

Pharmacokinetics of Psilocybin: A Systematic Review
 , , , , , , , ,
, , , , , , , ,  and
and
Abstract
1. Introduction
2. Methods
2.1. Search Strategy
2.2. Eligibility Criteria
2.3. Data Screening and Extraction
2.4. Risk of Bias Assessment
2.5. Results Synthesis
3. Results
3.1. Risk of Bias Assessment Results
3.2. Study and Sample Characteristics
3.3. Pharmacokinetics
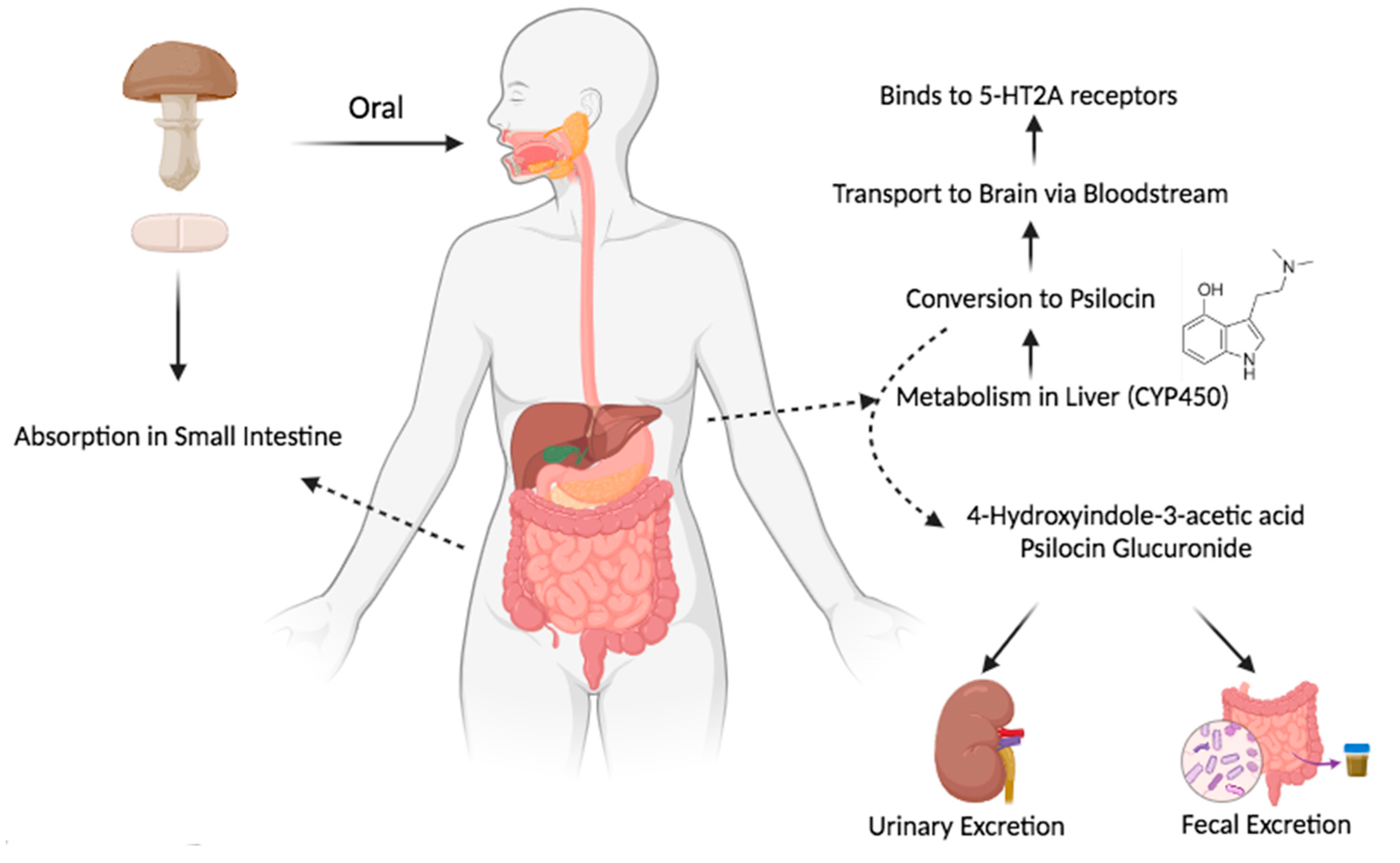
3.3.1. Absorption
3.3.2. Distribution
3.3.3. Metabolism
3.3.4. Half-Life and Excretion
4. Discussion
5. Conclusions
6. Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hofmann, A.; Heim, R.; Brack, A.; Kobel, H. Psilocybin, a psychotropic substance from the Mexican mushroom Psilicybe mexicana Heim. Experientia 1958, 14, 107–109. [Google Scholar] [PubMed]
- Passie, T.; Seifert, J.; Schneider, U.; Emrich, H.M. The pharmacology of psilocybin. Addict. Biol. 2002, 7, 357–364. [Google Scholar] [PubMed]
- Passie, T.; Metzner, R. A History of the Use of Psilocybin in Psychotherapy; Teonanacatl: Sacred Mushroom of Vision; Four Trees: El Verano, CA, USA, 2004; pp. 109–134. [Google Scholar]
- Carhart-Harris, R.L.; Goodwin, G.M. The therapeutic potential of psychedelic drugs: Past, present, and future. Neuropsychopharmacology 2017, 42, 2105–2113. [Google Scholar] [PubMed]
- Carter, O.L.; Burr, D.C.; Pettigrew, J.D.; Wallis, G.M.; Hasler, F.; Vollenweider, F.X. Using psilocybin to investigate the relationship between attention, working memory, and the serotonin 1A and 2A receptors. J. Cogn. Neurosci. 2005, 17, 1497–1508. [Google Scholar]
- Goldberg, S.B.; Pace, B.T.; Nicholas, C.R.; Raison, C.L.; Hutson, P.R. The experimental effects of psilocybin on symptoms of anxiety and depression: A meta-analysis. Psychiatry Res. 2020, 284, 112749. [Google Scholar] [CrossRef]
- Romeo, B.; Karila, L.; Martelli, C.; Benyamina, A. Efficacy of psychedelic treatments on depressive symptoms: A meta-analysis. J. Psychopharmacol. 2020, 34, 1079–1085. [Google Scholar]
- Carhart-Harris, R.L.; Roseman, L.; Bolstridge, M.; Demetriou, L.; Pannekoek, J.N.; Wall, M.B.; Tanner, M.; Kaelen, M.; McGonigle, J.; Murphy, K.; et al. Psilocybin for treatment-resistant depression: fMRI-measured brain mechanisms. Sci. Rep. 2017, 7, 13187. [Google Scholar]
- Mithoefer, M.C.; Grob, C.S.; Brewerton, T.D. Novel psychopharmacological therapies for psychiatric disorders: Psilocybin and MDMA. Lancet Psychiatry 2016, 3, 481–488. [Google Scholar]
- Benet, L.Z.; Kroetz, D.; Sheiner, L.; Hardman, J.; Limbird, L. Pharmacokinetics: The dynamics of drug absorption, distribution, metabolism, and elimination. Goodman Gilman’s Pharmacol. Basis Ther. 1996, 3, e27. [Google Scholar]
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in drug discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar]
- Dinis-Oliveira, R.J. Metabolism of psilocybin and psilocin: Clinical and forensic toxicological relevance. Drug Metab. Rev. 2017, 49, 84–91. [Google Scholar] [PubMed]
- Brown, R.T.; Nicholas, C.R.; Cozzi, N.V.; Gassman, M.C.; Cooper, K.M.; Muller, D.; Thomas, C.D.; Hetzel, S.J.; Henriquez, K.M.; Ribaudo, A.S.; et al. Pharmacokinetics of escalating doses of oral psilocybin in healthy adults. Clin. Pharmacokinet. 2017, 56, 1543–1554. [Google Scholar] [PubMed]
- Papaseit, E.; Torrens, M.; Pérez-Mañá, C.; Muga, R.; Farré, M. Key interindividual determinants in MDMA pharmacodynamics. Expert Opin. Drug Metab. Toxicol. 2018, 14, 183–195. [Google Scholar] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Int. J. Surg. 2021, 88, 105906. [Google Scholar]
