

Oral Metronomic Delivery of Atorvastatin and Docetaxel via Transporter-Targeted Nanoemulsions Enhances Antitumor Efficacy and Immune Modulation in Colon Cancer

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Preparation and Characterization of AT/DT-NE
2.4. In Vitro Artificial Intestinal Membrane Permeability of AT/DT-NE
2.5. In Vitro Caco-2 Cell Monolayer Permeability of AT/DT-NE
2.6. In Vitro Cellular Uptake Study in Caco-2 and MDCK Cells
2.7. In Vitro Cytotoxicity Study
2.8. In Vitro Drug Release Study
2.9. In Situ Single-Pass Rat Intestinal Perfusion Study
2.10. In Vivo Pharmacokinetics in Rats
2.11. In Vivo Antitumor Efficacy of Oral MCT AT/DT-NE in CT26.CL25 Cell-Bearing Mice
2.12. In Vivo Immune Modulatory Effects of ATV/DTX-NEs
2.13. Pharmacokinetics and Statistical Analyses
3. Results
3.1. Preparation and Characterization of AT/DT-NEs

3.2. In Vitro Permeability of AT/DT-NEs
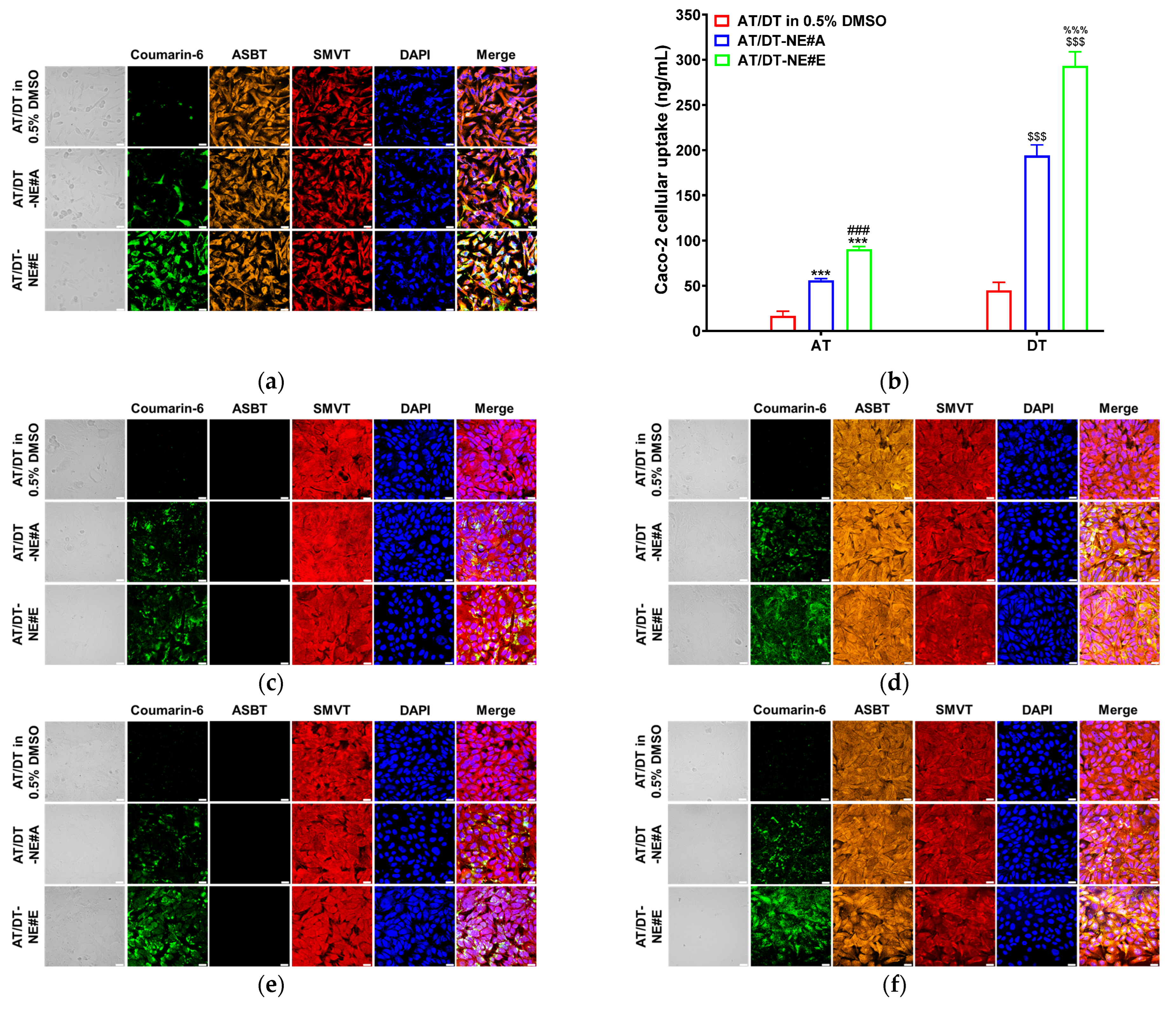
3.3. In Vitro Cellular Uptake Study
3.4. In Vitro Cytotoxicity Study of AT/DT-NEs
3.5. In Vitro Drug Dissolution of AT/DT-NE#E-Loaded Capsules
3.6. In Situ Single-Pass Intestinal Perfusion of AT/DT-NE in Rats
3.7. In Vivo Pharmacokinetic Study of AT/DT-NE in Rats
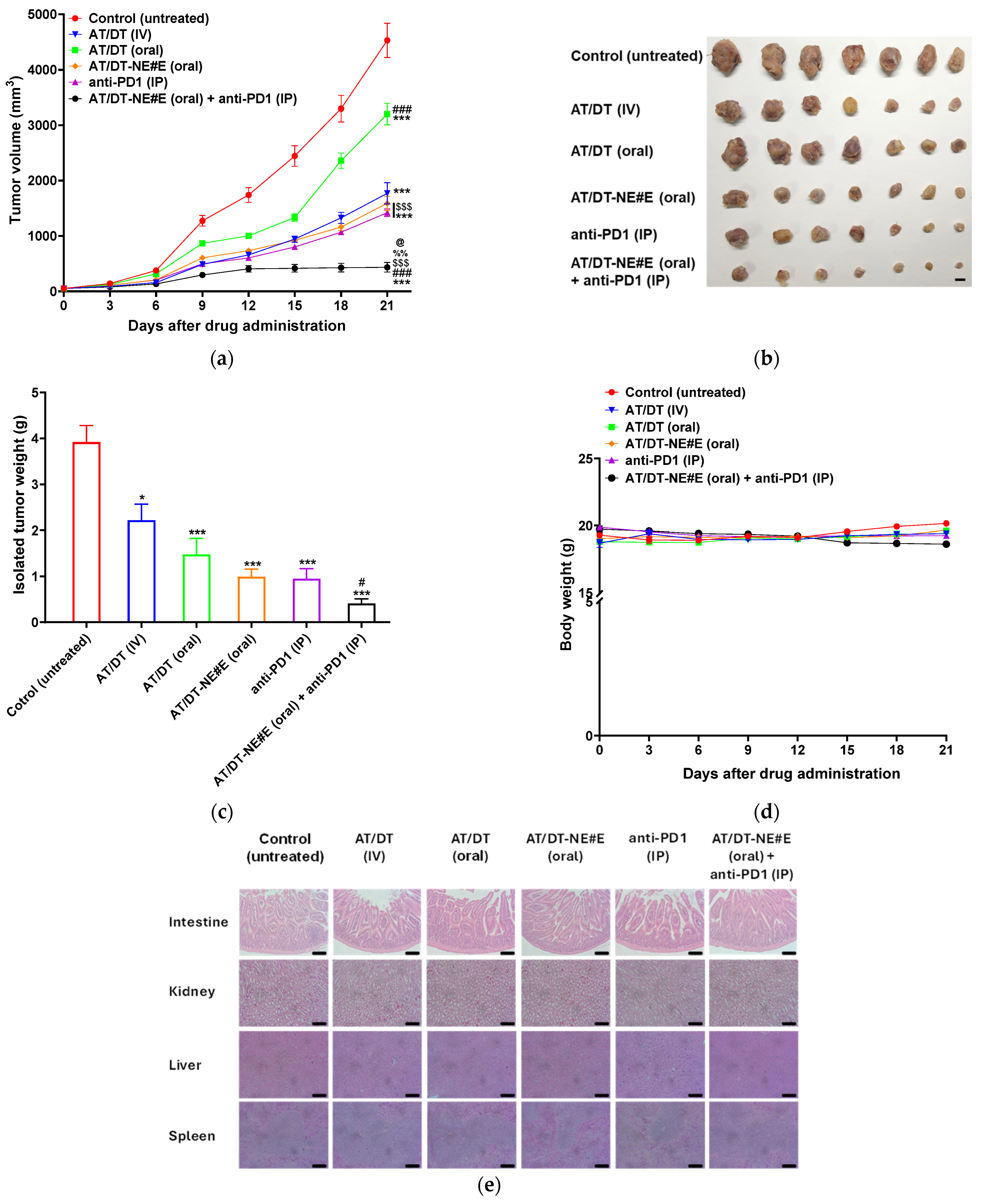
3.8. In Vivo Tumor Growth Inhibitory Effect of Metronomic Oral AT/DT-NEs in CT26.CL25 Cell-Bearing Mice
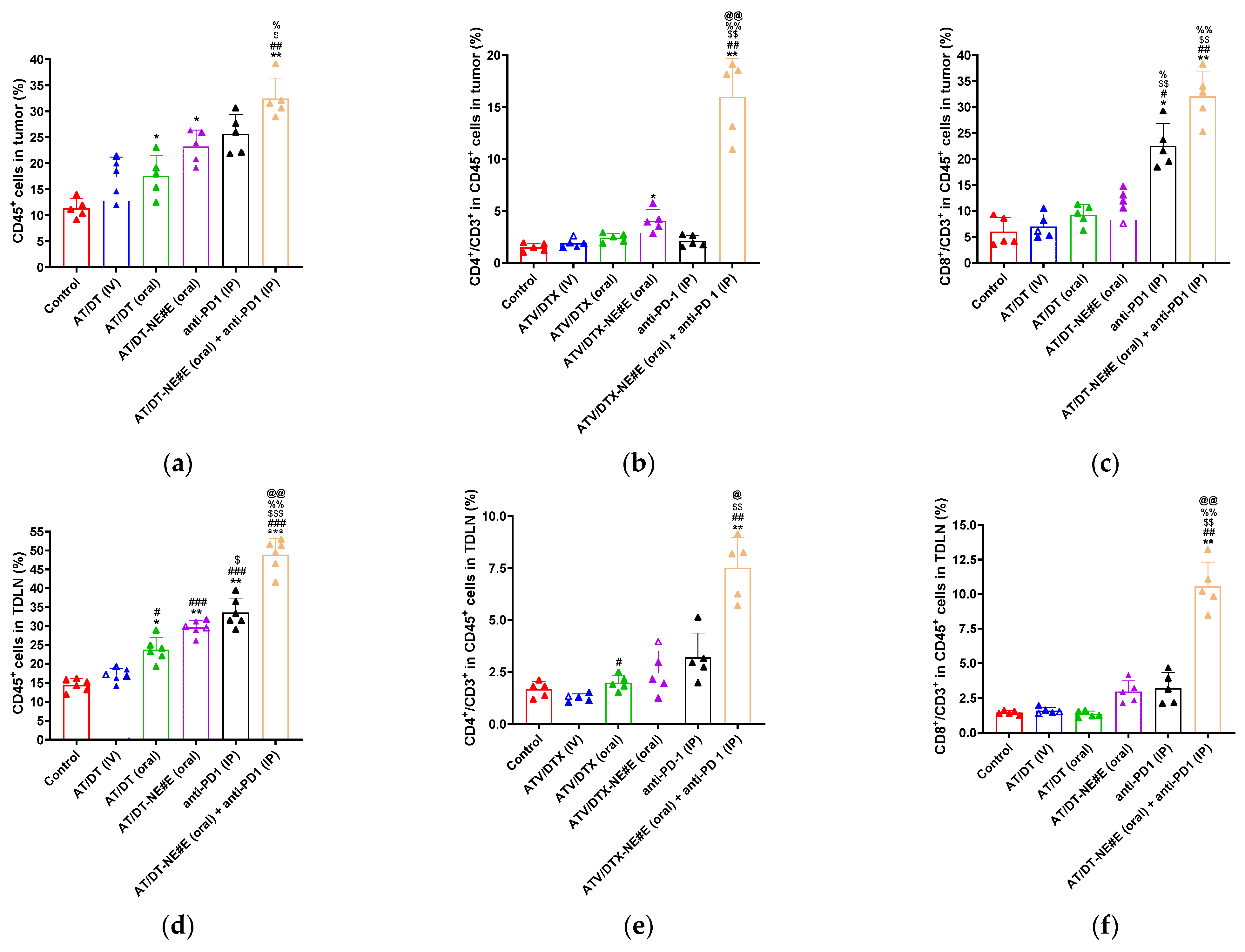
3.9. In Vivo Evaluation of Immune Modulatory Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AT | Atorvastatin |
| DT | Docetaxel |
| NE | Nanoemulsion |
| ASBT | Apical sodium-dependent bile acid transporter |
| SMVT | Sodium multivitamin transporter |
| DA | Deoxycholic acid |
| DOTAP | 1,2-dioleyl-3-trimethylammonium propane |
| D-TAP | Ionic complex of DA with DOTAP |
| Biotin-PE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) |
| TPGS | d-α-tocopherol polyethylene glycol succinate |
| TME | Tumor microenvironment |
| MCT | Metronomic chemotherapy |
| MTD | Maximum tolerated dose |
| P-gp | P-glycoprotein |
| DMSO | Dimethyl sulfoxide |
| PA | Pantothenic acid |
| DSC | Differential scanning calorimetry |
| FT-IR | Fourier transform infrared spectroscopy |
| Smix | Surfactant and co-surfactant mixture |
| DLS | Dynamic light scattering |
| TEM | Transmission electron microscopy |
| Pe | Effective permeability across the artificial membrane |
