
Exploring the Link Between Periodontitis and Alzheimer’s Disease—Could a Nanoparticulate Vaccine Break It?

 ,
,  and
and
Abstract
:1. Introduction
2. Alzheimer’s Disease and Pathogenesis Hypotheses
2.1. The Cholinergic Hypothesis
2.2. The Amyloid Cascade Hypothesis
2.3. The Tau Hypothesis
2.4. The Inflammation Hypothesis
2.5. The Infection and Antimicrobial Protection Hypotheses
3. Periodontitis and Porphyromonas gingivalis
4. Human Epidemiological and Genetic Evidence Linking PeD/Pg with AD
4.1. Evidence Linking Clinical PeD with AD
4.2. Evidence Linking Immunological Markers of PeD with AD
4.3. Evidence Linking PeD with AD Pathology
4.4. Presence of Pg and Periodontal Bacteria in AD Human Brains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Study Design | Main Objective | Sample Characterisation | Assessment | Findings |
|---|---|---|---|---|---|
| Kaye et al. (2010), USA [93] | Prospective cohort (32 y) | Determine whether rates of tooth loss, PeD progression, and caries incidence predict cognitive decline | 597 subjects (28–70 y) | Exposures: > rate of tooth loss > PeD progression (PPD and ABL) > caries incidence Outcome studied: incident cognitive decline (MMSE, SCT) | Higher risk of low cognitive scores among >45-year-old subjects with higher rates of tooth loss, ABL progression, PPD progression, and new caries |
| Ide et al. (2016), UK [132] | Observational cohort (6 mo) | Examine the effect of PeD on the cognitive status and systemic proinflammatory status of AD patients | 59 subjects (MA = 77.7 y) > w mild-to-moderate AD and PeD (n = 22) > w mild-to-moderate AD only (n = 37) | Exposure: PeD (CDC/AAP def.) Outcomes studied: > variation in cognitive decline (NINCDS-ADRDA, ADAS-cog, and sMMSE) > variation in serum proinflammatory state (serum CRP, TNF-α, and IL-10) | Concurrent PeD at baseline was associated with increases in the proinflammatory state and rate of cognitive decline (6-fold, ADAS-cog) |
| Iwasaki et al. (2016), Japan [94] | Retrospective cohort (3 y) | Examine the relationship between severe PeD and cognitive decline | 85 subjects (≥75 y, MA = 79.3 y) > w severe PeD (n = 21) > wo severe PeD (n = 64) | Exposure: severe PeD (CDC/AAP def.) Outcome studied: incident cognitive decline (MMSE) | Severe PeD was associated with cognitive decline |
| Iwasaki et al. (2019), Japan [95] | Prospective cohort (5 y) | Determine the effect of severe PeD and periodontal inflammation (PI) on the incidence of MCI | 179 subjects (≥75 y, MA = 80.1 y) > w severe PeD > wo severe PeD | Exposures: > severe PeD (CDC/AAP and EWP defs.) > PI (PISA) Outcome studied: incident MCI (diff. diagnostic criteria) | Severe PeD and PI were associated with MCI |
| Tzeng et al. (2016), Taiwan [96] | Retrospective cohort (10 y) | Determine the effect of chronic gingivitis/PeD on the risk of dementia | 8828 subjects (≥20 y) > w chronic gingivitis/PeD (n = 2207) > wo chronic gingivitis/PeD (n = 6621) | Exposure: chronic gingivitis/PeD (ICD-9-CM) Outcome studied: incident dementia (DSM-IV, DSM-IV-TR) | Chronic gingivitis/PeD patients with a higher risk for dementia |
| Lee YT et al. (2017), Taiwan [97] | Retrospective cohort (10 y) | Evaluate PeD as a modifiable risk factor for dementia | 6056 subjects (≥65 y, MA = 72.4 y) > w PeD (n = 3028) > wo PeD (n = 3028) | Exposure: PeD (ICD-9-CM) Outcome studied: incident dementia (ICD-9-CM) | PeD patients with a higher risk for dementia |
| Chen et al. (2017), Taiwan [13] | Retrospective cohort (16 y) | Determine the effect of PeD on the risk for AD | 27,963 subjects (≥50 y, MA = 54.2 y) > w PeD (n = 9291) > wo PeD (n = 18,672) | Exposure: PeD (ICD-9-CM) Outcome studied: incident AD (ICD-9-CM) > ≥1 y after exposure (excl. of diagnosed AD < 1 y) > ≥10 y after exposure (excl. of diagnosed AD < 10 y) | Only ≥ 10-year PeD-exposed patients with a higher risk for developing AD |
| Choi et al. (2019), South Korea [100] | Retrospective cohort (10 y) | Determine the effect of PeD on the risk for overall dementia, AD, and vascular dementia (VD) | 262,349 subjects (≥50 y, MA = 60.2 y) > w PeD (n = 46,344) > wo PeD (n = 216,005) | Exposure: PeD (ICD-10) Outcomes studied: incident overall dementia, AD, and VD (ICD-10) | PeD patients with a higher risk for overall dementia and AD |
| Nilsson et al. (2018), Sweden [105] | Prospective cohort (6 y) | Evaluate whether PeD is associated with cognitive decline | 704 subjects (≥60 y) > w PeD > wo PeD | Exposure: history of PeD (radiographic ABL-based criterion as indicator) Outcome studied: incident cognitive decline (MMSE) | History of PeD was associated with cognitive decline |
| Kim et al. (2020), South Korea [125] | Retrospective cohort (14 y) | Evaluate severe PeD with tooth loss as a modifiable risk factor for AD, VD, and mixed dementia (MD) | 20 230 subjects (40–79 y) > w severe PeD with tooth loss (n = 10,115) > wo severe PeD with tooth loss (n = 10,115) | Exposure: severe PeD (defined as requiring surgical intervention) with tooth loss Outcomes studied: incident AD, VD, and MD (KCD-7) | Severe PeD with 1–9 remaining teeth at higher risk of AD, VD, and MD |
| Demmer et al. (2020), USA [126] | Retrospective cohort (20 y) | Evaluate whether PeD is associated with increased risk for dementia and MCI | 8275 subjects (MA = 63 y) > w PeD > wo PeD | Exposure: PeD (PPC, CDC/APP, and others) Outcomes studied: incident dementia and MCI/dementia composite (NINCDS-ADRDA and DSM-V) | Several associations only between the most diseased PPC categories and increased risk for incident dementia and incident MCI/dementia |
| Nikitin et al. (2021), USA [103] | Prospective cohort (11 y) | Assess the association between clinical measures of PeD and subsequent cognitive decline | 967 subjects (70–79 y) | Exposure: PeD (GI, CAL, and PPD as indicators) Outcome studied: incident cognitive decline (3MS and DSST) | Only GI was consistently associated with higher cognitive decline |
| Malone et al. (2022), USA [102] | Retrospective cohort (4 y) | Examine whether PeD increases the risk of developing AD and related dementias (ADRD) among HCV patients | 439 760 subjects with HCV (44% ≥65 y) > HCV patients w PeD (n = 32,231) > HCV patients wo PeD (n = 407,529) | Exposure: PeD (ICD-9 and ICD-10) Outcome studied: incident ADRD (MBSF data) | PeD increased the risk for ADRD among HCV patients |
| Holmer et al. (2022), Sweden [118] | Retrospective cohort (mean 7.5 y) | Determine the effect of deep PPD, as a PeD proxy, on the incidence of dementia | 37 174 subjects (≥40 y, MA = 61 y) > w deep PPD (n = 7992) > wo deep PPD (n = 29,182) | Exposure: PeD (deep PPD as proxy) Outcome studied: incident dementia (ICD-10, others) | Deep PPD was not associated with dementia |
| Adam et al. (2022), USA [146] | Prospective cohort (max. 17 y) | Investigate whether PeD is prospectively associated with cerebrovascular and neurodegenerative markers of dementia and AD pathology | 1306 subjects for MRI cohort (MA = 61.8 y) 248 subjects for PET cohort (MA = 61.2 y) > w PeD > wo PeD | Exposure: PeD (modified PPC) Outcomes studied: > altered brain volumes (MRI) > incident microhaemorrhages (MRI) > incident brain β-amyloid positivity (PET scan) | PeD was not associated with altered brain volumes, microhaemorrhages, or β-amyloid positivity |
| Carballo et al. (2023), Spain [140] | Prospective cohort (2 y) | Evaluate whether PeD is associated with cognitive decline and blood-based markers of AD | 101 subjects with a history of hypertension (≥60 y, MA = 71 y) > w PeD (n = 63) > wo PeD (n = 38) | Exposure: PeD (CDC/AAP def.) Outcomes studied: > variation in cognitive decline (MMSE and ACE) > variation in plasma levels of Aβ1–40, Aβ1–42, p-tau, and t-tau | PeD was associated with cognitive decline, its progression, and increased plasma levels of Aβ1–40 and p-tau |
| Lee YL et al. (2017), Taiwan [98] | Retrospective cohort (10 y) | Assess the effect of PeD severity and PeD-related treatments on the incidence of dementia | 182,747 subjects with PeD (≥45 y) assigned to: > dental prophylaxis (n = 97,802) > PeD-intensive treatment (n = 5373) > PeD w tooth extraction (n = 59,898) > PeD wo treatment (n = 19,674) | Exposure: one of the four treatments (treatment received as a marker for PeD severity) Outcome studied: incident dementia (ICD-9-CM) | Higher risk of dementia for patients with untreated or more-severe PeD (tooth extraction) PeD prevention or treatment might reduce or delay the development of dementia |
| Schwahn et al. (2022), Germany [145] | Quasi-experimental—trial emulation approach (21 y) | Investigate the relationship between PeD treatment and the preclinical AD | 586 subjects with criteria for periodontal treatment (<60 y, MA = 45 y) > treated from GANI_MED (n = 177) > untreated from SHIP-TREND (n = 409) | Exposure: periodontal treatment Outcomes studied: > AD score [143] (brain atrophy MRI marker) > brain age gap [144] (MRI) | Periodontal treatment had a favourable effect on the preclinical AD-related AD score |