- Joanna Briggs Institute. Checklist for Systematic Reviews and Research Syntheses. 2017. Available online: https://jbi.global/critical-appraisal-tools (accessed on 30 December 2024).
- Schneider, K.; Schwarz, M.; Burkholder, I.; Kopp-Schneider, A.; Edler, L.; Kinsner-Ovaskainen, A.; Hartung, T.; Hoffmann, S. “ToxRTool”, a new tool to assess the reliability of toxicological data. Toxicol. Lett. 2009, 189, 138–144. [Google Scholar]
- Campbell, M.; McKenzie, J.E.; Sowden, A.; Katikireddi, S.V.; Brennan, S.E.; Ellis, S.; Hartmann-Boyce, J.; Ryan, R.; Shepperd, S.; Thomas, J.; et al. Synthesis without meta-analysis (SWiM) in systematic reviews: Reporting guideline. BMJ 2020, 368, l6890. [Google Scholar]
- Becker, A.M.; Holze, F.; Grandinetti, T.; Klaiber, A.; Toedtli, V.E.; Kolaczynska, K.E.; Duthaler, U.; Varghese, N.; Eckert, A.; Grünblatt, E.; et al. Acute Effects of Psilocybin After Escitalopram or Placebo Pretreatment in a Randomized, Double-Blind, Placebo-Controlled, Crossover Study in Healthy Subjects. Clin. Pharmacol. Ther. 2022, 111, 886–895. [Google Scholar] [CrossRef]
- Chen, J.; Li, M.; Yan, X.; Wu, E.; Zhu, H.; Lee, K.J.; Chu, V.M.; Zhan, L.; Lee, W.; Kang, J.S. Determining the pharmacokinetics of psilocin in rat plasma using ultra-performance liquid chromatography coupled with a photodiode array detector after orally administering an extract of Gymnopilus spectabilis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2669–2672. [Google Scholar]
- Donovan, L.L.; Johansen, J.V.; Ros, N.F.; Jaberi, E.; Linnet, K.; Johansen, S.S.; Ozenne, B.; Issazadeh-Navikas, S.; Hansen, H.D.; Knudsen, G.M. Effects of a single dose of psilocybin on behaviour, brain 5-HT2A receptor occupancy and gene expression in the pig. Eur. Neuropsychopharmacol. 2021, 42, 1–11. [Google Scholar]
- Hasler, F.; Bourquin, D.; Brenneisen, R.; Bär, T.; Vollenweider, F.X. Determination of psilocin and 4-hydroxyindole-3-acetic acid in plasma by HPLC-ECD and pharmacokinetic profiles of oral and intravenous psilocybin in man. Pharm. Acta Helv. 1997, 72, 175–184. [Google Scholar]
- Hasler, F.; Bourquin, D.; Brenneisen, R.; Vollenweider, F.X. Renal excretion profiles of psilocin following oral administration of psilocybin: A controlled study in man. J. Pharm. Biomed. Anal. 2002, 30, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Holze, F.; Ley, L.; Müller, F.; Becker, A.M.; Straumann, I.; Vizeli, P.; Kuehne, S.S.; Roder, M.A.; Duthaler, U.; Kolaczynska, K.E.; et al. Direct comparison of the acute effects of lysergic acid diethylamide and psilocybin in a double-blind placebo-controlled study in healthy subjects. Neuropsychopharmacology 2022, 47, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Horita, A.; Weber, L.J. The enzymic dephosphorylation and oxidation of psilocybin and psilocin by mammalian tissue homogenates. Biochem. Pharmacol. 1961, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kolaczynska, K.E.; Liechti, M.E.; Duthaler, U. Development and validation of an LC-MS/MS method for the bioanalysis of psilocybin’s main metabolites, psilocin and 4-hydroxyindole-3-acetic acid, in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2021, 1164, 122486. [Google Scholar] [CrossRef]
- Ley, L.; Holze, F.; Arikci, D.; Becker, A.M.; Straumann, I.; Klaiber, A.; Coviello, F.; Dierbach, S.; Thomann, J.; Duthaler, U.; et al. Comparative acute effects of mescaline, lysergic acid diethylamide, and psilocybin in a randomized, double-blind, placebo-controlled cross-over study in healthy participants. Neuropsychopharmacology 2023, 48, 1659–1667. [Google Scholar] [CrossRef]
