
Modulating NPC1L1 to Potentiate PARP Inhibitor-Induced Ferroptosis and Immune Response in Triple-Negative Breast Cancer


Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Absolute Quantitative Lipidomics
2.4. PCR Array and Quantitative Real-Time PCR Analysis
2.5. Western Blot
2.6. Dual-Luciferase Reporter Assay
2.7. Transcriptome Analysis
2.8. Total Cholesterol Measurement
2.9. ATP Levels Determination
2.10. GSH Measurement and MDA Levels Analysis
2.11. Flow Cytometry
2.12. Cell Viability Assay
2.13. Gene Silencing Experiments
2.14. T Cell Cytotoxic Assay
2.15. Syngeneic 4T1 Mammary Tumor Model
2.16. Paraffin Section Immunohistochemistry Experiment
2.17. Statistical Analysis
3. Results
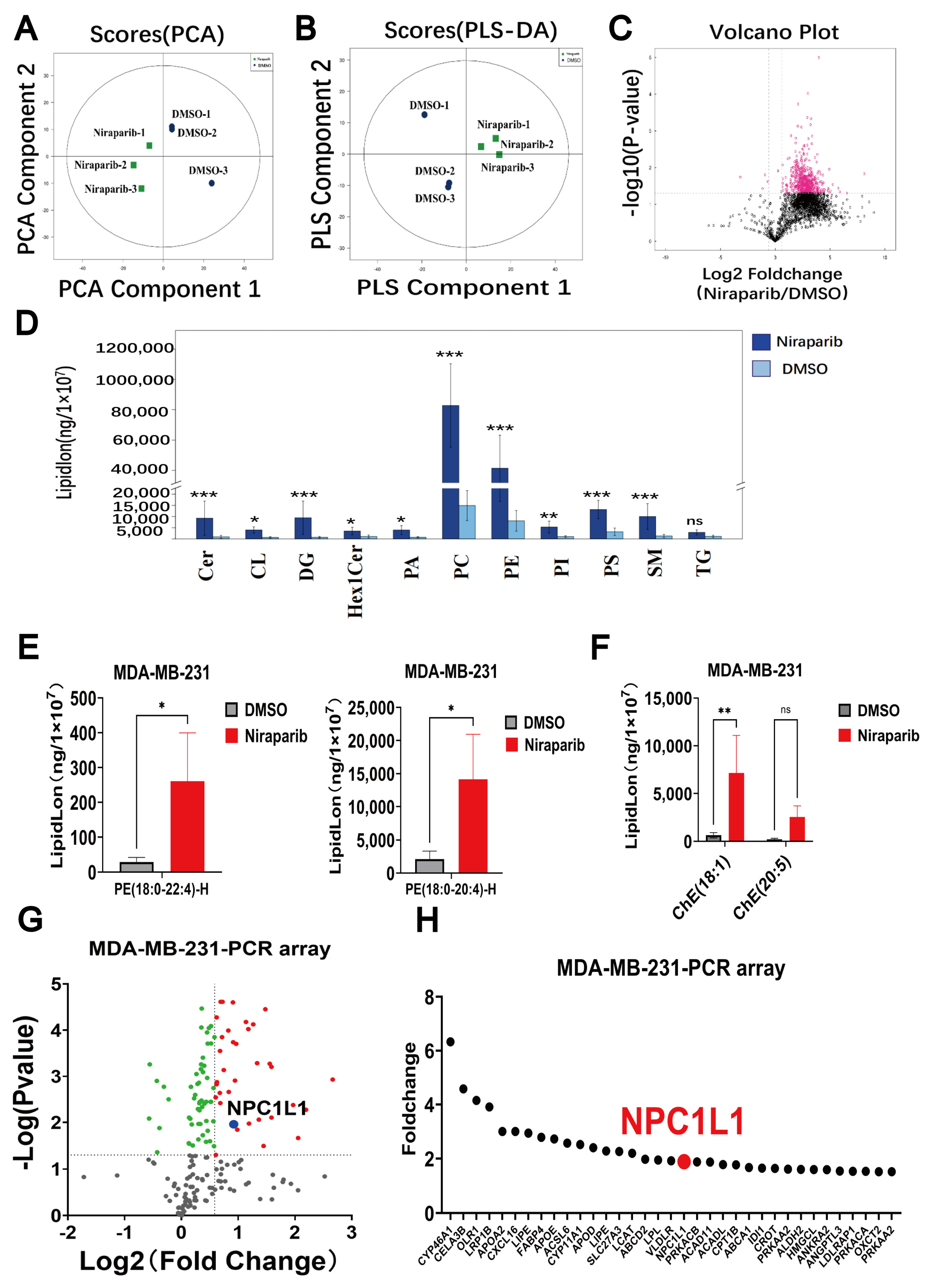
3.1. Lipid Metabolism Significantly Altered by PARP Inhibitors
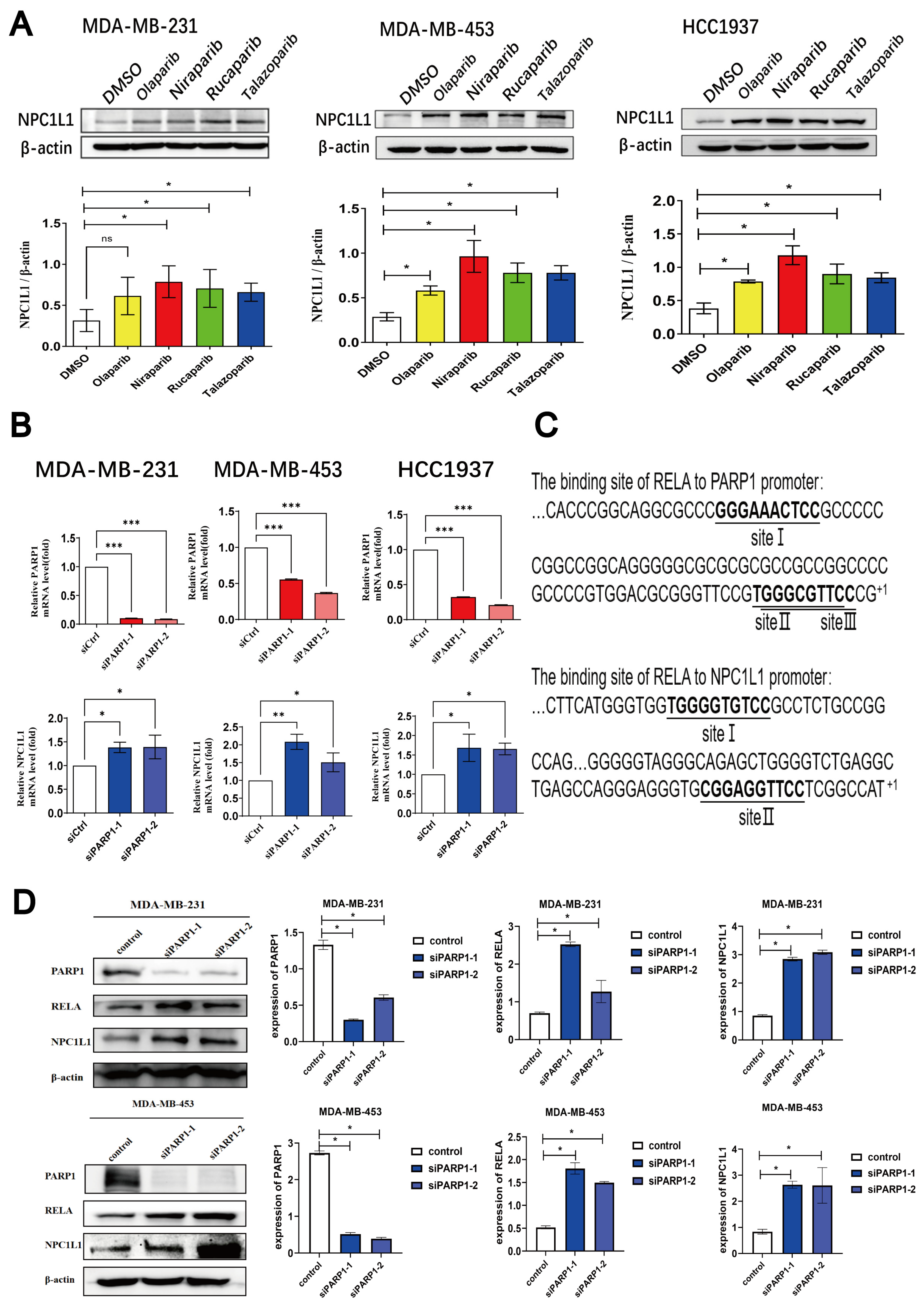
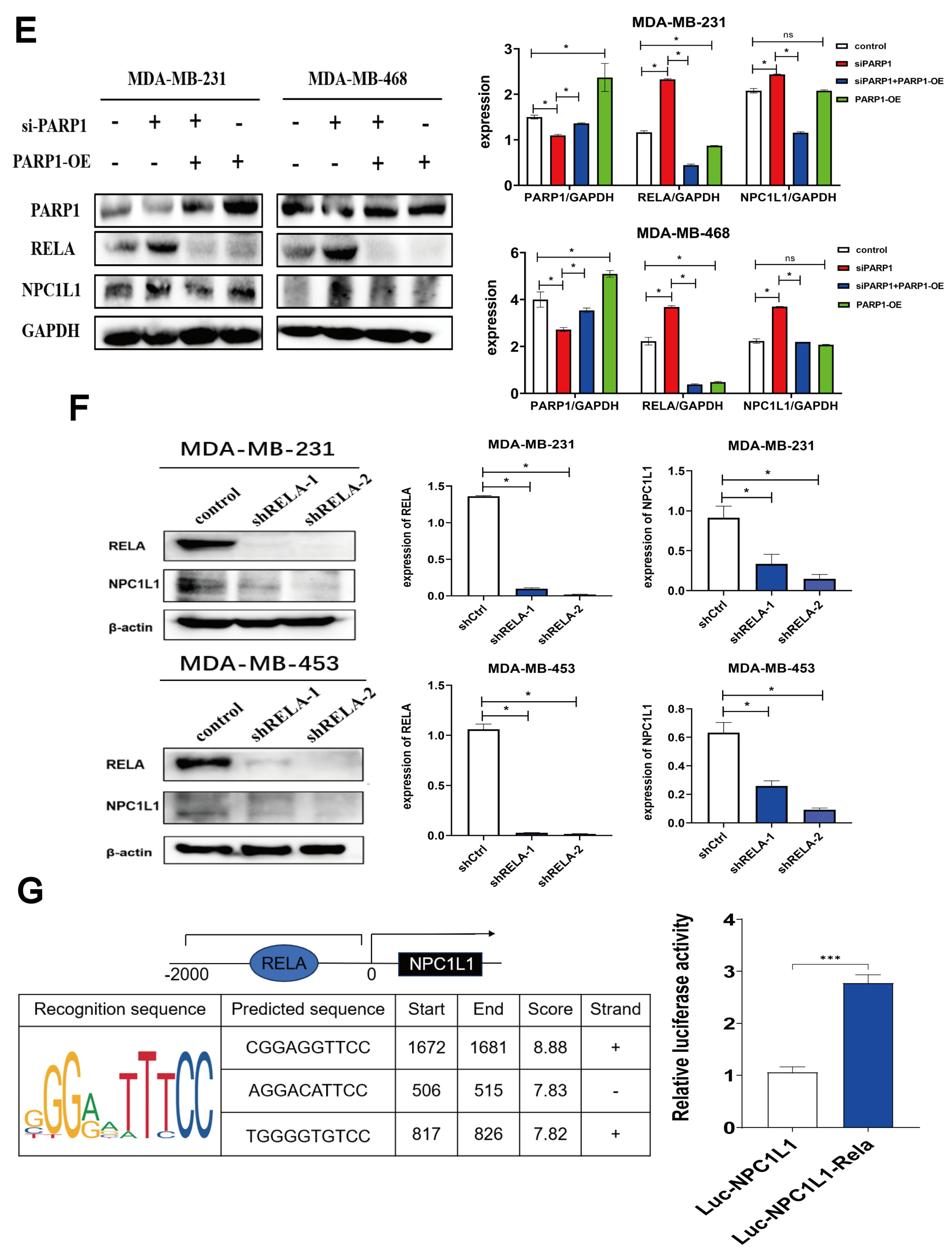
3.2. PARP Inhibitors Regulate the PARP1-RELA-NPC1L1 Signaling Axis
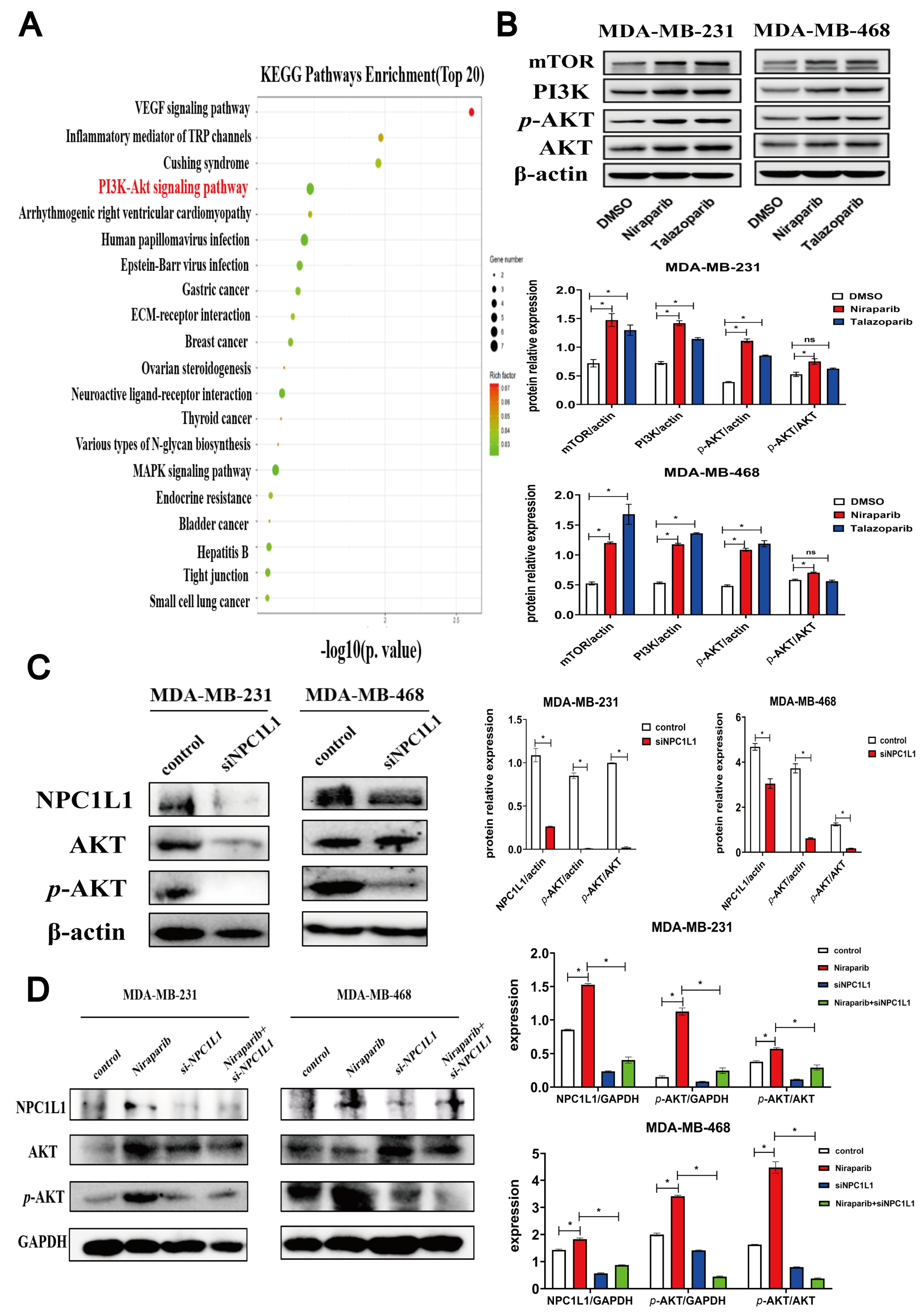
3.3. PARP Inhibitors Activate the AKT Signaling Pathway Through NPC1L1
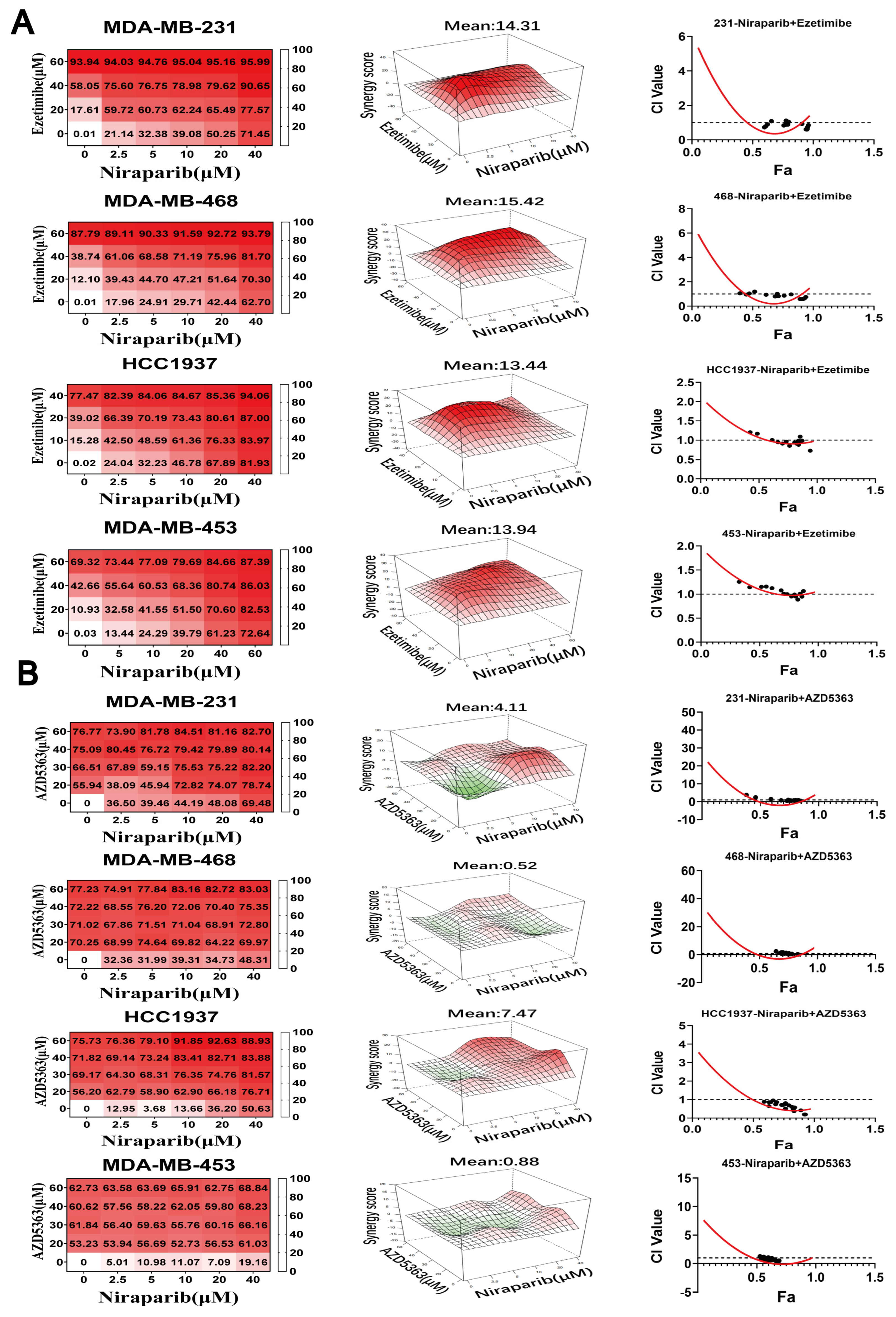
3.4. NPC1L1 or AKT Inhibition Synergizes with PARP Inhibitors to Suppress TNBC Proliferation
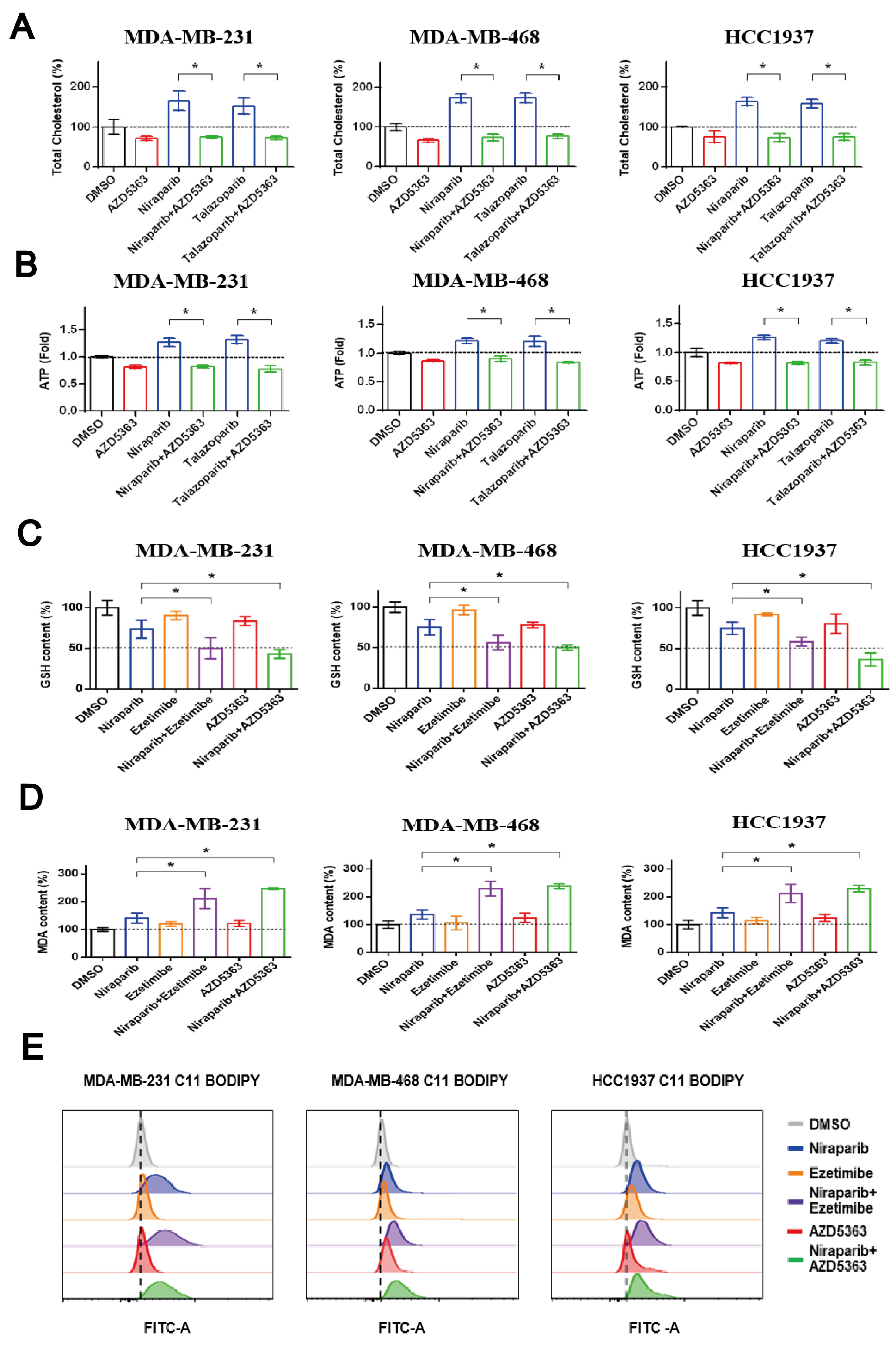
3.5. Co-Targeting PARP and NPC1L1 or AKT Signaling Vulnerabilities Triggers Synergistic Ferroptosis in TNBC
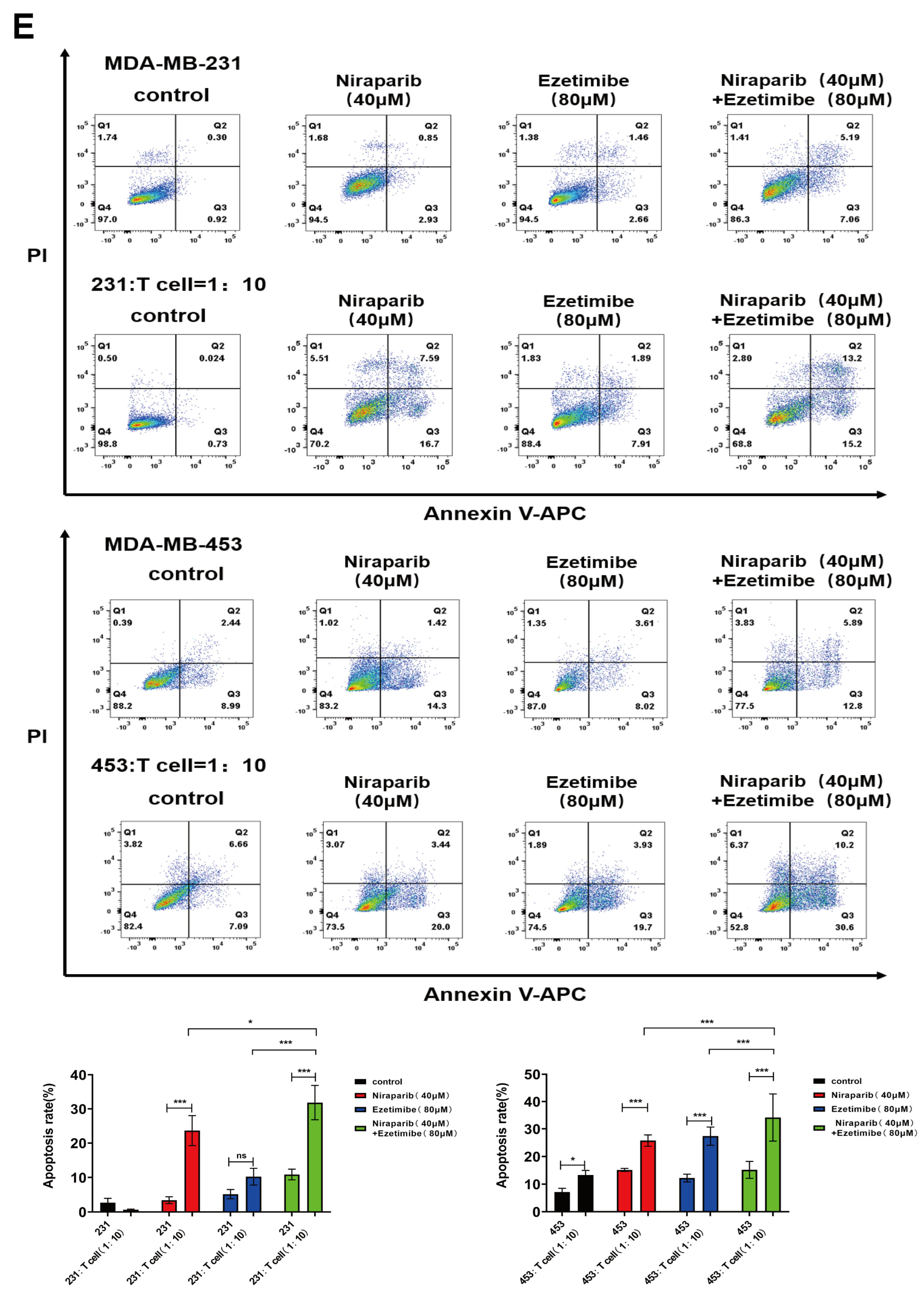
3.6. The Combination of PARP Inhibitor and NPC1L1 Inhibitor Enhances the Killing Effect of T Cells on Tumor Cells
3.7. Evaluation of the Antitumor Activity of PARP Inhibitor Niraparib in Combination with AZD5363 or Ezetimibe in a 4T1 Tumor-Bearing Mouse Model
