
Molecule Formats of B-Cell Targeting Biologics: Applications in Autoimmune Disease Treatment and Impacts on Manufacturability



Abstract
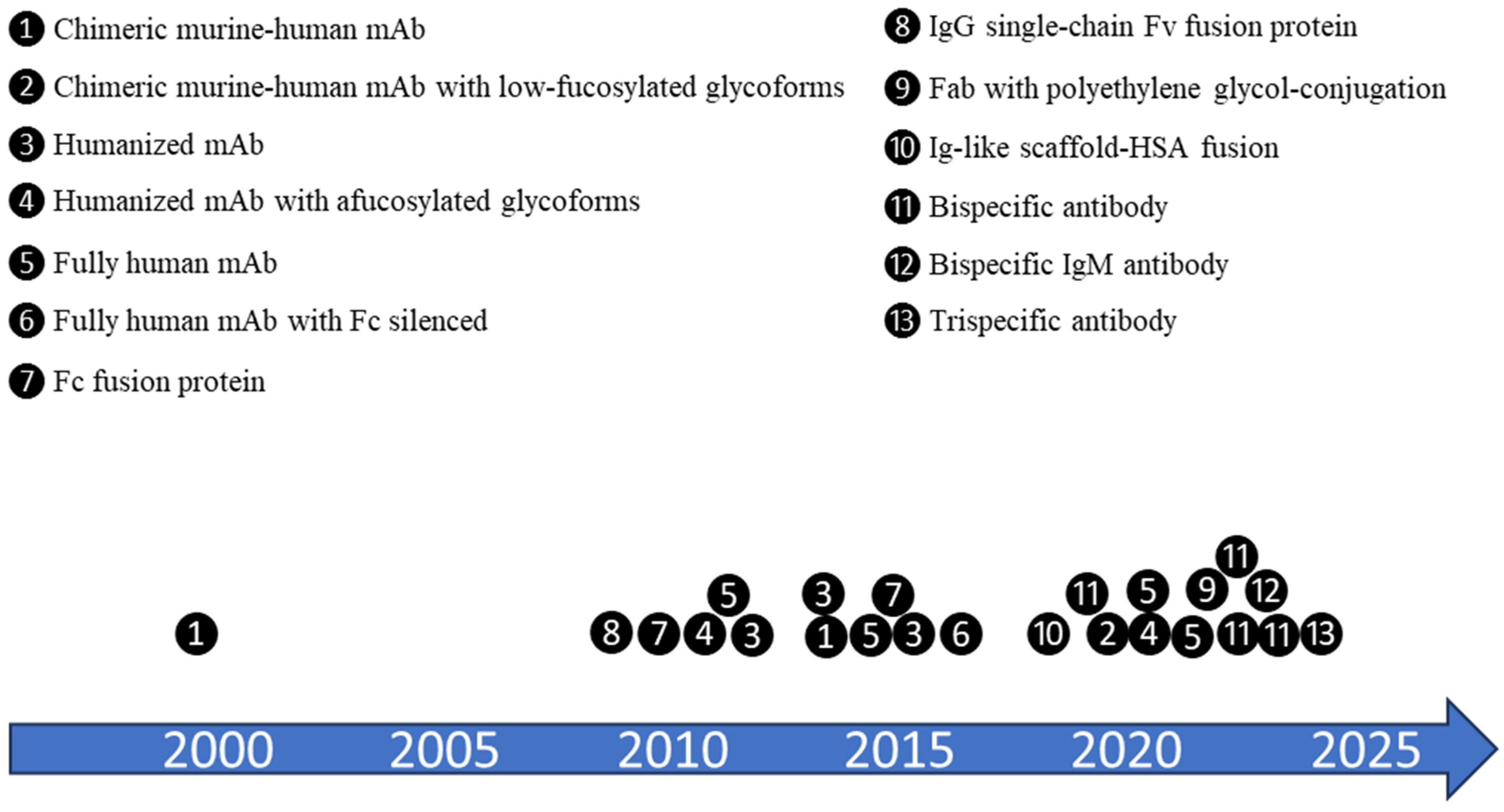
:1. Introduction
2. Different Molecule Formats and Features Applied in Biologics Development for Autoimmune Disease Treatments
2.1. Ocrelizumab and Ofatumumab—Next Generation Anti-CD20 Monoclonal Antibodies (mAbs)
2.2. Inebilizumab—Afucosylated Anti-CD19 mAb
2.3. Belimumab—mAb Indirectly Targeting B Cells
2.4. Ianalumab (VAY736)—mAb Directly Targeting B Cells
2.5. Dazodalibep (HZN4920/AMG611)—HSA-Fusion Protein Antagonizing CD40L
2.6. PRV-3279—Bispecific Antibody
3. Impact of Molecule Format on Manufacturability
3.1. Molecule Format Selection and Process Development
3.1.1. Afucosylated mAb
3.1.2. Fusion Protein
3.1.3. Bispecific Antibody
3.2. Scalability Considerations for Manufacturability Improvement
4. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurosaki, T. B-lymphocyte biology. Immunol. Rev. 2010, 237, 5–9. [Google Scholar] [CrossRef]
- Townsend, M.J.; Monroe, J.G.; Chan, A.C. B-cell targeted therapies in human autoimmune diseases: An updated perspective. Immunol. Rev. 2010, 237, 264–283. [Google Scholar] [CrossRef]
- Barnas, J.L.; Looney, R.J.; Anolik, J.H. B cell targeted therapies in autoimmune disease. Curr. Opin. Immunol. 2019, 61, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Stensland, Z.C.; Cambier, J.C.; Smith, M.J. Therapeutic Targeting of Autoreactive B Cells: Why, How, and When? Biomedicines 2021, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Merino-Vico, A.; Frazzei, G.; van Hamburg, J.P.; Tas, S.W. Targeting B cells and plasma cells in autoimmune diseases: From established treatments to novel therapeutic approaches. Eur. J. Immunol. 2023, 53, e2149675. [Google Scholar] [CrossRef]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto. Immun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Huda, R. New Approaches to Targeting B Cells for Myasthenia Gravis Therapy. Front. Immunol. 2020, 11, 240. [Google Scholar] [CrossRef]
- Hofmann, K.; Clauder, A.K.; Manz, R.A. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef]
- Gurcan, H.M.; Keskin, D.B.; Stern, J.N.; Nitzberg, M.A.; Shekhani, H.; Ahmed, A.R. A review of the current use of rituximab in autoimmune diseases. Int. Immunopharmacol. 2009, 9, 10–25. [Google Scholar] [CrossRef]
- Kanatas, P.; Stouras, I.; Stefanis, L.; Stathopoulos, P. B-Cell-Directed Therapies: A New Era in Multiple Sclerosis Treatment. Can. J. Neurol. Sci. 2022, 20, 355–364. [Google Scholar] [CrossRef]
- Rubbert-Roth, A. TRU-015, a fusion protein derived from an anti-CD20 antibody, for the treatment of rheumatoid arthritis. Curr. Opin. Mol. Ther. 2010, 12, 115–123. [Google Scholar] [PubMed]
- Robinson, W.H.; Fiorentino, D.; Chung, L.; Moreland, L.W.; Deodhar, M.; Harler, M.B.; Saulsbery, C.; Kunder, R. Cutting-edge approaches to B-cell depletion in autoimmune diseases. Front. Immunol. 2024, 15, 1454747. [Google Scholar] [CrossRef]
- Stone, J.H.; Khosroshahi, A.; Zhang, W.; Della Torre, E.; Okazaki, K.; Tanaka, Y.; Lohr, J.M.; Schleinitz, N.; Dong, L.; Umehara, H.; et al. Inebilizumab for Treatment of IgG4-Related Disease. N. Engl. J. Med. 2022, 132, 102873. [Google Scholar] [CrossRef]
