Liposomal Formulations in Clinical Use: An Updated Review



Abstract
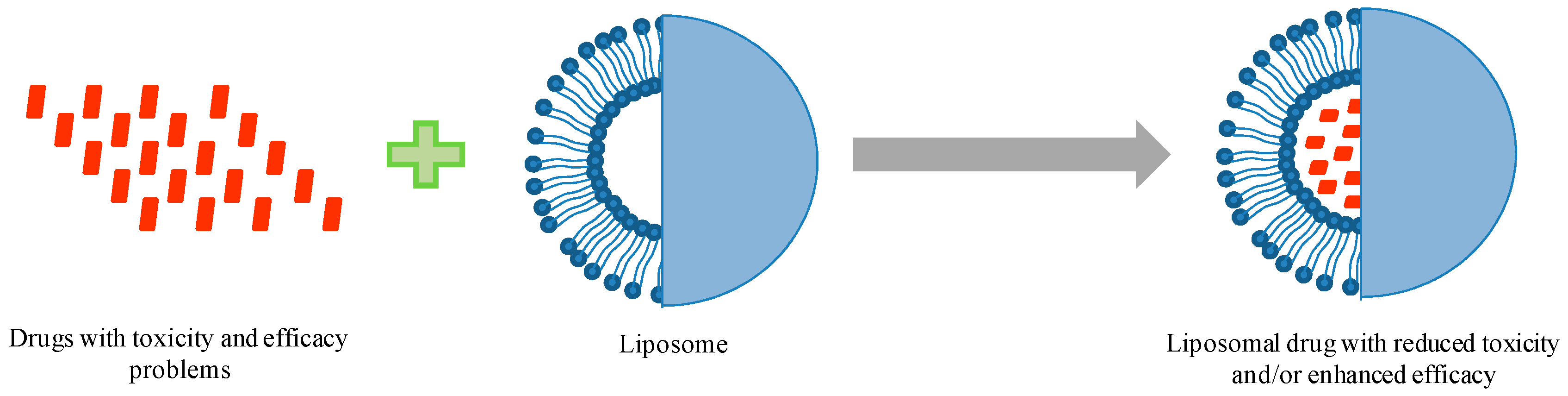
:1. Introduction
2. Liposome Technologies for Delivery of Therapeutics
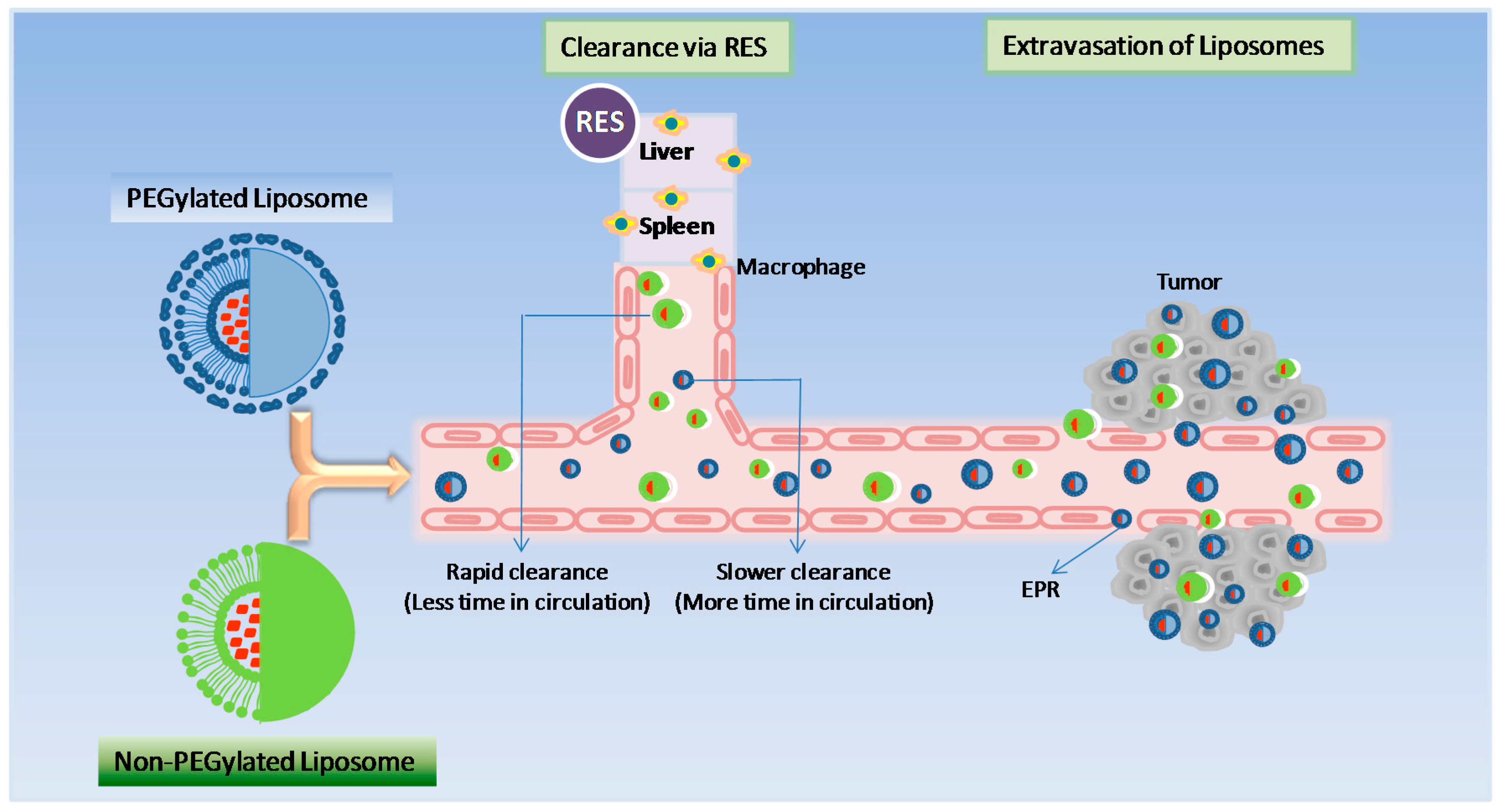
2.1. Stealth Liposome Technology
2.2. Non-PEGylated Liposome Technology
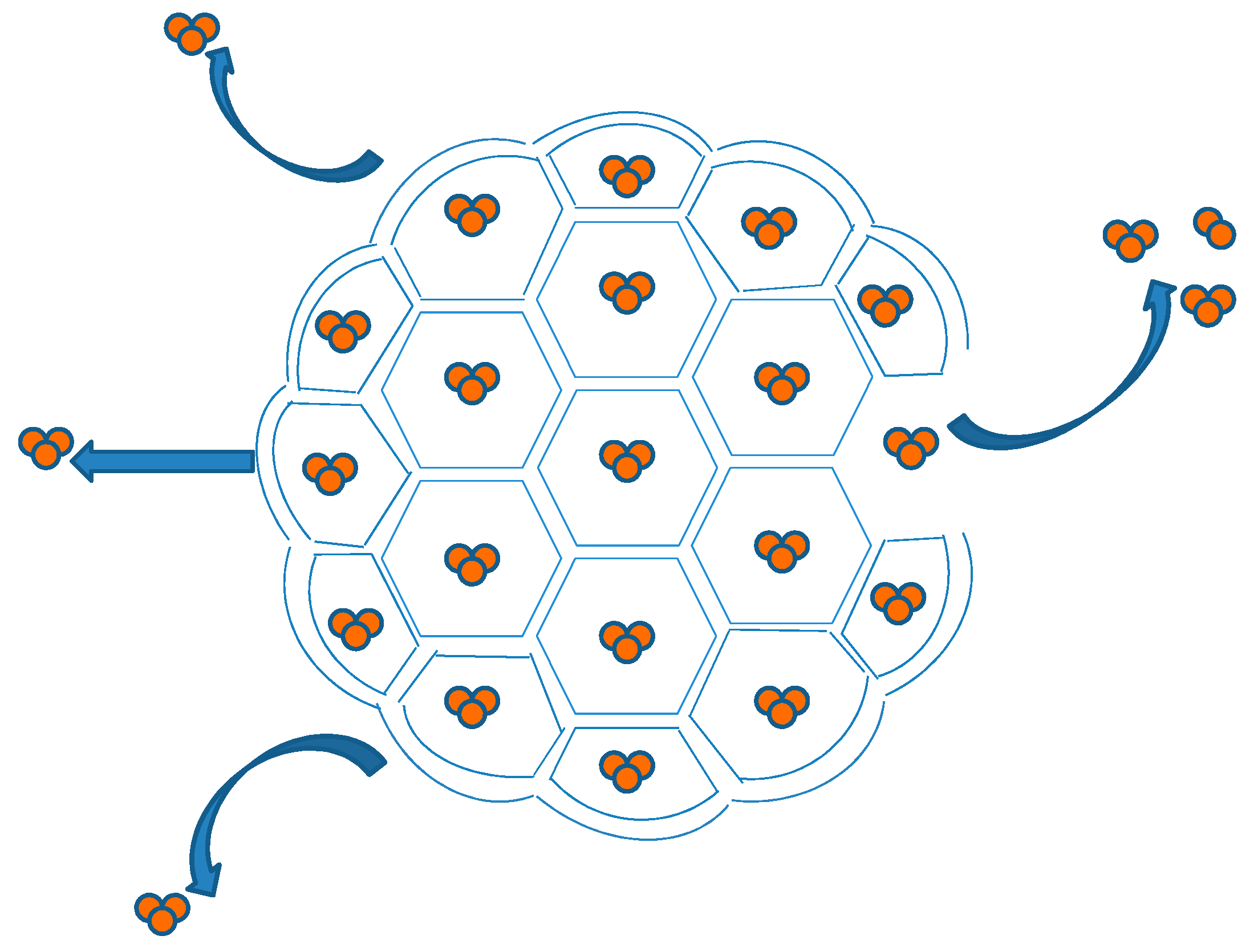
2.3. DepoFoam™ Liposome Technology
2.4. Lysolipid Thermally Sensitive Liposome (LTSL) Technology
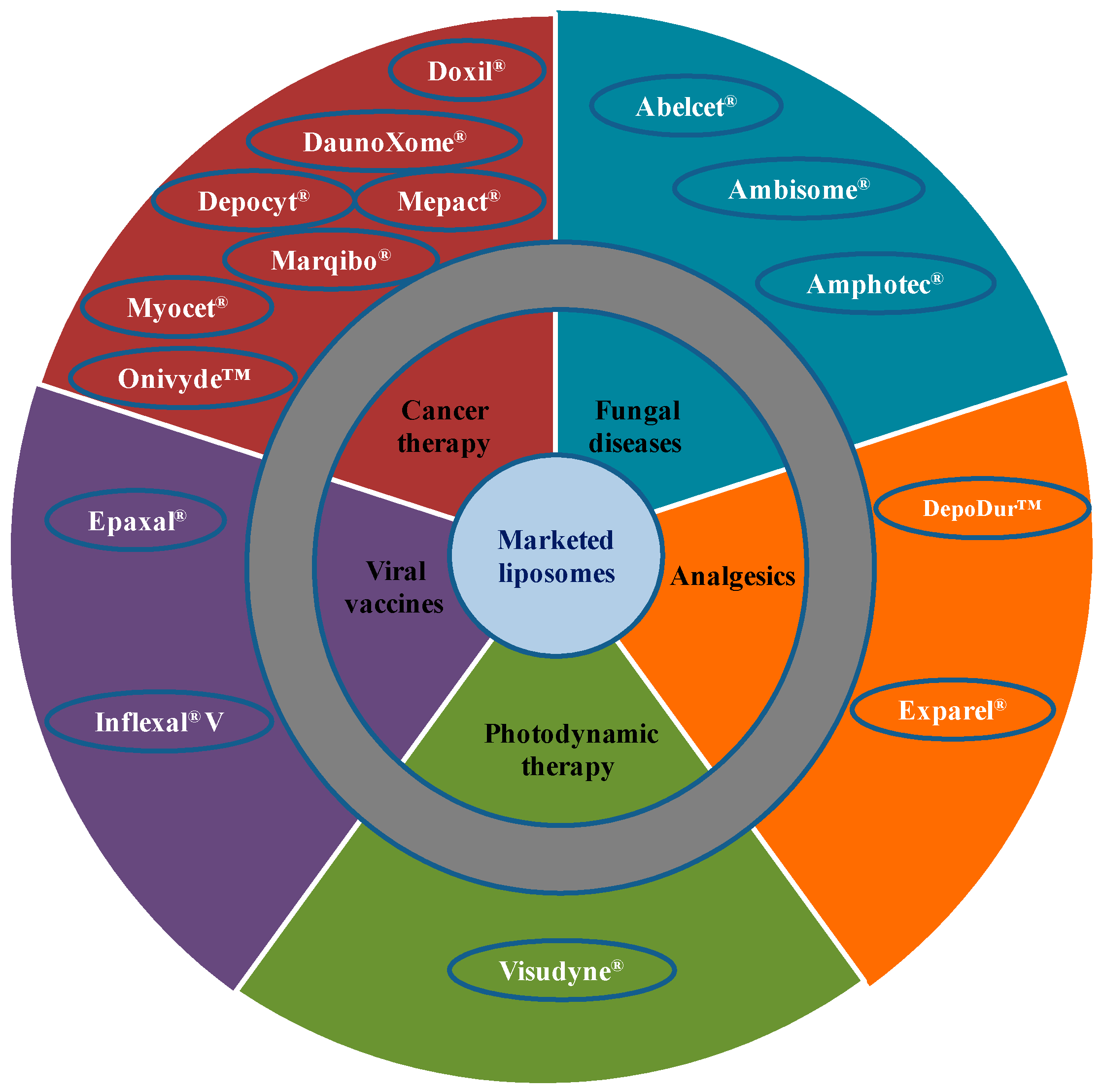
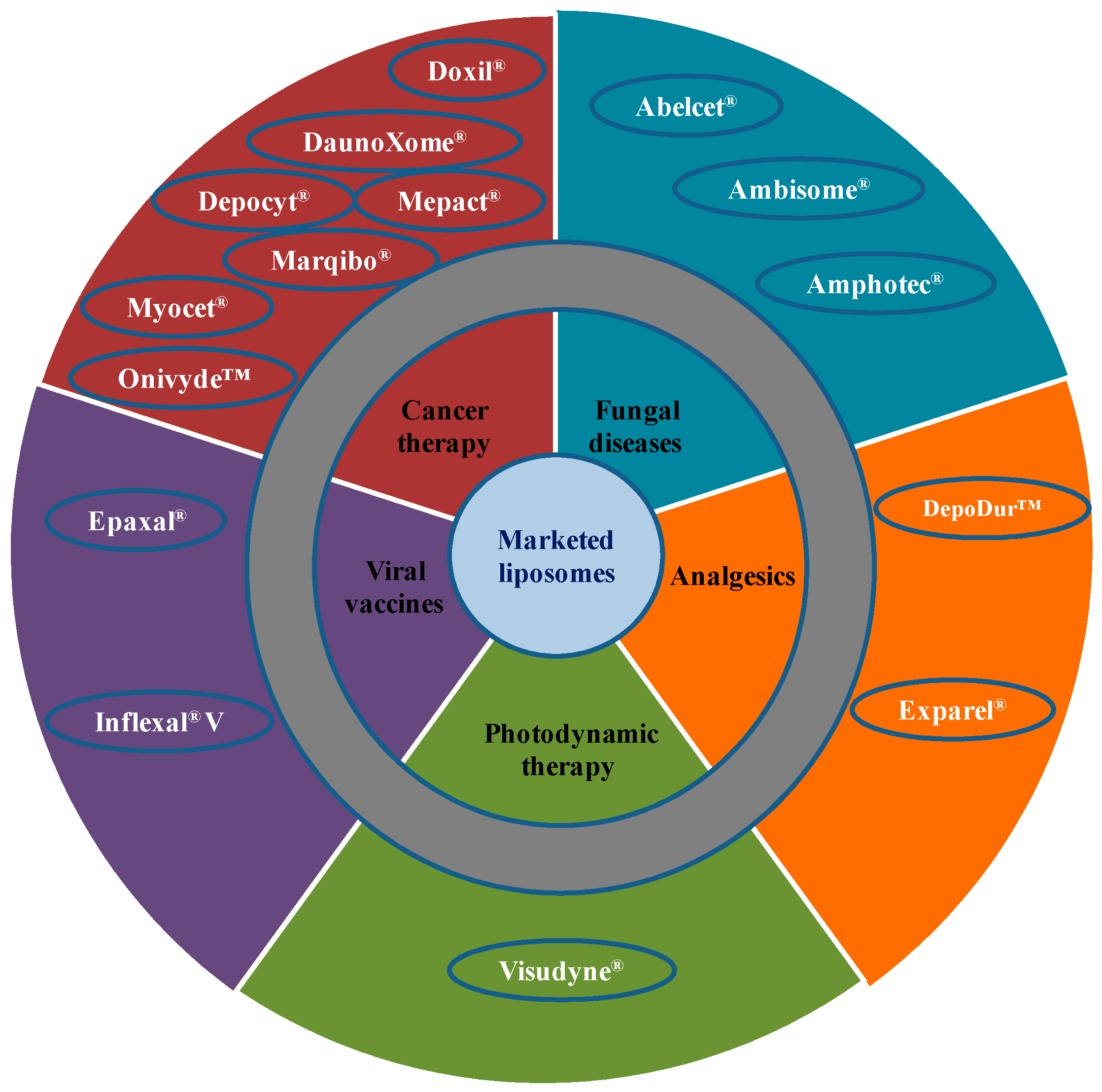
3. Clinically Available Liposome-Based Products
3.1. Liposomes for Cancer Therapy
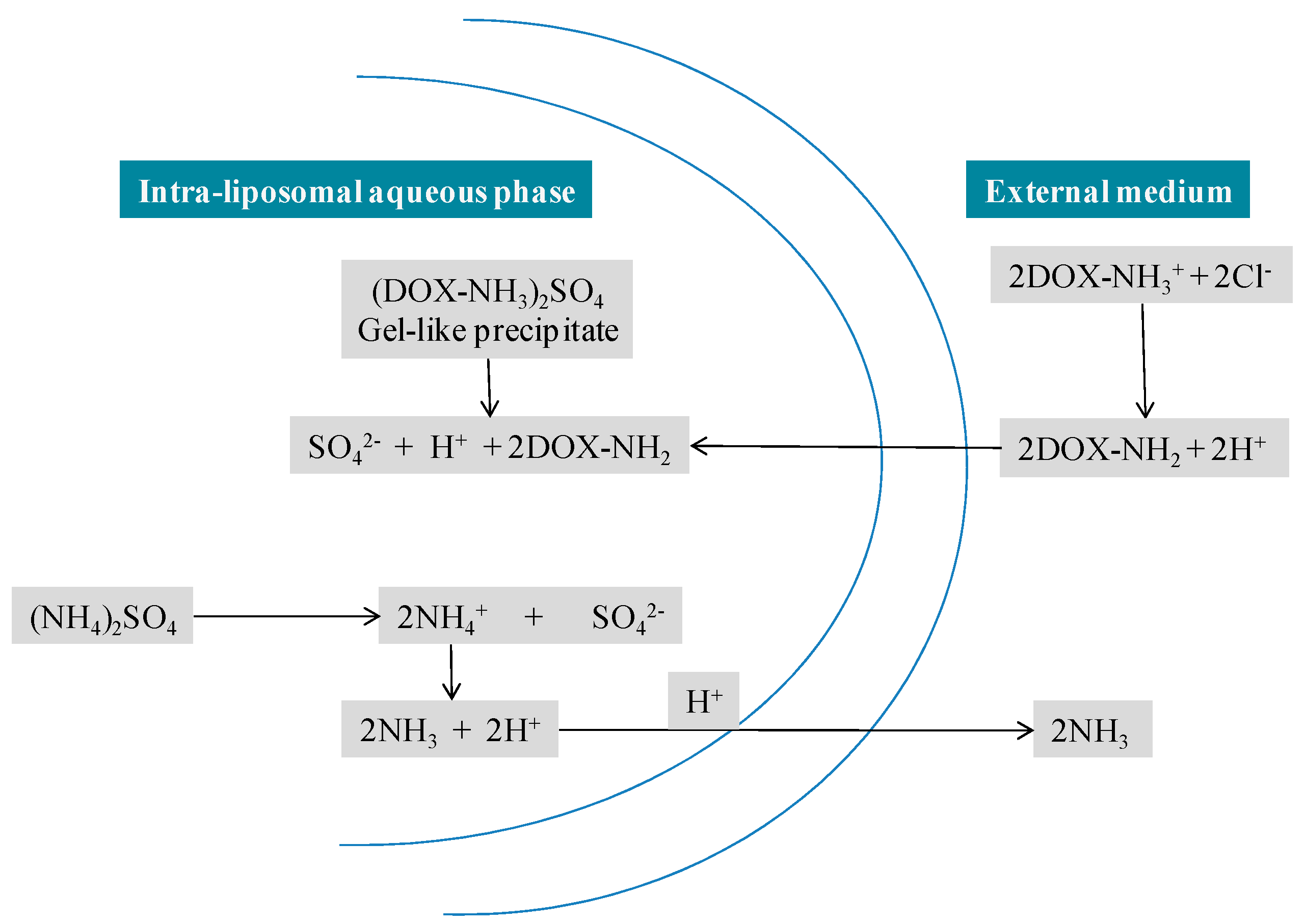
3.1.1. Doxil®
3.1.2. DaunoXome®
3.1.3. Depocyt®
3.1.4. Myocet®
3.1.5. Mepact®
3.1.6. Marqibo®
3.1.7. Onivyde™
3.2. Liposomes for Fungal Infections
3.2.1. Abelcet®
3.2.2. Ambisome®
3.2.3. Amphotec®
3.3. Liposomes for Photodynamic Therapy
Visudyne®
3.4. Liposomes for Pain Management
3.4.1. DepoDur™
3.4.2. Exparel®
3.5. Liposomes for Viral Infections
3.5.1. Epaxal®