| Papp | Apparent permeability across the Caco-2/HT29 MTXE12 cell monolayer |
| DAPI | 4′,6-diamidino-2-phenylindole |
| MDCKs | Madin–Darby canine kidney cells |
| ASBT (–) | ASBT-non-transfected MDCK cells |
| ASBT (+) | ASBT-transfected MDCK cells |
| SMVT (–) | SMVT-inhibited MDCK cells |
| SMVT (+) | SMVT-expressed MDCK cells |
| Peff | Intestinal effective permeability |
| IV | Intravenous |
| IP | Intraperitoneal |
| Anti-PD1 | Anti-programmed cell death protein 1 antibody |
| TDLN | Tumor-draining lymph node |
| ICD | Immunogenic cell death |
| DAMP | Damage-associated molecular pattern |
References
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.-Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Ricco, N.; Kron, S.J. Statins in Cancer Prevention and Therapy. Cancers 2023, 15, 3948. [Google Scholar] [CrossRef] [PubMed]
- Ferkel, S.A.M.; Holman, E.A.; Sojwal, R.S.; Rubin, S.J.S.; Rogalla, S. Tumor-Infiltrating Immune Cells in Colorectal Cancer. Neoplasia 2025, 59, 101091. [Google Scholar] [CrossRef]
- Meyiah, A.; Khan, F.I.; Alfaki, D.A.; Murshed, K.; Raza, A.; Elkord, E. The colorectal cancer microenvironment: Preclinical progress in identifying targets for cancer therapy. Transl. Oncol. 2025, 53, 102307. [Google Scholar] [CrossRef]
- Skavatsou, E.; Semitekolou, M.; Morianos, I.; Karampelas, T.; Lougiakis, N.; Xanthou, G.; Tamvakopoulos, C. Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC. Cancers 2021, 13, 1901. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, Y.; Si, C.; Cao, Y.; Shao, P.; Zhang, P.; Wang, N.; Su, G.; Qian, J.; Yang, L. Cholesterol: The driving force behind the remodeling of tumor microenvironment in colorectal cancer. Heliyon 2024, 10, e39425. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Tripathi, S.; Gupta, E.; Galande, S. Statins as anti-tumor agents: A paradigm for repurposed drugs. Cancer Rep. 2024, 7, e2078. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Reimers, M.S.; Bastiaannet, E.; Jacobs, R.J.; van Eijk, R.; Zanders, M.M.J.; Herings, R.M.C.; van Herk-Sukel, M.P.P.; Kodach, L.L.; van Wezel, T.; et al. Statin Use After Diagnosis of Colon Cancer and Patient Survival. J. Gastroenterol. 2017, 153, 470–479. [Google Scholar] [CrossRef]
- Liu, C.; Chen, H.; Hu, B.; Shi, J.; Chen, Y.; Huang, K. New insights into the therapeutic potentials of statins in cancer. Front. Pharmacol. 2023, 14, 1188926. [Google Scholar] [CrossRef] [PubMed]