- Arbitman, L.; Furie, R.; Vashistha, H. B cell-targeted therapies in systemic lupus erythematosus. J. Autoimmun. 2022, 132, 102873. [Google Scholar] [CrossRef] [PubMed]
- Subklewe, M.; Magno, G.; Gebhardt, C.; Bucklein, V.; Szelinski, F.; Arevalo, H.J.R.; Hanel, G.; Dorner, T.; Zugmaier, G.; von Bergwelt-Baildon, M.; et al. Application of blinatumomab, a bispecific anti-CD3/CD19 T-cell engager, in treating severe systemic sclerosis: A case study. Eur. J. Cancer 2024, 204, 114071. [Google Scholar] [CrossRef]
- Zugmaier, G.; Klinger, M.; Subklewe, M.; Zaman, F.; Locatelli, F. B-Cell-Depleting Immune Therapies as Potential New Treatment Options for Systemic Sclerosis. Sclerosis 2025, 3, 5. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Wu, D.; Zhou, J.; Leung, S.O.; Zhang, F. SM03, an anti-human CD22 monoclonal antibody, for active rheumatoid arthritis: A phase II, randomized, double-blind, placebo-controlled study. Rheumatology 2022, 61, 1841–1848. [Google Scholar] [CrossRef]
- Geh, D.; Gordon, C. Epratuzumab for the treatment of systemic lupus erythematosus. Expert. Rev. Clin. Immunol. 2018, 14, 245–258. [Google Scholar] [CrossRef]
- Runkel, L.; Stacey, J. Lupus clinical development: Will belimumab’s approval catalyse a new paradigm for SLE drug development? Expert. Opin. Biol. Ther. 2014, 14, 491–501. [Google Scholar] [CrossRef]
- Bowman, S.J.; Fox, R.; Dorner, T.; Mariette, X.; Papas, A.; Grader-Beck, T.; Fisher, B.A.; Barcelos, F.; De Vita, S.; Schulze-Koops, H.; et al. Safety and efficacy of subcutaneous ianalumab (VAY736) in patients with primary Sjogren’s syndrome: A randomised, double-blind, placebo-controlled, phase 2b dose-finding trial. Lancet 2022, 399, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.A.; Bruce, I.N.; Dorner, T.; Leon, M.G.; Leszczynski, P.; Urowitz, M.; Haier, B.; Jimenez, T.; Brittain, C.; Liu, J.; et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology 2021, 60, 5397–5407. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Stan, M.N.; Frommer, L.; Gergely, P.; Colin, L.; Amer, A.; Schuhmann, I.; Espie, P.; Rush, J.S.; Basson, C.; et al. A Novel Anti-CD40 Monoclonal Antibody, Iscalimab, for Control of Graves Hyperthyroidism-A Proof-of-Concept Trial. J. Clin. Endocrinol. Metab. 2020, 105, 696–704. [Google Scholar] [CrossRef]
- Fisher, B.A.; Szanto, A.; Ng, W.-F.; Bombardieri, M.; Gergely, P. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: A multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol. 2020, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, S.; Daniluk, S.; Ptaszynski, R.; Muller-Ladner, U.; Ramanujam, M.; Rosenstock, B.; Eleftheraki, A.G.; Vinisko, R.; Petrikova, A.; Kellner, H.; et al. Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase IIa study. Ann. Rheum. Dis. 2019, 78, 754–760. [Google Scholar] [CrossRef]
- Kivitz, A. A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Mechanistic Insight and Dosage Optimization Study of the Efficacy and Safety of Dazodalibep (VIB4920/HZN4920) in Patients with Rheumatoid Arthritis Having Inadequate Response to Conventional/Biological DMARDs. Arthritis Rheumatol. 2022, 74, 1045–1048. [Google Scholar]
- Oganesyan, V.; Ferguson, A.; Grinberg, L.; Wang, L.; Phipps, S.; Chacko, B.; Drabic, S.; Thisted, T.; Baca, M. Fibronectin type III domains engineered to bind CD40L: Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of two complexes. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 1045–1048. [Google Scholar] [CrossRef]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef]