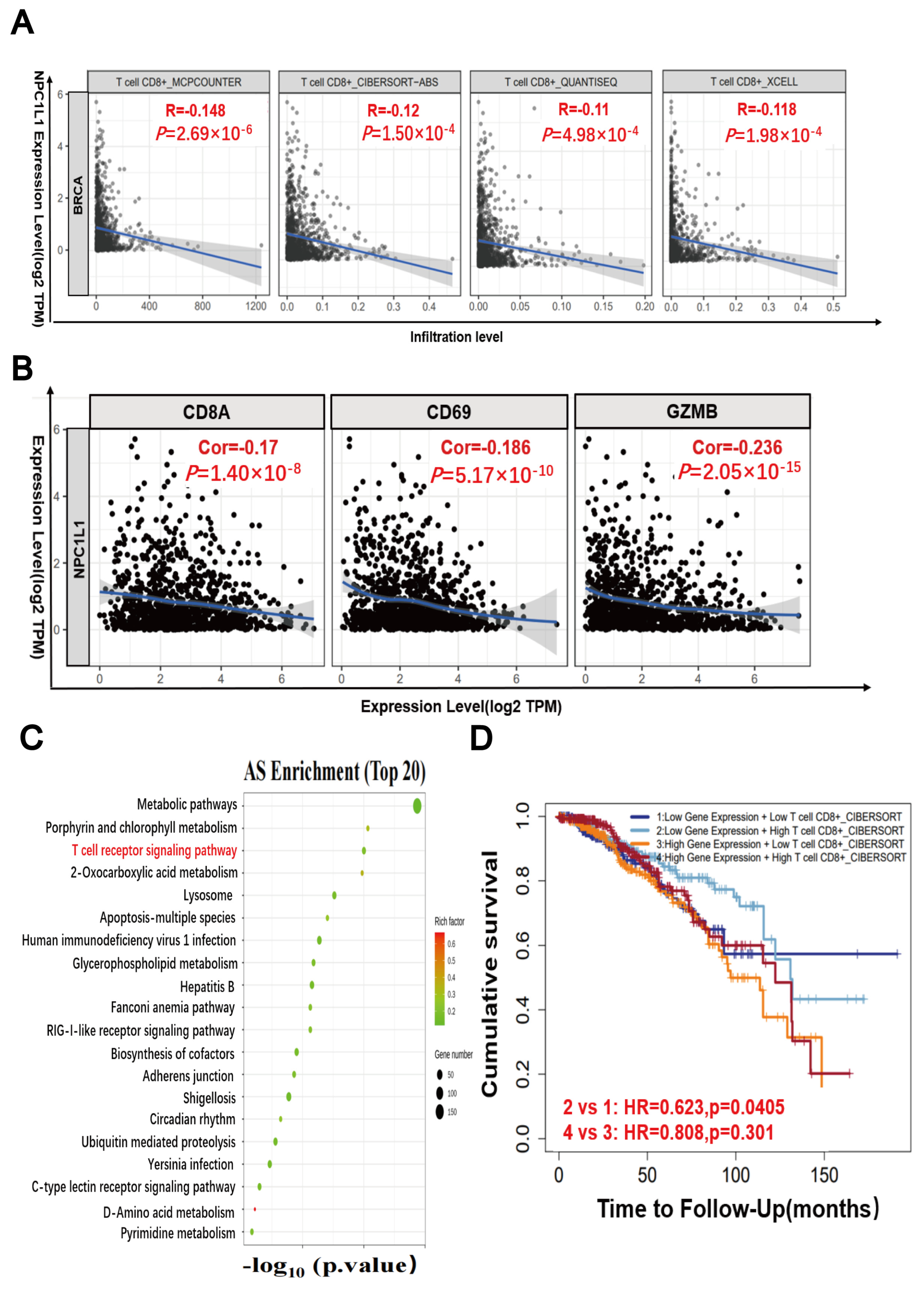
3.8. The Combined Use of PARP Inhibitors and NPC1L1 Inhibitors Activates the In Vivo Immune-Mediated Tumor-Killing Effect
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| poly (ADP-ribose) polymerase PARP | PARP |
| triple-negative breast cancer | TNBC |
| polyunsaturated fatty acid | PUFA |
| homologous recombination deficiency | HRD |
| 7-dehydrocholesterol | 7-DHC |
| 7-dehydrocholesterol reductase | DHCR7 |
| Niemann-Pick C1-Like 1 | NPC1L1 |
| tumor microenvironment | TME |
| lipid peroxidation | LPO |
| methyl tert-butyl ether | MTBE |
| glutathione | GSH |
| oxidized glutathione | GSSG |
| 5,5′-dithiobis (2-nitrobenzoic acid) | DTNB |
| 1-methyl-2-vinylpyridinium triflate | M2VP |
| thiobarbituric acid | TBA |
| malondialdehyde | MDA |
| phosphate-buffered saline | PBS |
| 3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide | MTT |
| peripheral blood mononuclear cells | PBMCs |
| fetal bovine serum | FBS |
| carboxyfluorescein succinimidyl ester | CFSE |
| Principal component analysis | PCA |
| partial least squares-discriminant analysis | PLS-DA |
| fold change | FC |
| phosphorylated AKT | p-AKT |
| Combination Index | CI |
| phosphatidylcholine | PC |
| phosphatidylethanolamine | PE |
| coding sequence | CDS |
| highest single agent | HSA |
| Tumor Immune Estimation Resource | TIMER |
| carboxyfluorescein diacetate succinimidyl ester | CFSE |
| reactive oxygen species | ROS |
| fold change | FC |
| cholesterol esters | ChE |
| endosomal recycling compartment | ERC |
| T cell immunoglobulin domain and mucin domain-3 | TIM-3 |
| Programmed death-ligand 1 | PD-L1 |