- Gligorov, J.; Lotz, J.-P. Preclinical Pharmacology of the Taxanes: Implications of the Differences. Oncol. 2004, 9, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Engels, F.K.; Sparreboom, A.; Mathot, R.A.A.; Verweij, J. Potential for improvement of docetaxel-based chemotherapy: A pharmacological review. Br. J. Cancer 2005, 93, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Manjappa, A.S.; Goel, P.N.; Vekataraju, M.P.; Rajesh, K.S.; Makwana, K.; Ukawala, M.; Nikam, Y.; Gude, R.P.; Murthy, R.S.R. Is an Alternative Drug Delivery System Needed for Docetaxel? The Role of Controlling Epimerization in Formulations and Beyond. Pharm. Res. 2013, 30, 2675–2693. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, J. Maximum tolerated dose and toxicity evaluation of orally administered docetaxel granule in mice. Toxicol. Rep. 2024, 12, 430–435. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Wu, J.; Huang, H.; Du, Z.; Zhang, K.; Zhou, D.; Hung, K.; Goodin, S.; Zheng, X. Mechanistic Study of Inhibitory Effects of Atorvastatin and Docetaxel in Combination on Prostate Cancer. Cancer Genom. Proteom. 2016, 13, 151–160. [Google Scholar]
- Jung, E.J.; Kim, C.W. 16P Simvastatin enhances docetaxel-induced cell death in DU145 prostate cancer cells. Ann. Oncol. 2015, 26, ix1. [Google Scholar] [CrossRef]
- Tilija Pun, N.; Jeong, C.H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals 2021, 14, 470. [Google Scholar] [CrossRef]
- Iannelli, F.; Roca, M.S.; Lombardi, R.; Ciardiello, C.; Grumetti, L.; De Rienzo, S.; Moccia, T.; Vitagliano, C.; Sorice, A.; Costantini, S.; et al. Synergistic antitumor interaction of valproic acid and simvastatin sensitizes prostate cancer to docetaxel by targeting CSCs compartment via YAP inhibition. J. Exp. Clin. Cancer Res. 2020, 39, 213. [Google Scholar] [CrossRef]
- Razak, S.A.A.; Mohd Gazzali, A.; Fisol, F.A.; Abdulbaqi, I.M.; Parumasivam, T.; Mohtar, N.; Wahab, H.A. Advances in nanocarriers for effective delivery of docetaxel in the treatment of lung cancer: An overview. Cancers 2021, 13, 400. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Chung, J.Y.; Pangeni, R.; Choi, H.S.; Subedi, L.; Kweon, S.; Choi, J.U.; Byun, Y.; Kim, Y.-H.; Park, J.W. Enhanced antitumor efficacy of bile acid-lipid complex-anchored docetaxel nanoemulsion via oral metronomic scheduling. J. Control. Release 2020, 328, 368–394. [Google Scholar] [CrossRef]