- Dhillon, S. Telitacicept: First Approval. Drugs 2021, 81, 1671–1675. [Google Scholar] [CrossRef]
- Bracewell, C.; Isaacs, J.D.; Emery, P.; Ng, W.F. Atacicept, a novel B cell-targeting biological therapy for the treatment of rheumatoid arthritis. Expert. Opin. Biol. Ther. 2009, 9, 909–919. [Google Scholar] [CrossRef]
- Dunford, P.; Comer, G.; Raymond, R.; Jung, D.; Moore, P.; Leon, F.; Merrill, J.T. PREVAIL 1: A Multiple Ascending Dose Study in Normal Healthy Volunteers of PRV-3279, a Novel Bispecific DART Molecule Targeting CD32B and CD79B on B Cells, with Potential for Treatment of SLE. Arthritis Rheumatol. 2020, 72, 13–25. [Google Scholar]
- Robak, T.; Robak, E. New anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoid malignancies. BioDrugs 2011, 25, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Ocrelizumab: First Global Approval. Drugs 2017, 77, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- McCool, R.; Wilson, K.; Arber, M.; Fleetwood, K.; Toupin, S.; Thom, H.; Bennett, I.; Edwards, S. Systematic review and network meta-analysis comparing ocrelizumab with other treatments for relapsing multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 29, 55–61. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.; Wang, Y.; Gallagher, S.; Mittereder, N.; Kuta, E.; Damschroder, M.; Woods, R.; Rowe, D.C.; Cheng, L.; Cook, K.; et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J. Pharmacol. Exp. Ther. 2010, 335, 213–222. [Google Scholar] [CrossRef]
- Gallagher, S.; Turman, S.; Yusuf, I.; Akhgar, A.; Wu, Y.; Roskos, L.K.; Herbst, R.; Wang, Y. Pharmacological profile of MEDI-551, a novel anti-CD19 antibody, in human CD19 transgenic mice. Int. Immunopharmacol. 2016, 36, 205–212. [Google Scholar] [CrossRef]
- Nie, T.; Blair, H.A. Inebilizumab: A Review in Neuromyelitis Optica Spectrum Disorder. CNS Drugs 2022, 36, 1133–1141. [Google Scholar] [CrossRef]
- Rensel, M.; Zabeti, A.; Mealy, M.A.; Cimbora, D.; She, D.; Drappa, J.; Katz, E. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: Analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for ⩾4 years in the N-MOmentum trial. Mult. Scler. 2022, 28, 925–932. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, J.; Xiao, F.; Zheng, L.; Zhu, X.; Wu, L.; Zhao, C.; Wang, S.; Rui, K.; Zou, H.; et al. B cell-activating factor and its targeted therapy in autoimmune diseases. Cytokine Growth Factor. Rev. 2022, 64, 57–70. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; James, L.K.; Schmierer, K.; Giovannoni, G. Failed B cell survival factor trials support the importance of memory B cells in multiple sclerosis. Eur. J. Neurol. 2020, 27, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Wiendl, H. What Have Failed, Interrupted, and Withdrawn Antibody Therapies in Multiple Sclerosis Taught Us? Neurotherapeutics 2022, 19, 785–807. [Google Scholar] [CrossRef]