| De Souza Rolim et al. (2014), Brazil [99] | Interventional pre–post (6 mo) | Compare the orofacial characteristics and functional/cognitive aspects of AD patients before and after dental treatment | 29 subjects with mild AD (age not specified) > no control group | Exposure: dental treatments (mainly periodontal treatment) Outcomes studied: > cognitive deficit (MMSE) > functional cognitive impairment (Pfeffer’s questionnaire [165]) > orofacial characteristics (diff. clinical parameters) | At last evaluation after dental treatment (6 mo): > overall improvement of orofacial co-morbidities > improvement in MMSE and functional cognitive impairment |
| Beydoun et al. (2020), USA [127] | Retrospective cohort (26 y) | Examine the association between PeD pathogens and incident all-cause/AD dementia, and AD mortality | 6650 subjects (≥45 y) from NHANES III, both phases 1988–94 3479 subjects (≥45 y) from NHANES III, phase 2 1991–94 | Exposures: > serum IgG to PeD bacteria at baseline > PPD > CAL Outcomes studied: > incident all-cause and AD dementia (ICD-9) > AD mortality (ICD-10) | Multiple associations between bacterial IgG titers, particularly Pg, and AD incidence/mortality PPD was associated with incident AD |
| Merchant et al. (2024), USA [135] | Prospective cohort (21 y) | Examine associations between clusters of serum IgG to PeD bacteria and AD mortality | 8153 subjects (≥40 y) from NHANES III | Exposure: serum IgG clusters to PeD bacteria at baseline Outcome studied: AD mortality | Clusters of serum IgG to PeD bacteria were not significantly associated with AD mortality |
| Sparks Stein et al. (2012), USA [123] | Nested case–control (mean 12.5 y for controls) | Compare baseline serum IgG levels to PeD bacteria between subjects who converted or not to AD during the follow-up | From 158 subjects (MA = 71.5 y) cognitively intact at baseline serum draw: > cases: w incident AD (n = 35) or MCI (n = 46) > control: cognitively healthy at last follow-up (n = 77) | Exposure studied: serum IgG to PeD bacteria at baseline (as PeD proxy) Outcomes: > AD (NINCDS-ADRDA and MMSE) > MCI (Petersen’s criteria [166,167]) | Increased anti-Fn and -Pi titers at baseline in the patients with incident AD |
| Noble et al. (2014), USA [79] | Case–cohort (mean 5 y) | Assess pre-morbid levels of serum IgG to PeD bacteria as possible predictors of incident AD | From 219 subjects (>65 y, MA = 75.6 y) cognitively intact at baseline serum draw: > cases: w incident AD (n = 110) > control: cognitively healthy at last follow-up (n = 109) | Exposure studied: serum IgG to PeD bacteria at baseline (as PeD proxy) Outcome: AD (diff. diagnostic criteria) | Increased anti-An titer at baseline was associated with higher risk of AD Serum IgG levels to common PeD bacteria may be predictors of incident AD |
| Kamer et al. (2009), USA [129] | Case–control | Determine whether elevated cytokine expression and plasma IgG levels to PeD bacteria are associated with AD | 24 subjects (≥40 y) > cases: w AD (n = 18) > control: cognitively healthy (n = 16) | Variables studied: > plasma IgG to PeD bacteria (as PeD proxy) > plasma TNF-α, IL-1β, and IL-6 (as systemic inflammation proxy) Outcome: AD (NINCDS-ADRDA, DSM-IV, and MMSE) | TNF-α levels and elevated numbers of IgG against PeD bacteria (Pg, Aa, and Tf) were associated with AD |
| Farhad et al. (2014), Iran [130] | Case–control | Evaluate the effect of PeD on serum levels of TNF-α in AD patients | 80 subjects with AD (40–70 y) > cases: w PeD (n = 40) > control: wo PeD (n = 40) | Variable studied: serum TNF-α Outcome: AD with concomitant PeD (dental examination, CAL) | Serum TNF-α levels were 3-fold higher in the AD patients with concomitant PeD |
| Cestari et al. (2016), Brazil [131] | Case–control | Investigate the prevalence of oral infections and serum IL-1β, IL-6, and TNF-α in patients with MCI or AD | 65 subjects (MA = 75.6 y) > cases: w MCI (n = 19) or AD (n = 25) > control: cognitively healthy (n = 21) | Variables studied: > serum levels of IL-1β, IL-6, and TNF-α > PeD (diff. clinical parameters) Outcomes: MCI and AD (NINCDS-ADRDA and MMSE) | Elevated IL-6 in AD/MCI patients was associated with high serum TNF-α in PeD patients |
| Laugisch et al. (2018), Germany [136] | Case–control | Investigate the presence of PeD pathogens and intrathecal pathogen-specific IgG in AD and non-AD demented patients | 40 subjects (30–70 y, MA = 59.7 y) > cases: w AD (n = 20) > control: non-AD demented (n = 20) | Variables studied: > periodontal destruction (number of teeth, PPD, CAL, BoP) > presence of PeD bacteria in periodontium, serum, and CSF > periodontium and serum levels of IL-1β and MCP1/CCL2 > serum and CSF IgG to PeD bacteria > CSF levels of Aβ1–42 and t-tau Outcome: AD (NIA-AA 2011 and MMSE) | Subjects with AD presented: > no differences in the control of periodontal destruction, IgG to PeD bacteria, and cytokine levels > presence of PeD bacteria only in periodontium > higher IgG to PeD bacteria in CSF than in serum > lower Aβ1–42 and higher t-tau levels in the CSF > association of CSF levels of t-tau with both serum levels of anti-Pg IgG and MCP1/CCL2 Local production of IgG to PeD bacteria in the CSF may occur in demented patients, but there was no association with AD |
| De Souza Rolim et al. (2014), Brazil [113] | Case–control | Compare the oral status, mandibular function, and orofacial pain between mild AD patients and healthy subjects | 59 subjects (59–91 y, MA = 68 y) > cases: w mild AD (n = 29) > control: wo AD (n = 30) | Variables studied: PeD (diff. clinical parameters), others Outcome: mild AD (NINCDS-ADRDA and MMSE) | Higher prevalence of PeD in subjects with AD than in healthy subjects |
| Gil-Montoya et al. (2015), Spain [110] | Case–control | Determine whether PeD is associated with the diagnosis of cognitive impairment/ dementia | 409 subjects (>50 y) > cases: w MCI/dementia (n = 180) > control: cognitively healthy (n = 229) | Variables studied: PeD (diff. clinical parameters) Outcomes: MCI/dementia (NINCDS-ADRDA, DSM-IV, and Robles et al. criteria [168]) | Associations were found between clinical PeD parameters (particularly CAL) and cognitive impairment/dementia |
| Gil-Montoya et al. (2017), Spain [111] | Case–control | Determine whether PeD relates with serum Aβ load and assess the role of such relationship in the association between Aβ and cognitive impairment/dementia | 288 subjects (MA = 76.6 y) > cases: w MCI/dementia (n = 166) > control: cognitively healthy (n = 122) | Variables studied: > serum levels of Aβ peptides > PeD (CAL-based criterion as severity indicator) Outcomes: MCI/dementia (NINCDS-ADRDA, DSM-IV-TR, and Robles et al. criteria [168]) | Severe PeD was associated with higher serum Aβ1–42 levels Serum Aβ1–42 levels were positively associated with cognitive impairment/dementia only in the severe PeD group |
| Shin et al. (2016), South Korea [106] | Case–control | Investigate the association between PeD and cognitive impairment | 189 subjects (≥60 y, MA = 69 y) > cases: w cognitive impairment (n = 65) > control: cognitively healthy (n = 124) | Variable studied: history of PeD (radiographic ABL) Outcome: cognitive impairment (MMSE-KC) | Subjects with a history of PeD were more likely to have cognitive impairment |
| Holmer et al. (2018), Sweden [112] | Case–control | Evaluate whether PeD increases the risk of MCI, subjective cognitive decline (SCD), and AD | 230 subjects (50–80 y, MA = 67.2 y) > cases: w MCI (n = 51), SCD (n = 51) or AD (n = 52) > control: cognitively healthy (n = 76) | Variables studied: PeD (marginal ABL, PPD, BoP, and others) Outcomes: MCI, SCD, and AD (ICD-10 and others) | Marginal PeD (generalised marginal ABL and increased PPD) was associated with the cases groups combined (MCI, SCD, and AD) |
| De Oliveira Araújo et al. (2021), Brazil [114] | Case–control | Determine whether PeD is associated with AD and its impact on the OHR-QoL perception | 102 subjects (n = 50, MA = 71.2 y) > cases: w mild-to-moderate AD (n = 50) > control: cognitively healthy (n = 52) | Variables studied: > PeD (PPD, CAL, BoP, others) > OHR-QoL (GOHAI) Outcome: mild-to-moderate AD (DSM, MMSE) | PeD was associated with AD, but not with OHR-QoL |
| Panzarella et al. (2022), Italy [115] | Case–control | Evaluate the relationship between measures of oral health and both amnestic MCI or AD | 60 subjects (MA = 80.0 y) > cases: w AD (n = 20) or aMCI (n = 20) > control: cognitively healthy (n = 20) | Variables studied: > dental status (DMFT score) > periodontal status (CPI and PSR scores) > subgingival plaque bacterial load > OHR-QoL (OHIP-14) Outcomes: > AD (NIA-AA 2011) > aMCI (modified Petersen’s criteria [167]) | Subjects with AD showed: > poor health status related to PeD > higher DMFT scores than aMCI and control > higher Fn bacterial load than aMCI and control > no statistically significant differences in OHR-QoL |