- Manevski, N.; Kurkela, M.; Höglund, C.; Mauriala, T.; Court, M.H.; Yli-Kauhaluoma, J.; Finel, M. Glucuronidation of psilocin and 4-hydroxyindole by the human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2010, 38, 386–395. [Google Scholar] [CrossRef]
- Raithatha, S.A.; Hagel, J.M.; Matinkhoo, K.; Yu, L.; Press, D.; Cook, S.G.; Sharma, G.; Dhananjaya, D.; Jensen, G.; Lee, J.B.; et al. Novel Psilocin Prodrugs with Altered Pharmacological Properties as Candidate Therapies for Treatment-Resistant Anxiety Disorders. J. Med. Chem. 2024, 67, 1024–1043. [Google Scholar] [CrossRef]
- Rakoczy, R.J.; Runge, G.N.; Sen, A.K.; Sandoval, O.; Wells, H.G.; Nguyen, Q.; Roberts, B.R.; Sciortino, J.H.; Gibbons WJJr Friedberg, L.M.; Jones, J.A.; et al. Pharmacological and behavioural effects of tryptamines present in psilocybin-containing mushrooms. Br. J. Pharmacol. 2024, 181, 3627–3641. [Google Scholar] [CrossRef]
- Thomann, J.; Kolaczynska, K.E.; Stoeckmann, O.V.; Rudin, D.; Vizeli, P.; Hoener, M.C.; Pryce, C.R.; Vollenweider, F.X.; Liechti, M.E.; Duthaler, U. In vitro and in vivo metabolism of psilocybin’s active metabolite psilocin. Front. Pharmacol. 2024, 15, 1391689. [Google Scholar] [CrossRef]
- Richter, L.M.; Al-Gousous, J.; de Araujo, G.L.; Davies, N.M.; Löbenberg, R. Assessing the utility of in silico tools in early drug development: The case of a pharmaceutically relevant formulation of the prodrug psilocybin. J. Drug Deliv. Sci. Technol. 2024, 92, 105305. [Google Scholar] [CrossRef]
- Sim, D.S.M. Drug absorption and bioavailability. In Pharmacological Basis of Acute Care; Springer: Cham, Switzerland, 2015; pp. 17–26. [Google Scholar]
- Stoner, K.L.; Harder, H.; Fallowfield, L.J.; Jenkins, V.A. Intravenous versus subcutaneous drug administration. Which do patients prefer? A systematic review. Patient-Patient-Centered Outcomes Res. 2015, 8, 145–153. [Google Scholar]
- Dodd, S.; Norman, T.R.; Eyre, H.A.; Stahl, S.M.; Phillips, A.; Carvalho, A.F.; Berk, M. Psilocybin in neuropsychiatry: A review of its pharmacology, safety, and efficacy. CNS Spectr. 2023, 28, 416–426. [Google Scholar] [PubMed]
- Roscoe, J.; Lozy, O. Can psilocybin be safely administered under medical supervision? A systematic review of adverse event reporting in clinical trials. Drug Sci. Policy Law 2022, 8, 20503245221085222. [Google Scholar]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Volume of distribution in drug design: Miniperspective. J. Med. Chem. 2015, 58, 5691–5698. [Google Scholar]
- Kroll, R.A.; Pagel, M.A.; Muldoon, L.L.; Roman-Goldstein, S.; Neuwelt, E.A. Increasing volume of distribution to the brain with interstitial infusion: Dose, rather than convection, might be the most important factor. Neurosurgery 1996, 38, 746–754. [Google Scholar]
- Otto, M.E.; van der Heijden, K.V.; Schoones, J.W.; van Esdonk, M.J.; Borghans, L.G.; Jacobs, G.E.; van Hasselt, C.J. Clinical Pharmacokinetics of Psilocin After Psilocybin Administration: A Systematic Review and Post-Hoc Analysis. Clin. Pharmacokinet. 2025, 64, 53–66. [Google Scholar]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Relevance of half-life in drug design: Miniperspective. J. Med. Chem. 2017, 61, 4273–4282. [Google Scholar]