3.5.2. Inflexal® V
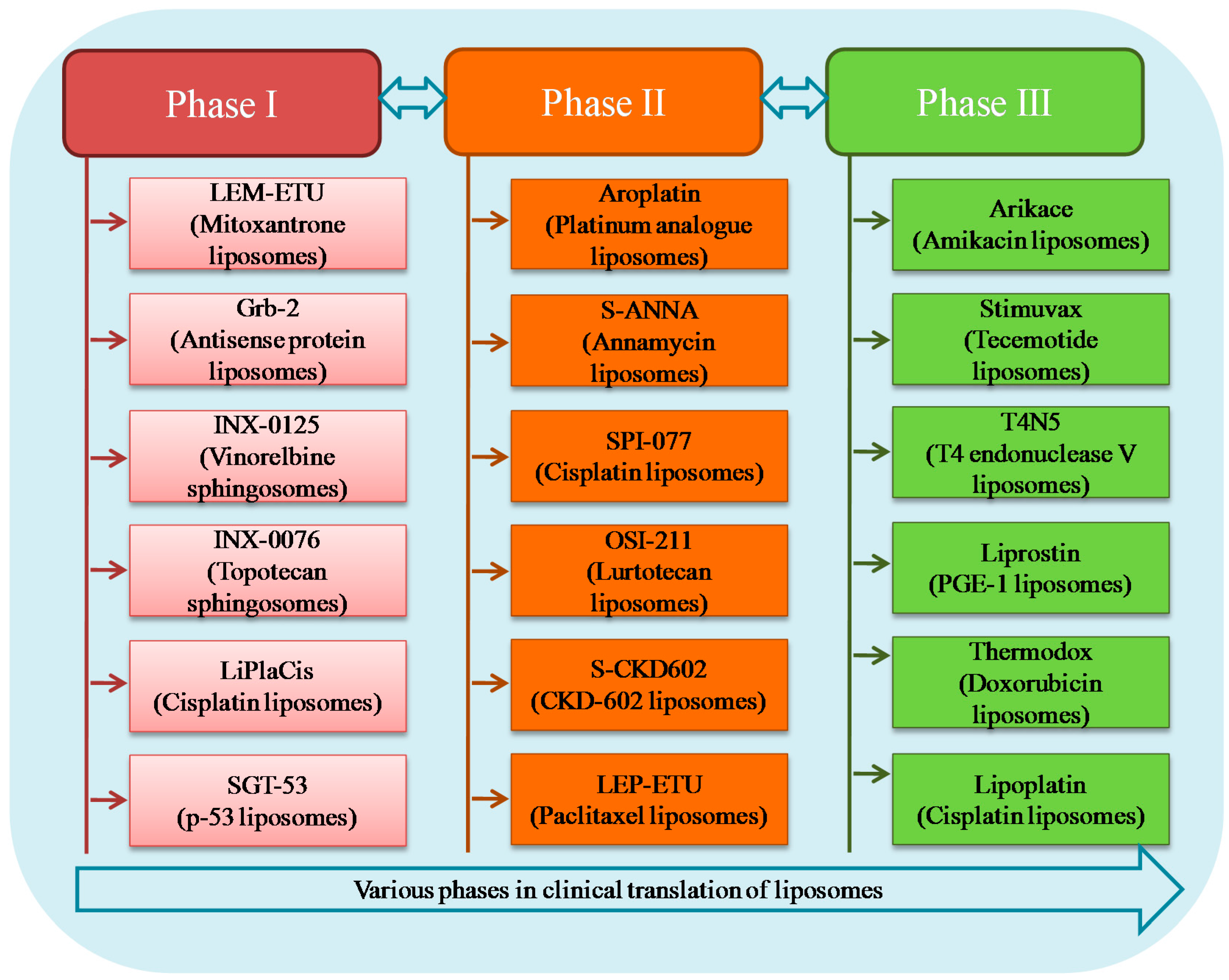
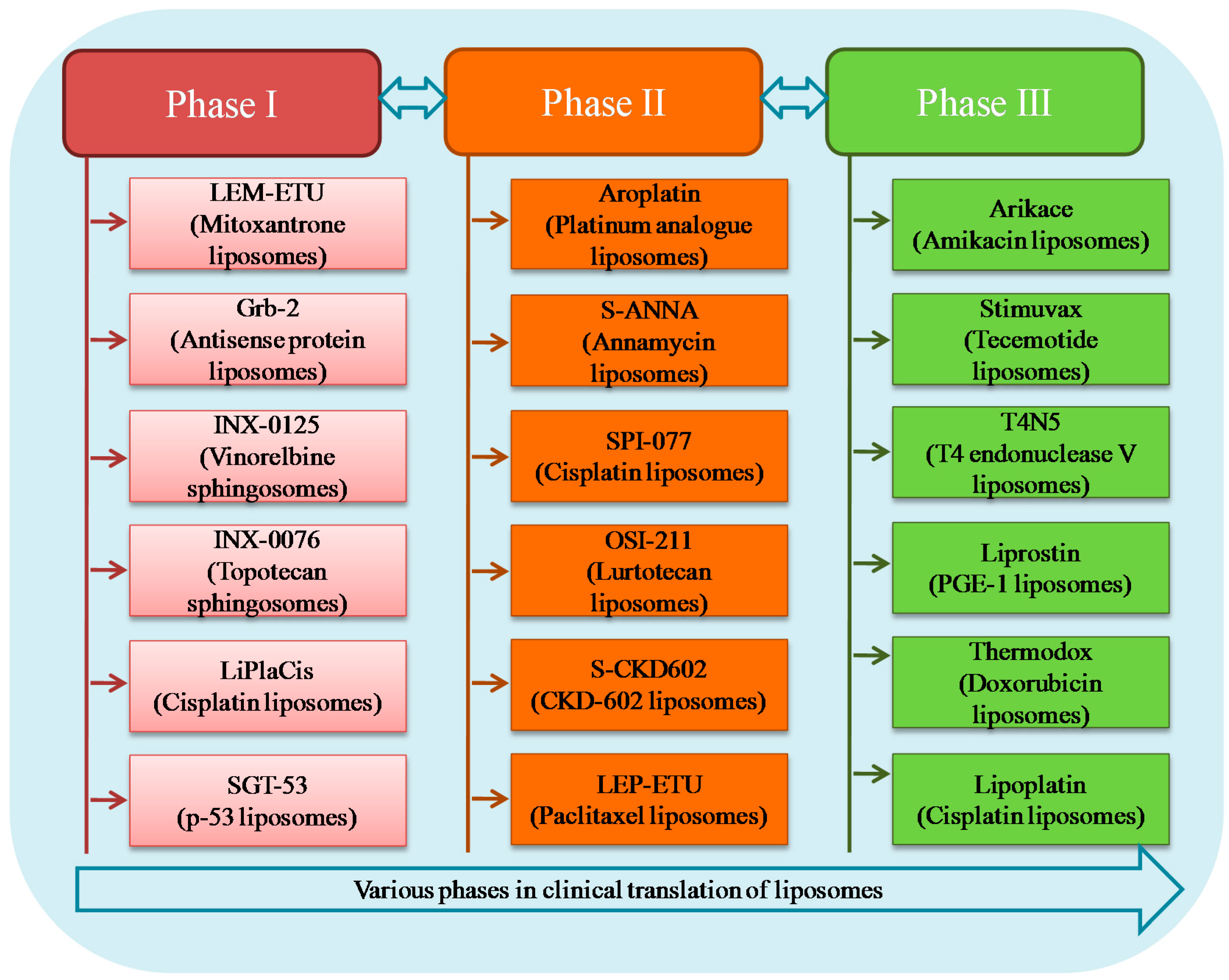
4. Liposomal Formulations in Clinical Trials
4.1. Phase III
4.1.1. Arikace™
4.1.2. Stimuvax®
4.1.3. T4N5
4.1.4. Liprostin™
4.1.5. ThermoDox®
4.1.6. Lipoplatin™
4.2. Phase II
4.2.1. Aroplatin™
4.2.2. Liposomal Annamycin
4.2.3. SPI-077
4.2.4. OSI-211
4.2.5. S-CKD602
4.2.6. LE-SN38
4.2.7. LEP-ETU
4.2.8. Endotag-I
4.2.9. Atragen®
4.3. Phase I
5. Conclusions
Conflicts of Interest
References
- Bangham, A.; Standish, M.M.; Watkins, J. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Torchilin, V.; Weissig, V. Liposomes: A Practical Approach; Oxford University Press: Kettering, UK, 2003; pp. 77–101. [Google Scholar]
- Barenholz, Y.C. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Harris, J.M. Introduction and overview of peptide and protein pegylation. Adv. Drug Deliv. Rev. 2002, 54, 453. [Google Scholar] [PubMed]
