
Synthesis of Arginase Inhibitors: An Overview

 , , , , , and
, , , , , and 
Abstract
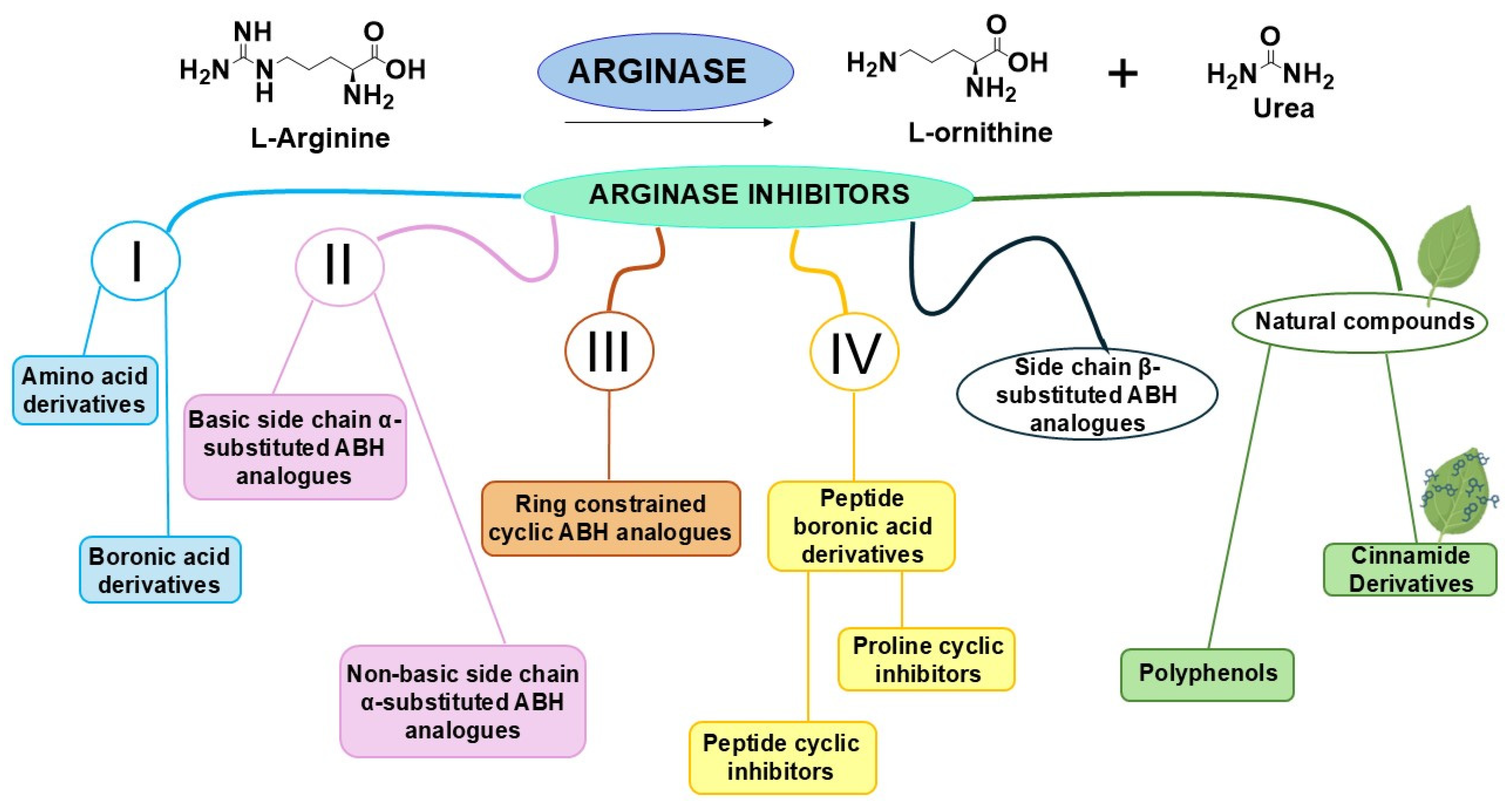
:1. Introduction
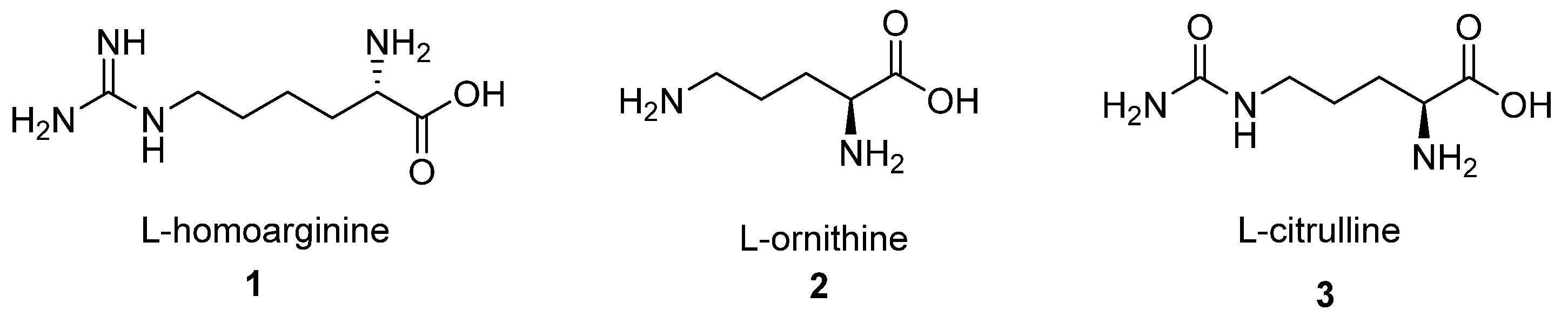
2. First-Generation Inhibitors
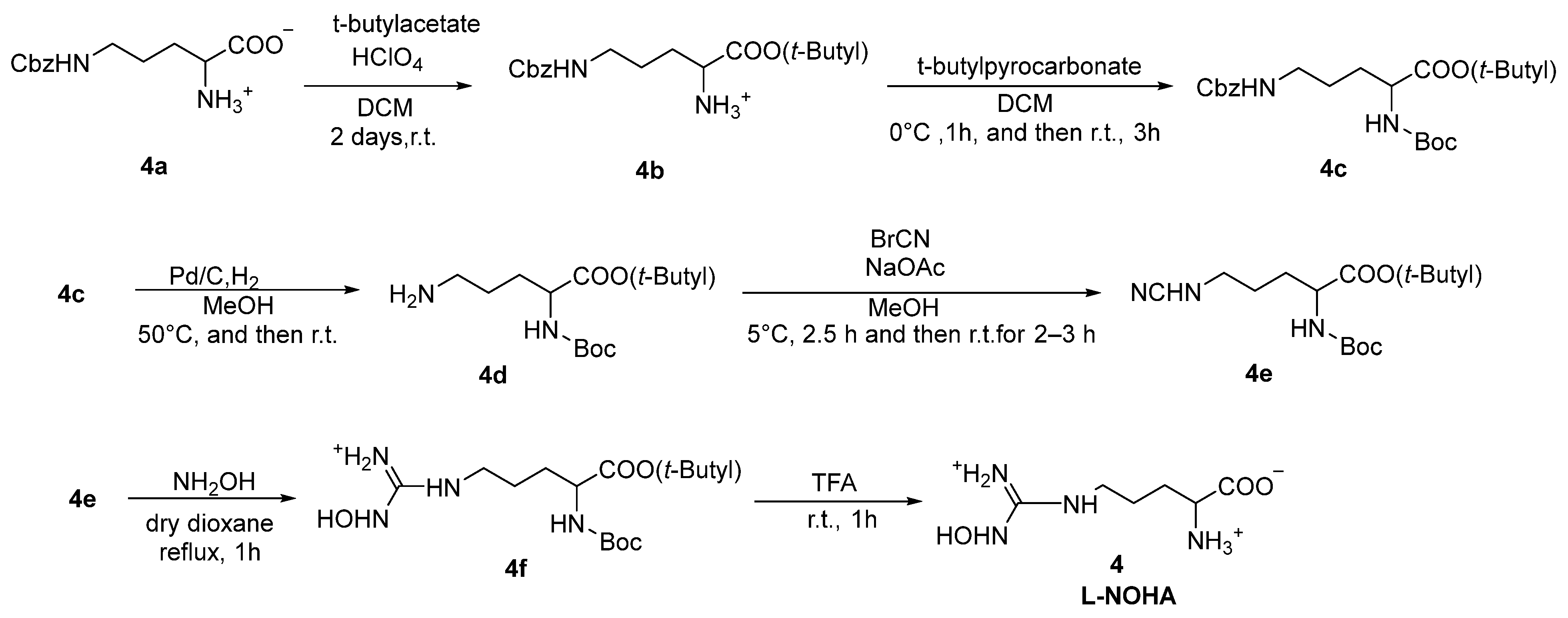
2.1. α-Amino Acid Derivatives
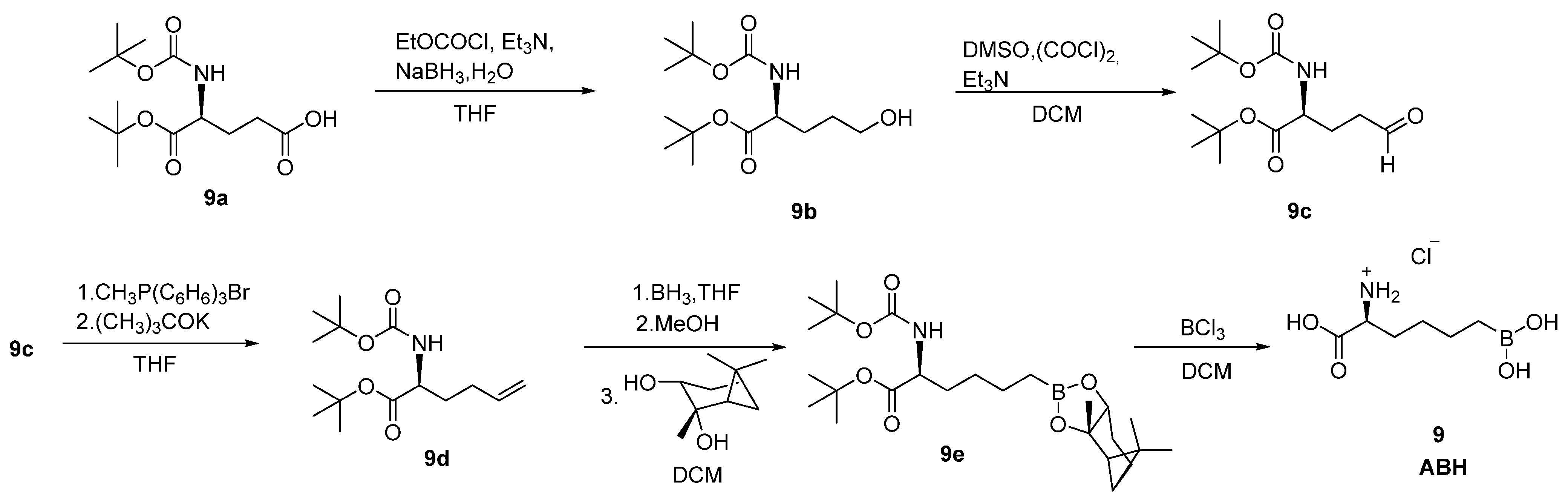
2.2. Boronic Acid Derivatives
3. Second-Generation Inhibitors
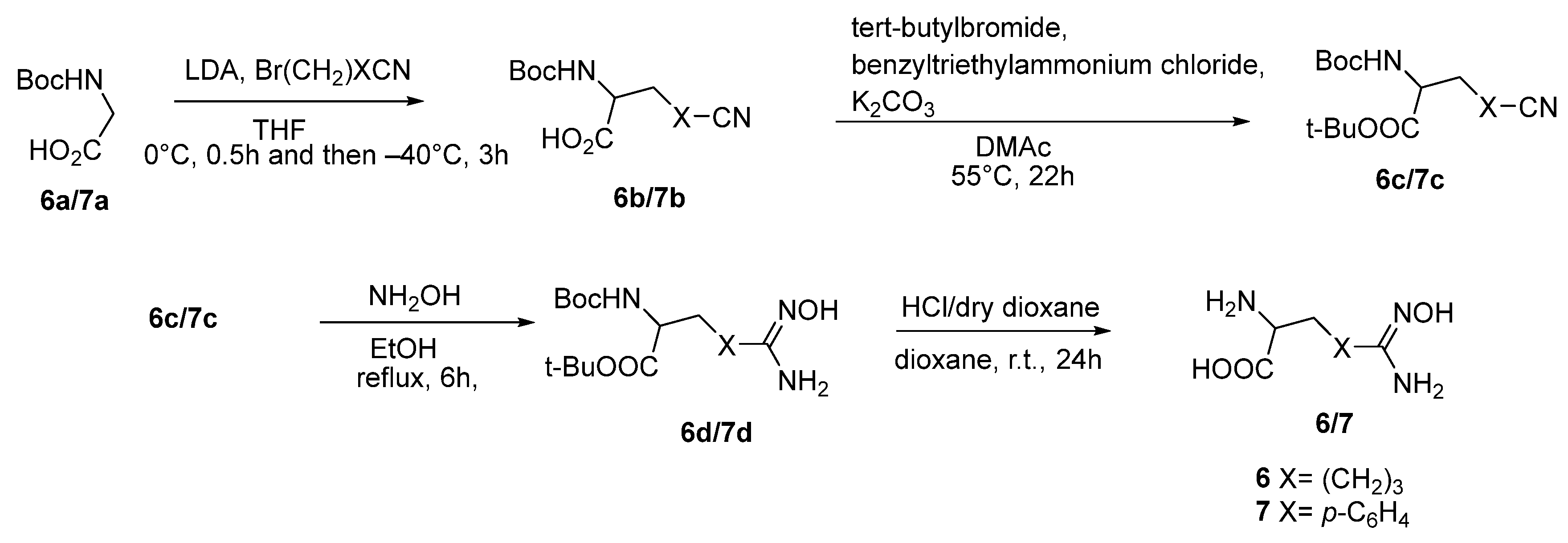
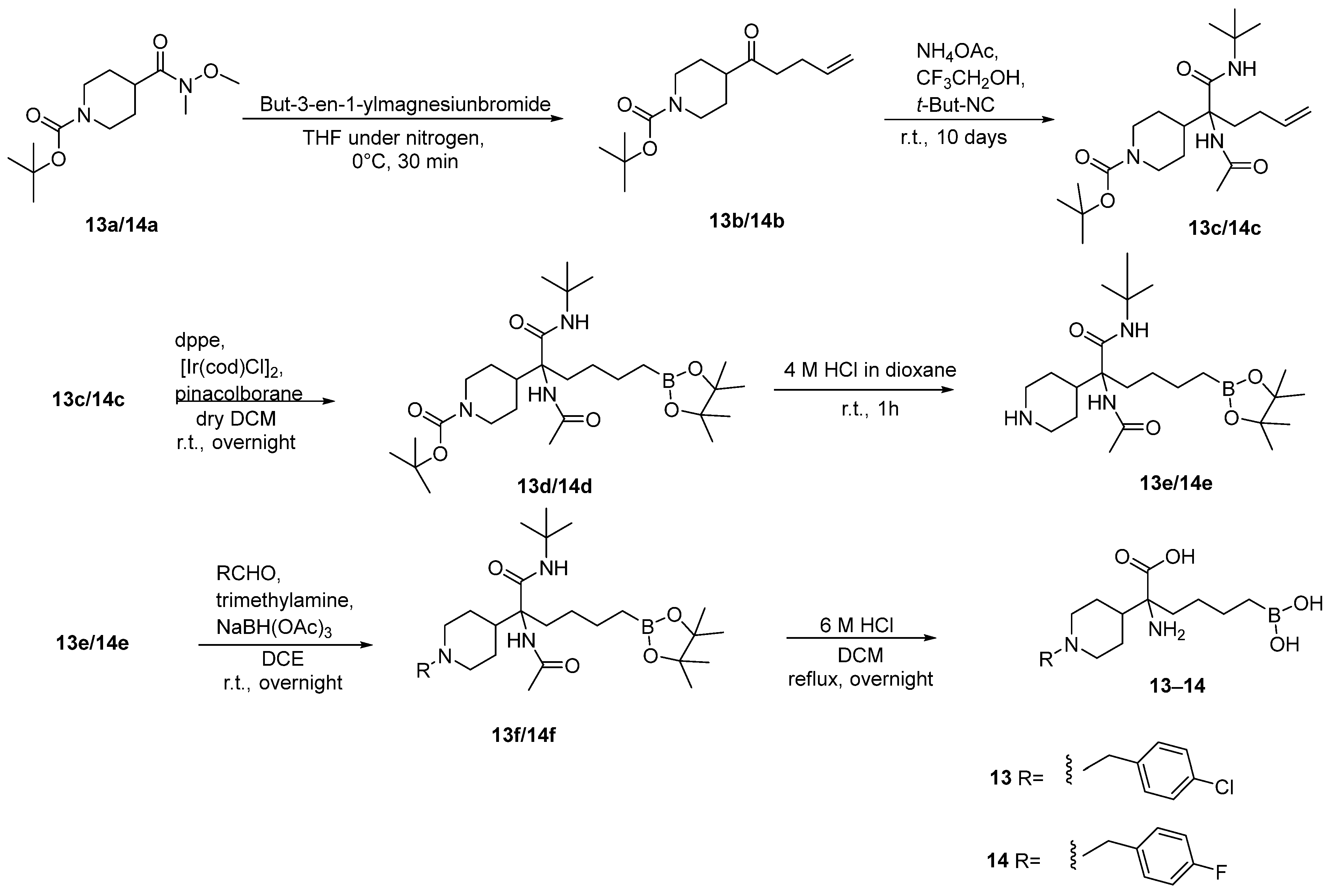
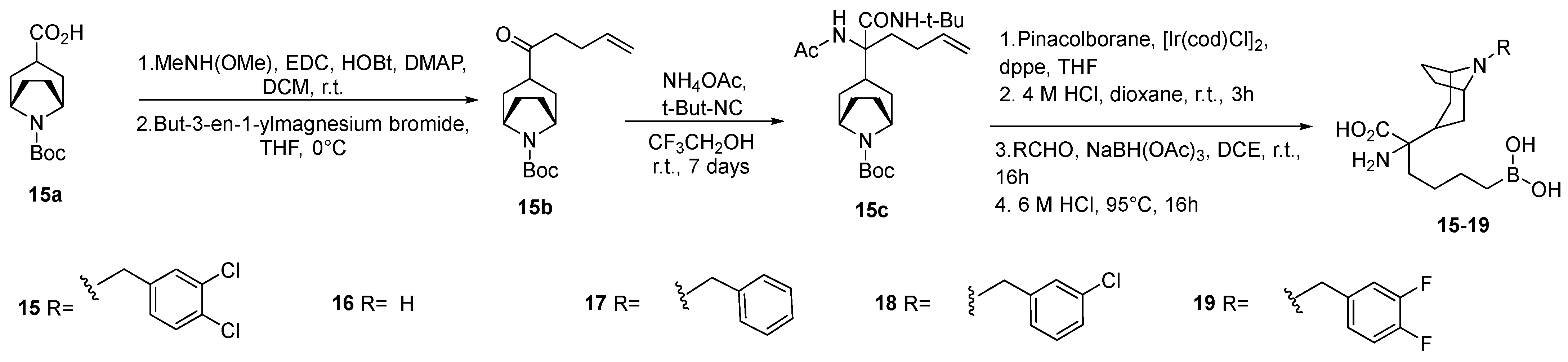
3.1. Basic Side Chain α-Substituted ABH Analogues
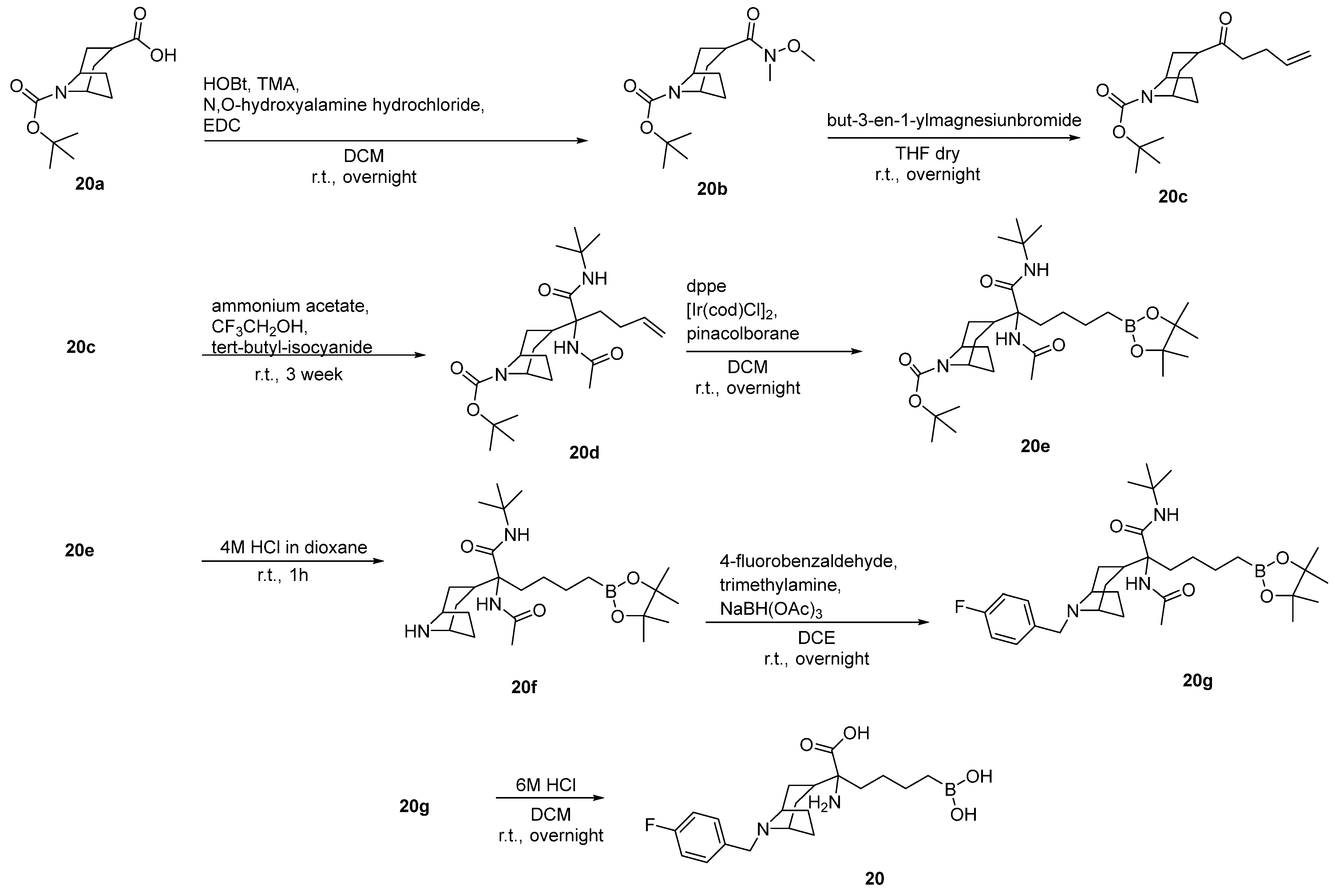
3.2. Non-Basic Side Chain α-Substituted ABH Analogues
4. Third-Generation Inhibitors
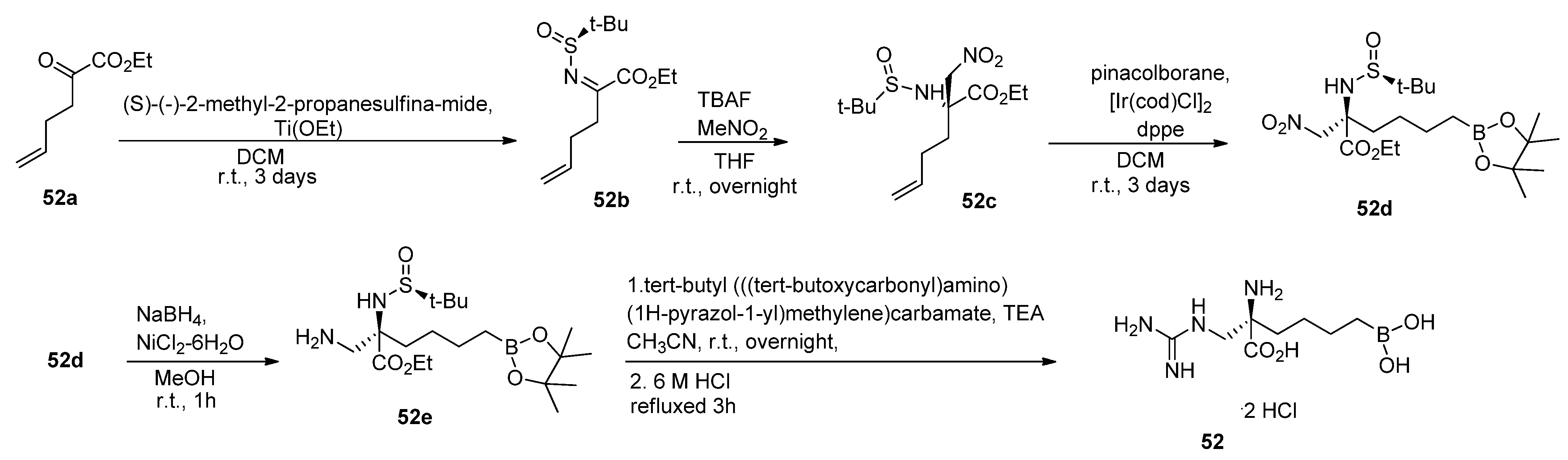
5. Fourth-Generation Inhibitors
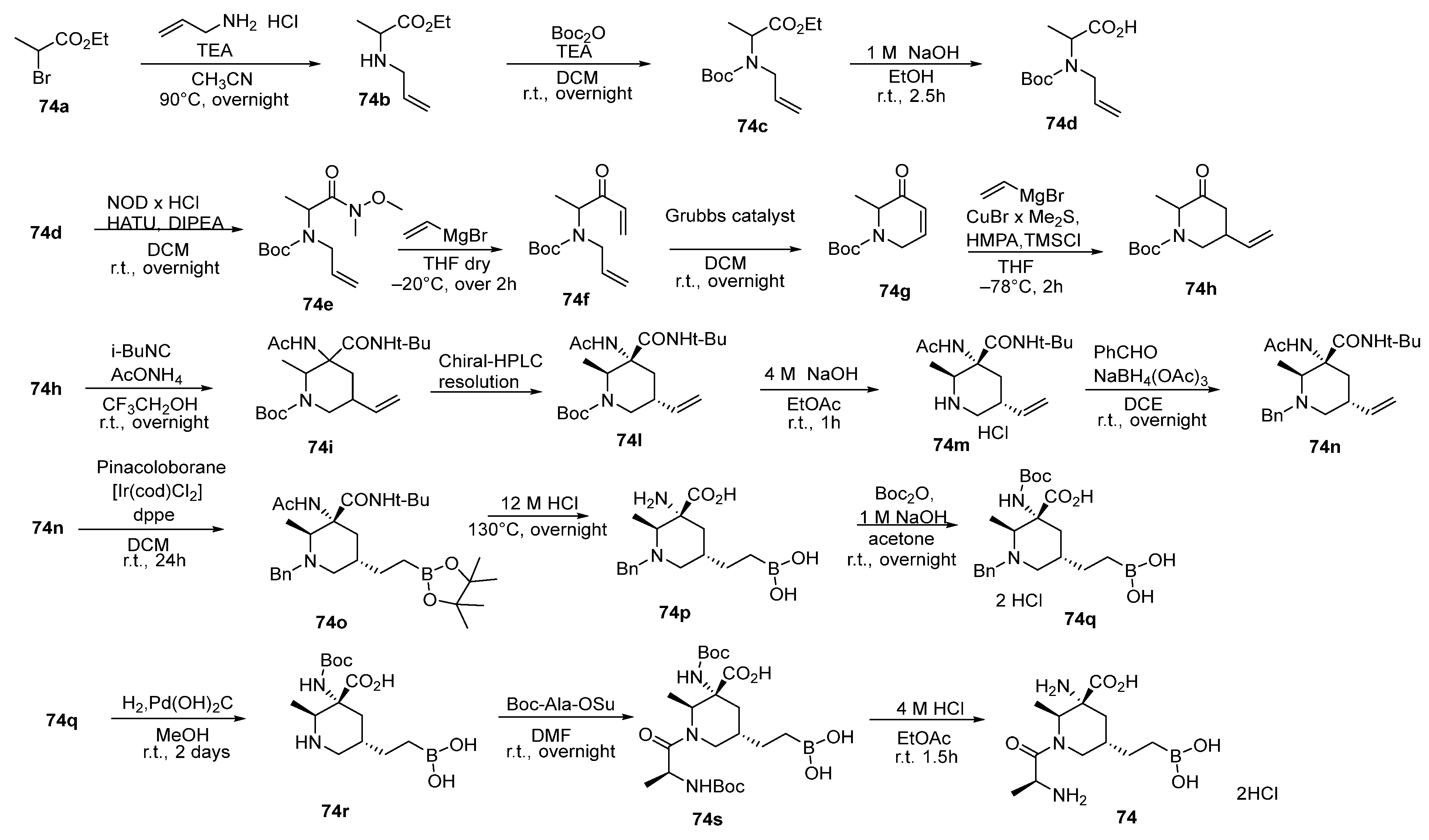
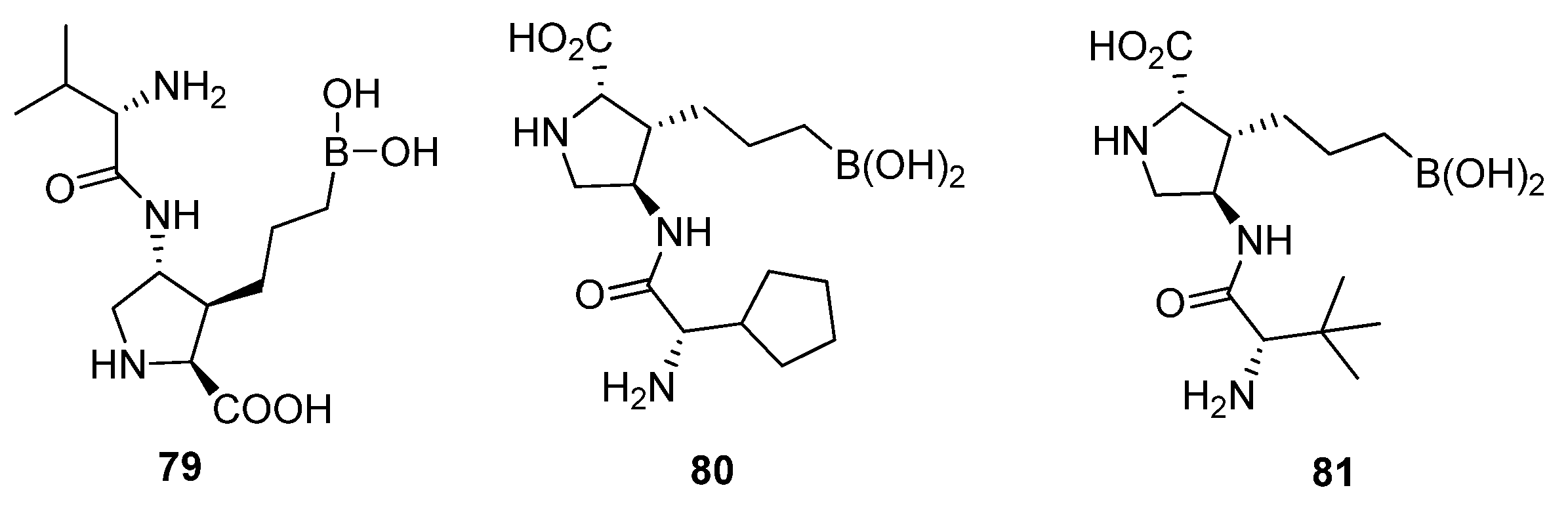
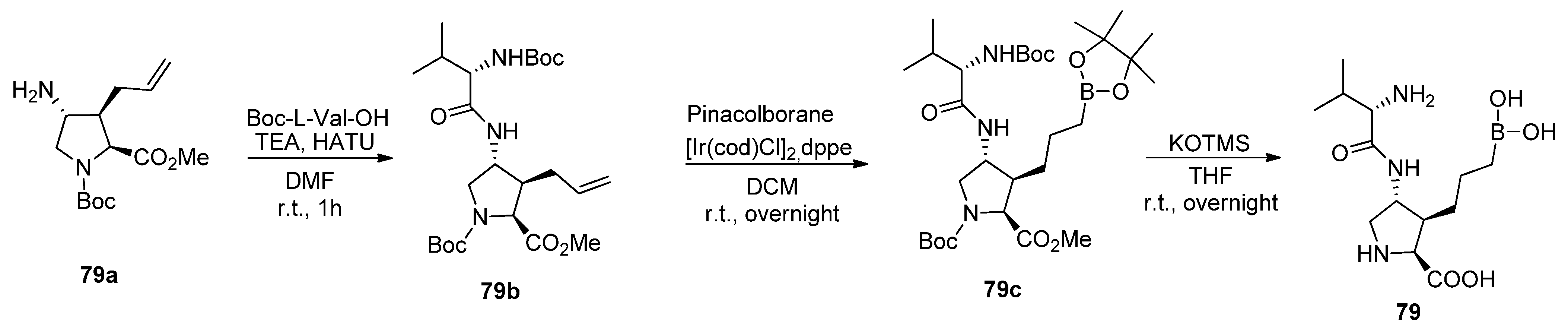
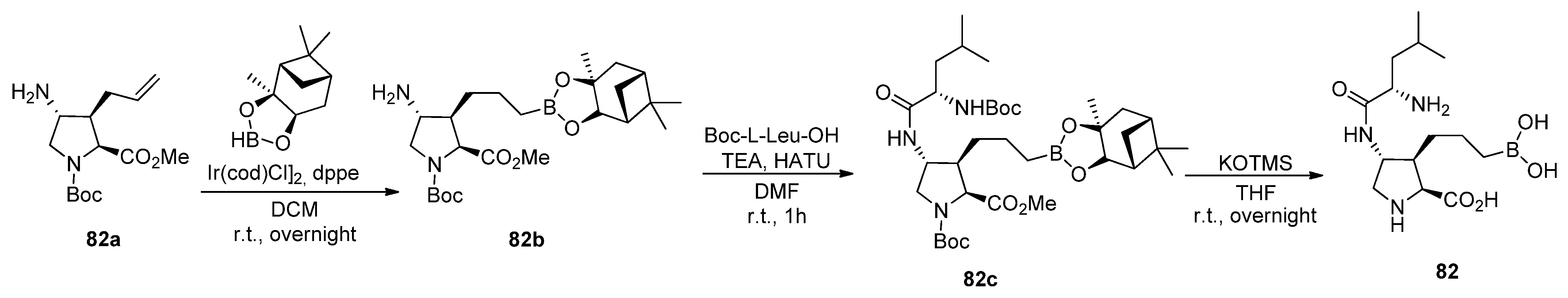
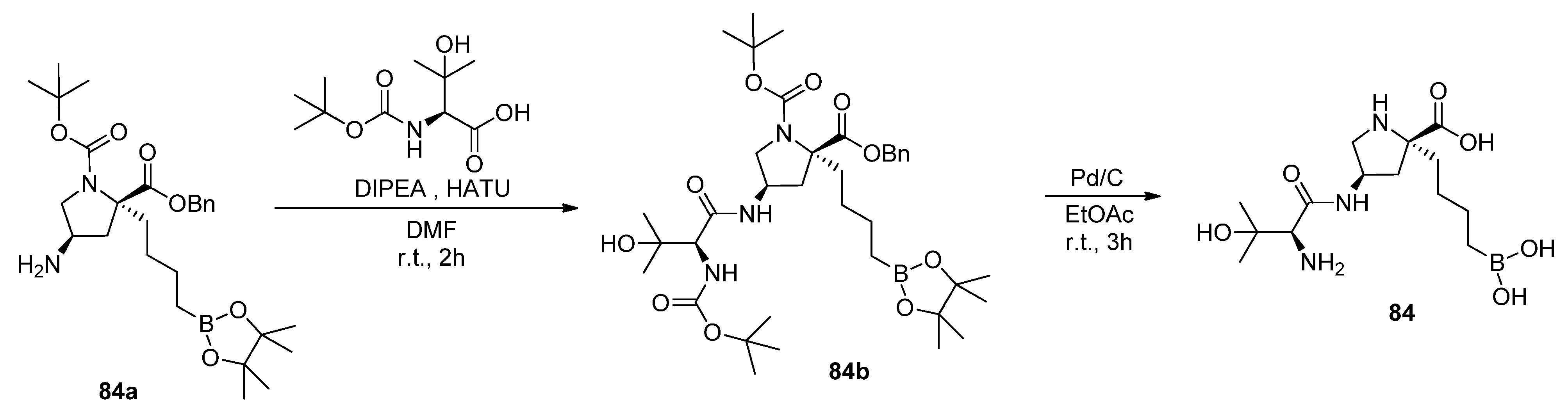
5.1. Peptide Boronic Acid Derivatives
5.1.1. Peptide Cyclic Inhibitors
5.1.2. Proline Cyclic Inhibitors
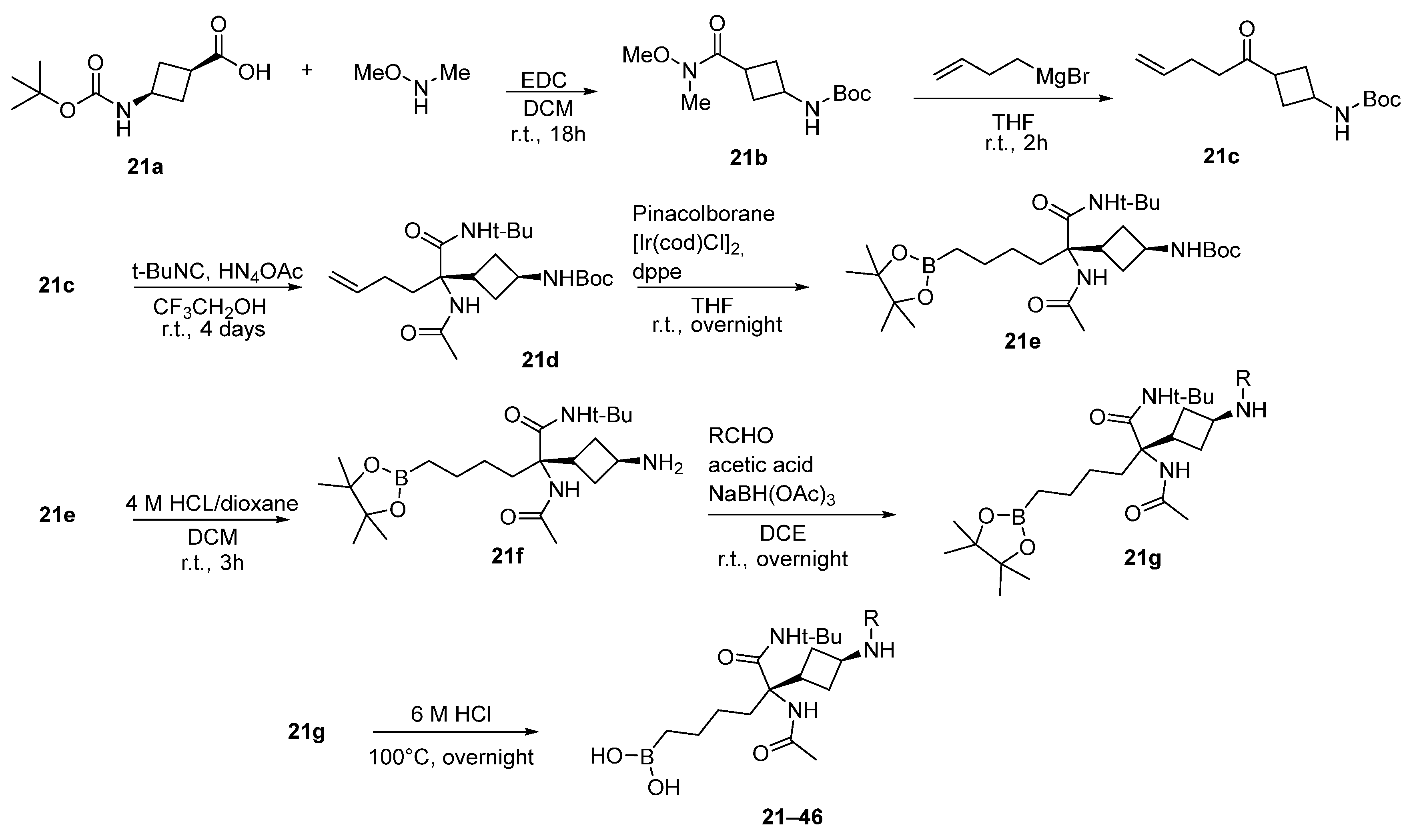
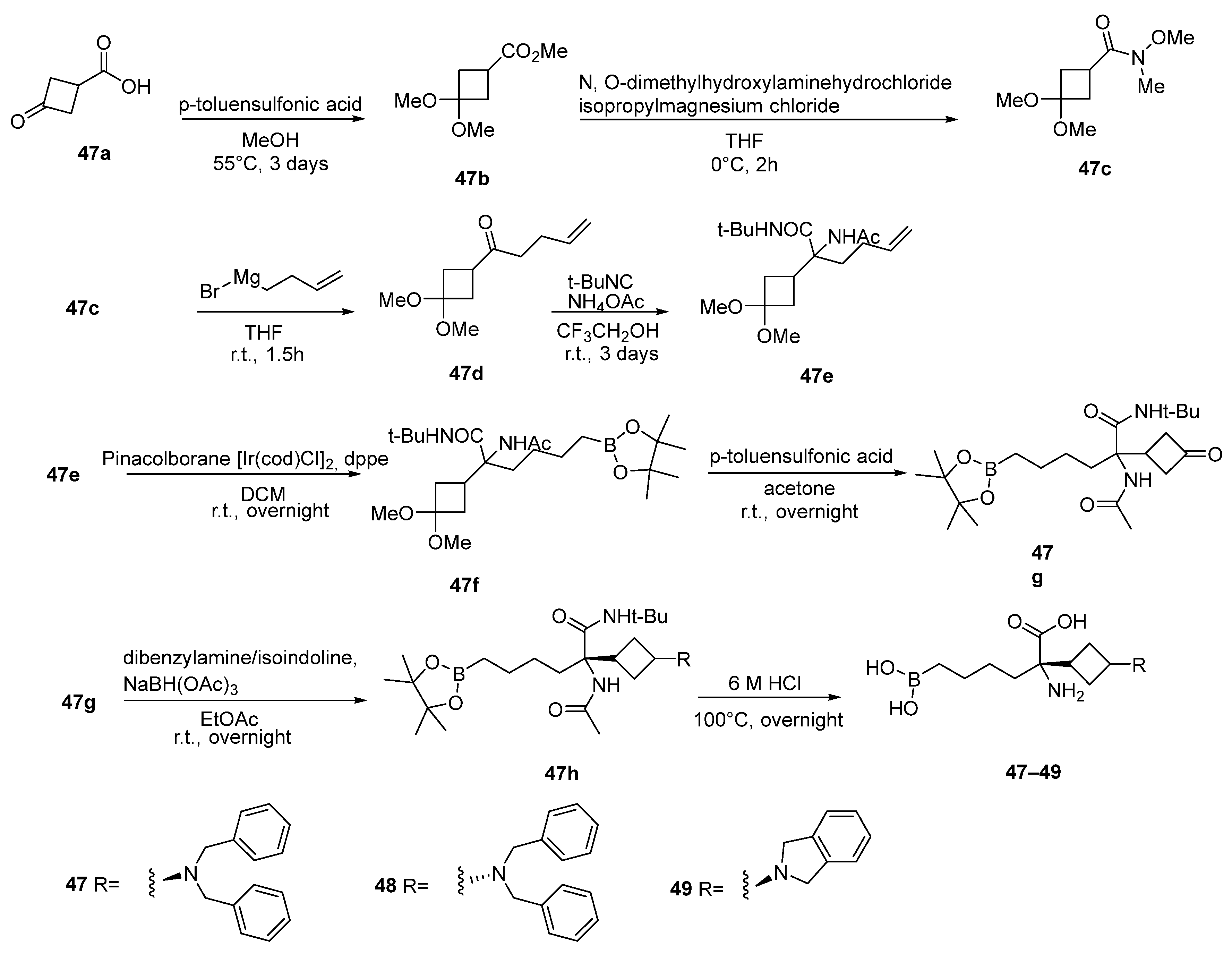
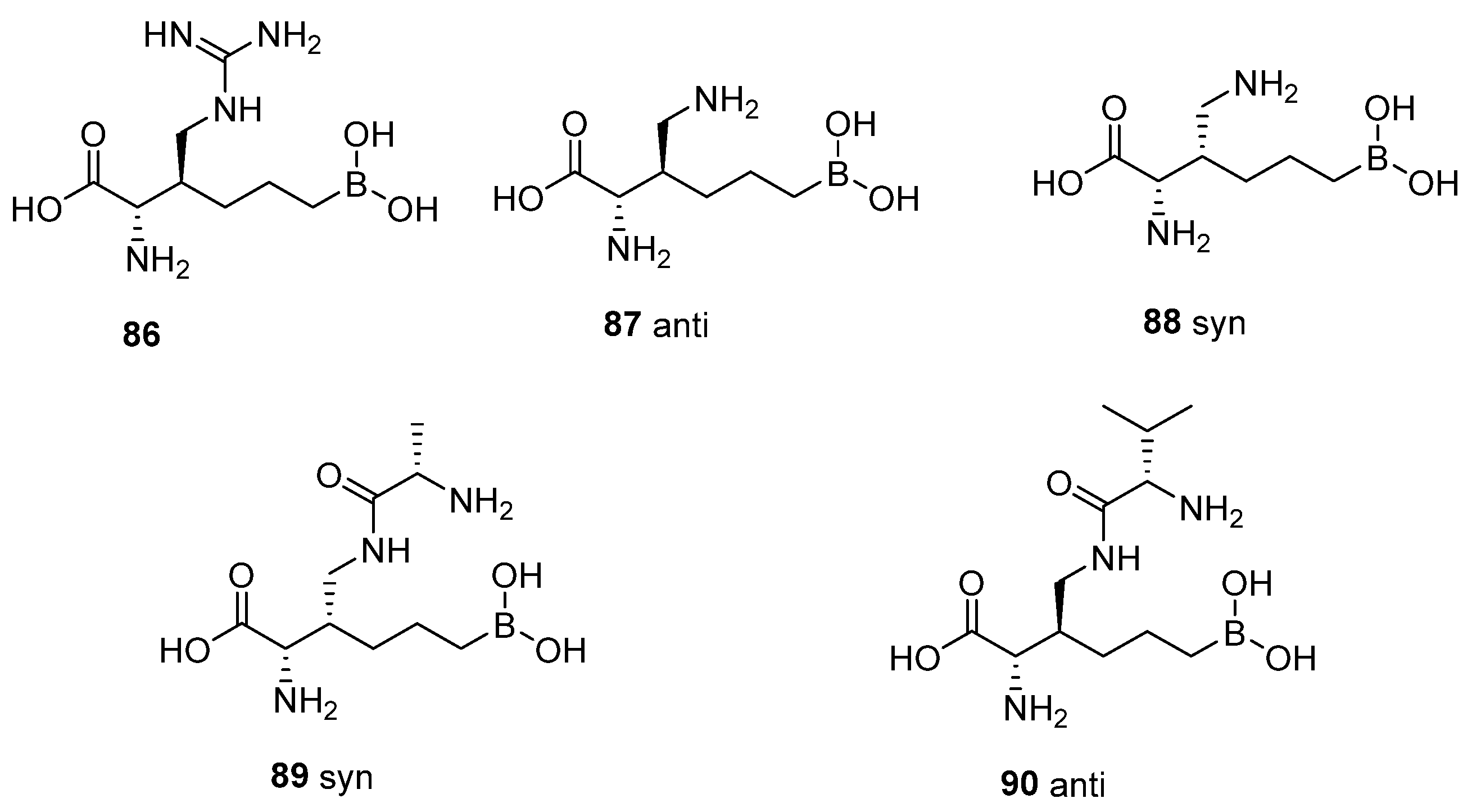
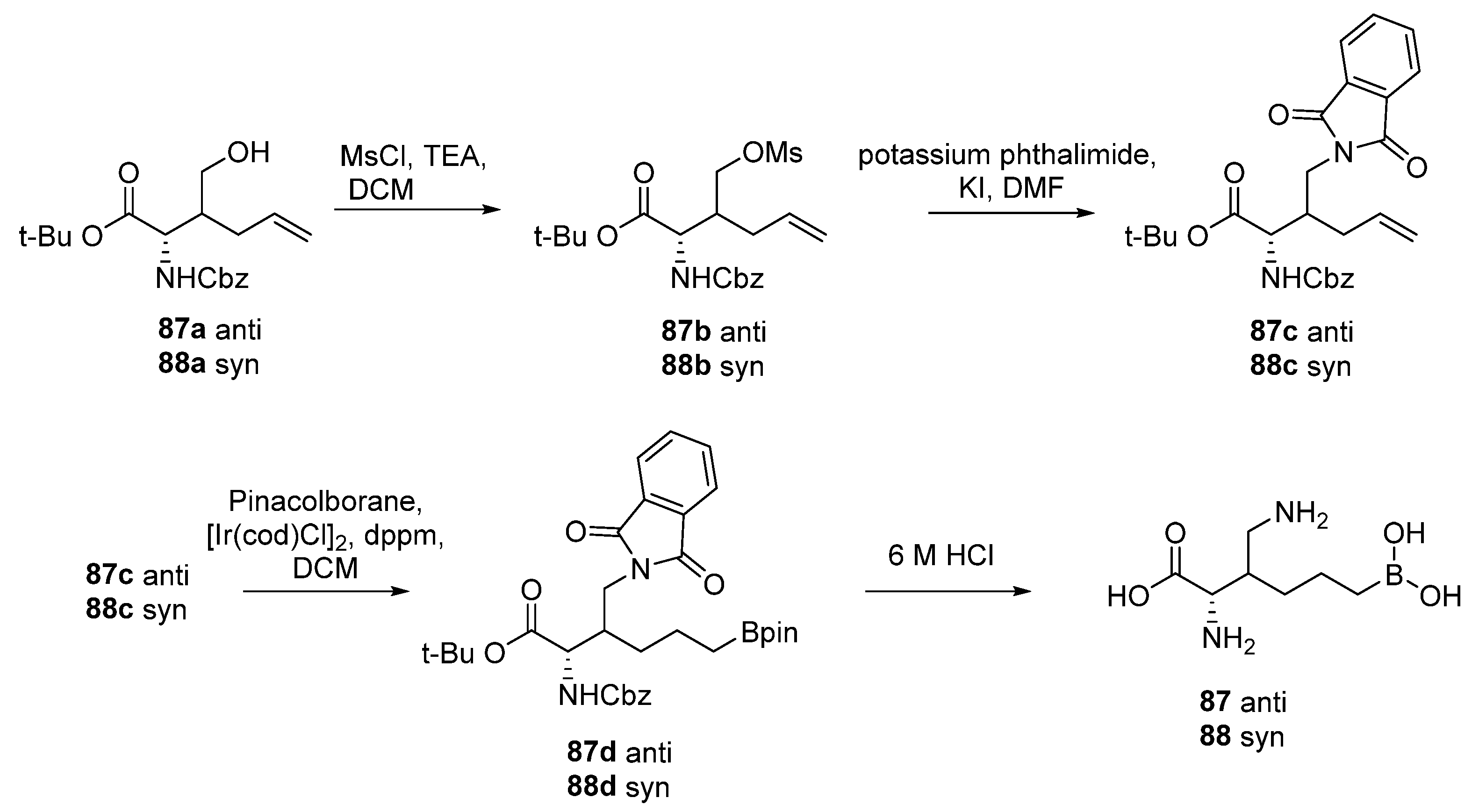
6. Side Chain β-Substituted ABH Analogues
7. Natural Compounds as ARG Inhibitors
7.1. Polyphenols
7.2. Cinnamide Derivatives
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An Old Enzyme with New Tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M. Arginine Metabolism: Nitric Oxide and Beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Morris, S.M. Recent Advances in Arginine Metabolism: Roles and Regulation of the Arginases. Br. J. Pharmacol. 2009, 157, 922–930. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Canè, S.; Geiger, R.; Bronte, V. The roles of arginases and arginine in immunity. Nat. Rev. Immunol. 2024; Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.M.; Boothby, I.; Clancy, S.; Ahn, R.S.; Liao, W.; Nguyen, D.N.; Schumann, K.; Marson, A.; Mahuron, K.M.; Kingsbury, G.A.; et al. Regulatory T Cells Use Arginase 2 to Enhance Their Metabolic Fitness in Tissues. JCI Insight 2019, 4, 129756. [Google Scholar] [CrossRef]
- Munder, M. Arginase: An Emerging Key Player in the Mammalian Immune System. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef]
- Durante, W. Role of Arginase in Vessel Wall Remodeling. Front. Immunol. 2013, 4, 111. [Google Scholar] [CrossRef]
- Wiesinger, H. Arginine Metabolism and the Synthesis of Nitric Oxide in the Nervous System. Prog. Neurobiol. 2001, 64, 365–391. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric Oxide and Redox Mechanisms in the Immune Response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef]
- Gotoh, T.; Mori, M. Arginase II Downregulates Nitric Oxide (NO) Production and Prevents NO-Mediated Apoptosis in Murine Macrophage-Derived RAW 264.7 Cells. J. Cell Biol. 1999, 144, 427–434. [Google Scholar] [CrossRef]
- Choudry, M.; Tang, X.; Santorian, T.; Wasnik, S.; Xiao, J.; Xing, W.; Lau, K.H.W.; Mohan, S.; Baylink, D.J.; Qin, X. Deficient Arginase II Expression without Alteration in Arginase I Expression Attenuated Experimental Autoimmune Encephalomyelitis in Mice. Immunology 2018, 155, 85–98. [Google Scholar] [CrossRef]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-Induced Vascular Dysfunction and Arterial Stiffening Requires Endothelial Cell Arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase Reciprocally Regulates Nitric Oxide Synthase Activity and Contributes to Endothelial Dysfunction in Aging Blood Vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef]
- Clemente, G.S.; van Waarde, A.; Antunes, I.F.; Dömling, A.; Elsinga, P.H. Arginase as a Potential Biomarker of Disease Progression: A Molecular Imaging Perspective. Int. J. Mol. Sci. 2020, 21, 5291. [Google Scholar] [CrossRef]
- Erdely, A.; Kepka-Lenhart, D.; Salmen-Muniz, R.; Chapman, R.; Hulderman, T.; Kashon, M.; Simeonova, P.P.; Morris, S.M., Jr. Arginase activities and global arginine bioavailability in wild-type and ApoE-deficient mice: Responses to high fat and high cholesterol diets. PLoS ONE 2010, 5, e15253. [Google Scholar] [CrossRef]
- Zhang, Y.; Higgins, C.B.; Fortune, H.M.; Chen, P.; Stothard, A.I.; Mayer, A.L.; Swarts, B.M.; DeBosch, B.J. Hepatic arginase 2 (Arg2) is sufficient to convey the therapeutic metabolic effects of fasting. Nat. Commun. 2019, 10, 1587. [Google Scholar] [CrossRef]