| Poole et al. (2013), UK [150] | Case–control | Establish a link between PeD and AD by identifying the major PeD bacteria and/or bacterial components in post mortem human brain specimens | 20 human post mortem (PM) brain tissue samples > cases: w AD diagnosis (n = 10) > control: wo AD diagnosis (n = 10) | Variables studied: > presence of major PeD bacteria (Td, Tf) in brain tissue > presence of Pg-LPS and/or Pg gingipains in brain tissue Outcome: AD | Statistically significant evidence was found to implicate the presence of Pg-LPS in AD cases |
| Dominy et al. (2019), USA [11] | Case–control | Demonstrate the presence of Pg DNA and gingipains in the brain of AD patients | Human post mortem (PM) brain tissue samples (n variable) > cases: w AD diagnosis > control: non-demented | Variables studied: > gingipains, tau, and ubiquitin load in brain tissue > presence of Pg DNA in brain tissue Outcome: AD | Gingipains load in the brain was correlated with AD diagnosis and pathology (tau and ubiquitin) RgpB gingipain colocalised with neurones, astrocytes, tau tangles, and intracellular Aβ in AD hippocampus Pg DNA and Kgp gingipain were identified in the AD cerebral cortex |
| Prospective pilot | Demonstrate the presence of Pg DNA in the CSF of living subjects diagnosed with probable AD | 10 CSF samples from subjects with probable AD (53–72 y) | Variable studied: presence of Pg DNA in CSF | Pg DNA was identified and quantified in the CSF of clinical AD patients | |
| Liu et al. (2019), China [59] | Case–control | Compare the composition of oral microbiota between AD patients and healthy subjects | 78 subjects (MA = 64 y) > cases: w mild (n = 13), moderate (n = 12) or severe AD (n = 14) > control: cognitively healthy (n = 39) | Variable studied: composition of salivary microbiota Outcome: AD (NINCDS-ADRDA and MMSE) | Although no particular bacteria were associated with AD severity, the richness and diversity of salivary microbiota flora were significantly reduced in AD patients |
| Leblhuber et al. (2020), Austria [147] | Cross-sectional | Investigate whether the presence of PeD pathogens in saliva is associated with cognitive impairment in patients with probable AD | 20 subjects with probable AD (MA = 78.1 y) | PeD-related measure: presence of PeD pathogens (saliva) AD-related outcomes: > cognitive tests (MMSE and CDT) > serum levels of neopterin, tryptophan, and kynurenine | Salivary presence of Pg was associated with lower MMSE and CDT scores Salivary presence of Td and Tf were associated with lower neopterin and kynurenine serum levels, respectively |
| Noble et al. (2009), USA [134] | Cross-sectional | Investigate the relationship between systemic exposure to PeD pathogens and cognitive test outcomes | 2355 subjects (≥60 y, MA = 70.8 y) from NHANES III | PeD-related measure: serum levels of anti-Pg IgG AD-related outcomes: cognitive tests (for verbal memory and subtraction) | Subjects with the highest anti-Pg IgG levels were associated with poor delayed verbal memory and impaired subtraction |
| Kamer et al. (2012), Denmark [107] | Cross-sectional | Assess the effect of periodontal inflammation (PI) or tooth loss on cognitive functioning | 152 subjects (70 y) | PeD-related measures: > PI (MCPI score) > tooth loss AD-related outcomes: cognitive tests (DSST and BDT) | The association of PI with DSST and BDT scores was dependent on the number of missing teeth Subjects with PI had lower adjusted mean DSST and BDT scores |
| Nilsson et al. (2018), Sweden [104] | Cross-sectional | Investigate the association between PeD or the number of teeth and cognitive functioning | 775 subjects (60–99 y) | PeD-related measures: > extent of ABL > prevalence of PeD pockets ≥5 mm on ≥30% of teeth > number of teeth AD-related outcomes: cognitive tests (MMSE and CDT) | Loss of alveolar bone and lower number of teeth were statistically associated with a lower outcome on the MMSE test |
| Zhang et al. (2020), China [108] | Cross-sectional | Examine the relationship between poor oral health conditions and cognitive decline | 102 subjects (52–101 y) | PeD-related measures: number of missing index teeth, BoP, and PPD AD-related outcome: cognitive test (MMSE) | Higher number of missing index teeth and higher average PPD were associated with lower cognitive MMSE scores |
| Marruganti et al. (2023), Spain [109] | Cross-sectional | Investigate the association between PeD and low cognitive performance | 2086 subjects (≥60 y, MA = 68.6 y) from NHANES 2011–2014 | PeD-related measures: PPD and CAL AD-related outcomes: cognitive tests (CERAD-WL, AFT, and DSST) | Severe or moderate PeD was associated with low DSST PPD and CAL were associated with low global cognition performance |
| Kamer et al. (2015), USA [137] | Cross-sectional | Investigate the association between PeD and Aβ brain load | 38 cognitively healthy subjects (44–79 y, MA = 61.3 y) | PeD-related measures: > CAL (primary exposure) > number of teeth, dental plaque, BoP, PPD AD-related outcome: brain Aβ load in AD-vulnerable areas (PIB-PET scan) | Clinical measures of PeD were associated with Aβ accumulation in AD-vulnerable brain areas |
| Kamer et al. (2021), USA [138] | Cross-sectional | Investigate whether periodontal dysbiosis is associated with CSF markers of AD pathology | 48 cognitively healthy subjects (MA = 69.2 y) | PeD-related measures: > dysbiotic index (DI—primary exposure) > subgingival bacterial species cluster (secondary exposure) AD-related outcomes: CSF levels of Aβ42 and p-tau181 | Higher periodontal dysbiosis was associated only with reduced CSF Aβ42 |
| Schwahn et al. (2022), Germany [145] | Cross-sectional | Investigate the association between PeD and preclinical AD | 1323 subjects (<60 y) | PeD-related measures: PPD, CAL, dental plaque, and calculus AD-related outcomes: > AD score [143] (brain atrophy MRI marker) > brain age (MRI) | Severe or moderate PeD involved in the continuum of preclinical AD severity: > dose–response relationship between PPD and AD score/brain age |
| Tiisanoja et al. (2019), Finland [116] | Cross-sectional | Investigate whether oral diseases and the related inflammatory burden are associated with diagnosed AD or dementia | 170 subjects (≥75 y, MA = 80.9 y) | Oral disease-related measures: > dental caries (number of carious teeth) > PeD (number of teeth with PeD pockets ≥4 mm) > stomatitis (visual inspection) > inflammatory burden (numeric score) AD-related outcomes: > diagnosed AD (DSM-IV) > all diagnosed dementias (DSM-IV and McKeith et al. criteria [169]) | Dental caries and inflammatory burden were associated with a higher likelihood of having AD Subjects with PeD and stomatitis had an increased, although not statistically significant, likelihood of having AD |
| Reference | Study Characterisation | Findings |
|---|---|---|
| Jiang et al. (2021) [91] | Bioinformatics study Functional enrichment (GO and KEGG pathway) and PPI network analyses of the crosstalk genes between: > DEGs of AD gene expression datasets (from GEO) > PeD gene set (from text mining) | Identification/characterisation of shared molecular linkages (core crosstalk genes, GO and KEGG overlapping functional terms) between PeD and AD |
| Jin et al. (2021) [92] | Bioinformatics study Series of bioinformatic analysis of the crosstalk genes between: > AD-related genes (from DisGeNET) > DEGs of PeD gene expression datasets (from GEO) Identification of core crosstalk genes by further overlapping: > feature selected crosstalk genes > with PeD-related genes (from DisGeNET) | Identification of shared molecular linkages (core crosstalk genes, transcription factors, and pathways) between PeD and AD |
| Sun et al. (2020) [90] | Bidirectional two-sample Mendelian randomisation Analysis of genetically predicted PeD on the risk of AD: > GWAS of PeD: 4.924 cases vs 7.301 controls (5 SNPs or 7 SNPs) > GWAS of AD: 21.982 cases vs 41.944 controls | Higher risk of AD (only if using five SNPs as instruments) |
| Carter et al. (2017) [12] | Bioinformatics study Analysis of gene/environment interactions between PeD/Pg and AD-associated genes, by comparing: > Pg/host interactome > with GWAS AD susceptibility genes AND > microarray data from PeD tissue or Pg-treated macrophages > with microarray datasets from AD hippocampus | > Pg/host interactome highly enriched in susceptibility genes of AD > KEGG analysis revealed pathways relevant to BBB and inflammation AND > misregulated genes in PeD/Pg microarrays matched those in the AD hippocampus > overlaps less significant in the Pg microarrays |
5. Opportunity for a Mucosal Pg Nanovaccine?