- Subedi, L.; Pandey, P.; Khadka, B.; Shim, J.H.; Cho, S.S.; Kweon, S.; Byun, Y.; Kim, K.T.; Park, J.W. Enhancement of the anticancer effect of atorvastatin-loaded nanoemulsions by improving oral absorption via multivalent intestinal transporter-targeting lipids. Drug Deliv. 2022, 29, 3397–3413. [Google Scholar] [CrossRef]
- Yeom, D.; Son, H.; Kim, J.; Kim, S.; Lee, S.; Song, S.; Chae, B.; Choi, Y. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int. J. Pharm. 2016, 506, 302–311. [Google Scholar] [CrossRef]
- Kim, C.H.; Kang, T.H.; Kim, B.D.; Lee, T.H.; Yoon, H.Y.; Goo, Y.T.; Choi, Y.S.; Kang, M.J.; Choi, Y.W. Enhanced docetaxel delivery using sterically stabilized RIPL peptide-conjugated nanostructured lipid carriers: In vitro and in vivo antitumor efficacy against SKOV3 ovarian cancer cells. Int. J. Pharm. 2020, 583, 119393. [Google Scholar] [CrossRef]
- Plaza-Oliver, M.; Santander-Ortega, M.J.; Lozano, M.V. Current approaches in lipid-based nanocarriers for oral drug delivery. Drug Deliv. Transl. Res. 2021, 11, 471–497. [Google Scholar] [CrossRef]
- Collnot, E.-M.; Baldes, C.; Schaefer, U.F.; Edgar, K.J.; Wempe, M.F.; Lehr, C.-M. Vitamin E TPGS P-Glycoprotein Inhibition Mechanism: Influence on Conformational Flexibility, Intracellular ATP Levels, and Role of Time and Site of Access. Mol. Pharm. 2010, 7, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Foucand, L.; Nakamura, Y.; Niot, I.; Besnard, P.; Fujita, M.; Sakai, Y.; Hatakeyama, K.; Ono, T.; Fujii, H. Regulation of expression of human intestinal bile acid-binding protein in Caco-2 cells. Biochem. J. 1998, 330, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, N.; Nigam, S.K. Drug transporters OAT1 and OAT3 have specific effects on multiple organs and gut microbiome as revealed by contextualized metabolic network reconstructions. Sci. Rep. 2022, 12, 18308. [Google Scholar] [CrossRef]
- Aapro, M.; Bruno, R. Early clinical studies with docetaxel. Docetaxel Investigators Group. Eur. J. Cancer 1995, 31 (Suppl. 4), S7–S10. [Google Scholar] [CrossRef]