| Cytotoxic T-lymphocyte-associated protein 4 | CTLA-4 |
| stimulator of interferon genes | STING |
| interferon | IFN |
References
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Kaye, S.; Banerjee, S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat. Rev. Clin. Oncol. 2017, 14, 284–296. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Szanto, M.; Gupte, R.; Kraus, W.L.; Pacher, P.; Bai, P. PARPs in lipid metabolism and related diseases. Prog. Lipid Res. 2021, 84, 101117. [Google Scholar] [CrossRef]
- Szanto, M.; Bai, P. The role of ADP-ribose metabolism in metabolic regulation, adipose tissue differentiation, and metabolism. Genes Dev. 2020, 34, 321–340. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.-Y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.P.; Alborzinia, H.; dos Santos, A.F.; Nepachalovich, P.; Pedrera, L.; Zilka, O.; Inague, A.; Klein, C.; Aroua, N.; Kaushal, K.; et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature 2024, 626, 401–410. [Google Scholar] [CrossRef]
- Li, Y.; Ran, Q.; Duan, Q.; Jin, J.; Wang, Y.; Yu, L.; Wang, C.; Zhu, Z.; Chen, X.; Weng, L.; et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature 2024, 626, 411–418. [Google Scholar] [CrossRef]
- Zhao, X.; Lian, X.; Xie, J.; Liu, G. Accumulated cholesterol protects tumors from elevated lipid peroxidation in the microenvironment. Redox Biol. 2023, 62, 102678. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.-L. Cholesterol-ferroptosis nexus: Unveiling novel cancer therapeutic avenues. Cancer Lett. 2024, 597, 217046. [Google Scholar] [CrossRef]
- Zhang, R.; Zeng, J.; Liu, W.; Meng, J.; Wang, C.; Shi, L.; Yang, S.; Chang, J.; Xing, D. The role of NPC1L1 in cancer. Front. Pharmacol. 2022, 13, 956619. [Google Scholar] [CrossRef]