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hippius, H.; Gabriele, N. The discovery of Alzheimer’s Disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, C.; Keipes, M. Geriatrics from the 19th to the 21st century. 150 years of geriatric medicine: From increasing life expectancy to improving quality of life for the very old. Bull. Soc. Sci. Med. Grand. Duche. Luxemb. 2014, 2, 69–78. [Google Scholar]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Lindemer, E.R.; Greve, D.N.; Fischl, B.R.; Augustinack, J.C.; Salat, D.H. Regional staging of white matter signal abnormalities in aging and Alzheimer’s disease. Neuroimage Clin. 2017, 14, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Tom, S.E.; Hubbard, R.A.; Crane, P.K.; Haneuse, S.J.; Bowen, J.; McCormick, W.C.; McCurry, S.; Larson, E.B. Characterization of dementia and Alzheimer’s disease in an older population: Updated incidence and life expectancy with and without dementia. Am. J. Public Health 2015, 105, 408–413. [Google Scholar] [CrossRef]
- Gauthier, S.; Webster, C.; Servaes, S.; Morais, J.A.; Rosa-Neto, P. World Alzheimer Report 2022: Life after Diagnosis: Navigating Treatment, Care and Support; Alzheimer’s Disease International: London, UK, 2022. [Google Scholar]
- Patterson, C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Baumgart, M.; Snyder, H.; Carrillo, M.; Fazio, S.; Kim, H.; Johns, H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimer’s Dement. 2015, 11, 718–726. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Liu, K.Y.; Costafreda, S.G.; Selbæk, G.; Alladi, S.; Ames, D.; Banerjee, S.; Burns, A.; Brayne, C.; et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet 2024, 404, 572–628. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Carter, C.J.; France, J.; Crean, S.; Singhrao, S.K. The Porphyromonas gingivalis/host interactome shows enrichment in GWASdb genes related to Alzheimer’s disease, diabetes and cardiovascular diseases. Front. Aging Neurosci. 2017, 9, 408. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Wu, Y.-T.; Chang, Y.-C. Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimer’s Res. Ther. 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Watling, M.; Imbimbo, B.P. Time to test antibacterial therapy in Alzheimer’s disease. Brain 2019, 142, 2905–2929. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Eratne, D.; Loi, S.M.; Farrand, S.; Kelso, W.; Velakoulis, D.; Looi, J.C.L. Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australas. Psychiatry 2018, 26, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef]
- Bateman, R.; Aisen, P.; Strooper, B.; Fox, N.; Lemere, C.; Ringman, J.; Salloway, S.; Sperling, R.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef]
- Pritchard, A.B.; Crean, S.; Olsen, I.; Singhrao, S.K. Periodontitis, microbiomes and their role in Alzheimer’s disease. Front. Aging Neurosci. 2017, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Paula, V.J.R.; Guimarães, F.M.; Diniz, B.S.; Forlenza, O.V. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, tau protein or both? Dement. Neuropsychol. 2009, 3, 188–194. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New insights into the pathogenesis of Alzheimer’s disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, S.; Hermkens, D.M.A.; van der Weerd, L.; De Vries, H.E.; Daemen, M.J.A.P. Vascular hypothesis of Alzheimer disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef]
- Chen, L.-L.; Fan, Y.-G.; Zhao, L.-X.; Zhang, Q.; Wang, Z.-Y. The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorg. Chem. 2023, 131, 106301. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Morgen, K.; Frölich, L. The metabolism hypothesis of Alzheimer’s disease: From the concept of central insulin resistance and associated consequences to insulin therapy. J. Neural. Transm. 2015, 122, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3β hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Ulland, T.K.; Wang, Y.; Tanzi, R.E.; Colonna, M. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimer’s Dement. 2017, 13, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef]
- Carter, C.J. Genetic, transcriptome, proteomic, and epidemiological evidence for blood-brain barrier disruption and polymicrobial brain invasion as determinant factors in Alzheimer’s disease. J. Alzheimer’s Dis. Rep. 2017, 1, 125–157. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J. Alzheimer’s disease—A neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflamm. 2011, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Rábano, A.; Horst, M.N.; Carrasco, L. Fungal enolase, β-tubulin, and chitin are detected in brain tissue from Alzheimer’s disease patients. Front. Microbiol. 2016, 7, 1772. [Google Scholar] [CrossRef]
- Seaks, C.E.; Wilcock, D.M. Infectious hypothesis of Alzheimer disease. PLoS Pathog. 2020, 16, e1008596. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Kandel, R.; Tripathi, S.; Condon, D.; Qi, L.; Taubenberger, J.; Hartshorn, K.L. Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS ONE 2014, 9, e101364. [Google Scholar] [CrossRef]
- Bourgade, K.; Le Page, A.; Bocti, C.; Witkowski, J.M.; Dupuis, G.; Frost, E.H.; Fülöp, T. Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimer’s Dis. 2016, 50, 1227–1241. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfiled, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Witkowski, J.M.; Bourgade, K.; Khalil, A.; Zerif, E.; Larbi, A.; Hirokawa, K.; Pawelec, G.; Bocti, C.; Lacombe, G.; et al. Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front. Aging Neurosci. 2018, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Herpes and Alzheimer’s disease: Subversion in the central nervous system and how it might be halted. J. Alzheimer’s Dis. 2016, 54, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Emery, D.C.; Shoemark, D.K.; Batstone, T.E.; Waterfall, C.M.; Coghill, J.A.; Cerajewska, T.L.; Davies, M.; West, N.X.; Allen, S.J. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front. Aging Neurosci. 2017, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Pisa, D.; Aguado, B.; Carrasco, L. Identification of fungal species in brain tissue from Alzheimer’s disease by next-generation sequencing. J. Alzheimer’s Dis. 2017, 58, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Cao, Z.-S.; Chang, K.-M.; Juang, J.-L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef]
- Liu, X.X.; Jiao, B.; Liao, X.-X.; Guo, L.-N.; Yuan, Z.-H.; Wang, X.; Xiao, X.-W.; Zhang, X.-Y.; Tang, B.-S.; Shen, L. Analysis of salivary microbiome in patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 72, 633–640. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Olsen, I. Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J. Oral Microbiol. 2019, 11, 1563405. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.; Al-Ansari, A.; Al-Khalifa, K.; Alhareky, M.; Gaffar, B.; Almas, K. Global prevalence of periodontal disease and lack of its surveillance. Sci. World J. 2020, 2020, 2146160. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N. The prevalence of periodontitis in the US. J. Dent. Res. 2012, 91, 907–908. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Mishra, A.; Chugh, S. Microbial role in periodontitis: Have we reached the top? Some unsung bacteria other than red complex. J. Indian Soc. Periodontol. 2014, 18, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.C.; Ebersole, J.; Felton, J.; Brunsvold, M.; Kornman, K.S. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science 1988, 239, 55–57. [Google Scholar] [CrossRef]
- Miller, D.P.; Hutcherson, J.A.; Wang, Y.; Nowakowska, Z.M.; Potempa, J.; Yoder-Himes, D.R.; Scott, D.A.; Whiteley, M.; Lamont, R.J. Genes contributing to Porphyromonas gingivalis fitness in abscess and epithelial cell colonization environments. Front. Cell. Infect. Microbiol. 2017, 7, 378. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur. J. Immunol. 2014, 44, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.C.; Kesavalu, L.; Walker, S.; Genco, C.A. Virulence factors of Porphyromonas gingivalis. Periodontol. 2000 1999, 20, 168–238. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Han, N.; Du, J.; Guo, L.; Luo, Z.; Liu, Y. Pathogenesis of important virulence factors of Porphyromonas gingivalis via toll-like receptors. Front. Cell. Infect. Microbiol. 2019, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Wyant, T.; Anaya-Bergman, C.; Aduse-Opoku, J.; Brunner, J.; Laine, M.L.; Curtis, M.A.; Lewis, J.P. The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect. Immun. 2011, 79, 4533–4542. [Google Scholar] [CrossRef] [PubMed]
- Enersen, M.; Nakano, K.; Amano, A. Porphyromonas gingivalis fimbriae. J. Oral Microbiol. 2013, 5, 20265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, D.; Liu, S.; Zhang, S.; Pan, Y. The role of Porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front. Cell. Infect. Microbiol. 2021, 10, 585917. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Ohara, N. Molecular mechanisms of Porphyromonas gingivalis-host cell interaction on periodontal diseases. Jpn. Dent. Sci. Rev. 2017, 53, 134–140. [Google Scholar] [CrossRef]
- Riviere, G.R.; Riviere, K.H.; Smith, K.S. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 2002, 17, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.V.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS ONE 2014, 9, e114959. [Google Scholar] [CrossRef]