- Nishiyama, M.; Wada, S. Docetaxel: Its role in current and future treatments for advanced gastric cancer. Gastric Cancer 2009, 12, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Komal; Kumar, D.; Khan, S.R.; Kaur, P.; Majumdar, K.; Chakraborty, N.; Singh, A. Advancements in Colon Cancer Therapy: A Review on Docetaxel and the Role of Nanotechnology in Enhancing Efficacy. Curr. Drug Saf. 2025; in press. [Google Scholar]
- Shaghaghi, Z.; Alvandi, M.; Farzipour, S.; Dehbanpour, M.R.; Nosrati, S. A review of effects of atorvastatin in cancer therapy. Med. Oncol. 2022, 40, 27. [Google Scholar] [CrossRef]
- Okita, Y.; Sobue, T.; Zha, L.; Kitamura, T.; Iwasaki, M.; Inoue, M.; Yamaji, T.; Tsugane, S.; Sawada, N. Long-term use of anti-cholesterol drugs and cancer risks in a Japanese population. Sci. Rep. 2024, 14, 2896. [Google Scholar] [CrossRef] [PubMed]
- Karimi, S.; Bakhshali, R.; Bolandi, S.; Zahed, Z.; Mojtaba Zadeh, S.S.; Kaveh Zenjanab, M.; Jahanban Esfahlan, R. For and against tumor microenvironment: Nanoparticle-based strategies for active cancer therapy. Mater. Today Bio 2025, 31, 101626. [Google Scholar] [CrossRef]
- Maharjan, R.; Choi, J.U.; Kweon, S.; Pangeni, R.; Lee, N.K.; Park, S.J.; Chang, K.-Y.; Park, J.W.; Byun, Y. A novel oral metronomic chemotherapy provokes tumor specific immunity resulting in colon cancer eradication in combination with anti-PD-1 therapy. Biomaterials 2022, 281, 121334. [Google Scholar] [CrossRef]
- Chen, Q.; Xia, R.; Zheng, W.; Zhang, L.; Li, P.; Sun, X.; Shi, J. Metronomic paclitaxel improves the efficacy of PD-1 monoclonal antibodies in breast cancer by transforming the tumor immune microenvironment. Am. J. Transl. Res. 2020, 12, 519–530. [Google Scholar]
- Koumarianou, A.; Christodoulou, M.-I.; Patapis, P.; Papadopoulos, I.; Liakata, E.; Giagini, A.; Stavropoulou, A.; Poulakaki, N.; Tountas, N.; Xiros, N.; et al. The effect of metronomic versus standard chemotherapy on the regulatory to effector T-cell equilibrium in cancer patients. Exp. Hematol. Oncol. 2014, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Mobarak, D.; Salah, S.; Ghorab, M. Improvement of dissolution of a class II poorly water-soluble drug, by developing a five-component self-nanoemulsifying drug delivery system. J. Drug Deliv. Sci. Technol. 2019, 50, 99–106. [Google Scholar] [CrossRef]
- Tong, L.; Zhou, Z.; Wu, C. A self-microemulsion enhances oral absorption of docetaxel by inhibiting P-glycoprotein and CYP metabolism. Drug Deliv. Transl. Res. 2023, 13, 983–993. [Google Scholar] [CrossRef]