- Lenert, A.; Niewold, T.B.; Lenert, P. Spotlight on blisibimod and its potential in the treatment of systemic lupus erythematosus: Evidence to date. Drug Des. Devel Ther. 2017, 11, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Al-Samkari, H.; Barcellini, W.; Cooper, N.; Ghanima, W.; Michel, M.; Wong, R.S.; Zaja, F.; Zhang, F.; Urban, P.; et al. Ianalumab, a Novel Anti-B-Cell Activating Factor (BAFF) Receptor (BAFF-R) Monoclonal Antibody (mAb) in Development for Immune Thrombocytopenia (ITP) and Warm Autoimmune Hemolytic Anemia (wAIHA), Has Demonstrated a Favorable Safety Profile in Sjögren’s Syndrome (SjS), Systemic Lupus Erythematosus (SLE) and Chronic Lymphocytic Leukemia (CLL). Blood 2023, 142, 5427. [Google Scholar] [CrossRef]
- St Clair, E.W.; Baer, A.N.; Ng, W.F.; Noaiseh, G.; Baldini, C.; Tarrant, T.K.; Papas, A.; Devauchelle-Pensec, V.; Wang, L.; Xu, W.; et al. CD40 ligand antagonist dazodalibep in Sjogren’s disease: A randomized, double-blinded, placebo-controlled, phase 2 trial. Nat. Med. 2024, 30, 1583–1592. [Google Scholar] [CrossRef]
- Amigorena, S.; Bonnerot, C.; Drake, J.R.; Choquet, D.; Hunziker, W.; Guillet, J.G.; Webster, P.; Sautes, C.; Mellman, I.; Fridman, W.H. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science 1992, 256, 1808–1812. [Google Scholar] [CrossRef]
- Renner, K.; Neumayer, S.; Talke, Y.; Buchtler, S.; Schmidbauer, K.; Nimmerjahn, F.; Lux, A.; Winter, F.; Salewski, J.N.; Mack, M. B-cell modulation with anti-CD79b antibodies ameliorates experimental autoimmune encephalitis in mice. Eur. J. Immunol. 2022, 52, 656–668. [Google Scholar] [CrossRef]
- Veri, M.C.; Burke, S.; Huang, L.; Li, H.; Gorlatov, S.; Tuaillon, N.; Rainey, G.J.; Ciccarone, V.; Zhang, T.; Shah, K.; et al. Therapeutic control of B cell activation via recruitment of Fcgamma receptor IIb (CD32B) inhibitory function with a novel bispecific antibody scaffold. Arthritis Rheum. 2010, 62, 1933–1943. [Google Scholar] [CrossRef]
- Chen, Z.; Qian, Y.; Song, Y.; Xu, X.; Tao, L.; Mussa, N.; Ghose, S.; Li, Z.J. Design of next-generation therapeutic IgG4 with improved manufacturability and bioanalytical characteristics. MAbs 2020, 12, 1829338. [Google Scholar] [CrossRef]
- Thomann, M.; Reckermann, K.; Reusch, D.; Prasser, J.; Tejada, M.L. Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol. Immunol. 2016, 73, 69–75. [Google Scholar] [CrossRef]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed]
- Olivier, S.; Jacoby, M.; Brillon, C.; Bouletreau, S.; Mollet, T.; Nerriere, O.; Angel, A.; Danet, S.; Souttou, B.; Guehenneux, F.; et al. EB66 cell line, a duck embryonic stem cell-derived substrate for the industrial production of therapeutic monoclonal antibodies with enhanced ADCC activity. MAbs 2010, 2, 405–415. [Google Scholar] [CrossRef]
- Okeley, N.M.; Alley, S.C.; Anderson, M.E.; Boursalian, T.E.; Burke, P.J.; Emmerton, K.M.; Jeffrey, S.C.; Klussman, K.; Law, C.L.; Sussman, D.; et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 2013, 110, 5404–5409. [Google Scholar] [CrossRef] [PubMed]
- Kilmartin, J.V.; Wright, B.; Milstein, C. Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. J. Cell Biol. 1982, 93, 576–582. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Ripka, J.; Adamany, A.; Stanley, P. Two Chinese hamster ovary glycosylation mutants affected in the conversion of GDP-mannose to GDP-fucose. Arch. Biochem. Biophys. 1986, 249, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Yamane-Ohnuki, N.; Kinoshita, S.; Inoue-Urakubo, M.; Kusunoki, M.; Iida, S.; Nakano, R.; Wakitani, M.; Niwa, R.; Sakurada, M.; Uchida, K.; et al. Establishment of FUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 2004, 87, 614–622. [Google Scholar] [CrossRef]