- Lange, P.S.; Langley, B.; Lu, P.; Ratan, R.R. Arginine Metabolism: Enzymology, Nutrition, and Clinical Significance Novel Roles for Arginase in Cell Survival, Regeneration, and Translation in the Central Nervous System. J. Nutr. 2004, 134, 2812S–2817S. [Google Scholar] [CrossRef]
- Polis, B.; Srikanth, K.D.; Gurevich, V.; Bloch, N.; Gil-Henn, H.; Samson, A.O. Arginase Inhibition Supports Survival and Differentiation of Neuronal Precursors in Adult Alzheimer’s Disease Mice. Int. J. Mol. Sci. 2020, 21, 1133. [Google Scholar] [CrossRef]
- Liu, P.; Fleete, M.S.; Jing, Y.; Collie, N.D.; Curtis, M.A.; Waldvogel, H.J.; Faull, R.L.M.; Abraham, W.C.; Zhang, H. Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol. Aging 2014, 35, 1992. [Google Scholar] [CrossRef]
- Borek, B.; Gajda, T.; Golebiowski, A.; Blaszczyk, R. Boronic Acid-Based Arginase Inhibitors in Cancer Immunotherapy. Bioorganic Med. Chem. 2020, 28, 115658. [Google Scholar] [CrossRef]
- Muller, J.; Cardey, B.; Zedet, A.; Desingle, C.; Grzybowski, M.; Pomper, P.; Foley, S.; Harakat, D.; Ramseyer, C.; Girard, C.; et al. Synthesis, Evaluation and Molecular Modelling of Piceatannol Analogues as Arginase Inhibitors. RSC Med. Chem. 2020, 11, 559–568. [Google Scholar] [CrossRef]
- Van Zandt, M.C.; Whitehouse, D.L.; Golebiowski, A.; Ji, M.K.; Zhang, M.; Beckett, R.P.; Jagdmann, G.E.; Ryder, T.R.; Sheeler, R.; Andreoli, M.; et al. Discovery of (R)-2-Amino-6-Borono-2-(2-(Piperidin-1-Yl)Ethyl)Hexanoic Acid and Congeners as Highly Potent Inhibitors of Human Arginases i and II for Treatment of Myocardial Reperfusion Injury. J. Med. Chem. 2013, 56, 2568–2580. [Google Scholar] [CrossRef]
- Blaszczyk, R.; Brzezinska, J.; Dymek, B.; Stanczak, P.S.; Mazurkiewicz, M.; Olczak, J.; Nowicka, J.; Dzwonek, K.; Zagozdzon, A.; Golab, J.; et al. Discovery and Pharmacokinetics of Sulfamides and Guanidines as Potent Human Arginase 1 Inhibitors. ACS Med. Chem. Lett. 2020, 11, 433–438. [Google Scholar] [CrossRef]
- Golebiowski, A.; Whitehouse, D.; Beckett, R.P.; Van Zandt, M.; Ji, M.K.; Ryder, T.R.; Jagdmann, E.; Andreoli, M.; Lee, Y.; Sheeler, R.; et al. Synthesis of Quaternary α-Amino Acid-Based Arginase Inhibitors via the Ugi Reaction. Bioorganic Med. Chem. Lett. 2013, 23, 4837–4841. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of Arginase by CB-1158 Blocks Myeloid Cell-Mediated Immune Suppression in the Tumor Microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Blaszczyk, R.; Nowicka, J.; Borek, B.; Brzezinska, J.; Gzik, A.; Dziegielewski, M.; Golebiowski, A.; Jedrzejczak, K.; Matyszewski, K.; Olczak, J. Arginase Inhibitors and Their Therapeutic Applications. World Intellectual Property Organization. WO2017191130A2, 9 November 2017. [Google Scholar]
- Grzybowski, M.M.; Stańczak, P.S.; Pomper, P.; Błaszczyk, R.; Borek, B.; Gzik, A.; Nowicka, J.; Jędrzejczak, K.; Brzezińska, J.; Rejczak, T.; et al. OATD-02 Validates the Benefits of Pharmacological Inhibition of Arginase 1 and 2 in Cancer. Cancers 2022, 14, 3967. [Google Scholar] [CrossRef]
- Niu, F.; Yu, Y.; Li, Z.; Ren, Y.; Li, Z.; Ye, Q.; Liu, P.; Ji, C.; Qian, L.; Xiong, Y. Arginase: An Emerging and Promising Therapeutic Target for Cancer Treatment. Biomed. Pharmacother. 2022, 149, 112840. [Google Scholar] [CrossRef]
- Failla, M.; Molaro, M.C.; Schiano, M.E.; Serafini, M.; Tiburtini, G.A.; Gianquinto, E.; Scoccia, R.; Battisegola, C.; Rimoli, M.G.; Chegaev, K.; et al. Opportunities and Challenges of Arginase Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2024, 67, 19988–20021. [Google Scholar] [CrossRef]
- Christianson, D.W. Arginase: Structure, Mechanism, and Physiological Role in Male and Female Sexual Arousal. Acc. Chem. Res. 2005, 38, 191–201. [Google Scholar] [CrossRef]
- Tommasi, S.; Elliot, D.J.; Da Boit, M.; Gray, S.R.; Lewis, B.C.; Mangoni, A.A. Homoarginine and Inhibition of Human Arginase Activity: Kinetic Characterization and Biological Relevance. Sci. Rep. 2018, 8, 3697. [Google Scholar] [CrossRef] [PubMed]