- Kondo, K.; Niino, M.; Shido, K. A case-control study of Alzheimer’s disease in Japan—Significance of life-styles. Dement. Geriatr. Cogn. Disord. 1994, 5, 314–326. [Google Scholar] [CrossRef]
- Ship, J.A.; Puckett, S.A. Longitudinal study on oral health in subjects with Alzheimer’s disease. J. Am. Geriatr. Soc. 1994, 42, 57–63. [Google Scholar] [CrossRef]
- Ma, K.S.; Hasturk, H.; Carreras, I.; Dedeoglu, A.; Veeravalli, J.J.; Huang, J.Y.; Kantarci, A.; Wei, J.C. Dementia and the risk of periodontitis: A population-based cohort study. J. Dent. Res. 2022, 101, 270–277. [Google Scholar] [CrossRef]
- Gatz, M.; Mortimer, J.A.; Fratiglioni, L.; Johansson, B.; Berg, S.; Reynolds, C.A.; Pedersen, N.L. Potentially modifiable risk factors for dementia in identical twins. Alzheimer’s Dement. 2006, 2, 110–117. [Google Scholar] [CrossRef]
- Sparks Stein, P.; Desrosiers, M.; Donegan, S.J.; Yepes, J.F.; Kryscio, R.J. Tooth loss, dementia and neuropathology in the Nun Study. J. Am. Dent. Assoc. 2007, 138, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Popovac, A.; Čelebić, A.; Peršić, S.; Stefanova, E.; Milić Lemić, A.; Stančić, I. Oral health status and nutritional habits as predictors for developing Alzheimer’s disease. Med. Princ. Pract. 2021, 30, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Alfotawi, R.; Alzahrani, S.; Alhefdhi, R.; Altamimi, A.; Alfadhel, A.; Alshareef, A.; Aldawsari, B.; Sonbol, S.; Alsubaie, F.; Alwahibi, A.; et al. The relation between teeth loss and cognitive decline among Saudi population in the city of Riyadh: A pilot study. Saudi Dent. J. 2020, 32, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Asher, S.; Stephen, R.; Ngandu, T.; Koskinen, S.; Suominen, A.L.; Solomon, A. Association between tooth loss and cognitive performance: 11-year follow-up cohort study. Alzheimer’s Dement. 2021, 17, e052069. [Google Scholar] [CrossRef]
- Chava, V.K.; Nuvvula, S.; Nuvvula, S. Primary culprit for tooth loss!! J. Indian Soc. Periodontol. 2016, 20, 222–224. [Google Scholar] [CrossRef]
- Kim, Y.-T.; Choi, J.-K.; Kim, D.-H.; Jeong, S.-N.; Lee, J.-H. Association between health status and tooth loss in Korean adults: Longitudinal results from the National Health Insurance Service-Health Examinee Cohort, 2002–2015. J. Periodontal Implant Sci. 2019, 49, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Q.; Richmond, R.C.; Chen, Y.; Mai, X.-M. Mixed evidence for the relationship between periodontitis and Alzheimer’s disease: A bidirectional Mendelian randomization study. PLoS ONE 2020, 15, e0228206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Shi, Y.; Zhao, W.; Zhou, L.; Zhang, B.; Xie, Y.; Zhang, Y.; Tan, G.; Wang, Z. Association between chronic periodontitis and the risk of Alzheimer’s disease: Combination of text mining and GEO dataset. BMC Oral Health 2021, 21, 466. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Guang, M.; Ogbuehi, A.C.; Li, S.; Zhang, K.; Ma, Y.; Acharya, A.; Guo, B.; Peng, Z.; Liu, X.; et al. Shared molecular mechanisms between Alzheimer’s disease and periodontitis revealed by transcriptomic analysis. Biomed. Res. Int. 2021, 2021, 6633563. [Google Scholar] [CrossRef] [PubMed]
- Kaye, E.K.; Valencia, A.; Baba, N.; Spiro, A.; Dietrich, T.; Garcia, R.I. Tooth loss and periodontal disease predict poor cognitive function in older men. J. Am. Geriatr. Soc. 2010, 58, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Yoshihara, A.; Kimura, Y.; Sato, M.; Wada, T.; Sakamoto, R.; Ishimoto, Y.; Fukutomi, E.; Chen, W.; Imai, H.; et al. Longitudinal relationship of severe periodontitis with cognitive decline in older Japanese. J. Periodontal Res. 2016, 51, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Kimura, Y.; Ogawa, H.; Yamaga, T.; Ansai, T.; Wada, T.; Sakamoto, R.; Ishimoto, Y.; Fujisawa, M.; Okumiya, K.; et al. Periodontitis, periodontal inflammation, and mild cognitive impairment: A 5-year cohort study. J. Periodontal Res. 2019, 54, 233–240. [Google Scholar] [CrossRef]
- Tzeng, N.-S.; Chung, C.-H.; Yeh, C.-B.; Huang, R.-Y.; Yuh, D.-Y.; Huang, S.-Y.; Lu, R.-B.; Chang, H.-A.; Kao, Y.-C.; Chiang, W.-S.; et al. Are chronic periodontitis and gingivitis associated with dementia? A nationwide, retrospective, matched-cohort study in Taiwan. Neuroepidemiology 2016, 47, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Lee, H.-C.; Hu, C.-J.; Huang, L.-K.; Chao, S.-P.; Lin, C.-P.; Su, E.C.-Y.; Lee, Y.-C.; Chen, C.-C. Periodontitis as a modifiable risk factor for dementia: A nationwide population-based cohort study. J. Am. Geriatr. Soc. 2017, 65, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Hu, H.-Y.; Huang, L.-Y.; Chou, P.; Chu, D. Periodontal disease associated with higher risk of dementia: Population-based cohort study in Taiwan. J. Am. Geriatr. Soc. 2017, 65, 1975–1980. [Google Scholar] [CrossRef] [PubMed]
- De Souza Rolim, T.; Fabri, G.M.C.; Nitrini, R.; Anghinah, R.; Teixeira, M.J.; De Siqueira, J.T.T.; Cestari, J.A.F.; De Siqueira, S.R.D.T. Evaluation of patients with Alzheimer’s disease before and after dental treatment. Arq. Neuropsiquiatr. 2014, 72, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, K.; Chang, J.; Kim, S.M.; Kim, S.J.; Cho, H.; Park, S.M. Association of chronic periodontitis on Alzheimer’s disease or vascular dementia. J. Am. Geriatr. Soc. 2019, 67, 1234–1239. [Google Scholar] [CrossRef]
- Chiu, W.-C.; Tsan, Y.-T.; Tsai, S.-L.; Chang, C.-J.; Wang, J.-D.; Chen, P.-C. Hepatitis C viral infection and the risk of dementia. Eur. J. Neurol. 2014, 21, 1068-e59. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.; Jung, J.; Tran, L.; Zhao, C. Periodontal disease and risk of dementia in Medicare patients with hepatitis C virus. J. Alzheimer’s Dis. 2022, 85, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, B.; Bahorik, A.L.; Mcevoy, C.T.; Yaffe, K. Markers of periodontal disease and cognitive decline in a diverse, older cohort. Alzheimer’s Dement. 2021, 17, e052296. [Google Scholar] [CrossRef]
- Nilsson, H.; Berglund, J.S.; Renvert, S. Periodontitis, tooth loss and cognitive functions among older adults. Clin. Oral Investig. 2018, 22, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, H.; Berglund, J.S.; Renvert, S. Longitudinal evaluation of periodontitis and development of cognitive decline among older adults. J. Clin. Periodontol. 2018, 45, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-S.; Shin, M.-S.; Ahn, Y.-B.; Choi, B.-Y.; Nam, J.-H.; Kim, H.-D. Periodontitis is associated with cognitive impairment in elderly Koreans: Results from the Yangpyeong cohort study. J. Am. Geriatr. Soc. 2016, 64, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Morse, D.E.; Holm-Pedersen, P.; Mortensen, E.L.; Avlund, K. Periodontal inflammation in relation to cognitive function in an older adult Danish population. J. Alzheimer’s Dis. 2012, 28, 613–624. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, F.; Wang, Z.; Qian, X.; Ji, Y.; Gong, L.; Ge, S.; Yan, F. Poor oral health conditions and cognitive decline: Studies in humans and rats. PLoS ONE 2020, 15, e0234659. [Google Scholar] [CrossRef] [PubMed]
- Marruganti, C.; Baima, G.; Aimetti, M.; Grandini, S.; Sanz, M.; Romandini, M. Periodontitis and low cognitive performance: A population-based study. J. Clin. Periodontol. 2023, 50, 418–429. [Google Scholar] [CrossRef]
- Gil-Montoya, J.A.; Sanchez-Lara, I.; Carnero-Pardo, C.; Fornieles, F.; Montes, J.; Vilchez, R.; Burgos, J.S.; González-Moles, M.A.; Barrios, R.; Bravo, M. Is periodontitis a risk factor for cognitive impairment and dementia? A case-control study. J. Periodontol. 2015, 86, 244–253. [Google Scholar] [CrossRef]
- Gil-Montoya, J.A.; Barrios, R.; Santana, S.; Sanchez-Lara, I.; Carnero-Pardo, C.; Francisco, F.; Montes, J.; Ramírez, C.; González-Moles, M.A.; Burgos, J.S. Association between periodontitis and amyloid β peptide in elderly people with and without cognitive impairment. J. Periodontol. 2017, 88, 1051–1058. [Google Scholar] [CrossRef]
- Holmer, J.; Eriksdotter, M.; Schultzberg, M.; Pussinen, P.J.; Buhlin, K. Association between periodontitis and risk of Alzheimer’s disease, mild cognitive impairment and subjective cognitive decline: A case-control study. J. Clin. Periodontol. 2018, 45, 1287–1298. [Google Scholar] [CrossRef]