- Malphettes, L.; Freyvert, Y.; Chang, J.; Liu, P.Q.; Chan, E.; Miller, J.C.; Zhou, Z.; Nguyen, T.; Tsai, C.; Snowden, A.W.; et al. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol. Bioeng. 2010, 106, 774–783. [Google Scholar] [CrossRef]
- Chan, K.F.; Shahreel, W.; Wan, C.; Teo, G.; Hayati, N.; Tay, S.J.; Tong, W.H.; Yang, Y.; Rudd, P.M.; Zhang, P.; et al. Inactivation of GDP-fucose transporter gene (Slc35c1) in CHO cells by ZFNs, TALENs and CRISPR-Cas9 for production of fucose-free antibodies. Biotechnol. J. 2016, 11, 399–414. [Google Scholar] [CrossRef]
- von Horsten, H.H.; Ogorek, C.; Blanchard, V.; Demmler, C.; Giese, C.; Winkler, K.; Kaup, M.; Berger, M.; Jordan, I.; Sandig, V. Production of non-fucosylated antibodies by co-expression of heterologous GDP-6-deoxy-D-lyxo-4-hexulose reductase. Glycobiology 2010, 20, 1607–1618. [Google Scholar] [CrossRef]
- Ferrara, C.; Brunker, P.; Suter, T.; Moser, S.; Puntener, U.; Umana, P. Modulation of therapeutic antibody effector functions by glycosylation engineering: Influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol. Bioeng. 2006, 93, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Imai-Nishiya, H.; Kuni-Kamochi, R.; Mori, K.; Inoue, M.; Kitajima-Miyama, K.; Okazaki, A.; Iida, S.; Shitara, K.; Satoh, M. Establishment of a GDP-mannose 4,6-dehydratase (GMD) knockout host cell line: A new strategy for generating completely non-fucosylated recombinant therapeutics. J. Biotechnol. 2007, 130, 300–310. [Google Scholar] [CrossRef]
- Nanda, S.; Bathon, J.M. Etanercept: A clinical review of current and emerging indications. Expert. Opin. Pharmacother. 2004, 5, 1175–1186. [Google Scholar] [CrossRef]
- Song, Y.; Qian, Y.; Huang, Z.; Khattak, S.F.; Li, Z.J. Computational insights into O-glycosylation in a CTLA4 Fc-fusion protein linker and its impact on protein quality attributes. Comput. Struct. Biotechnol. J. 2020, 18, 3925–3935. [Google Scholar] [CrossRef] [PubMed]
- Way, J.C.; Lauder, S.; Brunkhorst, B.; Kong, S.M.; Qi, A.; Webster, G.; Campbell, I.; McKenzie, S.; Lan, Y.; Marelli, B.; et al. Improvement of Fc-erythropoietin structure and pharmacokinetics by modification at a disulfide bond. Protein Eng. Des. Sel. 2005, 18, 111–118. [Google Scholar] [CrossRef]
- Trummer, E.; Fauland, K.; Seidinger, S.; Schriebl, K.; Lattenmayer, C.; Kunert, R.; Vorauer-Uhl, K.; Weik, R.; Borth, N.; Katinger, H.; et al. Process parameter shifting: Part I. Effect of DOT, pH, and temperature on the performance of Epo-Fc expressing CHO cells cultivated in controlled batch bioreactors. Biotechnol. Bioeng. 2006, 94, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Hossler, P.; Khattak, S.F.; Li, Z.J. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009, 19, 936–949. [Google Scholar] [CrossRef]
- Ha, T.K.; Lee, G.M. Effect of glutamine substitution by TCA cycle intermediates on the production and sialylation of Fc-fusion protein in Chinese hamster ovary cell culture. J. Biotechnol. 2014, 180, 23–29. [Google Scholar] [CrossRef]
- Taschwer, M.; Hackl, M.; Hernandez Bort, J.A.; Leitner, C.; Kumar, N.; Puc, U.; Grass, J.; Papst, M.; Kunert, R.; Altmann, F.; et al. Growth, productivity and protein glycosylation in a CHO EpoFc producer cell line adapted to glutamine-free growth. J. Biotechnol. 2012, 157, 295–303. [Google Scholar] [CrossRef]