- Sun, L.; Ding, H.; Jia, Y.; Shi, M.; Guo, D.; Yang, P.; Wang, Y.; Liu, F.; Zhang, Y.; Zhu, Z. Associations of genetically proxied inhibition of HMG-CoA reductase, NPC1L1, and PCSK9 with breast cancer and prostate cancer. Breast Cancer Res. 2022, 24, 12. [Google Scholar] [CrossRef]
- He, Q.; Kong, L.; Shi, W.; Ma, D.; Liu, K.; Yang, S.; Xin, Q.; Jiang, C.; Wu, J. Ezetimibe inhibits triple-negative breast cancer proliferation and promotes cell cycle arrest by targeting the PDGFR/AKT pathway. Heliyon 2023, 9, e21343. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Ji, F.; Zhang, F.; Zhang, M.; Long, K.; Xia, M.; Lu, F.; Li, E.; Chen, J.; Li, J.; Chen, Z.; et al. Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J. Hematol. Oncol. 2021, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lin, S.-S.; Yu, Z.-L.; Wu, X.-H.; Liu, J.-W.; Tu, G.-H.; Liu, Q.-Y.; Tang, Y.-L.; Jiang, Q.-N.; Xu, J.-H.; et al. A PARP1 PROTAC as a novel strategy against PARP inhibitor resistance via promotion of ferroptosis in p53-positive breast cancer. Biochem. Pharmacol. 2022, 206, 115329. [Google Scholar] [CrossRef]
- Song, L.; Liu, Z.; Hu, H.-H.; Yang, Y.; Li, T.Y.; Lin, Z.-Z.; Ye, J.; Chen, J.; Huang, X.; Liu, D.-T.; et al. Proto-oncogene Src links lipogenesis via lipin-1 to breast cancer malignancy. Nat. Commun. 2020, 11, 5842. [Google Scholar] [CrossRef]