- Pudlo, M.; Demougeot, C.; Girard-Thernier, C. Arginase Inhibitors: A Rational Approach Over One Century. Med. Res. Rev. 2017, 37, 475–513. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.L.; Custot, J.; Vadon, S.; Delaforge, M.; Lepoivre, M.; Tenu, J.P.; Yapo, A.; Mansuy, D. N Omega-Hydroxyl-L-Arginine, an Intermediate in the L-Arginine to Nitric Oxide Pathway, Is a Strong Inhibitor of Liver and Macrophage Arginase. Biochem. Biophys. Res. Commun. 1994, 203, 1614–1621. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Ilies, M.; Thorn, K.J.; Christianson, D.W. Inhibition of Human Arginase I by Substrate and Product Analogues. Arch. Biochem. Biophys. 2010, 496, 101–108. [Google Scholar] [CrossRef]
- Colleluori, D.M.; Ash, D.E. Classical and Slow-Binding Inhibitors of Human Type II Arginase. Biochemistry 2001, 40, 9356–9362. [Google Scholar] [CrossRef]
- Wallace, G.C.; Fukuto, J.M. Synthesis and Bioactivity of N Omega-Hydroxyarginine: A Possible Intermediate in the Biosynthesis of Nitric Oxide from Arginine. J. Med. Chem. 1991, 34, 1746–1748. [Google Scholar] [CrossRef]
- Bodanszky, M.; Bodanszky, A. In The Practice of Peptide Synthesis, 1st ed.; Springer: New York, NY, USA, 1984. [Google Scholar]
- Bailey, D.M.; Degrazia, C.G.; Lape, H.E.; Frering, R.; Fort, D.; Skulan, T. Hydroxyguanidines. A New Class of Antihypertensive Agents. J. Med. Chem. 1973, 16, 151–156. [Google Scholar] [CrossRef]
- Moali, C.; Brollo, M.; Custot, J.; Sari, M.A.; Boucher, J.L.; Stuehr, D.J.; Mansuy, D. Recognition of α-Amino Acids Bearing Various C=NOH Functions by Nitric Oxide Synthase and Arginase Involves Very Different Structural Determinants. Biochemistry 2000, 39, 8208–8218. [Google Scholar] [CrossRef]
- Teng, H.B. Synthesis of Nω-Hydroxy-nor-L-Arginine. Chin. J. Appl. Chem. 2010, 27, 1111–1113. [Google Scholar]
- Custot, J.; Moali, C.; Brollo, M.; Boucher, J.L.; Delaforge, M.; Mansuy, D.; Tenu, J.P.; Zimmermann, J.L. Nω-Hydroxy-nor-L-Arginine: A High-Affinity Inhibitor of Arginase Well Adapted To Bind to Its Manganese Cluster. J. Am. Chem. Soc. 1997, 119, 4086–4087. [Google Scholar] [CrossRef]
- Vadon, S.; Custot, J.; Boucher, J.L.; Mansuy, D. Synthesis and Effects on Arginase and Nitric Oxide Synthase of Two Novel Analogues of N"-Hydroxyarginine, Nu-Hydroxyindospicine and p-h y Drox y Amidinophen y Lalanine. J. Chem. Soc. 1996, 1, 645–648. [Google Scholar]
- Metcalf, B.W.; Bey, P.; Danzin, C.; Jung, M.J.; Casara, P.; Vevert, J.P. Catalytic Irreversible Inhibition of Mammalian Ornithine Decarboxylase (E.C.4.1.1.17) by Substrate and Product Analogs. J. Am. Chem. Soc. 1978, 100, 2551–2553. [Google Scholar] [CrossRef]
- Selamnia, M.; Mayeur, C.; Robert, V.; Blachier, F. Difluoromethylornithine (DFMO) as a Potent Arginase Activity Inhibitor in Human Colon Carcinoma Cells. Biochem. Pharmacol. 1998, 55, 1241–1245. [Google Scholar] [CrossRef]
- Zhu, J.; Chadwick, S.T.; Price, B.A.; Zhao, S.X.; Costello, C.A.; Vemishetti, P. Processes for the Production of Alpha-Difluoromethyl Ornithine (DFMO). WO2003020209A2, 4 May 2004. [Google Scholar]
- Baggio, R.; Elbaum, D.; Kanyo, Z.F.; Carroll, P.J.; Christopher Cavalli, R.; Ash, D.E.; Christianson, D.W. Inhibition of Mn2+2-Arginase by Borate Leads to the Design of a Transition State Analogue Inhibitor, 2(S)-Amino-6-Boronohexanoic Acid. J. Am. Chem. Soc. 1932, 119, 8107–8108. [Google Scholar] [CrossRef]
- Preite, M.D.; Manriquez Mujica, J.M.; Correa Vargas, J.M.; Iturriaga Aguera, R.M.; Casanello Toledo, P.C.; Krause Leyton, B.J. Method for the Enantioselective Synthesis of 2(s)-Amino-6-Boronohexanoic Acid (ABH) and Purification Thereof. WO2016037298A1, 17 May 2016. [Google Scholar]
- Matteson, D.S.; Soloway, A.H.; Tomlinson, D.W.; Campbell, J.D.; Nixon, G.A. Synthesis and Biological Evaluation of Water-Soluble 2-Boronoethylthio Compounds. J. Med. Chem. 1964, 7, 640–643. [Google Scholar] [CrossRef]
- Van Zandt, M.C.; Golebiowski, A.; Ji, M.K.; Whitehouse, D.; Ryder, T.; Beckett, P. Inhibitor Arginase and Their Therapeutic Application. US20120083469A1, 5 April 2012. [Google Scholar]
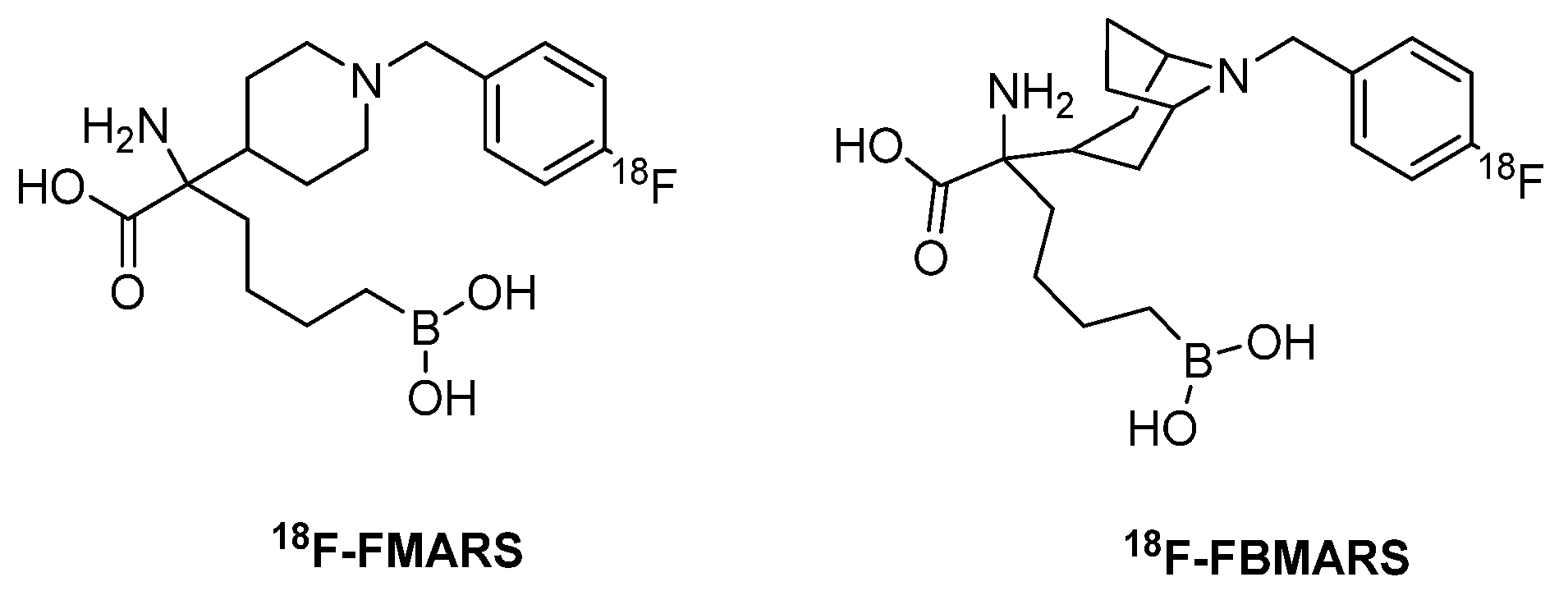
- Clemente, G.S.; Antunes, I.F.; Kurhade, S.; Van Den Berg, M.P.M.; Sijbesma, J.W.A.; Van Waarde, A.; Buijsman, R.C.; Willemsen-Seegers, N.; Gosens, R.; Meurs, H.; et al. Mapping Arginase Expression with 18F-Fluorinated Late-Generation Arginase Inhibitors Derived from Quaternary a-Amino Acids. J. Nucl. Med. 2021, 62, 1163–1170. [Google Scholar] [CrossRef]
- Moretto, J.; Pudlo, M.; Demougeot, C. Human-Based Evidence for the Therapeutic Potential of Arginase Inhibitors in Cardiovascular Diseases. Drug Discov. Today 2021, 26, 138–147. [Google Scholar] [CrossRef]
