-Yvelines cedex, France) | ||
| LC pump | Acquity UPLC I-Class (Waters) | ||
| Column oven | Acquity UPLC Column Heater (Waters) | ||
| Detector | Acquity PDA Detector (Waters) | ||
| Software | MassLynx 4.2 & TargetLynx 4.2 (Waters) | ||
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | Mobile Phase B (%) |
| 0.0 | 0.5 | 60 | 40 |
| 5.0 | 0.5 | 10 | 90 |
| 7.0 | 0.5 | 10 | 90 |
| 7.1 | 0.5 | 60 | 40 |
| 9.0 | 0.5 | 60 | 40 |
| Formulation | Zero Order 1 R2 | First Order 2 R2 | Higuchi 3 R2 | Hixson-Crowell 4 R2 | Korsmeyer–Peppas 5 R2 |
|---|---|---|---|---|---|
| MitoF1 | 0.9378 | 0.9635 | 0.9833 | 0.9556 | 0.9774 |
| Lysodren® | 0.9762 | 0.9751 | 0.8647 | 0.9757 | 0.8867 |
| Pure drug | 0.6892 | 0.6917 | 0.9028 | 0.6851 | 0.8517 |
| Animal ID | Tmax (h) | Cmax (µg/mL) | AUC0–t (µg·h/mL) | Rsq² | t1/2 (h) | Lz (1/h) | Lz Lower (h) | Lz Upper (h) |
| 1 | 5 | 0.4975 | 2.635 | - | - | - | - | - |
| 2 | 4 | 0.7330 | 3.761 | 0.95 | 1.839 | 0.377 | 5 | 12.02 |
| 3 | 3 | 1.399 | 6.779 | 0.95 | 2.055 | 0.337 | 4 | 12 |
| 4 | 5 | 2.551 | 21.34 | 0.82 | 8.269 | 0.0838 | 6 | 48 |
| 5 | 4 | 3.684 | 17.32 | 0.98 | 4.977 | 0.139 | 8.983 | 23.98 |
| 6 | 3 | 1.302 | 7.103 | 0.91 | 2.169 | 0.320 | 5 | 12 |
| 7 | 3 | 1.212 | 6.584 | 1.00 | 2.997 | 0.231 | 6 | 12 |
| 8 | 3 | 1.552 | 11.43 | 0.99 | 5.092 | 0.136 | 9 | 24 |
| N | 8 | 8 | 8 | - | 7 | 7 | - | - |
| Mean (1) | - | 1.616 | 9.619 | - | 2.987 | 0.204 | - | - |
| SD | - | 1.035 | 6.618 | - | - | - | - | - |
| CV% | - | 64 | 69 | |||||
| Min | 3 | 0.4975 | 2.635 | - | - | - | - | - |
| Median | 3.5 | 1.351 | 6.941 | - | - | - | - | |
| Animal ID | Tmax (h) | Cmax (µg/mL) | AUC0–t (µg·h/mL) | Rsq² | t1/2 (h) | Lz (1/h) | Lz Lower (h) | Lz Upper (h) |
| 9 | 4 | 3.871 | 31.41 | 0.81 | 7.007 | 0.0989 | 5 | 48 |
| 10 | 6 | 4.350 | 32.71 | 0.70 | - (2) | - (2) | - | - |
| 11 | 4 | 4.243 | 40.40 | 0.88 | 7.608 | 0.0911 | 5 | 48 |
| 12 | 4 | 6.991 | 37.19 | 0.84 | 13.90 | 0.0499 | 12 | 48.02 |
| 13 | 4 | 7.815 | 38.06 | 0.84 | 9.935 | 0.0698 | 9 | 48 |
| 14 | 5 | 5.267 | 42.2 | 0.84 | 7.311 | 0.0948 | 6 | 48 |
| 15 | 3 | 5.895 | 38.99 | 0.75 | - (2) | - (2) | - | - |
| 16 | 3 | 7.254 | 59.41 | 0.82 | 7.500 | 0.0924 | 4 | 47.98 |
| N | 8 | 8 | 8 | - | 6 | 6 | - | - |
| Mean | - | 5.711 | 40.05 | - | 8.370 | 0.0806 | - | - |
| SD | - | 1.515 | 8.631 | - | - | - | - | - |
| CV% | - | 27 | 22 | - | - | - | - | - |