- Jing, Y.; Qian, Y.; Li, Z.J. Sialylation enhancement of CTLA4-Ig fusion protein in Chinese hamster ovary cells by dexamethasone. Biotechnol. Bioeng. 2010, 107, 488–496. [Google Scholar] [CrossRef]
- Qian, Y.; Lewis, A.M.; Sidnam, S.M.; Bergeron, A.; Abu-Absi, N.R.; Vaidyanathan, N.; Deresienski, A.; Qian, N.-X.; Borys, M.C.; Li, Z.J. LongR3 enhances Fc-fusion protein N-linked glycosylation while improving protein productivity in an industrial CHO cell line. Process Biochem. 2017, 53, 9. [Google Scholar] [CrossRef]
- Ying, J.; Borys, B.C.; Samiksha, N.; Egan, S.; Qian, Y.; Pan, S.-H.; Li, Z.J. Identification of cell culture conditions to control protein aggregation of IgG fusion proteins expressed in Chinese hamster ovary cells. Process Biochem. 2012, 47, 6. [Google Scholar] [CrossRef]
- Qian, Y.; Jing, Y.; Li, Z.J. Glucocorticoid receptor-mediated reduction of IgG-fusion protein aggregation in Chinese hamster ovary cells. Biotechnol. Prog. 2010, 26, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.A.; Gupta, P.; Han, X. Protein aggregation kinetics during Protein A chromatography. Case study for an Fc fusion protein. J. Chromatogr. A 2007, 1171, 22–28. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Purdie, J.L.; Kowle, R.L.; Langland, A.L.; Patel, C.N.; Ouyang, A.; Olson, D.J. Cell culture media impact on drug product solution stability. Biotechnol. Prog. 2016, 32, 998–1008. [Google Scholar] [CrossRef]
- Gomez, N.; Wieczorek, A.; Lu, F.; Bruno, R.; Diaz, L.; Agrawal, N.J.; Daris, K. Culture temperature modulates half antibody and aggregate formation in a Chinese hamster ovary cell line expressing a bispecific antibody. Biotechnol. Bioeng. 2018, 115, 2930–2940. [Google Scholar] [CrossRef]
- Tustian, A.D.; Laurin, L.; Ihre, H.; Tran, T.; Stairs, R.; Bak, H. Development of a novel affinity chromatography resin for platform purification of bispecific antibodies with modified protein a binding avidity. Biotechnol. Prog. 2018, 34, 650–658. [Google Scholar] [CrossRef]
- Brantley, T.; Moore, B.; Grinnell, C.; Khattak, S. Investigating trace metal precipitation in highly concentrated cell culture media with Pourbaix diagrams. Biotechnol. Bioeng. 2021, 118, 3888–3897. [Google Scholar] [CrossRef]
- Qian, Y.; Khattak, S.F.; Xing, Z.; He, A.; Kayne, P.S.; Qian, N.X.; Pan, S.H.; Li, Z.J. Cell culture and gene transcription effects of copper sulfate on Chinese hamster ovary cells. Biotechnol. Prog. 2011, 27, 1190–1194. [Google Scholar] [CrossRef]
- Qian, Y.; Xing, Z.; Lee, S.; Mackin, N.A.; He, A.; Kayne, P.S.; He, Q.; Qian, N.X.; Li, Z.J. Hypoxia influences protein transport and epigenetic repression of CHO cell cultures in shake flasks. Biotechnol. J. 2014, 9, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Sowa, S.W.; Aron, K.L.; Xu, P.; Langsdorf, E.; Warrack, B.; Aranibar, N.; Tremml, G.; Xu, J.; Duncan, M.; et al. New insights into genetic instability of an industrial CHO cell line by orthogonal omics. Biochem. Eng. J. 2020, 164, 12. [Google Scholar] [CrossRef]