- Ilies, M.; Di Costanzo, L.; Dowling, D.P.; Thorn, K.J.; Christianson, D.W. Binding of α,α-Disubstituted Amino Acids to Arginase Suggests New Avenues for Inhibitor Design. J. Med. Chem. 2011, 54, 5432–5443. [Google Scholar] [CrossRef]
- Van Zandt, M.; Golebiowski, A.; Koo Ji, M.; Whitehouse, D.; Ryder, T.; Beckett, R.P. Inhibitor Arginase and Their Therapeutic Application. WO 2013/059437A1, 25 April 2013. [Google Scholar]
- Tomczuk, B.E.; Olson, G.L.; Pottorf, R.S.; Wang, J.; Nallaganchu, B.R. Arginase Inhibitors and Methods of Use Thereof. WO2012091757A1, 5 July 2012. [Google Scholar]
- Błaszczyk, R.; Brzezinska, J.; Golebiowski, A.; Olczak, J. Arginase Inhibitors and Their Therapeutic Applications. WO2016108707A1, 7 July 2016. [Google Scholar]
- Golebiowski, A.; Paul Beckett, R.; Van Zandt, M.; Ji, M.K.; Whitehouse, D.; Ryder, T.R.; Jagdmann, E.; Andreoli, M.; Mazur, A.; Padmanilayam, M.; et al. 2-Substituted-2-Amino-6-Boronohexanoic Acids as Arginase Inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 2027–2030. [Google Scholar] [CrossRef]
- Van Zandt, M.; Jagdmann, G.E., Jr. Boronates as Arginase Inhibitors. WO2012058065A1, 3 March 2012. [Google Scholar]
- Van Zandt, M.C.; Jagdmann, G.E.; Whitehouse, D.L.; Ji, M.; Savoy, J.; Potapova, O.; Cousido-Siah, A.; Mitschler, A.; Howard, E.I.; Pyle, A.M.; et al. Discovery of N-Substituted 3-Amino-4-(3-Boronopropyl)Pyrrolidine-3-Carboxylic Acids as Highly Potent Third-Generation Inhibitors of Human Arginase i and II. J. Med. Chem. 2019, 62, 8164–8177. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Ma, J.; Shao, Y. Heterocyclic Compounds as Arginase Inhibitors. WO2019120296A1, 27 June 2019. [Google Scholar]
- Foley, C.N.; Grange, R.L.; Guney, T.; Kalisiak, J.; Newcomb, E.T.; Tran, A.T. Arginase Inhibitors. WO2019173188A1, 19 September 2019. [Google Scholar]
- Mitcheltree, M.J.; Li, D.; Achab, A.; Beard, A.; Chakravarthy, K.; Cheng, M.; Cho, H.; Eangoor, P.; Fan, P.; Gathiaka, S.; et al. Discovery and Optimization of Rationally Designed Bicyclic Inhibitors of Human Arginase to Enhance Cancer Immunotherapy. ACS Med. Chem. Lett. 2020, 11, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, R.; Brzezinska, J.; Gzik, A.; Golebiowski, A.; Nowicka, J.; Borek, B.; Dziegielewski, M.; Jedrzejczak, K.; Matyszewski, K.; Olczak, J. Arginase Inhibitors and Their Therapeutic Applications. US 10391077, 27 August 2019. [Google Scholar]
- Marques, F.A.; Lenz, C.A.; Simonelli, F.; Noronha Sales Maia, B.H.L.; Vellasco, A.P.; Eberlin, M.N. Structure Confirmation of a Bioactive Lactone Isolated from Otoba Parvifolia through the Synthesis of a Model Compound. J. Nat. Prod. 2004, 67, 1939–1941. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, H.; Li, D.; Childers, M.; Pu, Q.; Palte, R.L.; Gathiaka, S.; Lyons, T.W.; Palani, A.; Fan, P.W.; et al. Structure-Based Discovery of Proline-Derived Arginase Inhibitors with Improved Oral Bioavailability for Immuno-Oncology. ACS Med. Chem. Lett. 2021, 12, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Achab, A.A.; Childers, M.L.; Cumming, J.N.; Fischer, C.A.; Gathiaka, S.; Gunadyn, H.; Lesburg, C.A.; Li, D.; Lu, M.; Palani, A.; et al. Arginase Inhibitors and Methods of Use. US20210040127A1, 11 February 2021. [Google Scholar]
- Mlynarski, S.N.; Grebe, T.; Kawatkar, S.; Verschoyle Finley, M.R.; Simpson, I.; Wang, J.; Cook, S. Arginase Inhibitors and Methods of Use Thereof. WO2019159120A1, 22 August 2019. [Google Scholar]
- Gross, M.; Chen, J.; Emberley, E.; Janes, J.; Li, W.; Mackinnon, A.; Pan, A.; Parlati, F.; Rodriguez, M.; Steggerda, S.; et al. Abstract A195: CB-1158 Inhibits the Immuno-Oncology Target Arginase and Causes an Immune Mediated Anti-Tumor Response. Mol. Cancer Ther. 2015, 14, A195. [Google Scholar] [CrossRef]
- Sjogren, E.B.; Li, J.; Van Zandt, M.; Whitehouse, D. Compositions and Methods for Inhibiting Arginase Activity. WO2017075363A1, 4 May 2017. [Google Scholar]
- Blaszczyk, R.; Gzik, A.; Borek, B.; Dziegielewski, M.; Jedrzejczak, K.; Nowicka, J.; Chrzanowski, J.; Brzezinska, J.; Golebiowski, A.; Olczak, J.; et al. Dipeptide Piperidine Derivatives. US20190300525A1, 3 October 2019. [Google Scholar]