- Gong, Y.; Ji, P.; Yang, Y.-S.; Xie, S.; Yu, T.-J.; Xiao, Y.; Jin, M.-L.; Ma, D.; Guo, L.-W.; Pei, Y.-C.; et al. Metabolic-Pathway-Based Subtyping of Triple-Negative Breast Cancer Reveals Potential Therapeutic Targets. Cell Metab. 2021, 33, 51–64.e9. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Field, F.J.; Watt, K.; Mathur, S.N. Ezetimibe interferes with cholesterol trafficking from the plasma membrane to the endoplasmic reticulum in CaCo-2 cells. J. Lipid Res. 2007, 48, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 2013, 61, 95–110. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Westin, S.N.; Labrie, M.; Litton, J.K.; Blucher, A.; Fang, Y.; Vellano, C.P.; Marszalek, J.R.; Feng, N.; Ma, X.; Creason, A.; et al. Phase Ib Dose Expansion and Translational Analyses of Olaparib in Combination with Capivasertib in Recurrent Endometrial, Triple-Negative Breast, and Ovarian Cancer. Clin. Cancer Res. 2021, 27, 6354–6365. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Wang, X.Q.; Danenberg, E.; Huang, C.-S.; Egle, D.; Callari, M.; Bermejo, B.; Dugo, M.; Zamagni, C.; Thill, M.; Anton, A.; et al. Spatial predictors of immunotherapy response in triple-negative breast cancer. Nature 2023, 621, 868–876. [Google Scholar] [CrossRef]
- Qin, S.; Dong, B.; Yi, M.; Chu, Q.; Wu, K. Prognostic Values of TIM-3 Expression in Patients With Solid Tumors: A Meta-Analysis and Database Evaluation. Front. Oncol. 2020, 10, 1288. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Li, Q.; Liu, J.; Zhang, Q.; Ouyang, Q.; Zhang, Y.; Liu, Q.; Sun, T.; Ye, F.; Zhang, B.; Xia, S.; et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 in combination with nab-paclitaxel in first-line treatment of metastatic triple-negative breast cancer: A multicenter phase II trial. Nat. Commun. 2024, 15, 1015. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef]