- De Souza Rolim, T.; Fabri, G.M.C.; Nitrini, R.; Anghinah, R.; Teixeira, M.J.; De Siqueira, J.T.T.; Cestari, J.A.F.; De Siqueira, S.R.D.T. Oral infections and orofacial pain in Alzheimer’s disease: A case-control study. J. Alzheimer’s Dis. 2014, 38, 823–829. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Araújo, R.; Villoria, G.E.M.; Luiz, R.R.; Esteves, J.C.; Leão, A.T.T.; Feres-Filho, E.J. Association between periodontitis and Alzheimer’s disease and its impact on the self-perceived oral health status: A case-control study. Clin. Oral Investig. 2021, 25, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Panzarella, V.; Mauceri, R.; Baschi, R.; Maniscalco, L.; Campisi, G.; Monastero, R. Oral health status in subjects with amnestic mild cognitive impairment and Alzheimer’s disease: Data from the Zabút Aging Project. J. Alzheimer’s Dis. 2022, 87, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Tiisanoja, A.; Syrjälä, A.-M.; Tertsonen, M.; Komulainen, K.; Pesonen, P.; Knuuttila, M.; Hartikainen, S.; Ylöstalo, P. Oral diseases and inflammatory burden and Alzheimer’s disease among subjects aged 75 years or older. Spec. Care Dentist. 2019, 39, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Weyant, R.J.; Garcia, M.E.; Harris, T.; Launer, L.J.; Satterfield, S.; Simonsick, E.M.; Yaffe, K.; Newman, A.B. Adverse oral health and cognitive decline: The health, aging and body composition study. J. Am. Geriatr. Soc. 2013, 61, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Holmer, J.; Eriksdotter, M.; Häbel, H.; Hed Myrberg, I.; Jonsson, A.; Pussinen, P.J.; Garcia-Ptacek, S.; Jansson, L.; Sandborgh-Englund, G.; Buhlin, K. Periodontal conditions and incident dementia: A nationwide Swedish cohort study. J. Periodontol. 2022, 93, 1378–1386. [Google Scholar] [CrossRef]
- Arrivé, E.; Letenneur, L.; Matharan, F.; Laporte, C.; Helmer, C.; Barberger-Gateau, P.; Miquel, J.L.; Dartigues, J.F. Oral health condition of French elderly and risk of dementia: A longitudinal cohort study. Community Dent. Oral Epidemiol. 2012, 40, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Asher, S.; Stephen, R.; Suominen, A.L.; Mäntylä, P.; Solomon, A. Association of periodontitis and cognitive impairment: A systematic review and meta-analysis. Alzheimer’s Dement. 2020, 16, e042580. [Google Scholar] [CrossRef]
- Torrealba-García, D.; Garcia-Morales, P.; Torrealba, E.; Cejudo, J.C.; Silvestre-Rangil, J. Is there a relationship between periodontitis and Alzheimer’s disease? Systematic review and comparative analysis. Alzheimer’s Dement. 2021, 17, e051470. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; Qiu, Y.; Liu, Z. Periodontal disease and the risk of Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Psychogeriatrics 2021, 21, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Sparks Stein, P.; Steffen, M.J.; Smith, C.; Jicha, G.; Ebersole, J.L.; Abner, E.; Dawson, D. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]
- Kim, D.-H.; Jeong, S.-N.; Lee, J.-H. Severe periodontitis with tooth loss as a modifiable risk factor for the development of Alzheimer, vascular, and mixed dementia: National Health Insurance Service-National Health Screening Retrospective Cohort 2002–2015. J. Periodontal Implant Sci. 2020, 50, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Demmer, R.T.; Norby, F.L.; Lakshminarayan, K.; Walker, K.A.; Pankow, J.S.; Folsom, A.R.; Mosley, T.; Beck, J.; Lutsey, P.L. Periodontal disease and incident dementia: The Atherosclerosis Risk in Communities Study (ARIC). Neurology 2020, 95, e1660–e1671. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Hossain, S.; El-Hajj, Z.W.; Weiss, J.; Zonderman, A.B. Clinical and bacterial markers of periodontitis and their association with incident all-cause and Alzheimer’s disease dementia in a large national survey. J. Alzheimer’s Dis. 2020, 75, 157–172. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Niederman, R.; Fortea, J.; De Leon, M.J. Periodontal disease as a possible cause for Alzheimer’s disease. Periodontol. 2000 2020, 83, 242–271. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Craig, R.G.; Pirraglia, E.; Dasanayake, A.P.; Norman, R.G.; Boylan, R.J.; Nehorayoff, A.; Glodzik, L.; Brys, M.; De Leon, M.J. TNF-α and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J. Neuroimmunol. 2009, 216, 92–97. [Google Scholar] [CrossRef]
- Farhad, S.Z.; Amini, S.; Khalilian, A.; Barekatain, M.; Mafi, M.; Barekatain, M.; Rafei, E. The effect of chronic periodontitis on serum levels of tumor necrosis factor-alpha in Alzheimer disease. Dent. Res. J. (Isfahan) 2014, 11, 549–552. [Google Scholar] [PubMed]
- Cestari, J.A.F.; Fabri, G.M.C.; Kalil, J.; Nitrini, R.; Jacob-Filho, W.; De Siqueira, J.T.T.; De Siqueira, S.R.D.T. Oral infections and cytokine levels in patients with Alzheimer’s disease and mild cognitive impairment compared with controls. J. Alzheimer’s Dis. 2016, 52, 1479–1485. [Google Scholar] [CrossRef]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and cognitive decline in Alzheimer’s disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef]
- Dye, B.A.; Herrera-Abreu, M.; Lerche-Sehm, J.; Vlachojannis, C.; Pikdoken, L.; Pretzl, B.; Schwartz, A.; Papapanou, P.N. Serum antibodies to periodontal bacteria as diagnostic markers of periodontitis. J. Periodontol. 2009, 80, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.M.; Borrell, L.N.; Papapanou, P.N.; Elkind, M.S.V.; Scarmeas, N.; Wright, C.B. Periodontitis is associated with cognitive impairment among older adults: Analysis of NHANES-III. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.T.; Zhao, L.; Bawa, E.M.; Yi, F.; Vidanapathirana, N.P.; Lohman, M.; Zhang, J. Association between clusters of antibodies against periodontal microorganisms and Alzheimer disease mortality: Evidence from a nationally representative survey in the USA. J. Periodontol. 2024, 95, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Laugisch, O.; Johnen, A.; Maldonado, A.; Ehmke, B.; Bürgin, W.; Olsen, I.; Potempa, J.; Sculean, A.; Duning, T.; Eick, S. Periodontal pathogens and associated intrathecal antibodies in early stages of Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 66, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Pirraglia, E.; Tsui, W.; Rusinek, H.; Vallabhajosula, S.; Mosconi, L.; Yi, L.; McHugh, P.; Craig, R.G.; Svetcov, S.; et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 2015, 36, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Pushalkar, S.; Gulivindala, D.; Butler, T.; Li, Y.; Annam, K.R.C.; Glodzik, L.; Ballman, K.V.; Corby, P.M.; Blennow, K.; et al. Periodontal dysbiosis associates with reduced CSF Aβ42 in cognitively normal elderly. Alzheimer’s Dement. 2021, 13, e12172. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M. Cerebrospinal fluid and plasma tau as a biomarker for brain tauopathy. Adv. Exp. Med. Biol. 2019, 1184, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Carballo, Á.; López-Dequidt, I.; Custodia, A.; Botelho, J.; Aramburu-Núñez, M.; Machado, V.; Pías-Peleteiro, J.M.; Ouro, A.; Romaus-Sanjurjo, D.; Vázquez-Vázquez, L.; et al. Association of periodontitis with cognitive decline and its progression: Contribution of blood-based biomarkers of Alzheimer’s disease to this relationship. J. Clin. Periodontol. 2023, 50, 1444–1454. [Google Scholar] [CrossRef]
- Kubota, T.; Maruyama, S.; Abe, D.; Tomita, T.; Morozumi, T.; Nakasone, N.; Saku, T.; Yoshie, H. Amyloid beta (A4) precursor protein expression in human periodontitis-affected gingival tissues. Arch. Oral Biol. 2014, 59, 586–594. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; McCue, L.; Jasielec, M.S.; Su, Y.; Gordon, B.A.; Xiong, C.; Holtzman, D.M.; Benzinger, T.L.S.; Morris, J.C.; Fagan, A.M. Imaging and cerebrospinal fluid biomarkers in early preclinical Alzheimer disease. Ann. Neurol. 2016, 80, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, S.; Wittfeld, K.; Habes, M.; Klinger-König, J.; Bülow, R.; Völzke, H.; Grabe, H.J. A biomarker for Alzheimer’s disease based on patterns of regional brain atrophy. Front. Psychiatry 2020, 10, 953. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Gaser, C. Ten years of BrainAGE as a neuroimaging biomarker of brain aging: What insights have we gained? Front. Neurol. 2019, 10, 789. [Google Scholar] [CrossRef]
- Schwahn, C.; Frenzel, S.; Holtfreter, B.; van der Auwera, S.; Pink, C.; Bülow, R.; Friedrich, N.; Völzke, H.; Biffar, R.; Kocher, T.; et al. Effect of periodontal treatment on preclinical Alzheimer’s disease—Results of a trial emulation approach. Alzheimer’s Dement. 2022, 18, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Adam, H.S.; Lakshminarayan, K.; Wang, W.; Norby, F.L.; Mosley, T.; Walker, K.A.; Gottesman, R.F.; Meyer, K.; Hughes, T.M.; Pankow, J.S.; et al. The prospective association between periodontal disease and brain imaging outcomes: The Atherosclerosis Risk in Communities Study. J. Clin. Periodontol. 2022, 49, 322–334. [Google Scholar] [CrossRef]
- Leblhuber, F.; Huemer, J.; Steiner, K.; Gostner, J.M.; Fuchs, D. Knock-on effect of periodontitis to the pathogenesis of Alzheimer’s disease? Wien. Klin. Wochenschr. 2020, 132, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Bathini, P.; Foucras, S.; Dupanloup, I.; Imeri, H.; Perna, A.; Berruex, J.; Doucey, M.; Annoni, J.; Auber Alberi, L. Classifying dementia progression using microbial profiling of saliva. Alzheimer’s Dement. 2020, 12, e12000. [Google Scholar] [CrossRef]