- Achab, A.A.; Childers, M.L.; Cumming, J.N.; Fischer, C.; Gathiaka, S.; Gunaydin, H.; Lesburg, C.A.; Li, D.; Lu, M.; Palani, A.; et al. Arginase Inhibitors and Methods of Use. WO2019177873A1, 19 September 2019. [Google Scholar]
- Mlynarski, S.N.; Aquila, B.M.; Cantin, S.; Cook, S.; Doshi, A.; Finlay, M.R.V.; Gangl, E.T.; Grebe, T.; Gu, C.; Kawatkar, S.P.; et al. Discovery of (2R,4R)-4-((S)-2-Amino-3-methylbutanamido)-2-(4-boronobutyl)pyrrolidine-2-carboxylic Acid (AZD0011), an Actively Transported Prodrug of a Potent Arginase Inhibitor to Treat Cancer. J. Med. Chem. 2024, 67, 20827–20841. [Google Scholar] [CrossRef]
- Grzybowski, M.M.; Stańczak, P.S.; Pęczkowicz-Szyszka, J.; Wolska, P.; Zdziarska, A.M.; Mazurkiewicz, M.; Brzezińska, J.; Blaszczyk, R.; Gołębiowski, A.; Dobrzański, P.; et al. Abstract P71: Novel Dual Arginase 1/2 Inhibitor OATD-02 (OAT-1746) Improves the Efficacy of Immune Checkpoint Inhibitors. Ann. Oncol. 2017, 28, xi20–xi21. [Google Scholar] [CrossRef]
- Borek, B.; Nowicka, J.; Gzik, A.; Dziegielewski, M.; Jedrzejczak, K.; Brzezinska, J.; Grzybowski, M.; Stanczak, P.; Pomper, P.; Zagozdzon, A.; et al. Arginase 1/2 Inhibitor OATD-02: From Discovery to First-in-Man Setup in Cancer Immunotherapy. Mol. Cancer Ther. 2023, 22, 807–817. [Google Scholar] [CrossRef]
- Shields, J.D.; Aquila, B.M.; Emmons, D.; Finlay, M.R.V.; Gangl, E.T.; Gu, C.; Mlynarski, S.N.; Petersen, J.; Pop-Damkov, P.; Sha, L.; et al. Design and Synthesis of Acyclic Boronic Acid Arginase Inhibitors. J. Med. Chem. 2024, 67, 20799–20826. [Google Scholar] [CrossRef]
- Girard-Thernier, C.; Pham, T.N.; Demougeot, C. The Promise of Plant-Derived Substances as Inhibitors of Arginase. Mini Rev. Med. Chem. 2015, 15, 798–808. [Google Scholar] [CrossRef]
- Bordage, S.; Pham, T.N.; Zedet, A.; Gugglielmetti, A.S.; Nappey, M.; Demougeot, C.; Girard-Thernier, C. Investigation of Mammal Arginase Inhibitory Properties of Natural Ubiquitous Polyphenols by Using an Optimized Colorimetric Microplate Assay. Planta Med. 2017, 83, 647–653. [Google Scholar] [CrossRef]
- Woo, A.; Min, B.; Ryoo, S. Piceatannol-3’-O-β-D-Glucopyranoside as an Active Component of Rhubarb Activates Endothelial Nitric Oxide Synthase through Inhibition of Arginase Activity. Exp. Mol. Med. 2010, 42, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Arraki, K.; Totoson, P.; Attia, R.; Zedet, A.; Pudlo, M.; Messaoud, C.; Demougeot, C.; Girard, C. Arginase Inhibitory Properties of Flavonoid Compounds from the Leaves of Mulberry (Morus Alba, Moraceae). J. Pharm. Pharmacol. 2020, 72, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Cuong, T.D.; Hung, T.M.; Ryoo, S.; Lee, J.H.; Kim, E.H.; Woo, M.H.; Choi, J.S.; Min, B.S. Arginase II Inhibitory Activity of Phenolic Compounds from Saururus Chinensis. Bull. Korean Chem. Soc. 2012, 33, 3079–3082. [Google Scholar] [CrossRef]
- Kim, S.W.; Cuong, T.D.; Hung, T.M.; Ryoo, S.; Lee, J.H.; Min, B.S. Arginase II Inhibitory Activity of Flavonoid Compounds from Scutellaria Indica. Arch. Pharm. Res. 2013, 36, 922–926. [Google Scholar] [CrossRef]
- Pham, T.N.; Bordage, S.; Pudlo, M.; Demougeot, C.; Thai, K.M.; Girard-Thernier, C. Cinnamide Derivatives as Mammalian Arginase Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Int. J. Mol. Sci. 2016, 17, 1656. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. National Library of Medicine. 2024. Available online: https://clinicaltrials.gov (accessed on 7 January 2025).
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar]
- Naing, A.; Bauer, T.; Papadopoulos, K.P.; Rahma, O.; Tsai, F.; Garralda, E.; Naidoo, J.; Pai, S.; Gibson, M.K.; Rybkin, I.; et al. Phase I study of the arginase inhibitor INCB001158 (1158) alone and in combination with pembrolizumab (PEM) in patients (Pts) with advanced/metastatic (adv/met) solid tumours. Ann. Oncol. 2019, 30, v160. [Google Scholar]
- Naing, A.; Papadopoulos, K.P.; Pishvaian, M.; Rahma, O.; Hanna, G.J.; Garralda, E.; Saavedra, O.; Gogov, S.; Kallender, H.; Cheng, L.; et al. First-in-human phase 1 study of the arginase inhibitor INCB001158 alone or combined with pembrolizumab in patients with advanced or metastatic solid tumours. BMJ Oncol. 2024, 3, e000249. [Google Scholar] [CrossRef]
- National Library of Medicine, Study to Evaluate the Safety of CB-280 in Patients with Cystic Fibrosis-NCT04279769. Available online: https://clinicaltrials.gov/study/NCT04279769 (accessed on 17 June 2024).