- Leonard, R.; Williams, S.; Tulpule, A.; Levine, A.; Oliveros, S. Improving the therapeutic index of anthracycline chemotherapy: Focus on liposomal doxorubicin (Myocet™). Breast 2009, 18, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Murry, D.J.; Blaney, S.M. Clinical pharmacology of encapsulated sustained-release cytarabine. Ann. Pharmacother. 2000, 34, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H.-J.; Gelderblom, H. Liposomal drug formulations in cancer therapy: 15 Years along the road. Drug Discov. Today 2012, 17, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Working, P.; Dayan, A. Pharmacological-toxicological expert report. CAELYX.(Stealth liposomal doxorubicin HCl). Hum. Exp. Toxicol. 1996, 15, 751. [Google Scholar] [PubMed]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal doxorubicin. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid interface Sci. 2001, 6, 66–77. [Google Scholar] [CrossRef]
- Barenholz, Y.; Haran, G. Efficient Loading and Controlled Release of Amphipathic Molecules. U.S. Patent No. 5,316,771, 31 May 1994. [Google Scholar]
- Lasic, D.; Frederik, P.; Stuart, M.; Barenholz, Y.; McIntosh, T. Gelation of liposome interior A novel method for drug encapsulation. FEBS Lett. 1992, 312, 255–258. [Google Scholar] [CrossRef]
- Lasic, D.; Čeh, B.; Stuart, M.; Guo, L.; Frederik, P.; Barenholz, Y. Transmembrane gradient driven phase transitions within vesicles: Lessons for drug delivery. Biochim. Biophys. Acta (BBA) Biomembr. 1995, 1239, 145–156. [Google Scholar] [CrossRef]
- Gabizon, A.A. Liposome circulation time and tumor targeting: Implications for cancer chemotherapy. Adv. Drug Deliv. Rev. 1995, 16, 285–294. [Google Scholar] [CrossRef]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar] [PubMed]
- Batist, G. Cardiac safety of liposomal anthracyclines. Cardiovasc. Toxicol. 2007, 7, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Dittmer, D.P. Liposomal daunorubicin as treatment for Kaposi’s sarcoma. Int. J. Nanomed. 2007, 2, 277. [Google Scholar]