- Qian, Y.; Rehmann, M.S.; Qian, N.X.; He, A.; Borys, M.C.; Kayne, P.S.; Li, Z.J. Hypoxia and transforming growth factor-beta1 pathway activation promote Chinese Hamster Ovary cell aggregation. Biotechnol. Bioeng. 2018, 115, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; He, Q.; Oliveira, C.; Qian, Y.; Egan, S.; Xu, J.; Qian, N.X.; Langsdorf, E.; Warrack, B.; Aranibar, N.; et al. Increased MSX level improves biological productivity and production stability in multiple recombinant GS CHO cell lines. Eng. Life Sci. 2020, 20, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Luna, G.; Alping, P.; Burman, J.; Fink, K.; Fogdell-Hahn, A.; Gunnarsson, M.; Hillert, J.; Langer-Gould, A.; Lycke, J.; Nilsson, P.; et al. Infection Risks Among Patients with Multiple Sclerosis Treated With Fingolimod, Natalizumab, Rituximab, and Injectable Therapies. JAMA Neurol. 2020, 77, 184–191. [Google Scholar] [CrossRef]
- Safavi, F.; Nourbakhsh, B.; Azimi, A.R. B-cell depleting therapies may affect susceptibility to acute respiratory illness among patients with multiple sclerosis during the early COVID-19 epidemic in Iran. Mult. Scler. Relat. Disord. 2020, 43, 102195. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Target | Drug Name | Molecule Format/Features | Autoimmune Indications | References |
|---|---|---|---|---|
| CD20 | Rituximab | Chimeric murine-human IgG1k mAb/targeting CD20 on pro-B cells and all mature B cells, but not long-lived plasma or plasmablast cells. | Approved: RA, GPA, MPA, PV Clinical trials: ITP, MG | [10] |
| Ocrelizumab | Humanized mAb/with afucosylated glycoforms enhancing ADCC | Approved: RRMS and PPMS | [11] | |
| Ublituximab | Chimeric murine-human IgG mAb/with low-fucosylated glycoforms enhancing ADCC | Approved: RRMS, CIS, SPMS | [11] | |
| Ofatumumab | Fully human monoclonal antibody/first B-cell-targeting therapy that is intended for self-administration at home | Approved: RRMS, CIS, SPMS Clinical trial: RA | [11] | |
| Veltuzumab | Humanized mAb/epratuzumab framework and rituximab CDRs | FDA granted orphan status designation for ITP and pemphigus Clinical trial: RA | [7] | |
| TRU-015 | Fully human IgG fusion protein/a single-chain Fv specific for CD20 linked to human IgG1 hinge, CH2, and CH3 domains but devoid of CH1 and CL domains | Clinical trials: active seropositive RA on a stable background of methotrexate | [12] | |
| Mosunetuzumab | Bispecific antibody/IgG, anti-CD20 and anti-CD3 | Clinical trials: SLE | [13] | |
| Imvotamab | Bispecific antibody/IgM, anti-CD20 and anti-CD3 | Clinical trials: RA, SLE | [13] | |
| CD19 | Inebilizumab | Humanized IgG1k mAb/with afucosylated glycoforms enhancing ADCC | Approved: NMOSD with AQP4-IgG+ Clinical trials: GM, IgG4-RD | [4,14] NCT04524273 |
| Obexelimab | Bispecific antibody/simultaneously binds CD19 and FcγRIIb, resulting in down-regulation of B cell activity | Clinical trials: GM; IgG4-RD | [15] | |
| Blinatumomab | Bispecific antibody/anti-CD19 and anti-CD3 | Clinical trials: RA, system sclerosis | [16,17] | |
| PIT565 | Trispecific antibody/anti-CD19, anti-CD3, and anti-CD2 | Clinical trials: SLE | NCT06335979 | |
| CD22 | SM03 | Chimeric murine-human mAb/targeting the extracellular portion of CD22 | Clinical trials: SLE, RA | [18] |
| Epratuzumab | Humanized mAb/targeting CD22 with modest ADCC activity | Clinical trials: SLE | [19] | |
| CD38 | Daratumumab | Fully human mAb/targeting CD38 on long-lived plasma cells | Clinical trials: SLE | [15] |
| BAFF/BAFF-R | Belimumab | Fully human mAb/neutralizing biologically active soluble form of BAFF | Approved: SLE and lupus nephritis | [20] |