- Yan, Y.-D.; Marasini, N.; Choi, Y.; Kim, J.; Woo, J.; Yong, C.; Han, C. Effect of dose and dosage interval on the oral bioavailability of docetaxel in combination with a curcumin self-emulsifying drug delivery system (SEDDS). Eur. J. Drug Metab. Pharmacokinet. 2011, 37, 217–224. [Google Scholar] [CrossRef]
- Deng, F.; Bae, Y.H. Bile acid transporter-mediated oral drug delivery. J. Control. Release 2020, 327, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.G.; Rapp, S.R.; Beaudry, J.A.; Napawan, N.; Butteiger, D.N.; Hall, K.A.; Null, C.L.; Luo, Y.; Keller, B.T. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE-/- mice by SC-435. J. Lipid Res. 2003, 44, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Vokral, I.; Evers, B.; de Graaf, I.A.M.; de Jager, M.H.; Groothuis, G.M.M. Human and rat precision-cut intestinal slices as ex vivo models to study bile acid uptake by the apical sodium-dependent bile acid transporter. Eur. J. Pharm. Sci. 2018, 121, 65–73. [Google Scholar] [CrossRef]
- Balamurugan, K.; Ortiz, A.; Said, H.M. Biotin uptake by human intestinal and liver epithelial cells: Role of the SMVT system. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 285, G73–G77. [Google Scholar] [CrossRef] [PubMed]
- Giram, P.S.; Nimma, R.; Bulbule, A.; Yadav, A.S.; Gorain, M.; Venkata Radharani, N.N.; Kundu, G.C.; Garnaik, B. Poly(d,l-lactide-co-glycolide) Surface-Anchored Biotin-Loaded Irinotecan Nanoparticles for Active Targeting of Colon Cancer. ACS Omega 2024, 9, 3807–3826. [Google Scholar] [CrossRef]
- Babu, L.T.; Roy, N.; Dasgupta, T.; Ghosh, S.; Tamizhselvi, R.; Paira, P. Engineering biotin anchored-MWCNTs as a superb carrier for facile delivery of the potent Ru(ii)-N^N scaffold in breast cancer cells. Chem. Commun. 2024, 60, 13376–13379. [Google Scholar] [CrossRef]
- Mooney, R.; Weng, Y.; Garcia, E.; Bhojane, S.; Smith-Powell, L.; Kim, S.U.; Annala, A.J.; Aboody, K.S.; Berlin, J.M. Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. J. Control. Release 2014, 191, 82–89. [Google Scholar] [CrossRef]
- Muraro, E.; Vinante, L.; Fratta, E.; Bearz, A.; Höfler, D.; Steffan, A.; Baboci, L. Metronomic Chemotherapy: Anti-Tumor Pathways and Combination with Immune Checkpoint Inhibitors. Cancers 2023, 15, 2471. [Google Scholar] [CrossRef]