- Meshkat, S.J.; Zeifman, R.; Stewart, K.; Janssen-Aguilar, R.; Lou, W.; Jetly, R.; Monson, C.M.; Bhat, V. Psilocybin-assisted massed cognitive processing therapy for chronic posttraumatic stress disorder: Protocol for an open-label pilot feasibility trial. PLoS ONE 2025, 20, e0313741. [Google Scholar]
- Husain, M.I.; Blumberger, D.M.; Castle, D.J.; Ledwos, N.; Fellows, E.; Jones, B.D.; Ortiz, A.; Kloiber, S.; Wang, W.; Rosenblat, J.D.; et al. Psilocybin for treatment-resistant depression without psychedelic effects: Study protocol for a 4-week, double-blind, proof-of-concept randomised controlled trial. BJPsych Open 2023, 9, e134. [Google Scholar]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 inhibition: Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A.M. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]
- Halman, A.; Kong, G.; Sarris, J.; Perkins, D. Drug-drug interactions between classic psychedelics and psychoactive drugs: A systematic review. medRxiv 2023. medRxiv:2023-06. [Google Scholar] [CrossRef]
- Boyd-Kimball, D.; Gonczy, K.; Lewis, B.; Mason, T.; Siliko, N.; Wolfe, J. Classics in chemical neuroscience: Chlorpromazine. ACS Chem. Neurosci. 2018, 10, 79–88. [Google Scholar] [PubMed]
- Vollenweider, F.X.; Vollenweider-Scherpenhuyzen, M.F.; Bäbler, A.; Vogel, H.; Hell, D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport 1998, 9, 3897–3902. [Google Scholar]
- LESSES Psychodynamic relationships between the degree of anxiety and other clinical symptoms. J. Nerv. Ment. Dis. 1958, 127, 124–130.
- Erkizia-Santamaría, I.; Alles-Pascual, R.; Horrillo, I.; Meana, J.J.; Ortega, J.E. Serotonin 5-HT2A, 5-HT2c and 5-HT1A receptor involvement in the acute effects of psilocybin in mice. In vitro pharmacological profile and modulation of thermoregulation and head-twich response. Biomed. Pharmacother. 2022, 154, 113612. [Google Scholar]
- Goodwin, G.M.; Croal, M.; Feifel, D.; Kelly, J.R.; Marwood, L.; Mistry, S.; O’Keane, V.; Peck, S.K.; Simmons, H.; Sisa, C.; et al. Psilocybin for treatment resistant depression in patients taking a concomitant SSRI medication. Neuropsychopharmacology 2023, 48, 1492–1499. [Google Scholar]
- Simonsson, O.; Hendricks, P.S.; Chambers, R.; Osika, W.; Goldberg, S.B. Prevalence and associations of challenging, difficult or distressing experiences using classic psychedelics. J. Affect. Disord. 2023, 326, 105–110. [Google Scholar]
- Nayak, S.M.; Gukasyan, N.; Barrett, F.S.; Erowid, E.; Erowid, F.; Griffiths, R.R. Classic psychedelic coadministration with lithium, but not lamotrigine, is associated with seizures: An analysis of online psychedelic experience reports. Pharmacopsychiatry 2021, 54, 240–245. [Google Scholar]
- Ali, A.; Gifford, M.E.; Lowe, H.; Gordon, L.; Grant, J. Natural vs. Synthetic Psilocybin: The Same or Completely Different? In Mushrooms with Therapeutic Potentials: Recent Advances in Research and Development; Springer Nature: Singapore, 2023; pp. 479–492. [Google Scholar]
- Lowe, H.; Toyang, N.; Steele, B.; Valentine, H.; Grant, J.; Ali, A.; Ngwa, W.; Gordon, L. The therapeutic potential of psilocybin. Molecules 2021, 26, 2948. [Google Scholar] [CrossRef]
- Johnson, M.W.; Griffiths, R.R. Potential therapeutic effects of psilocybin. Neurotherapeutics 2017, 14, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Smigielski, L.; Scheidegger, M.; Kometer, M.; Vollenweider, F.X. Psilocybin-assisted mindfulness training modulates self-consciousness and brain default mode network connectivity with lasting effects. NeuroImage 2019, 196, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, J.J.; Perkins, D.; Ruffell, S.; Lawrence, A.J.; Hoyer, D.; Jacobson, L.H.; Timmermann, C.; Castle, D.; Rossell, S.L.; Downey, L.A.; et al. Default mode network modulation by psychedelics: A systematic review. Int. J. Neuropsychopharmacol. 2023, 26, 155–188. [Google Scholar] [PubMed]