- Allen, T.M.; Martin, F.J. Advantages of liposomal delivery systems for anthracyclines. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Forssen, E.A.; Ross, M.E. Daunoxome® treatment of solid tumors: Preclinical and clinical investigations. J. Liposome Res. 1994, 4, 481–512. [Google Scholar] [CrossRef]
- Forssen, E.A.; Coulter, D.M.; Proffitt, R.T. Selective in vivo localization of daunorubicin small unilamellar vesicles in solid tumors. Cancer Res. 1992, 52, 3255–3261. [Google Scholar] [PubMed]
- Gill, P.S.; Espina, B.M.; Muggia, F.; Cabriales, S.; Tulpule, A.; Esplin, J.A.; Liebman, H.A.; Forssen, E.; Ross, M.E.; Levine, A.M. Phase I/II clinical and pharmacokinetic evaluation of liposomal daunorubicin. J. Clin. Oncol. 1995, 13, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, L.; Zucchetti, M.; Parisi, I.; Viganò, M.G.; Zecca, B.; Careddu, A.; D’Incalci, M.; Lazzarin, A. The pharmacokinetics of liposomal encapsulated daunorubicin are not modified by HAART in patients with HIV-associated Kaposi’s sarcoma. Cancer Chemother. Pharmacol. 2000, 45, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Alberts, D.S.; Bachur, N.R.; Holtzman, J.L. The pharmacokinetics of daunomycin in man. Clin. Pharmacol. Ther. 1971, 12, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C.; Kormanik, P.; Howell, S.B.; Kim, S. Pharmacokinetics of intralumbar DTC-101 for the treatment of leptomeningeal metastases. Arch. Neurol. 1995, 52, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chatelut, E.; Kim, J.C.; Howell, S.B.; Cates, C.; Kormanik, P.A.; Chamberlain, M.C. Extended CSF cytarabine exposure following intrathecal administration of DTC 101. J. Clin. Oncol. 1993, 11, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Glantz, M.J.; Jaeckle, K.A.; Chamberlain, M.C.; Phuphanich, S.; Recht, L.; Swinnen, L.J.; Maria, B.; LaFollette, S.; Schumann, G.B.; Cole, B.F. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin. Cancer Res. 1999, 5, 3394–3402. [Google Scholar] [PubMed]