- Boas, S.; Donaldson, S.; McBennett, K.; Liou, T.; Howrylak, J.; Johnson, L.; Teneback, C.; Dozor, A.; Sawicki, G.; Dumlao, J.; et al. 529: A phase 1b, randomized, double-blind, placebo-controlled, dose-escalation trial of CB-280, an arginase inhibitor, in patients with cystic fibrosis. J. Cyst. Fibros. 2021, 20, S250. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Martinenaite, E.; Kjeldsen, J.W.; Holmstroem, R.B.; Mørk, S.K.; Pedersen, A.W.; Ehrnrooth, E.; An-dersen, M.H.; Svane, I.M. Arginase-1 targeting peptide vaccine in patients with metastatic solid tumors—A phase I trial. Front. Immunol. 2022, 13, 1023023. [Google Scholar] [CrossRef]
- Sodano, F.; Cristiano, C.; Rolando, B.; Marini, E.; Lazzarato, L.; Cuozzo, M.; Albrizio, S.; Russo, R.; Rimoli, M.G. Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs. Pharmaceuticals 2022, 15, 552. [Google Scholar] [CrossRef] [PubMed]
- Sodano, F.; Gazzano, E.; Fraix, A.; Rolando, B.; Lazzarato, L.; Russo, M.; Blangetti, M.; Riganti, C.; Fruttero, R.; Gasco, A.; et al. A Molecular Hybrid for Mitochondria-Targeted NO Photodelivery. ChemMedChem 2018, 13, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Sodano, F.; Rolando, B.; Spyrakis, F.; Failla, M.; Lazzarato, L.; Gazzano, E.; Riganti, C.; Fruttero, R.; Gasco, A.; Sortino, S. Tuning the Hydrophobicity of a Mitochondria-Targeted NO Photodonor. ChemMedChem 2018, 13, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Sodano, F.; Gazzano, E.; Rolando, B.; Marini, E.; Lazzarato, L.; Fruttero, R.; Riganti, C.; Gasco, A. Tuning NO release of organelle-targeted furoxan derivatives and their cytotoxicity against lung cancer cells. Bioorg Chem. 2021, 111, 104911. [Google Scholar] [CrossRef] [PubMed]






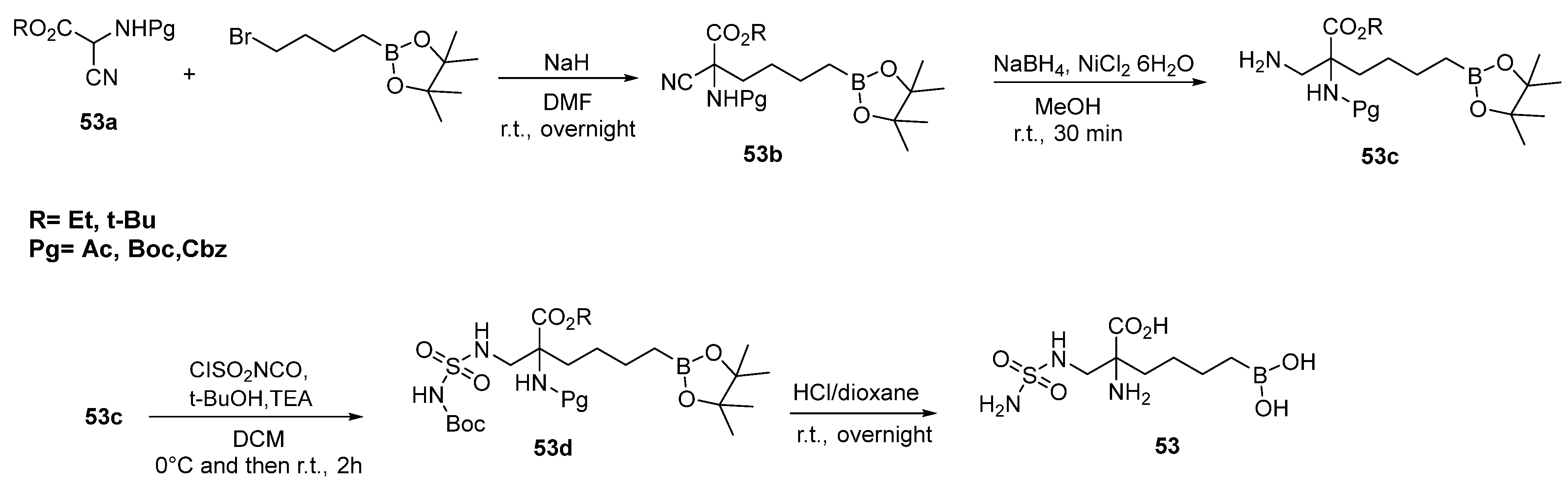
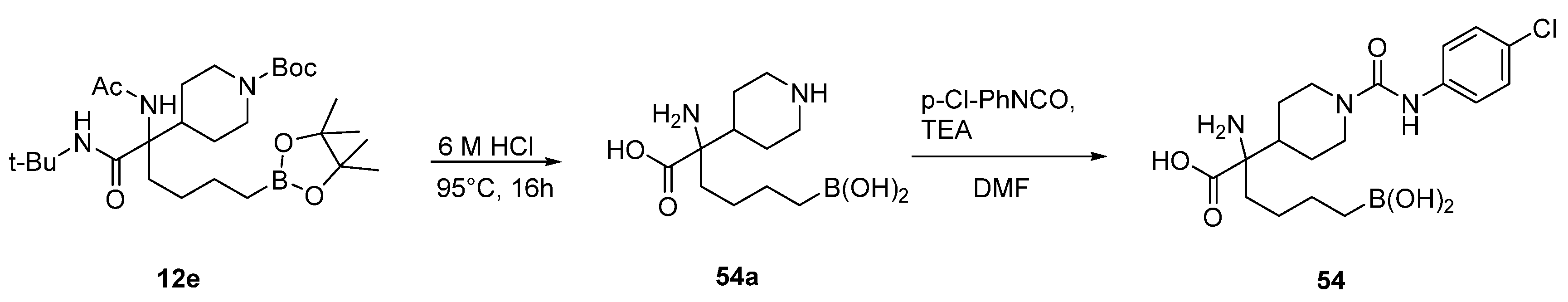
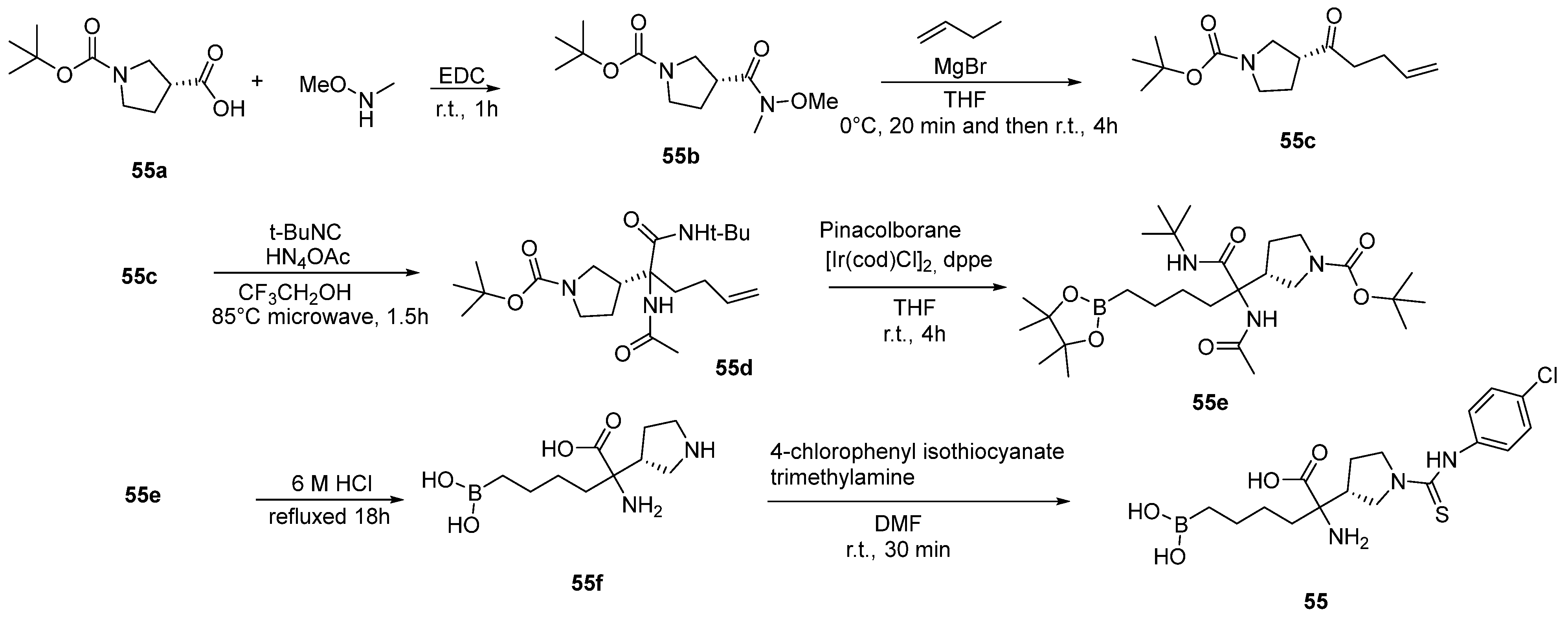

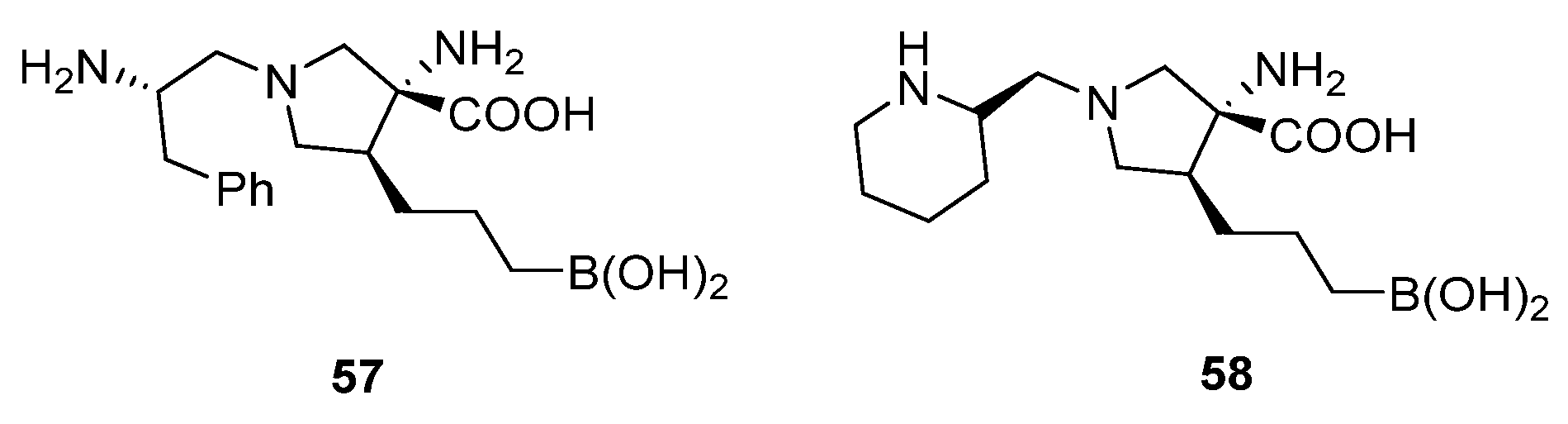
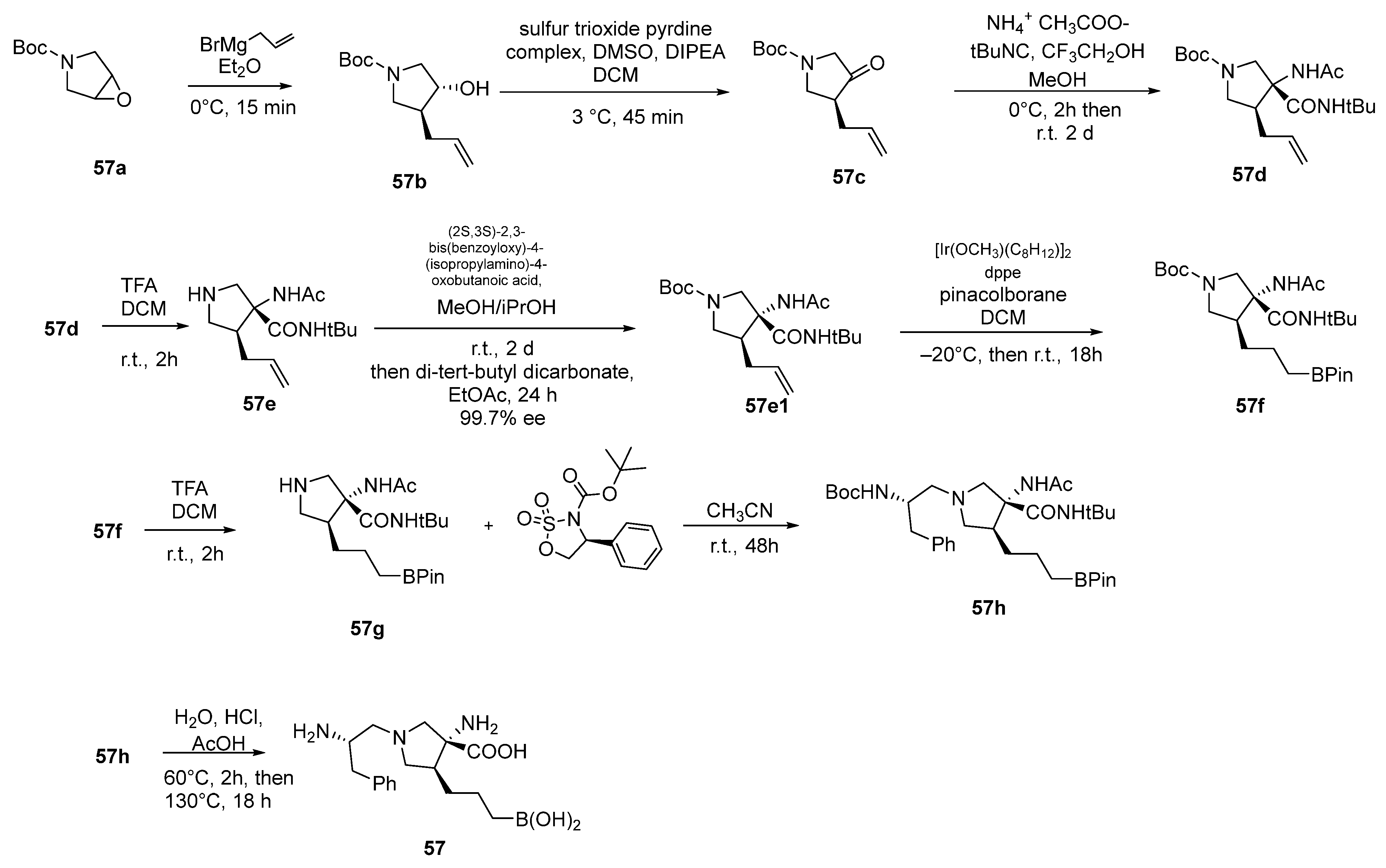

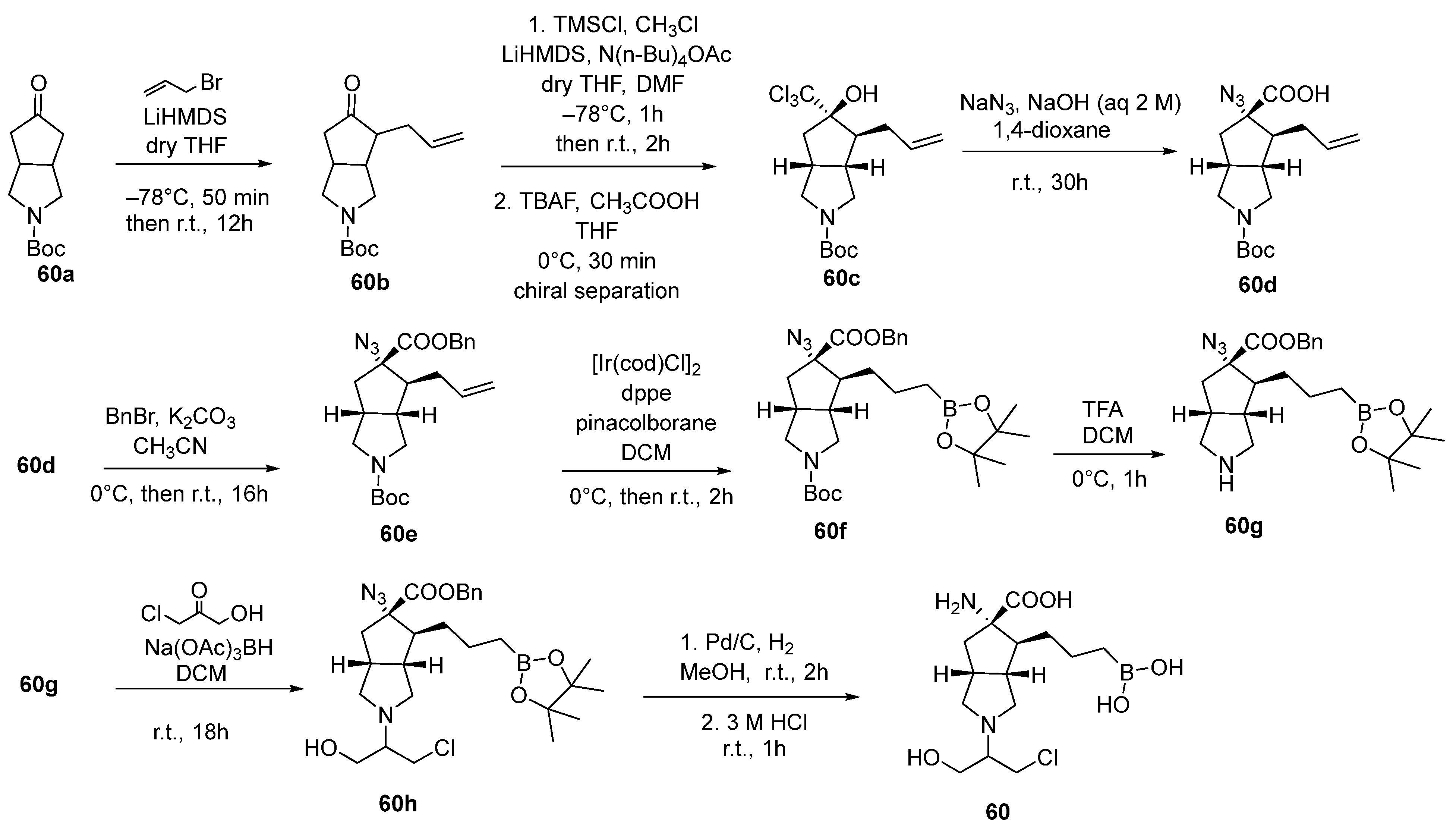
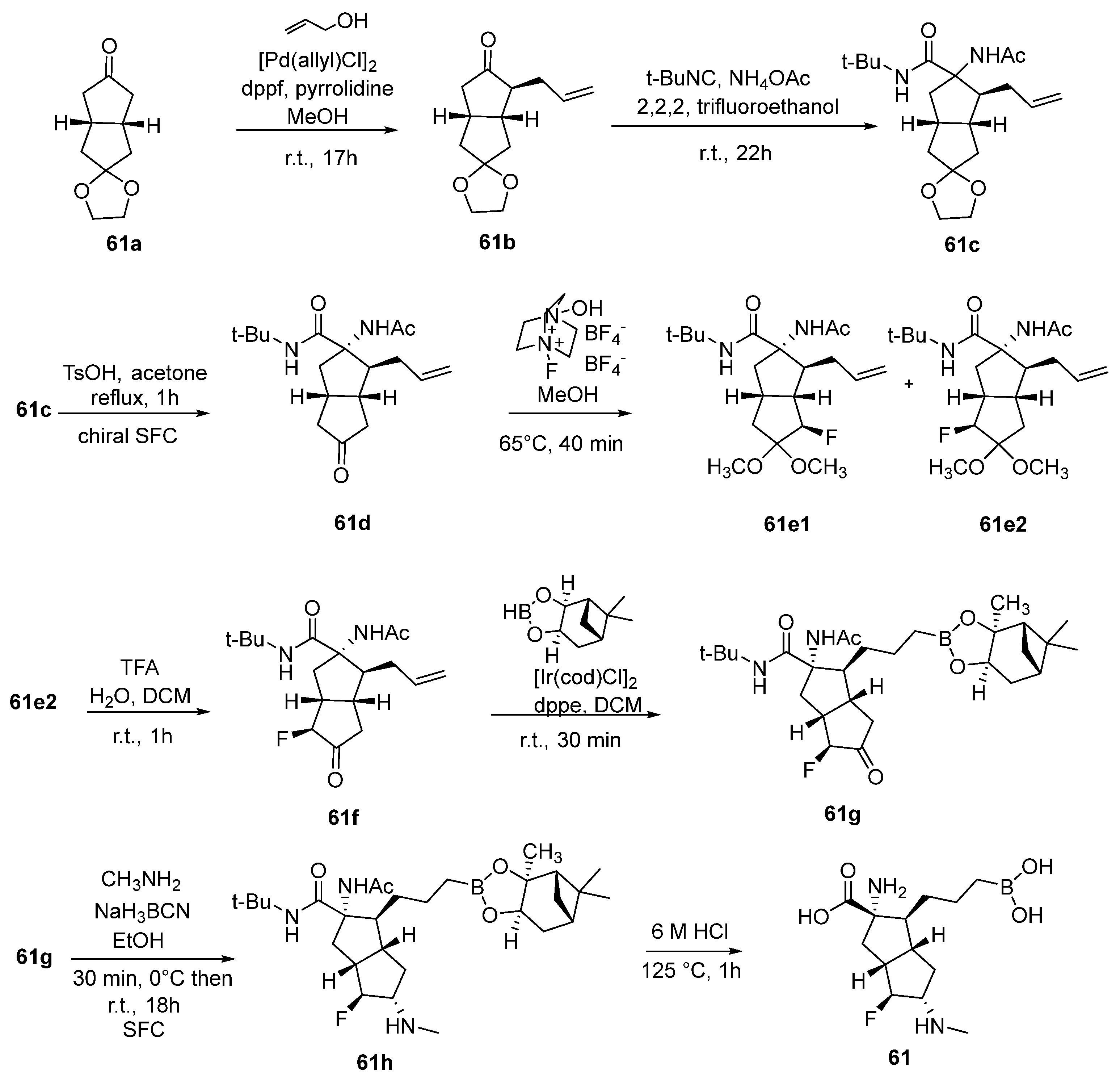

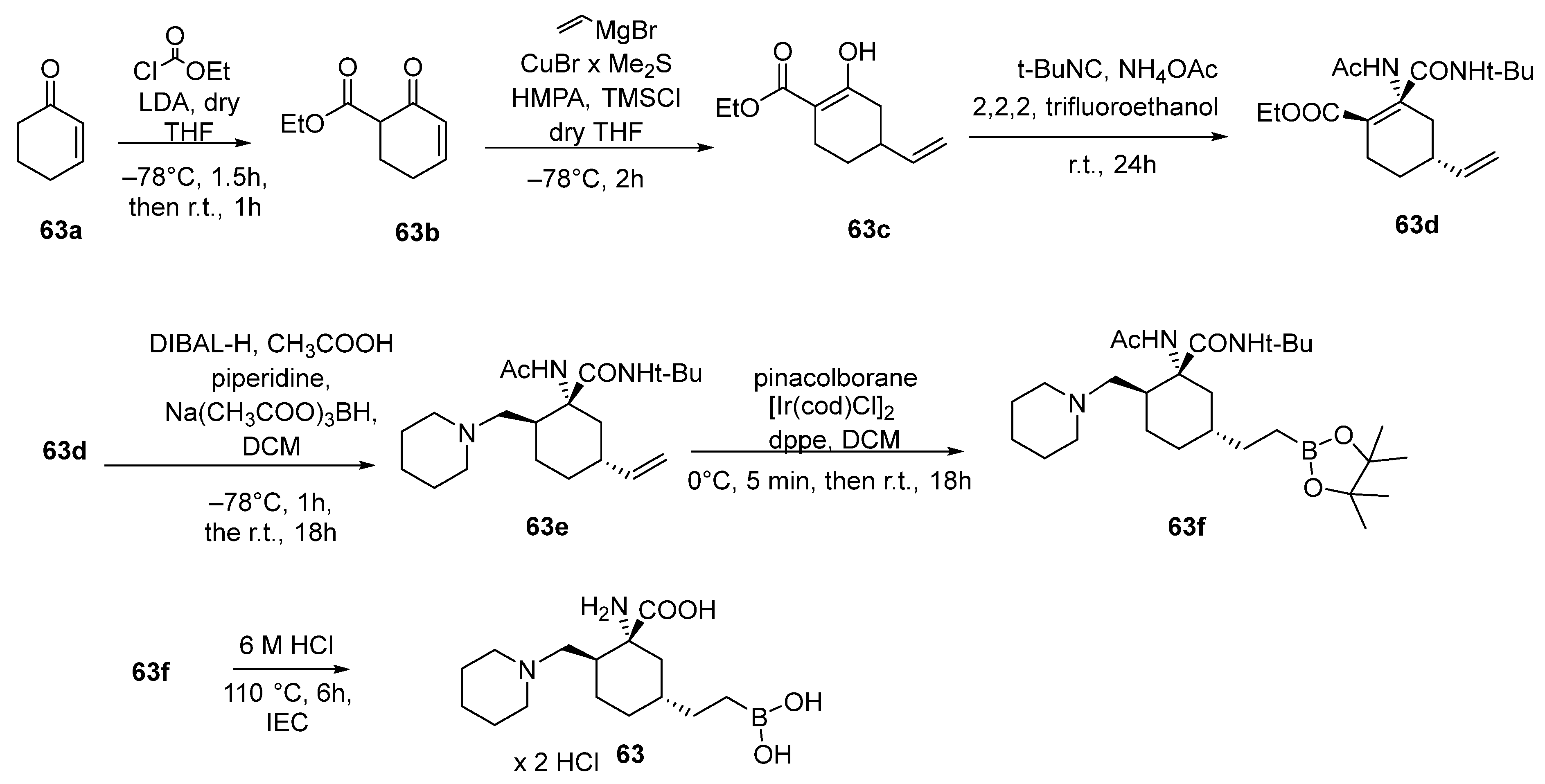
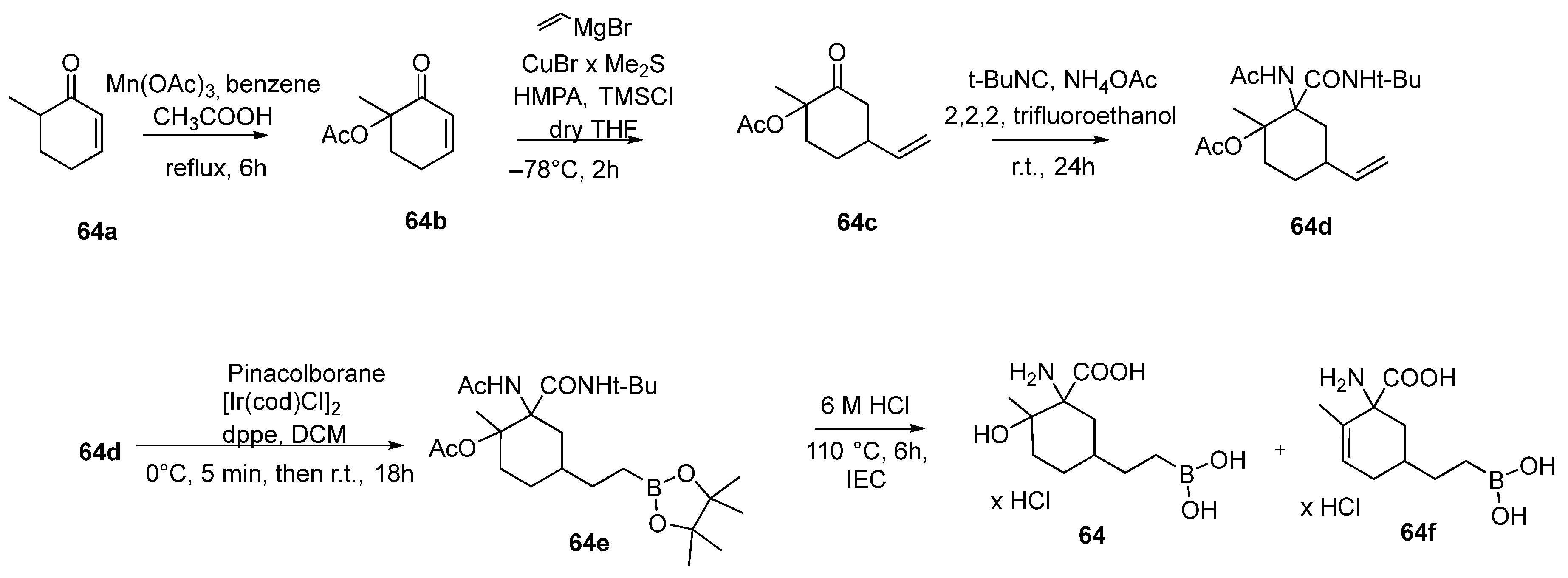
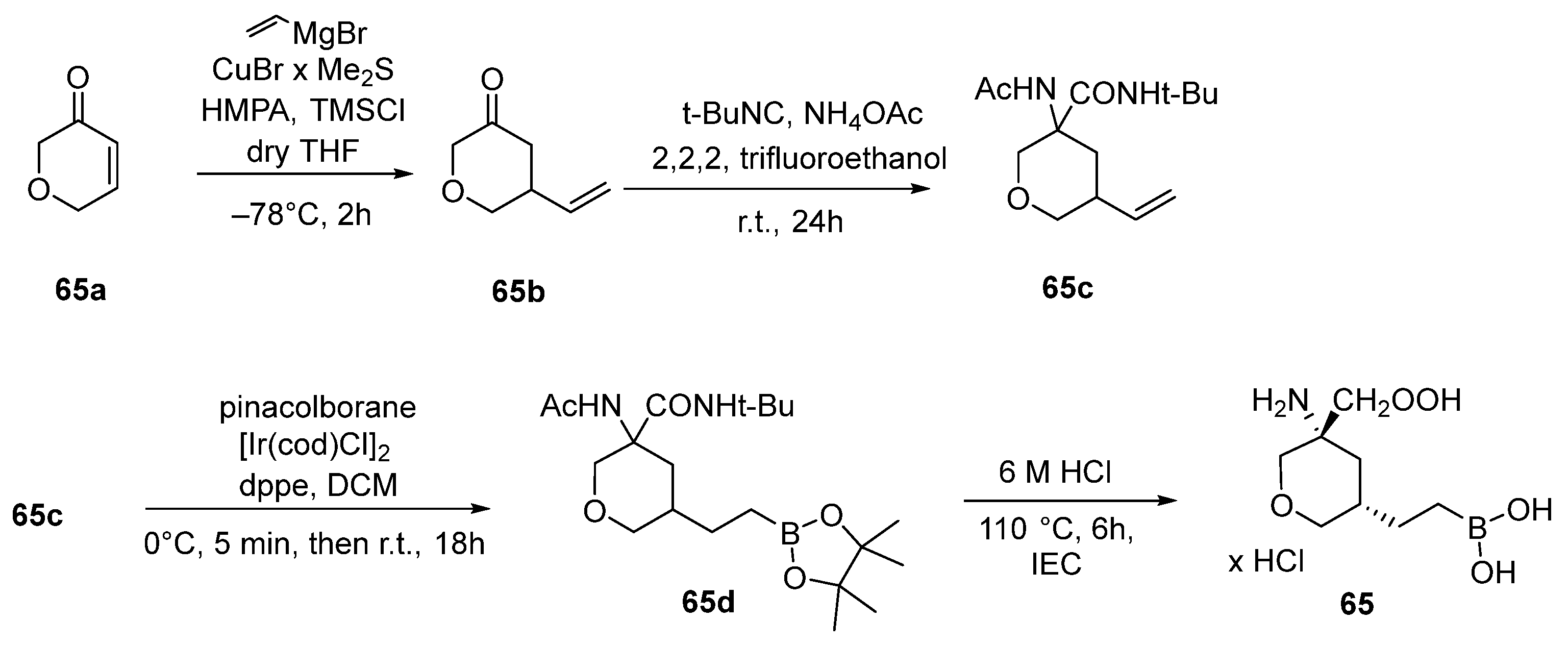

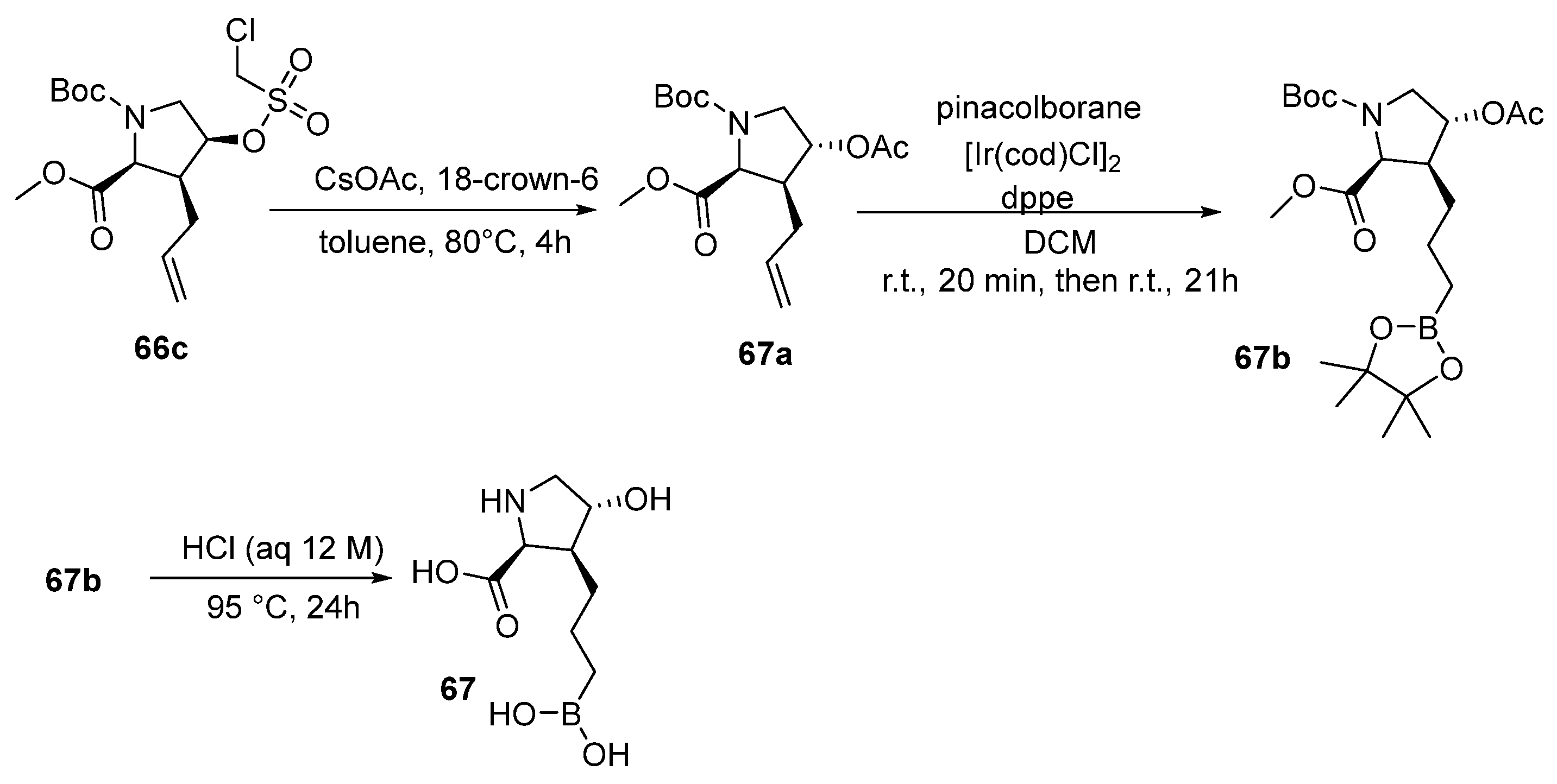

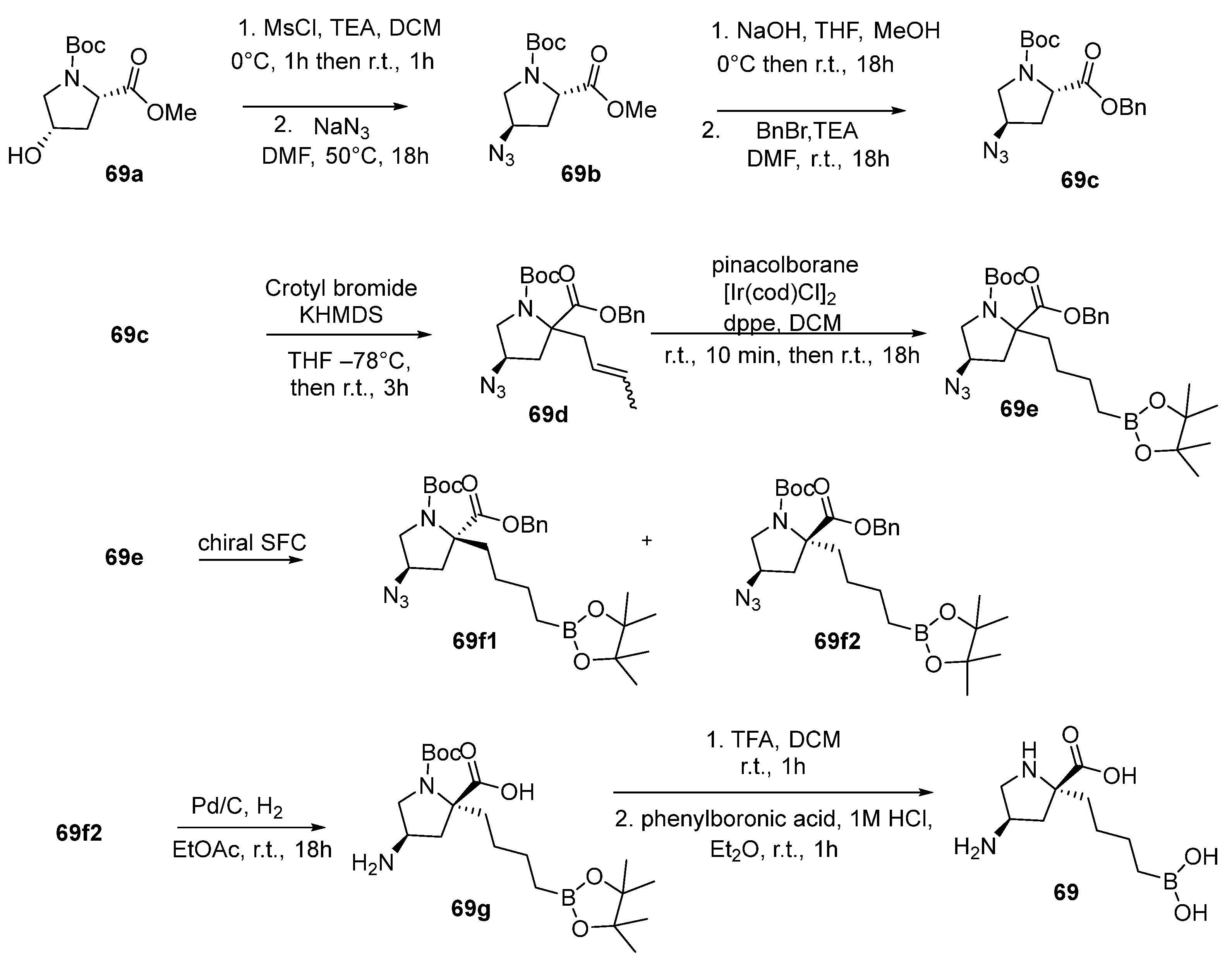







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Compound | R | IC50 |
| 21 (S)-2-amino-6-borono-2-(1S,3R)-3-(3-phenylpropylamino)cyclobutyl)hexanoic acid |  | 0.1–25 nM (hARG-1) 26–100 nM (hARG-2) |
| 22 (S)-2-amino-6-borono-2-((1S,3R)-3-(3-(3-chloro-5fluorophenyl)propylamino) cyclobutyl)hexanoic acid | 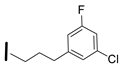 | 26–100 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 23 (S)-2-amino-6-borono-2-((1s,3R)-3-(3-(3,4-difluorophenyl)propylamino)cyclobutyl) hexanoic acid |  | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 24 (S)-2-amino-6-borono-2-((1S,3R)-3-(3-(2,4-dichlorophenyl)propylamino)cyclobutyl)hexanoic acid | 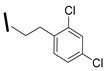 | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 25 (S)-2-amino-6-borono-2-((1S,3R)-3-(2,3-dihydro-1h-inden-2yl-amino)cyclobutyl)hexanoic acid |  | 0.1–25 nM (hARG-1) 26–100 nM (hARG-2) |