- Basar, O.Y.; Mohammed, S.; Qoronfleh, M.W.; Acar, A. Optimizing cancer therapy: A review of the multifaceted effects of metronomic chemotherapy. Front. Cell Dev. Biol. 2024, 12, 1369597. [Google Scholar] [CrossRef]
- Jan, N.; Sofi, S.; Qayoom, H.; Shabir, A.; Haq, B.U.; Macha, M.A.; Almilaibary, A.; Mir, M.A. Metronomic chemotherapy and drug repurposing: A paradigm shift in oncology. Heliyon 2024, 10, e24670. [Google Scholar] [CrossRef]
- Mavroeidis, L.; Sheldon, H.; Briasoulis, E.; Marselos, M.; Pappas, P.; Harris, A.L. Metronomic vinorelbine: Anti-angiogenic activity in vitro in normoxic and severe hypoxic conditions, and severe hypoxia-induced resistance to its anti-proliferative effect with reversal by Akt inhibition. Int. J. Oncol. 2015, 47, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Browder, T.; Butterfield, C.E.; Kräling, B.M.; Shi, B.; Marshall, B.; O’Reilly, M.S.; Folkman, J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000, 60, 1878–1886. [Google Scholar] [PubMed]
- Hong, J.H.; Woo, I.S. Metronomic chemotherapy as a potential partner of immune checkpoint inhibitors for metastatic colorectal cancer treatment. Cancer Lett. 2023, 565, 216236. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | AT (mg) | DT (mg) | Capryol 90 (mg) | Tween 80 (mg) | Transcutol HP (mg) | TPGS (mg) | D-TAP (mg) | Biotin-PE (mg) |
|---|---|---|---|---|---|---|---|---|
| AT/DT-NE#A | 2.5 | 10 | 42 | 250 | 500 | |||
| AT/DT-NE#B | 2.5 | 10 | 42 | 250 | 500 | 20 | ||
| AT/DT-NE#C | 2.5 | 10 | 42 | 250 | 500 | 20 | 16.83 | |
| AT/DT-NE#D | 2.5 | 10 | 42 | 250 | 500 | 20 | 15 | |
| AT/DT-NE#E | 2.5 | 10 | 42 | 250 | 500 | 20 | 16.83 | 15 |
| Test Material | Particle Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) | Aqueous Solubility (mg/mL) | |
|---|---|---|---|---|---|
| AT | DT | ||||
| AT/DT in water | 0.52 ± 0.021 | 3.06 ± 0.016 | |||
| AT/DT in 0.5% DMSO | 2.81 ± 0.011 | 12.4 ± 0.301 | |||
| AT/DT-NE#A | 12.9 ± 0.087 | 0.114 ± 0.001 | −2.87 ± 0.161 | 3.16 ± 0.025 | 12.6 ± 0.061 |
| AT/DT-NE#B | 13.0 ± 0.021 | 0.112 ± 0.008 | −3.35 ± 0.017 | 3.15 ± 0.010 | 12.6 ± 0.145 |
| AT/DT-NE#C | 13.1 ± 0.072 | 0.100 ± 0.004 | 1.93 ± 0.046 | 3.15 ± 0.011 | 12.6 ± 0.046 |
| AT/DT-NE#D | 13.1 ± 0.070 | 0.104 ± 0.012 | −6.62 ± 0.117 | 3.16 ± 0.038 | 12.6 ± 0.055 |
| AT/DT-NE#E | 13.0 ± 0.035 | 0.112 ± 0.003 | −4.08 ± 0.076 | 3.16 ± 0.023 | 12.6 ± 0.099 |