| Min | 3 | 3.871 | 31.41 | - | - | - | - | - |
| Median | 4 | 5.581 | 38.53 | - | - | - | - | - |
| Animal ID | Tmax (h) | Cmax (µg/mL) | AUC0–t (µg·h/mL) | Rsq² | t1/2 (h) | Lz (1/h) | Lz Lower (h) | Lz Upper (h) |
| 18 | 4 | 5.590 | 35.70 | 0.90 | 5.646 | 0.123 | 9 | 24 |
| 19 | 4 | 5.235 | 35.08 | 0.96 | 4.938 | 0.140 | 9 | 24 |
| 20 | 3 | 7.297 | 45.27 | 0.84 | 10.14 | 0.0684 | 9 | 47.9 |
| 21 | 3 | 5.325 | 28.08 | 0.80 | 12.33 | 0.0562 | 9 | 47.88 |
| 22 | 3 | 3.890 | 35.06 | 0.81 | 7.830 | 0.0885 | 4 | 47.87 |
| 23 | 12 | 1.361 | 19.19 | - | - | - | - | - |
| 24 | 6 | 5.678 | 34.51 | 0.69 | - (2) | - (2) | - | - |
| N | 7 | 7 | 7 | - | 5 | 5 | - | - |
| Mean | - | 4.911 | 33.27 | - | 7.277 | 0.0899 | - | - |
| SD | - | 1.856 | 7.989 | - | - | - | - | - |
| CV% | - | 38 | 24 | - | - | - | - | - |
| Min | 3 | 1.361 | 19.19 | - | - | - | - | - |
| Median | 4 | 5.325 | 35.06 | - | - | - | - | - |
| Max | 12 | 7.297 | 45.27 | - | - | - | - | - |
| Cmax | Lysodren | MitoF1 | MitoF2 | AUC0–t | Lysodren | MitoF1 | MitoF2 |
| Lysodren | 0.0001554 | 0.00373 | Lysodren | 0.0001554 | 0.0006216 | ||
| MitoF1 | 0.6126 | MitoF1 | 0.152 | ||||
| MitoF2 | MitoF2 | ||||||
| t1/2 | Lysodren | MitoF1 | MitoF2 | Lz | Lysodren | MitoF1 | MitoF2 |
| Lysodren | 0.01399 | 0.04798 | Lysodren | 0.01399 | 0.04798 | ||
| MitoF1 | 0.9307 | MitoF1 | 0.9307 | ||||
| MitoF2 | MitoF2 | ||||||
| AUCinf | Lysodren | MitoF1 | MitoF2 | ||||
| Lysodren | 0.001166 | 0.002525 | |||||
| MitoF1 | 0.2468 | ||||||
| MitoF2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skiba, M.; Lefébure, V.; Bounoure, F.; Milon, N.; Thomas, M.; Lefebvre, H.; Malika, L.-S. Powder Self-Emulsifying Drug Delivery System for Mitotane: In Vitro and In Vivo Evaluation. Pharmaceutics 2024, 16, 1194. https://doi.org/10.3390/pharmaceutics16091194
Skiba M, Lefébure V, Bounoure F, Milon N, Thomas M, Lefebvre H, Malika L-S. Powder Self-Emulsifying Drug Delivery System for Mitotane: In Vitro and In Vivo Evaluation. Pharmaceutics. 2024; 16(9):1194. https://doi.org/10.3390/pharmaceutics16091194
Chicago/Turabian StyleSkiba, Mohamed, Valentin Lefébure, Frederic Bounoure, Nicolas Milon, Michael Thomas, Herve Lefebvre, and Lahiani-Skiba Malika. 2024. "Powder Self-Emulsifying Drug Delivery System for Mitotane: In Vitro and In Vivo Evaluation" Pharmaceutics 16, no. 9: 1194. https://doi.org/10.3390/pharmaceutics16091194