- Wu, Y.-F.; Lee, W.-F.; Salamanca, E.; Yao, W.-L.; Su, J.-N.; Wang, S.-Y.; Hu, C.-J.; Chang, W.-J. Oral microbiota changes in elderly patients, an indicator of Alzheimer’s disease. Int. J. Environ. Res. Public Health 2021, 18, 4211. [Google Scholar] [CrossRef] [PubMed]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimer’s Dis. 2013, 36, 665–677. [Google Scholar] [CrossRef]
- Bennett, J.P.; Keeney, P.M.; Brohawn, D.G. RNA sequencing reveals small and variable contributions of infectious agents to transcriptomes of postmortem nervous tissues from amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson’s disease subjects, and increased expression of genes from disease-activated microglia. Front. Neurosci. 2019, 13, 235. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Harding, A.; Poole, S.; Kesavalu, L.; Crean, S. Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediat. Inflamm. 2015, 2015, 137357. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.B.; Morales, D.; Jones, K.; Joshi, S. Alzheimer’s disease: A novel hypothesis integrating spirochetes, biofilm, and the immune system. J. Neuroinfect. Dis. 2016, 7, 200. [Google Scholar] [CrossRef]
- Allen, H.B. Alzheimer’s disease: Assessing the role of spirochetes, biofilms, the immune system, and amyloid-β with regard to potential treatment and prevention. J. Alzheimer’s Dis. 2016, 53, 1271–1276. [Google Scholar] [CrossRef]
- Miklossy, J. Bacterial amyloid and DNA are important constituents of senile plaques: Further evidence of the spirochetal and biofilm nature of senile plaques. J. Alzheimer’s Dis. 2016, 53, 1459–1473. [Google Scholar] [CrossRef]
- Allen, H.B.; Morales, D.; Jones, K.; Joshi, S. Alzheimer’s disease: A novel hypothesis for the development and the subsequent role of beta amyloid. J. Neuroinfect. Dis. 2016, 7, 211. [Google Scholar] [CrossRef]
- Senejani, A.G.; Maghsoudlou, J.; El-Zohiry, D.; Gaur, G.; Wawrzeniak, K.; Caravaglia, C.; Khatri, V.A.; MacDonald, A.; Sapi, E. Borrelia burgdorferi co-localizing with amyloid markers in Alzheimer’s disease brain tissues. J. Alzheimer’s Dis. 2022, 85, 889–903. [Google Scholar] [CrossRef]
- Ng, H.M.; Slakeski, N.; Butler, C.A.; Veith, P.D.; Chen, Y.-Y.; Liu, S.W.; Hoffmann, B.; Dashper, S.G.; Reynolds, E.C. The role of Treponema denticola motility in synergistic biofilm formation with Porphyromonas gingivalis. Front. Cell. Infect. Microbiol. 2019, 9, 432. [Google Scholar] [CrossRef]
- Wozniak, M.; Mee, A.; Itzhaki, R. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Chen, C.; Feng, P.; Slots, J. Herpesvirus-bacteria synergistic interaction in periodontitis. Periodontol. 2000 2020, 82, 42–64. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol. 2000 2010, 54, 15–44. [Google Scholar] [CrossRef]
- Olsen, I.; Singhrao, S.K. Poor oral health and its neurological consequences: Mechanisms of Porphyromonas gingivalis involvement in cognitive dysfunction. Curr. Oral Health Rep. 2019, 6, 120–129. [Google Scholar] [CrossRef]
- Lobmaier, I.V.K.; Vege, Å.; Gaustad, P.; Rognum, T.O. Bacteriological investigation—Significance of time lapse after death. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1191–1198. [Google Scholar] [CrossRef]
- Pfeffer, R.I.; Kurosaki, T.T.; Harrah, C.H.; Chance, J.M.; Filos, S. Measurement of functional activities in older adults in the community. J. Gerontol. 1982, 37, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Stevens, J.C.; Ganguli, M.; Tangalos, E.G.; Cummings, J.L.; DeKosky, S.T. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review). Neurology 2001, 56, 1133–1142. [Google Scholar] [CrossRef]
- Winblad, B.; Palmer, K.; Kivipelto, M.; Jelic, V.; Fratiglioni, L.; Wahlund, L.-O.; Nordberg, A.; Backman, L.; Albert, M.; Almkvist, O.; et al. Mild cognitive impairment—Beyond controversies, towards a consensus: Report of the international working group on mild cognitive impairment. J. Intern. Med. 2004, 256, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Robles-Bayón, A.; Del Ser, T.; Alom, J.; Peña-Casanova, J. Proposal of criteria for clinical diagnosis of mild cognitive impairment, dementia and Alzheimer’s disease. Neurologia 2002, 17, 17–32. [Google Scholar]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, L.A.; Salmon, D.P.; Lowe, J.; Mirra, S.S.; Byrne, E.J.; et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB). Neurology 1996, 47, 1113–1124. [Google Scholar] [CrossRef]
- Abbayya, K.; Puthanakar, N.Y.; Naduwinmani, S.; Chidambar, Y.S. Association between periodontitis and Alzheimer’s disease. N. Am. J. Med. Sci. 2015, 7, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.P.-H.; Huang, J.; Chan, K.W.Y.; Leung, W.K.; Goto, T.; Ho, J.Y.S.; Chang, R.C.-C. IL-1β and TNF-α play an important role in modulating the risk of periodontitis and Alzheimer’s disease. J. Neuroinflamm. 2023, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, Z.; Zhang, X.; Ni, J.; Yu, W.; Zhou, Y.; Nakanishi, H. Leptomeningeal cells transduce peripheral macrophages inflammatory signal to microglia in response to Porphyromonas gingivalis LPS. Mediat. Inflamm. 2013, 2013, 407562. [Google Scholar] [CrossRef]
- Sato, N.; Matsumoto, T.; Kawaguchi, S.; Seya, K.; Matsumiya, T.; Ding, J.; Aizawa, T.; Imaizumi, T. Porphyromonas gingivalis lipopolysaccharide induces interleukin-6 and c-c motif chemokine ligand 2 expression in cultured hCMEC/D3 human brain microvascular endothelial cells. Gerodontology 2022, 39, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shin, Y.-J.; Yoo, J.-W.; Park, H.-S.; Kim, D.-H. Extracellular vesicles derived from Porphyromonas gingivalis induce trigeminal nerve-mediated cognitive impairment. J. Adv. Res. 2023, 54, 293–303. [Google Scholar] [CrossRef]
- Olsen, I.; Singhrao, S.K. Can oral infection be a risk factor for Alzheimer’s disease? J. Oral Microbiol. 2015, 7, 29143. [Google Scholar] [CrossRef]
- Dando, S.J.; Mackay-Sim, A.; Norton, R.; Currie, B.J.; St. John, J.A.; Ekberg, J.A.K.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev. 2014, 27, 691–726. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I. Possible effects of Porphyromonas gingivalis on the blood-brain barrier in Alzheimer’s disease. Expert Rev. Anti Infect. Ther. 2021, 19, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Singhrao, S.K.; Olsen, I. Are Porphyromonas gingivalis outer membrane vesicles microbullets for sporadic Alzheimer’s disease manifestation? J. Alzheimer’s Dis. Rep. 2018, 2, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Nara, P.L.; Sindelar, D.; Penn, M.S.; Potempa, J.; Griffin, W.S.T. Porphyromonas gingivalis outer membrane vesicles as the major driver of and explanation for neuropathogenesis, the cholinergic hypothesis, iron dyshomeostasis, and salivary lactoferrin in Alzheimer’s disease. J. Alzheimer’s Dis. 2021, 82, 1417–1450. [Google Scholar] [CrossRef]
- Yoshida, K.; Yoshida, K.; Seyama, M.; Hiroshima, Y.; Mekata, M.; Fujiwara, N.; Kudo, Y.; Ozaki, K. Porphyromonas gingivalis outer membrane vesicles in cerebral ventricles activate microglia in mice. Oral Dis. 2023, 29, 3688–3697. [Google Scholar] [CrossRef]
- Hao, X.; Li, Z.; Li, W.; Katz, J.; Michalek, S.M.; Barnum, S.R.; Pozzo-Miller, L.; Saito, T.; Saido, T.C.; Wang, Q.; et al. Periodontal infection aggravates C1q-mediated microglial activation and synapse pruning in Alzheimer’s mice. Front. Immunol. 2022, 13, 816640. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ni, J.; Liu, Y.; Teeling, J.L.; Takayama, F.; Collcutt, A.; Ibbett, P.; Nakanishi, H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 2017, 65, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.; Lu, W.; Song, Z. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflamm. 2018, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Chi, L.; Lin, T.; Liang, F.; Pei, Z.; Sun, J.; Teng, W. Exogenous monocyte myeloid-derived suppressor cells ameliorate immune imbalance, neuroinflammation and cognitive impairment in 5xFAD mice infected with Porphyromonas gingivalis. J. Neuroinflamm. 2023, 20, 55. [Google Scholar] [CrossRef]
- Ishida, N.; Ishihara, Y.; Ishida, K.; Tada, H.; Funaki-Kato, Y.; Hagiwara, M.; Ferdous, T.; Abdullah, M.; Mitani, A.; Michikawa, M.; et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech. Dis. 2017, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wu, Z.; Zeng, F.; Jiang, M.; Teeling, J.L.; Ni, J.; Takahashi, I. Systemic exposure to lipopolysaccharide from Porphyromonas gingivalis induces bone loss-correlated Alzheimer’s disease-like pathologies in middle-aged mice. J. Alzheimer’s Dis. 2020, 78, 61–74. [Google Scholar] [CrossRef]
- Lei, S.; Li, J.; Yu, J.; Li, F.; Pan, Y.; Chen, X.; Ma, C.; Zhao, W.; Tang, X. Porphyromonas gingivalis bacteremia increases the permeability of the blood-brain barrier via the Mfsd2a/Caveolin-1 mediated transcytosis pathway. Int. J. Oral Sci. 2023, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.B.; Fabian, Z.; Lawrence, C.L.; Morton, G.; Crean, S.; Alder, J.E. An investigation into the effects of outer membrane vesicles and lipopolysaccharide of Porphyromonas gingivalis on blood-brain barrier integrity, permeability, and disruption of scaffolding proteins in a human in vitro model. J. Alzheimer’s Dis. 2022, 86, 343–364. [Google Scholar] [CrossRef]