- Zhao, X.; Du, Y.; Yao, Y.; Dai, W.; Yin, Y.; Wang, G.; Li, Y.; Zhang, L. Psilocybin promotes neuroplasticity and induces rapid and sustained antidepressant-like effects in mice. J. Psychopharmacol. 2024, 38, 489–499. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; Meshkat, S.; Doyle, Z.; Kaczmarek, E.; Brudner, R.M.; Kratiuk, K.; Mansur, R.B.; Schulz-Quach, C.; Sethi, R.; Abate, A.; et al. Psilocybin-assisted psychotherapy for treatment resistant depression: A randomized clinical trial evaluating repeated doses of psilocybin. Med 2024, 5, 190–200. [Google Scholar] [CrossRef]
- Haikazian, S.; Chen-Li, D.C.; Johnson, D.E.; Fancy, F.; Levinta, A.; Husain, M.I.; Mansur, R.B.; McIntyre, R.S.; Rosenblat, J.D. Psilocybin-assisted therapy for depression: A systematic review and meta-analysis. Psychiatry Res. 2023, 329, 115531. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Participant Information | Pharmacokinetics | Main Results | |||||
|---|---|---|---|---|---|---|---|---|
| Author | Study Design | Participants | Psilocybin | Absorption | Distribution | Metabolism | Excretion | |
| Becker et al., 2022 [19] | Clinical: RCT Crossover | n = 23 Human (healthy) 48% female Mean age = 34 ± 10 yrs | Synthetic 25 mg, single dose administered twice (14 days apart), oral dose | Tmax (h): Escitalopram: Psilocin Unconjugated = 2 Psilocin Glucuronide = 4 Psilocin Total = 3 4-HIAA = 2 Placebo: Psilocin Unconjugated = 2 Psilocin Glucuronide = 4 Psilocin Total = 3 4-HIAA = 2 Cmax (ng/mL) in Plasma: Escitalopram: Psilocin Unconjugated = 22 ± 8.5 Psilocin Glucuronide = 82 ± 30 Psilocin Total = 97 ± 33 4-HIAA = 106 ± 37 Placebo: Psilocin Unconjugated = 20 ± 5.4 Psilocin Glucuronide = 82 ± 28 Psilocin Total = 96 ± 28 4-HIAA = 105 ± 30 | N/R | Enzyme = N/R Metabolites = psilocin, psilocin glucuronide, or 4-HIAA | t1/2 (h): Escitalopram: Psilocin Unconjugated = 2.0 ± 0.5 Psilocin Glucuronide = 5.7 ± 2.4 Psilocin total = 4.8 ± 1.8 4-HIAA = 1.7 ± 0.5 Placebo: Psilocin Unconjugated = 1.8 ± 0.3 Psilocin Glucuronide = 4.7 ± 1.6 Psilocin total = 4.3 ± 1.3 4-HIAA = 1.6 ± 0.3 | Escitalopram pretreatment did not significantly affect psilocybin’s positive mood effects but reduced its adverse effects (e.g., anxiety and cardiovascular reactions), without altering psilocin pharmacokinetics. |
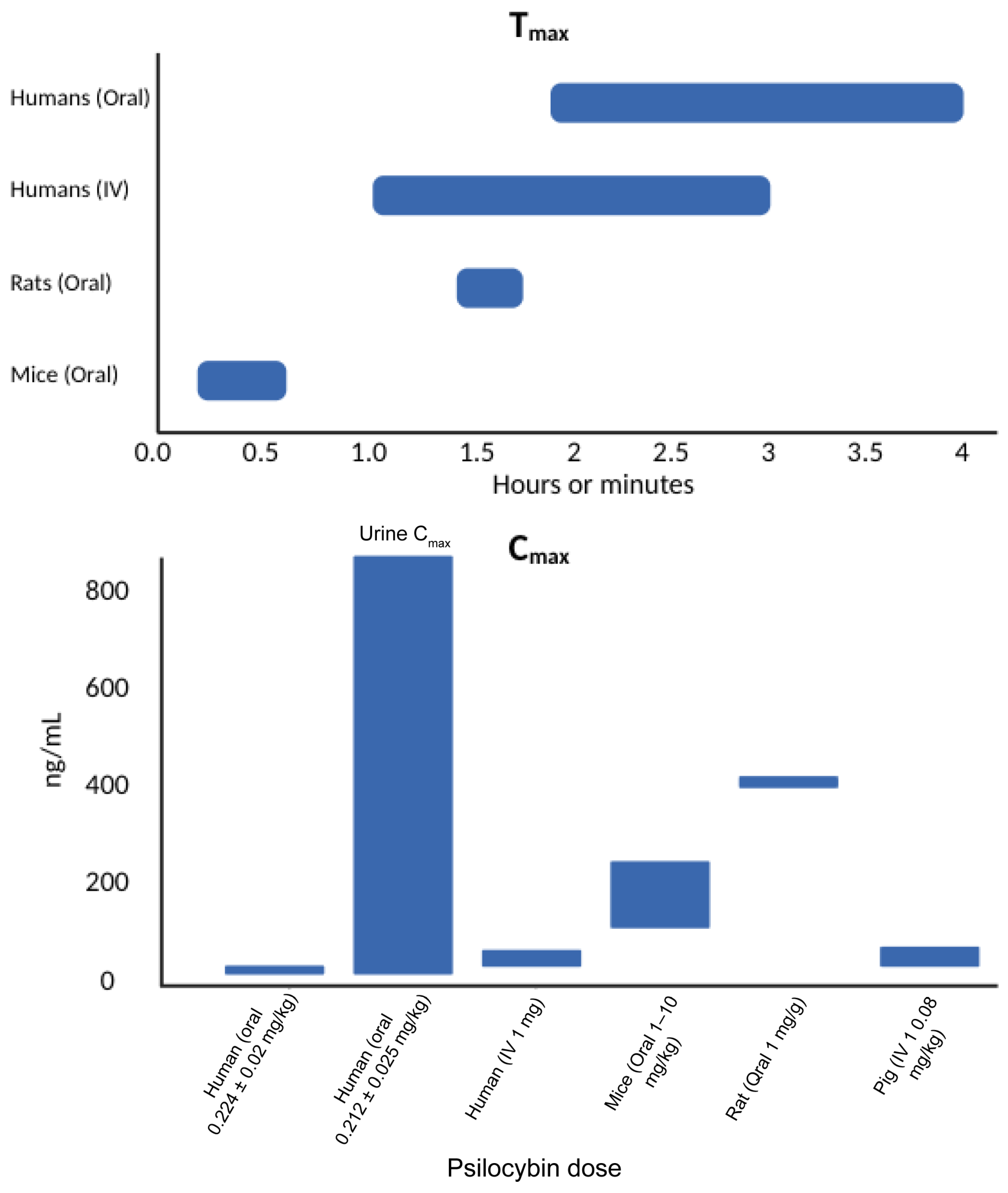
| Brown et al., 2017 [13] | Clinical: pre–post study design | n = 12 Human (healthy) 17% female Mean age = 43 yrs | Synthetic Escalating oral dose of 0.3, 0.45, and 0.6 mg/kg | Tmax (h): 0.3 mg/kg = 2.03 0.45 mg/kg = 2.03 0.6 mg/kg = 2.05 Cmax (ng/mL) in Plasma: 0.3 mg/kg = 16 0.45 mg/kg = 26 0.6 mg/kg = 37.6 | Vd (L) = 298 | Enzyme = N/R Metabolites = psilocin and psilocin glucuronide | t1/2 (h) = 3 ± 1.1 Clearance (L/h) = 164 ± 23.2 Route of Elimination = Renal | Oral psilocybin demonstrates linear pharmacokinetics over the dose range of 0.3–0.6 mg/kg, with psilocin having a rapid onset, a half-life of approximately 3 h, and minimal renal excretion, suggesting no need for dose adjustment in mild to moderate renal impairment. A fixed 25 mg oral dose approximates the exposure of 0.3 mg/kg. |