| Test Material | AT-IV | AT-Oral | AT/DT-NE#E | DT-IV | DT-Oral | AT/DT-NE#E |
|---|---|---|---|---|---|---|
| Administration route | IV | Oral | Oral | IV | Oral | Oral |
| AT dose (mg/kg) | 2.5 | 2.5 | 2.5 | |||
| DT dose (mg/kg) | 5 | 10 | 10 | |||
| Tmax (h) | 6.00 ± 0.000 | 4.00 ± 0.000 | 8.00 ± 0.000 | 6.67 ± 1.15 | ||
| T1/2 (h) | 0.703 ± 0.406 | 28.0 ± 11.7 | 23.5 ± 13.0 | 8.33 ± 7.84 | 8.67 ± 0.881 | 7.90 ± 0.921 |
| Cmax (ng/mL) | 250 ± 62.2 | 2.78 ± 0.200 | 11.5 ± 0.637 | 479 ± 45.3 | 21.0 ± 5.17 | 122 ± 12.4 |
| AUClast (ng·h/mL) | 204 ± 45.6 | 39.7 ± 2.96 | 86.4 ± 12.5 | 1450 ± 109 | 210 ± 26.8 | 997 ± 77.9 |
| AUCinf (ng·h/mL) | 205 ± 45.7 | 90.7 ± 19.9 | 136 ± 12.7 | 4191 ± 264 | 264 ± 38.3 | 1229 ± 74.8 |
| Bioavailability (%) | 100 | 19.4 ± 1.45 | 42.3 ± 6.11 | 100 | 7.22 ± 0.924 | 34.4 ± 2.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, L.; Bamjan, A.D.; Phuyal, S.; Khadka, B.; Chaudhary, M.; Kim, K.-T.; Kim, K.H.; Shim, J.-H.; Cho, S.-S.; Yu, J.E.; et al. Oral Metronomic Delivery of Atorvastatin and Docetaxel via Transporter-Targeted Nanoemulsions Enhances Antitumor Efficacy and Immune Modulation in Colon Cancer. Pharmaceutics 2025, 17, 872. https://doi.org/10.3390/pharmaceutics17070872
Subedi L, Bamjan AD, Phuyal S, Khadka B, Chaudhary M, Kim K-T, Kim KH, Shim J-H, Cho S-S, Yu JE, et al. Oral Metronomic Delivery of Atorvastatin and Docetaxel via Transporter-Targeted Nanoemulsions Enhances Antitumor Efficacy and Immune Modulation in Colon Cancer. Pharmaceutics. 2025; 17(7):872. https://doi.org/10.3390/pharmaceutics17070872
Chicago/Turabian StyleSubedi, Laxman, Arjun Dhwoj Bamjan, Susmita Phuyal, Bikram Khadka, Mansingh Chaudhary, Ki-Taek Kim, Ki Hyun Kim, Jung-Hyun Shim, Seung-Sik Cho, Ji Eun Yu, and et al. 2025. "Oral Metronomic Delivery of Atorvastatin and Docetaxel via Transporter-Targeted Nanoemulsions Enhances Antitumor Efficacy and Immune Modulation in Colon Cancer" Pharmaceutics 17, no. 7: 872. https://doi.org/10.3390/pharmaceutics17070872
APA StyleSubedi, L., Bamjan, A. D., Phuyal, S., Khadka, B., Chaudhary, M., Kim, K.-T., Kim, K. H., Shim, J.-H., Cho, S.-S., Yu, J. E., & Park, J. W. (2025). Oral Metronomic Delivery of Atorvastatin and Docetaxel via Transporter-Targeted Nanoemulsions Enhances Antitumor Efficacy and Immune Modulation in Colon Cancer. Pharmaceutics, 17(7), 872. https://doi.org/10.3390/pharmaceutics17070872