- Nelson, E.R.; Chang, C.-Y.; McDonnell, D.P. Cholesterol and breast cancer pathophysiology. Trends Endocrinol. Metab. 2014, 25, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Huang, C.-H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P., IV; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.W.; Yu, S.-Y.; Teng, T.-H.K.; Li, X.; Cheung, K.-S.; Wu, M.-Z.; Li, H.-L.; Wong, P.-F.; Tse, H.-F.; Lam, C.S.P.; et al. Statin associated lower cancer risk and related mortality in patients with heart failure. Eur. Heart J. 2021, 42, 3049–3059. [Google Scholar] [CrossRef]
- Nowakowska, M.K.; Lei, X.; Thompson, M.T.; Shaitelman, S.F.; Wehner, M.R.; Woodward, W.A.; Giordano, S.H.; Nead, K.T. Association of statin use with clinical outcomes in patients with triple-negative breast cancer. Cancer 2021, 127, 4142–4150. [Google Scholar] [CrossRef]
- Ahern, T.P.; Lash, T.L.; Damkier, P.; Christiansen, P.M.; Cronin-Fenton, D.P. Statins and breast cancer prognosis: Evidence and opportunities. Lancet Oncol. 2014, 15, e461–e468. [Google Scholar] [CrossRef]
- Kobbero Lauridsen, B.; Stender, S.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Using genetics to explore whether the cholesterol-lowering drug ezetimibe may cause an increased risk of cancer. Int. J. Epidemiol. 2017, 46, 1777–1785. [Google Scholar] [CrossRef]
- Ferrari, P.; Scatena, C.; Ghilli, M.; Bargagna, I.; Lorenzini, G.; Nicolini, A. Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. Int. J. Mol. Sci. 2022, 23, 1665. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Bil, J. Statins and Cancer: A Complex Relationship Worth Exploring. Pharmaceuticals 2023, 16, 1570. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef] [PubMed]