| Chen et al., 2011 [20] | Laboratory: experimental study | n = 10 Sprague Dawley rats | Natural (from G. spectabilis) 1 mL/g, single dose, oral gavage | Tmax (h): Psilocin = 1.5 ± 0.03 Cmax (ng/mL) in Plasma: Psilocin = 430 ± 120 | Vd (L) = 0.0032 ± 0.0017 | Enzyme = N/R Metabolite = psilocin | Distribution t1/2 (h) = 1.95 ± 0.67 Elimination t1/2 (h) = 2.5 ± 1 Clearance (L/h) = 0.132 ± 0.054 | The study developed a UPLC-PDA detector to assess psilocin pharmacokinetics in rat plasma, showing rapid absorption post oral administration of Gymnopilus spectabilis extract. |
| Donovan et al., 2021 [21] | Laboratory: dose–response experimental study | n = 25 Danish slaughter pigs 100% female Mean age = 9 weeks | Unclear 0.08 mg/kg, single dose, IV | Cmax (ng/mL) in Plasma = 12–19 | Non-displaceable Vd (mL/cm3) = 4.2 | Enzyme = N/R Metabolite = psilocin | t1/2 (h) = 0.3 | Psilocybin in pigs can induce transient behavioral changes (i.e., headshaking and scratching), achieves 67% occupancy of cerebral 5-HT2A receptors, results in small changes in PFC gene expression, and modulates immune-related gene expression pathways in pigs. |
| Hasler et al., 1997 [22] | Clinical: controlled clinical trial | n = 9 Human (healthy) 13% female Mean age = 31 ± 6 | Synthetic Six participants, single dose (0.224 ± 0.02 mg/kg), orally; six participants, single IV dose (1 mg) | Oral: Tmax (h): Psilocin = 1.75 ± 0.62 4-HIAA = 1.88 ± 0.68 Cmax (ng/mL) in Plasma: Psilocin = 8.2 ± 2.8 4-HIAA = 150 ± 61 Bioavailability (%): Psilocin = 52.7 ± 20 IV: Tmax (h): Psilocin = 0.0317 (1.9 min) Cmax (ng/mL): Psilocin = 12.9 ± 5.6 | IV: Vd (L) = 277 ± 92 | Enzyme = N/R Metabolites = psilocin and 4-HIAA | Oral: t1/2 (h) = Psilocin = 2.7 ± 1.06 4-HIAA = 2.4 ± 1.61 IV: t1/2 (h) = Psilocin = 1.2 ± 0.33 Clearance (L/h) = 187.56 ± 43.14 | The study revealed differences between oral and IV psilocybin administration. IV resulted in a rapid peak of psilocin levels, while oral dosing delayed the peak with a longer half-life. 4HIAA was detected only after oral administration, highlighting first-pass metabolism. |
| Hasler et al., 2002 [23] | Clinical: controlled clinical trial | n = 8 Human (healthy) 50% female Mean age = 33 ± 6 | Synthetic 0.212 ± 0.025 (mg/kg), single oral dose | Tmax (h): Psilocin Unconjugated = 2–4 Cmax (ng/mL) in urine: Psilocin Unconjugated = 871 | N/R | Enzyme = enzymatic glucuronide Metabolite = psilocin | t1/2 (h) = 3.29 ± 0.57 Route of Elimination = Renal | Psilocybin is rapidly metabolized to psilocin. Psilocin undergoes partial glucuronidation, extending its detectability, and 3.4% of the administered psilocybin dose is excreted as unconjugated psilocin within 24 h. |
| Holze et al., 2022 [24] | Clinical: RCT crossover | n = 28 Human (healthy) 50% female Mean age = 35 ± 9.4 yrs | Synthetic 15 mg or 30 mg, single, oral dose | Tmax (h): Psilocin Unconjugated 15 mg = 2.3 30 mg = 2.5 Cmax (ng/mL) in Plasma: Psilocin Unconjugated 15 mg = 13 30 mg = 25 | Vd (L): 15 mg = 925 30 mg = 1016 | Enzyme = N/R Metabolite = psilocin | t1/2 (h): 15 mg = 2.4 30 mg = 2.7 Clearance (L/h): 15 mg = 262 30 mg = 263 | Psilocybin produced dose-dependent effects on mood and consciousness comparable to LSD but with a shorter duration of action. While both substances exhibit cardiostimulatory effects, psilocybin increases blood pressure more significantly, whereas LSD has a greater impact on heart rate. |