| 26 (S)-2-amino-6-borono-2-((1S,3R)-3-(4-tert-butylbenzylamino)cyclobutyl)hexanoic acid |  | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 27 (S)-2-amino-2-((1S,3R)-3-(biphenyl-3-ylmethylamino)cyclobutyl)-6-boronohexanoic acid |  | 0.1–25 nM (hARG-1) 26–100 nM (hARG-2) |
| 28 (S)-2-amino-6-borono-2-((1S,3R)-3-((4′-(trifluoromethyl)biphenyl-3-yl) methylamino)cyclobutyl)hexanoic acid | 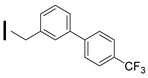 | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 29 (S)-2-amino-6-borono-2-((1S,3R)-3-((4′-chlorobiphenyl-3yl)methylamino)cyclobutyl)hexanoic acid | 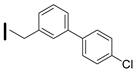 | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 30 (S)-2-amino-6-borono-2-((1S,3R)-3-((4-fluoronaphthalen-1yl)methylamino)cyclobutyl)hexanoic acid | 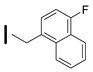 | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 31 (S)-2-amino-6-borono-2-((1S,3R)-3-((5-fluoronaphthalen-1yl)methylaminocyclobutyl)hexanoic acid |  | 0.1–25 nM (hARG-1) 26–100 nM (hARG-2) |
| 32 (S)-2-amino-2-(1S,3R)-3-(anthracen-9-ylmethylamino)cyclobutyl)-6-borono hexanoic acid |  | 26–100 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 33 (S)-2-amino-6-borono-2-((1S,3R)-3-(2-morpholinobenzylamino)cyclobutyl)hexanoic acid |  | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 34 (S)-2-amino-6-borono-2-((1R,3R)-3-(((S)-1,2,3,4-tetrahydroisoquinolin-3-yl)methylamino)cyclobutyl)hexanoic acid | 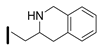 | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 35 (S)-2-amino-6-borono-2-((1S,3R)-3-((2,3-dihydrobenzofuran-5-yl)methylamino)cyclobutyl)hexanoic acid |  | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 36 (S)-2-amino-6-borono-2-((1S,3R)-3-((3′,4′-dichlorobiphenyl-4yl)methylamino)cyclobutyl)hexanoic acid |  | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 37 (S)-2-amino-6-borono-2-((1S,3R)-3-((4′-chlorobiphenyl-4yl)methylamino)cyclobutyl)hexanoic acid | 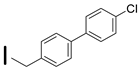 | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 38 (S)-2-amino-6-borono-2-((1S,3R)-3-((4′-(trifluoromethyl)biphenyl-4-yl)methylamino)cyclobutyl) hexanoic acid | 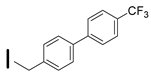 | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 39 (S)-2-amino-6-borono-2-((1S,3R)-3-((4′-fluorobiphenyl-4yl)methylamino)cyclobutyl)hecanoic acid | 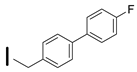 | 0.1–25 nM (hARG-1) 26–100 nM (hARG-2) |
| 40 (S)-2-amino-6-borono-2-((1S,3R)-3-(4-hydroxybenzylamino)cyclobutyl)hexanoic acid | 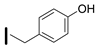 | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 41 (S)-2-amino-6-borono-2-((1S,3R)-3-(4-(4-chlorophenoxy)benzylamino)cyclobutyl)hexanoic acid | 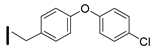 | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 42 (S)-2-amino-6-borono-2-(1S,3R)-3-((4′-chlorobiphenyl-2-yl)methylamino)cyclobutyl)hexanoic acid |  | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 43 (S)-2-amino-6-borono-2-((1S,3R)-3-((6-phenylpyridin-3-yl)methylamino)cyclobutyl)hexanoic acid | 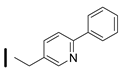 | 26–100 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 44 (S)-2-((1S,3R)-3-((9h-fluoren-2-yl)methylamino)cyclobutyl)-2-amino-6-borono-hexanoic acid |  | 0.1–25 nM (hARG-1) 0.1–25 nM (hARG-2) |