- de Souza, J.C.; Pimenta, T.M.; Martins, B.d.S.; Junior, R.S.R.; Butzene, S.M.S.; Tessarolo, N.G.; Cilas, P.M.L.; Silva, I.V.; Rangel, L.B.A. The impact of lipid metabolism on breast cancer: A review about its role in tumorigenesis and immune escape. Cell Commun. Signal. 2023, 21, 161. [Google Scholar]
- Wang, Y.; You, S.; Su, S.; Yeon, A.; Lo, E.M.; Kim, S.; Mohler, J.L.; Freeman, M.R.; Kim, H.L. Cholesterol-Lowering Intervention Decreases mTOR Complex 2 Signaling and Enhances Antitumor Immunity. Clin. Cancer Res. 2022, 28, 414–424. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, Y.; Shen, L.; Xiao, T.; Cao, R.; Wei, S.; Tang, M.; Du, L.; Wu, H.; Wu, B.; et al. Regulation of PD-L1 through direct binding of cholesterol to CRAC motifs. Sci. Adv. 2022, 8, eabq4722. [Google Scholar] [CrossRef]
- Lim, W.J.; Lee, M.; Oh, Y.; Fang, X.-Q.; Lee, S.; Lim, C.-H.; Park, J.; Lim, J.-H. Statins Decrease Programmed Death-Ligand 1 (PD-L1) by Inhibiting AKT and beta-Catenin Signaling. Cells 2021, 10, 2488. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Direction | Sequence (5′→3′) |
|---|---|---|
| PARP1-(homo) | Forward | CTTGACAACCTGCTGGACAT |
| Reverse | CCTGATGATCTCGGCTTCTTC | |
| NPC1L1-(homo) | Forward | GAGCTGCATGGCTGACTACG |
| Reverse | CCGCAGGGTAATTGTTGAG |
| Gene | Primer Direction | Sequence (5′→3′) |
|---|---|---|
| PARP1-Homo-1 | sense | GGAUGGGUUCUCUGAGCUUTT |
| antisense | AAGCUCAGAGAACCCAUCCTT | |
| PARP1-Homo-2 | sense | GAGGAAGGUAUCAACAAAUTT |
| antisense | AUUUGUUGAUACCUUCCUCTT | |
| NPC1L1-Homo | sense | GAGGCCUUCUUAGAGGAAATT |
| antisense | UUUCCUCUAAGAAGGCCUCTT |