| Horita et al., 1961 [25] | Laboratory: experimental study | n = N/R Sprague Dawley rats 100% male | Synthetic 2 micromoles/mL, single addition | N/R | N/R | Enzymes = oxidase enzyme and phosphatase enzyme Metabolite = psilocin | N/R | Psilocybin dephosphorylation was most active in the kidneys of rats and mice and the small intestine mucosa of guinea pigs and rabbits. Oxidase activity peaked in the heart across species and in the kidneys of rats and mice. These findings suggest that psilocybin is rapidly converted to its active form, psilocin, with its effect duration potentially regulated by psilocin oxidation to an o-quinone structure. |
| Kolaczynska et al., 2021 [26] | Laboratory: experimental study | Plasma from n = 3 | Synthetic 25 mg single dose, oral | Tmax (h): Psilocin = 2.3 ± 0.77 Psilocin Glucuronide = 3.67 ± 1.53 4-HIAA = 2 ± 1 Cmax (ng/mL) in Plasma: Psilocin = 19.2 ± 4.0 Psilocin Glucuronide = 78.3 ± 7.9 4-HIAA = 137 ± 22 | N/R | N/R | t1/2 (h): Psilocin = 2.1 ± 0.3 Psilocin Glucuronide = 3.58 ± 1.2 4-HIAA = 2.3 ± 1.05 Route of Elimination = Renal | There was rapid metabolism of psilocybin into its active form, psilocin, with efficient glucuronidation and renal elimination. A reliable LC-MS/MS method for quantifying psilocin and its metabolites was developed, providing valuable insights into psilocybin’s pharmacokinetics and supporting its potential for therapeutic use and future research on efficacy and safety. |
| Ley et al., 2023 [27] | Clinical: RCT | n = 32 Human (healthy) 50% female Mean age = 29 ± 4 yrs | Synthetic 20 mg (four oral capsules of 5 mg each), single dose | Tmax (h): Psilocin = 2.1 Psilocin Glucuronide = 4.4 4-HIAA = 1.8 h Cmax (ng/mL) in Plasma: Psilocin = 17 Psilocin Glucuronide = 70 4-HIAA = 86 | Vd (L): Psilocin = 505 Psilocin glucuronide = 190 4-HIAA = 116 | Enzyme = N/R Metabolites = psilocin, psilocin glucuronide, and 4-HIAA | t1/2 (h): Psilocin = 2.3 Psilocin Glucuronide = 3.2 4-HIAA = 2.1 Clearance Rate (L/h): Psilocin = 155 Psilocin Glucuronide = 41 4-HIAA = 37 Route of Elimination = Renal | No qualitative differences in altered states of consciousness were observed between 500 mg mescaline, 100 µg LSD, and 20 mg psilocybin, though their durations of action differed. The findings support dose optimization for research and psychedelic-assisted therapy. |
| Manevski et al., 2010 [28] | Laboratory: experimental study | 19 UGTs | Synthetic 50–5000 uM, single dose | N/R | N/R | Enzymes = UDP-Glucuronosyltransferases (UGTs; UGT1A10, UGT1A9, UGT1A6, and UGT1A8) Metabolites = psilocin and 4-hydroxyindole | N/R | The study revealed that psilocin undergoes extensive glucuronidation, with UGT1A10 playing a key role in first-pass metabolism in the small intestine and UGT1A9, contributing to liver clearance. Substrate specificity was observed, with psilocin primarily metabolized by UGT1A10 and 4-hydroxyindole by UGT1A6, highlighting the tissue-specific roles of UGTs. |
| Raithatha et al., 2023 [29] | Laboratory: experimental study | Pharmacokinetic studies: n = 12 mice, (n = 3 per dose level group) Head twitch: N = 6 mice Marble burying: n = 36 | Synthetic Pharmacokinetics: 1 mg/kg IV, 1,3, or 10 mg/kg oral gavage Behavioral test: 1 mg/kg oral gavage, single dose | Tmax = 0.25 h Cmax (oral; ng/mL) in Plasma: 1 mg/kg = 52.9 10 mg/kg = 243 High Bioavailability | N/R | Enzymes = alkalinephosphatase and nonspecific esterases Metabolite: psilocin | Plasma Psilocin Levels Detectable for up to 24 h | Tailored prodrugs (novel psilocin drugs) may be more effective than psilocybin for treating depression and anxiety without unwanted psychedelic effects. |