| 45 (S)-2-amino-6-borono-2-((1S,3R)-3-((4′-(trifluoromethyl)biphenyl-2 yl)methylamino)cyclobutyl)hexanoic acid |  | 26–100 nM (hARG-1) 26–100 nM (hARG-2) |
| 46 (S)-2-amino-6-borono-2-((1S,3R)-3-(4-cyclohexylbenzylamino)cyclobutyl)hexanoic acid |  | 0.1–25 nM (hARG-1) 26–100 nM (hARG-2) |
| Compound | Structure | ARG-1 Activity | ARG-2 Activity |
|---|---|---|---|
| 91 Chlorogenic acid | 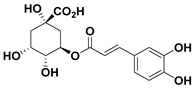 | IC50 = 10.6 µM a | |
| 92 Picetannol | 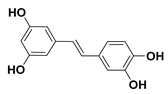 | IC50 = 12.1 µM a | |
| 93 Resveratrol | 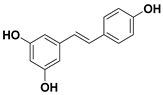 | IC50 = 18.2 µM a | |
| 94 Taxifolin | 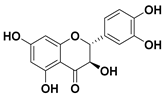 | IC50 = 23.2 µM a | |
| 95 Piceatannol-3′-O-β-D-glucopyranoside | 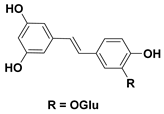 | IC50 = 11.22 µM b | IC50 = 11.06 µM c |
| 96 Ellagic acid |  | IC50 = 78.9 µM a | |
| 97 Luteolin-7-diglucoside | 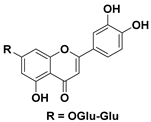 | IC50 = 152.4 µM a | |
| 98 Luteolin-7-glucoside | 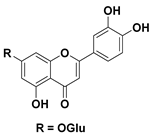 | IC50 = 99.4 µM a | |
| 99 Luteolin |  | IC50 = 95.3 µM a | |
| 100 Scirpusin B |  | IC50 = 22.6 µM a | |
| 101 ε-Viniferin | 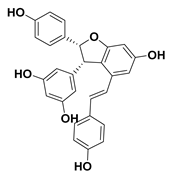 | IC50 = 27.8 µM a | |
| 102 Cyperusphenol B | 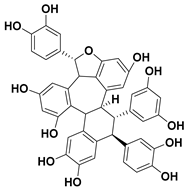 | IC50 = 12.2 µM a | |
| 103 Carexinol A |  | IC50 = 25.3 µM a | |
| 104 Virgatanol | 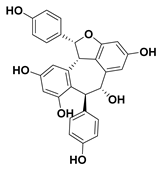 | IC50 = 182.1 µM a | |
| 105 Sauchinone |  | IC50 = 61.4 µM d | |
| 106 (2S)-5,7-dihydroxy-8,20-dimethoxyflavanone | 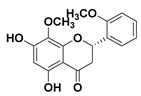 | IC50 = 25.1 µM c | |
| 107 (2S)-5,20,50-trihydroxy-7, 8-dimethoxyflavanone |  | IC50 = 11.6 µM c | |
| 108 Caffeic acid phenylamide (CAPA) |  | IC50 = 6.9 µM a IC50 = 60.3 µM e |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molaro, M.C.; Battisegola, C.; Schiano, M.E.; Failla, M.; Rimoli, M.G.; Lazzarato, L.; Chegaev, K.; Sodano, F. Synthesis of Arginase Inhibitors: An Overview. Pharmaceutics 2025, 17, 117. https://doi.org/10.3390/pharmaceutics17010117
Molaro MC, Battisegola C, Schiano ME, Failla M, Rimoli MG, Lazzarato L, Chegaev K, Sodano F. Synthesis of Arginase Inhibitors: An Overview. Pharmaceutics. 2025; 17(1):117. https://doi.org/10.3390/pharmaceutics17010117
Chicago/Turabian StyleMolaro, Maria Cristina, Chiara Battisegola, Marica Erminia Schiano, Mariacristina Failla, Maria Grazia Rimoli, Loretta Lazzarato, Konstantin Chegaev, and Federica Sodano. 2025. "Synthesis of Arginase Inhibitors: An Overview" Pharmaceutics 17, no. 1: 117. https://doi.org/10.3390/pharmaceutics17010117
APA StyleMolaro, M. C., Battisegola, C., Schiano, M. E., Failla, M., Rimoli, M. G., Lazzarato, L., Chegaev, K., & Sodano, F. (2025). Synthesis of Arginase Inhibitors: An Overview. Pharmaceutics, 17(1), 117. https://doi.org/10.3390/pharmaceutics17010117