| Gene | Primer Direction | Sequence (5′→3′) |
|---|---|---|
| RELA (103374-1) | sense | GATTGAGGAGAAACGTAAA |
| antisense | TTTACGTTTCTCCTCAATC | |
| RELA (103375-1) | sense | CTTAATAGTAGGGTAAGTT |
| antisense | AACTTACCCTACTATTAAG |
| Cell Lines | Niraparib (μM) | Ezetimibe (μM) | AZD5363 (μM) | Niraparib (μM) + Ezetimibe (μM) | Niraparib (μM) + AZD5363 (μM) |
|---|---|---|---|---|---|
| MDA-MB-231 | 20.52 | 31.76 | 14.26 | 9.18/13.77 | 7.84/11.76 |
| MDA-MB-453 | 30.56 | 44.34 | 10.28 | 18.76/18.76 | 7.27/10.90 |
| MDA-MB-468 | 23.79 | 38.03 | 10.57 | 12.36/18.54 | 2.04/3.06 |
| HCC1937 | 49.37 | 23.12 | 12.79 | 7.42/7.42 | 7.73/11.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Yuan, Y.; Wu, X.; Wu, L. Modulating NPC1L1 to Potentiate PARP Inhibitor-Induced Ferroptosis and Immune Response in Triple-Negative Breast Cancer. Pharmaceutics 2025, 17, 554. https://doi.org/10.3390/pharmaceutics17050554
Li G, Yuan Y, Wu X, Wu L. Modulating NPC1L1 to Potentiate PARP Inhibitor-Induced Ferroptosis and Immune Response in Triple-Negative Breast Cancer. Pharmaceutics. 2025; 17(5):554. https://doi.org/10.3390/pharmaceutics17050554
Chicago/Turabian StyleLi, Ge, Yuxia Yuan, Xinhua Wu, and Lixian Wu. 2025. "Modulating NPC1L1 to Potentiate PARP Inhibitor-Induced Ferroptosis and Immune Response in Triple-Negative Breast Cancer" Pharmaceutics 17, no. 5: 554. https://doi.org/10.3390/pharmaceutics17050554
APA StyleLi, G., Yuan, Y., Wu, X., & Wu, L. (2025). Modulating NPC1L1 to Potentiate PARP Inhibitor-Induced Ferroptosis and Immune Response in Triple-Negative Breast Cancer. Pharmaceutics, 17(5), 554. https://doi.org/10.3390/pharmaceutics17050554