| Ianalumab (VAY736) | Fully human mAb/antagonizing BAFF-R | Clinical trials: MS, SLE, Sjögren’s syndrome, Diffuse Cutaneous Systemic Sclerosis | [21] | |
| CD40/CD40L | Dapirolizumab pegol | Fab/polyethylene glycol-conjugated, anti-CD40L, lacking the Fc-portion to avoid platelet activation | Clinical trials: SLE | [22] |
| Iscalimab (CFZ533) | Fully human mAb/Fc-silenced, antagonizing CD40 | Clinical trials: Graves disease (GD); Sjögren’s syndrome | [23,24] | |
| BI 655064 | Humanized mAb/anti-CD40 blocking CD40-CD40L interaction | Clinical trials: RA | [25] | |
| Dazodalibep (AMG611, HZN-4920) | Ig-like scaffold-HSA fusion protein/Tn3 scaffolds derived from the 3rd fibronectin type III domain of human tenascin-C, structurally analogous to antibody CDRs and functionally blocking CD40-CD40L interaction | Clinical trials: RA, Sjögren’s syndrome | [26,27,28] | |
| BAFF/APRIL | Telitacicept | Fc fusion protein/fused with extracellular domain (amino acids 13-118) of TACI binding to and neutralizing BAFF and APRIL | Approved: SLE (in China) Clinical trials: IgA nephropathy, MS, RA, MG, Sjögren’s syndrome | [29] |
| Atacicept | Fc fusion protein/fused with extracellular domain (amino acids 30-110) of TACI binding to and neutralizing BAFF and APRIL | Clinical trials: SLE, RA, IgA nephropathy | [30] | |
| CD32B/CD79B | PRV-3279 (MGD010) | Bispecific antibody/simultaneously targeting B-cell surface proteins CD32B and CD79B | Clinical trials: SLE | [31] |
| Cell Line | Affected Biosynthesis Pathway | Reference |
|---|---|---|
| CHO Lec13 (Pro-Lec13.6a) | Natural deficiency in endogenous GDP-mannose 4,6-dehydratase (GMD) | [51,56] |
| CHO-DG44 FUT8−/− (BioWa) | FUT8 knockout by homologous recombination | [57] Patent# US6946292B2 |
| CHO-K1 FUT8−/− | FUT8 deletion by ZFN | [58] |
| CHO-gmt3 (CHO-glycosylation mutant3) | GDP-fucose transporter (SLC35C1) inactivation | [59] |
| CHO-RMD | Heterologous expression of GDP-6 deoxy-d-lyxo-4-hexulose reductase (RMD) in the cytosol of CHO cells to deflect the GDP-fucose de novo pathway | [60] |
| CHO-GnT-III | Overexpressed GnTIII catalyzes the formation of a bisecting GlcNAc to reduce Fc core fucosylation | [61] |
| CHO-SM | GDP-fucose 4,6-dehydratase (GMD) knockout, which makes the cell unable to produce intracellular GDP-fucose and fucosylated glycoproteins in the absence of L-fucose | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Y.; Mahmoud, T.I. Molecule Formats of B-Cell Targeting Biologics: Applications in Autoimmune Disease Treatment and Impacts on Manufacturability. Pharmaceutics 2025, 17, 495. https://doi.org/10.3390/pharmaceutics17040495
Qian Y, Mahmoud TI. Molecule Formats of B-Cell Targeting Biologics: Applications in Autoimmune Disease Treatment and Impacts on Manufacturability. Pharmaceutics. 2025; 17(4):495. https://doi.org/10.3390/pharmaceutics17040495
Chicago/Turabian StyleQian, Yueming, and Tamer I. Mahmoud. 2025. "Molecule Formats of B-Cell Targeting Biologics: Applications in Autoimmune Disease Treatment and Impacts on Manufacturability" Pharmaceutics 17, no. 4: 495. https://doi.org/10.3390/pharmaceutics17040495
APA StyleQian, Y., & Mahmoud, T. I. (2025). Molecule Formats of B-Cell Targeting Biologics: Applications in Autoimmune Disease Treatment and Impacts on Manufacturability. Pharmaceutics, 17(4), 495. https://doi.org/10.3390/pharmaceutics17040495