| Rakoczy et al., 2023 [30] | Laboratory: experimental study | Head-twitch response: n = 67, Long Evans rats Forced swim: n = 60 Toxicology: n = 15 | Synthetic Head-twitch response: psilocybin dosages tested: 0.1, 0.2, 1.0, and 2.0 mg/kg; intraoral gavage; single dose Forced swim test: psilocybin dosage: 1.0 mg/kg; intragastric infusion; 3 times over 24 h Toxicology: psilocybin dosage: 1.0 mg/kg; oral gavage; single dose | N/R | N/R | Enzymes = alkaline phosphatase, and MAO-A Metabolites: psilocin, 4-hydroxyindole-3-acetaldehyde, and 4-HIAA | N/R | In vitro assays revealed similar dephosphorylation and metabolism rates across compounds. Dephosphorylated baeocystin and norbaeocystin crossed a blood–brain barrier mimetic and activated the 5-HT2A receptor with efficacy comparable to psilocin. Only psilocybin induced head-twitch responses in rats, indicating psychedelic effects, while norbaeocystin improved forced swim test outcomes. |
| Thomann et al., 2024 [31] | Laboratory: controlled experimental study Clinical: secondary analysis (RCT) | Mouse: n = 10 (5 experimental, 5 control) Adult C57BL/6J mice 100% male Human: N = 5 See Holze et al., 2022 [24] | Synthetic Mouse: 3 mg/kg, single, oral gavage needle dose | Mouse: Tmax(h): Psilocin = 0.30 ± 0.11 Psilocin-O-Glucuronide = 0.35 ± 0.14 4-HIAA = 0.30 ± 0.11 4-HIAA-Glucuronide = 0.45 ± 0.11 Cmax (ng/mL) in Plasma: Psilocin = 198 ± 28 Psilocin-O-Glucuronide = 521 ± 57 4-HIAA = 84.9 ± 17.7 4-HIAA-Glucuronide = 30.0 ± 6.7 | N/R | Enzymes = CYP, MAO-A, and UGT Metabolites = psilocin, psilocin-O-glucuronide, 4-HIAA, 4-HTP, oxidized psilocin metabolite, and norpsilocin | Mouse: t1/2 (h): Psilocin = 0.91 ± 0.11 Psilocin-O-Glucuronide = 0.97 ± 0.06 4-HIAA = 0.75 ± 0.11 4-HIAA-Glucuronide = 1.38 ± 0.27 | Six psilocin metabolites were identified, confirming in vivo glucuronidation and highlighting interspecies differences, such as 4-HIAA glucuronidation and norpsilocin detection in mice but not humans. MAO-A plays a key role in converting psilocin to 4-HIAA and 4-HTP, while the roles of ALDH and ADH remain unclear. CYP2D6 minimally contributes to psilocin metabolism, producing norpsilocin and an oxidized metabolite, while CYP3A4’s role is uncertain. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meshkat, S.; Al-Shamali, H.; Perivolaris, A.; Tullu, T.; Zeifman, R.J.; Zhang, Y.; Burback, L.; Winkler, O.; Greenshaw, A.; Husain, M.I.; et al. Pharmacokinetics of Psilocybin: A Systematic Review. Pharmaceutics 2025, 17, 411. https://doi.org/10.3390/pharmaceutics17040411
Meshkat S, Al-Shamali H, Perivolaris A, Tullu T, Zeifman RJ, Zhang Y, Burback L, Winkler O, Greenshaw A, Husain MI, et al. Pharmacokinetics of Psilocybin: A Systematic Review. Pharmaceutics. 2025; 17(4):411. https://doi.org/10.3390/pharmaceutics17040411
Chicago/Turabian StyleMeshkat, Shakila, Huda Al-Shamali, Argyrios Perivolaris, Trusha Tullu, Richard J. Zeifman, Yanbo Zhang, Lisa Burback, Olga Winkler, Andrew Greenshaw, Muhammad Ishrat Husain, and et al. 2025. "Pharmacokinetics of Psilocybin: A Systematic Review" Pharmaceutics 17, no. 4: 411. https://doi.org/10.3390/pharmaceutics17040411
APA StyleMeshkat, S., Al-Shamali, H., Perivolaris, A., Tullu, T., Zeifman, R. J., Zhang, Y., Burback, L., Winkler, O., Greenshaw, A., Husain, M. I., C. Reichelt, A., Vermetten, E., Jha, M. K., Jetly, R., Loebenberg, R., & Bhat, V. (2025). Pharmacokinetics of Psilocybin: A Systematic Review. Pharmaceutics, 17(4), 411. https://doi.org/10.3390/pharmaceutics17040411