- Nonaka, S.; Kadowaki, T.; Nakanishi, H. Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins. Neurochem. Int. 2022, 154, 105282. [Google Scholar] [CrossRef]
- Qian, X.; Zhang, S.; Duan, L.; Yang, F.; Zhang, K.; Yan, F.; Ge, S. Periodontitis deteriorates cognitive function and impairs neurons and glia in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2021, 79, 1785–1800. [Google Scholar] [CrossRef] [PubMed]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef]
- Hu, Y.; Li, H.; Zhang, J.; Zhang, X.; Xia, X.; Qiu, C.; Liao, Y.; Chen, H.; Song, Z.; Zhou, W. Periodontitis induced by P. gingivalis-LPS is associated with neuroinflammation and learning and memory impairment in Sprague-Dawley rats. Front. Neurosci. 2020, 14, 658. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Zúñiga, J.; More, J.; Melgar-Rodríguez, S.; Jiménez-Unión, M.; Villalobos-Orchard, F.; Muñoz-Manríquez, C.; Monasterio, G.; Valdés, J.L.; Vernal, R.; Paula-Lima, A. Alzheimer’s disease-like pathology triggered by Porphyromonas gingivalis in wild type rats is serotype dependent. Front. Immunol. 2020, 11, 588036. [Google Scholar] [CrossRef] [PubMed]
- Bahar, B.; Kanagasingam, S.; Tambuwala, M.M.; Aljabali, A.A.A.; Dillon, S.A.; Doaei, S.; Welbury, R.; Chukkapalli, S.S.; Singhrao, S.K. Porphyromonas gingivalis (W83) infection induces Alzheimer’s disease-like pathophysiology in obese and diabetic mice. J. Alzheimer’s Dis. 2021, 82, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Liang, D.; Cheng, M.; Su, X.; Liu, R.; Zhang, Y.; Wu, H. Effects of Porphyromonas gingivalis and its underlying mechanisms on Alzheimer-like tau hyperphosphorylation in Sprague-Dawley rats. J. Mol. Neurosci. 2021, 71, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Kuraji, R.; Ye, C.; Gao, L.; Radaic, A.; Kamarajan, P.; Taketani, Y.; Kapila, Y.L. Nisin a probiotic bacteriocin mitigates brain microbiome dysbiosis and Alzheimer’s disease-like neuroinflammation triggered by periodontal disease. J. Neuroinflamm. 2023, 20, 228. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Guo, J.; Sun, J.; Sun, Q. Salvianolic acid B improves cognitive impairment by inhibiting neuroinflammation and decreasing Aβ level in Porphyromonas gingivalis-infected mice. Aging (Albany NY) 2020, 12, 10117–10128. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.; Wu, Z.; Ni, J.; Zeng, F.; Yu, W.; Zhang, Y.; Kadowaki, T.; Kashiwazaki, H.; Teeling, J.L.; Zhou, Y. Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J. Alzheimer’s Dis. 2019, 72, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Leira, Y.; Iglesias-Rey, R.; Gómez-Lado, N.; Aguiar, P.; Campos, F.; D’Aiuto, F.; Castillo, J.; Blanco, J.; Sobrino, T. Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch. Oral Biol. 2019, 99, 120–125. [Google Scholar] [CrossRef]
- Zeng, F.; Liu, Y.; Huang, W.; Qing, H.; Kadowaki, T.; Kashiwazaki, H.; Ni, J.; Wu, Z. Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection. J. Neurochem. 2021, 158, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Singhrao, S.K. Porphyromonas gingivalis infection may contribute to systemic and intracerebral amyloid-beta: Implications for Alzheimer’s disease onset. Expert Rev. Anti-Infect. Ther. 2020, 18, 1063–1066. [Google Scholar] [CrossRef]
- Xue, L.; Zou, X.; Yang, X.-Q.; Peng, F.; Yu, D.-K.; Du, J.-R. Chronic periodontitis induces microbiota-gut-brain axis disorders and cognitive impairment in mice. Exp. Neurol. 2020, 326, 113176. [Google Scholar] [CrossRef] [PubMed]
- Shoemark, D.K.; Allen, S.J. The microbiome and disease: Reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 725–738. [Google Scholar] [CrossRef]
- Fulop, T.; Tripathi, S.; Rodrigues, S.; Desroches, M.; Bunt, T.; Eiser, A.; Bernier, F.; Beauregard, P.B.; Barron, A.E.; Khalil, A.; et al. Targeting impaired antimicrobial immunity in the brain for the treatment of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2021, 17, 1311–1339. [Google Scholar] [CrossRef]
- French, P.W. Unfolded p53 in non-neuronal cells supports bacterial etiology of Alzheimer’s disease. Neural Regen. Res. 2022, 17, 2619–2622. [Google Scholar] [CrossRef] [PubMed]
- Sadrameli, M.; Bathini, P.; Alberi, L. Linking mechanisms of periodontitis to Alzheimer’s disease. Curr. Opin. Neurol. 2020, 33, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A. Bacteriological Etiology of Alzheimer’s Disease: Possibility of a Vaccine? Master’s Thesis, Faculdade de Farmácia da Universidade de Lisboa, Lisbon, Portugal, 2021. [Google Scholar]
- Jong, R.A.; van der Reijden, W.A. Feasibility and therapeutic strategies of vaccines against Porphyromonas gingivalis. Expert Rev. Vaccines 2014, 9, 193–208. [Google Scholar] [CrossRef]
- O’Brien-Simpson, N.M.; Holden, J.A.; Lenzo, J.C.; Tan, Y.; Brammar, G.C.; Walsh, K.A.; Singleton, W.; Orth, R.K.H.; Slakeski, N.; Cross, K.J.; et al. A therapeutic Porphyromonas gingivalis gingipain vaccine induces neutralising IgG1 antibodies that protect against experimental periodontitis. npj Vaccines 2016, 1, 16022. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Peres, C.; Florindo, H.F.; Almeida, A.J. Mucosal immunization using polyester-based particulate systems. In Handbook of Polyester Drug Delivery Systems; Ravi Kumar, M.N.V., Ed.; Pan Stanford Publishing Pte Ltd.: Singapore, 2016; pp. 521–561. ISBN 978-9814669658. [Google Scholar]
- Bhardwaj, P.; Bhatia, E.; Sharma, S.; Ahamad, N.; Banerjee, R. Advancements in prophylactic and therapeutic nanovaccines. Acta Biomater. 2020, 108, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Gao, S.; Cui, X.; Sun, D.; Zhao, K. Adjuvants and delivery systems based on polymeric nanoparticles for mucosal vaccines. Int. J. Pharm. 2019, 572, 118731. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, A.; Galhardas, J.; Cunha, A.; Cruz, P.; Gonçalves, L.M.D.; Almeida, A.J. Microencapsulation of Streptococcus equi antigens in biodegradable microspheres and preliminary immunisation studies. Eur. J. Pharm. Biopharm. 2006, 64, 131–137. [Google Scholar] [CrossRef]
- Florindo, H.F.; Pandit, S.; Gonçalves, L.M.D.; Videira, M.; Alpar, H.O.; Almeida, A.J. Antibody and cytokine-associated immune responses to S. equi antigens entrapped in PLA nanospheres. Biomaterials 2009, 30, 5161–5169. [Google Scholar] [CrossRef]
- Florindo, H.F.; Pandit, S.; Gonçalves, L.M.D.; Alpar, H.O.; Almeida, A.J. Streptococcus equi antigens adsorbed onto surface modified poly-ɛ-caprolactone microspheres induce humoral and cellular specific immune responses. Vaccine 2008, 26, 4168–4177. [Google Scholar] [CrossRef]
- Florindo, H.F.; Pandit, S.; Gonçalves, L.M.D.; Alpar, H.O.; Almeida, A.J. New approach on the development of a mucosal vaccine against strangles: Systemic and mucosal immune responses in a mouse model. Vaccine 2009, 27, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Caetano, L.A.; Figueiredo, L.; Almeida, A.J.; Gonçalves, L.M.D. BCG-loaded chitosan microparticles: Interaction with macrophages and preliminary in vivo studies. J. Microencapsul. 2017, 34, 203–217. [Google Scholar] [CrossRef]
- Wang, S.; Yan, T.; Zhang, B.; Chen, Y.; Li, Z. Porphyromonas gingivalis vaccine: Antigens and mucosal adjuvants. Vaccines 2024, 12, 619. [Google Scholar] [CrossRef] [PubMed]
- Ferreira da Silva, A.; Gonçalves, L.M.D.; Fernandes, A.; Almeida, A.J. Optimization and evaluation of a chitosan-coated PLGA nanocarrier for mucosal delivery of Porphyromonas gingivalis antigens. Eur. J. Pharm. Sci. 2024, 202, 106896. [Google Scholar] [CrossRef] [PubMed]
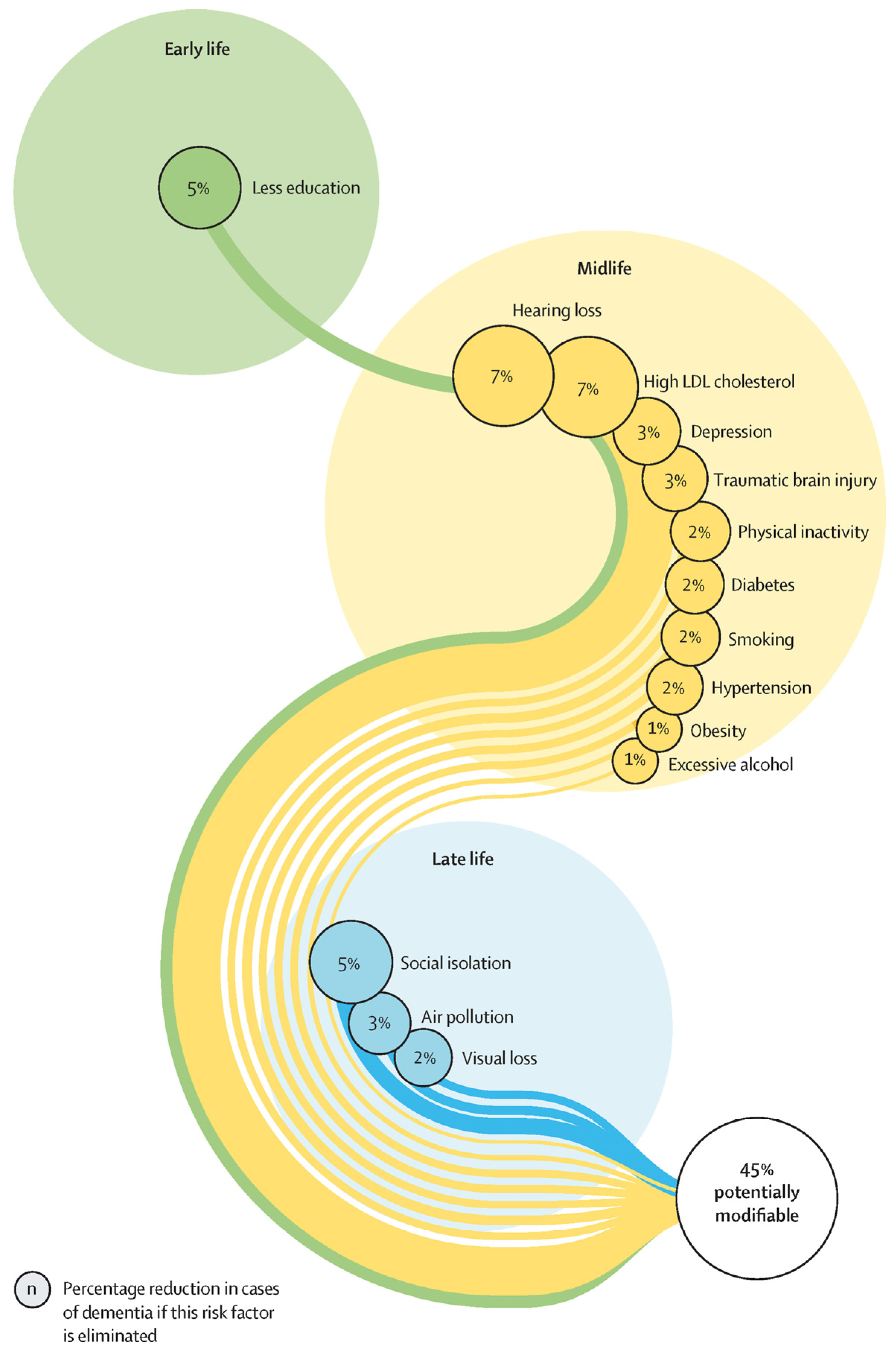
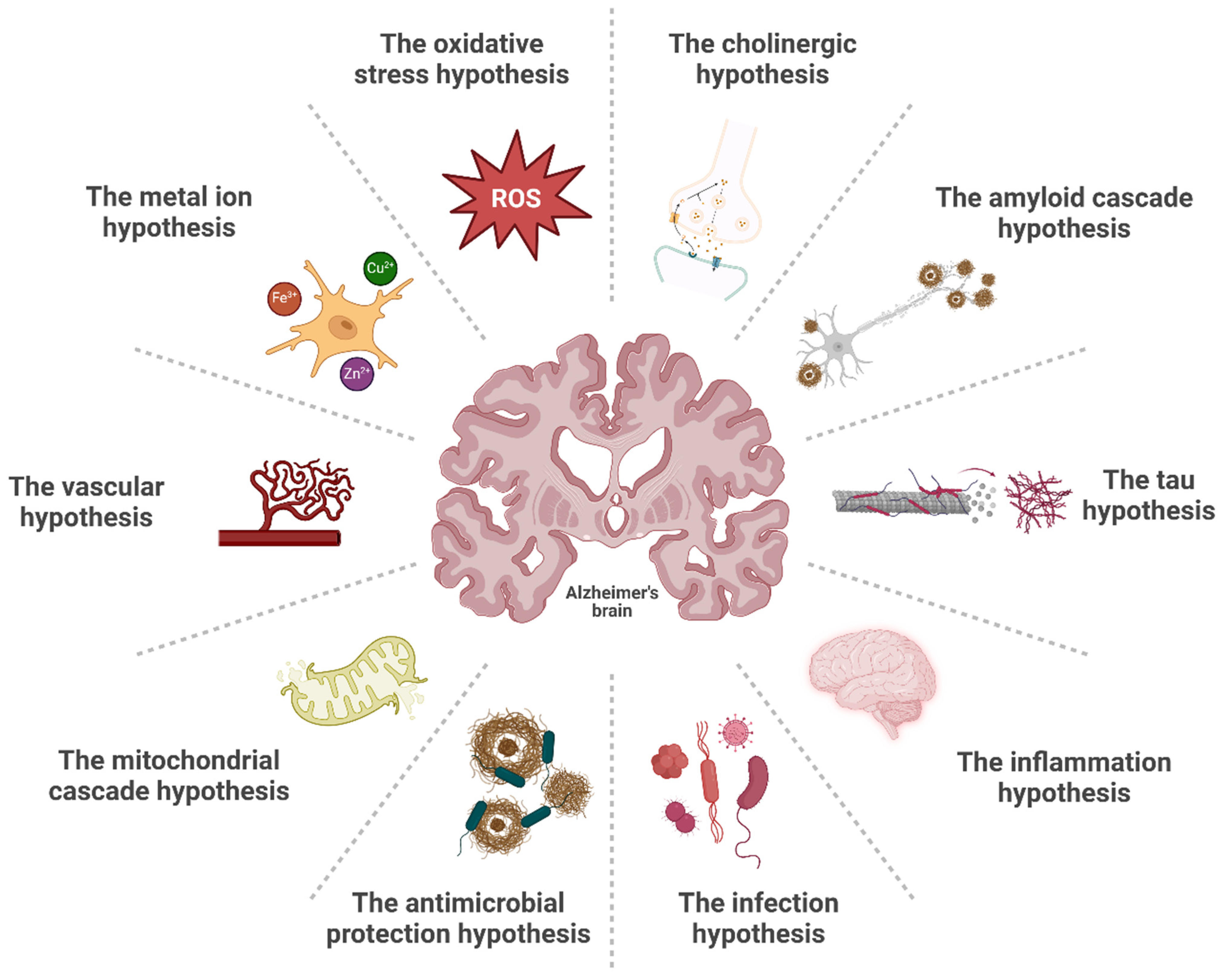
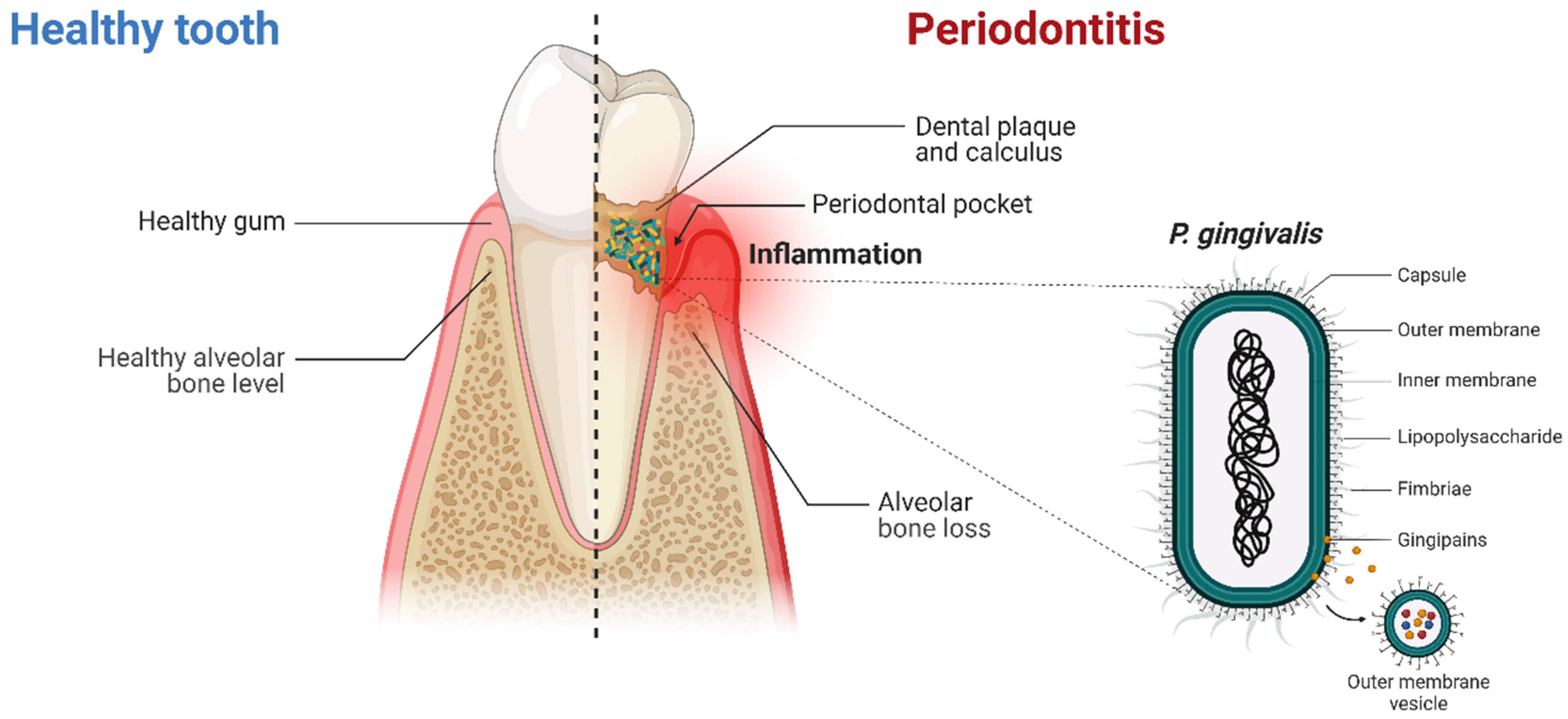
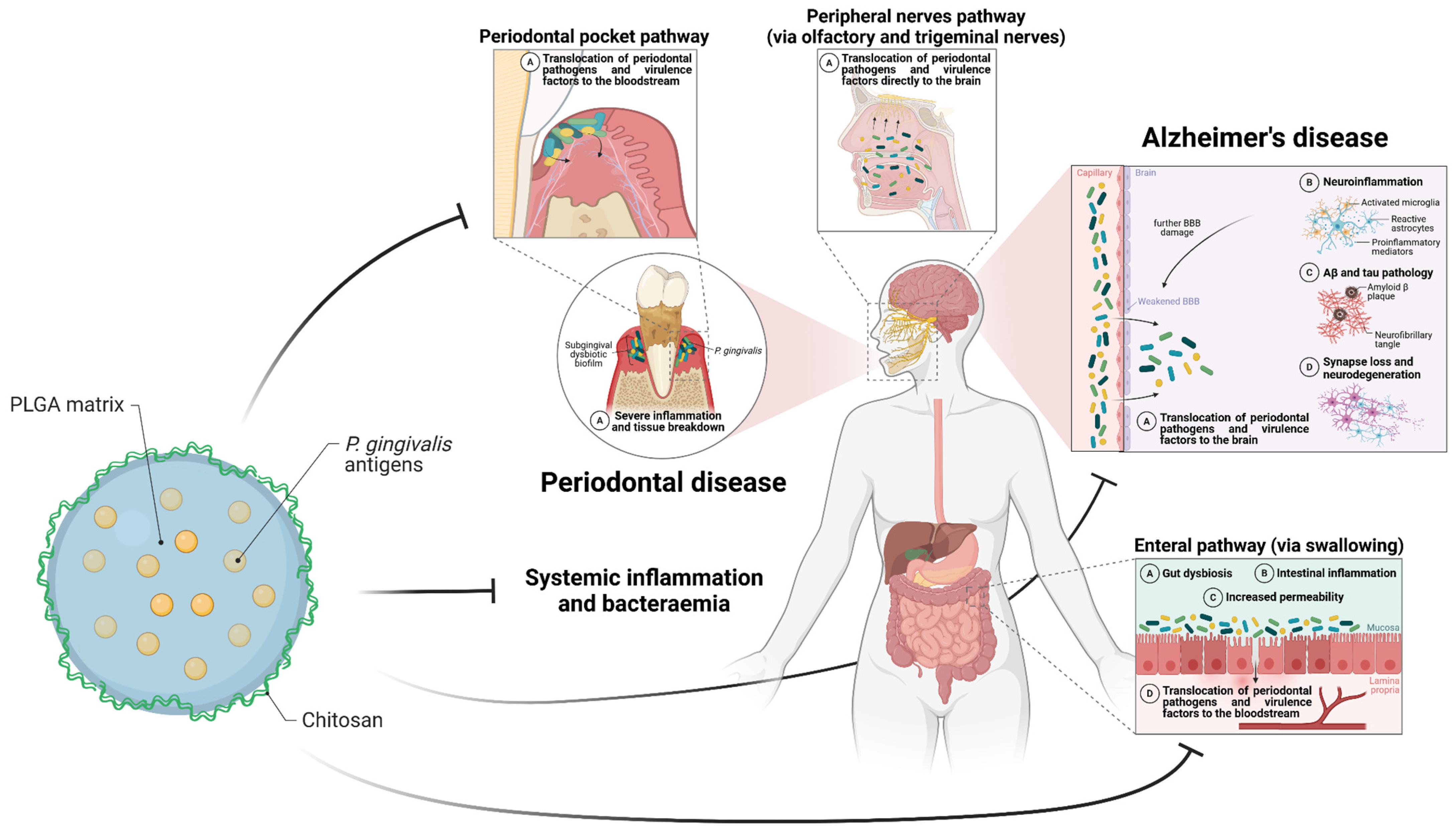
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira da Silva, A.; Gomes, A.; Gonçalves, L.M.D.; Fernandes, A.; Almeida, A.J. Exploring the Link Between Periodontitis and Alzheimer’s Disease—Could a Nanoparticulate Vaccine Break It? Pharmaceutics 2025, 17, 141. https://doi.org/10.3390/pharmaceutics17020141
Ferreira da Silva A, Gomes A, Gonçalves LMD, Fernandes A, Almeida AJ. Exploring the Link Between Periodontitis and Alzheimer’s Disease—Could a Nanoparticulate Vaccine Break It? Pharmaceutics. 2025; 17(2):141. https://doi.org/10.3390/pharmaceutics17020141
Chicago/Turabian StyleFerreira da Silva, André, Alexandra Gomes, Lídia M. D. Gonçalves, Adelaide Fernandes, and António J. Almeida. 2025. "Exploring the Link Between Periodontitis and Alzheimer’s Disease—Could a Nanoparticulate Vaccine Break It?" Pharmaceutics 17, no. 2: 141. https://doi.org/10.3390/pharmaceutics17020141
APA StyleFerreira da Silva, A., Gomes, A., Gonçalves, L. M. D., Fernandes, A., & Almeida, A. J. (2025). Exploring the Link Between Periodontitis and Alzheimer’s Disease—Could a Nanoparticulate Vaccine Break It? Pharmaceutics, 17(2), 141. https://doi.org/10.3390/pharmaceutics17020141