- Balazsovits, J.; Mayer, L.; Bally, M.; Cullis, P.; McDonell, M.; Ginsberg, R.; Falk, R. Analysis of the effect of liposome encapsulation on the vesicant properties, acute and cardiac toxicities, and antitumor efficacy of doxorubicin. Cancer Chemother. Pharmacol. 1989, 23, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kanter, P.; Bullard, G.; Pilkiewicz, F.; Mayer, L.; Cullis, P.; Pavelic, Z. Preclinical toxicology study of liposome encapsulated doxorubicin (TLC D-99): Comparison with doxorubicin and empty liposomes in mice and dogs. In Vivo 1992, 7, 85–95. [Google Scholar]
- Sparano, J.A.; Winer, E.P. Liposomal anthracyclines for breast cancer. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Cowens, J.; Creaven, P.; Greco, W.; Brenner, D.; Tung, Y.; Ostro, M.; Pilkiewicz, F.; Ginsberg, R.; Petrelli, N. Initial clinical (phase I) trial of TLC D-99 (doxorubicin encapsulated in liposomes). Cancer Res. 1993, 53, 2796–2802. [Google Scholar] [PubMed]
- Harasym, T.O.; Cullis, P.R.; Bally, M.B. Intratumor distribution of doxorubicin following iv administration of drug encapsulated in egg phosphatidylcholine/cholesterol liposomes. Cancer Chemother. Pharmacol. 1997, 40, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.; Batist, G.; Belt, R.; Rovira, D.; Navari, R.; Azarnia, N.; Welles, L.; Winer, E. Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer 2002, 94, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Alphandéry, E.; Grand-Dewyse, P.; Lefèvre, R.; Mandawala, C.; Durand-Dubief, M. Cancer therapy using nanoformulated substances: Scientific, regulatory and financial aspects. Expert Rev. Anticancer ther. 2015, 15, 1233–1255. [Google Scholar] [CrossRef] [PubMed]
- Vail, D.M.; MacEwen, E.G.; Kurzman, I.D.; Dubielzig, R.R.; Helfand, S.C.; Kisseberth, W.C.; London, C.A.; Obradovich, J.E.; Madewell, B.R.; Rodriguez, C.O. Liposome-encapsulated muramyl tripeptide phosphatidylethanolamine adjuvant immunotherapy for splenic hemangiosarcoma in the dog: A randomized multi-institutional clinical trial. Clin. Cancer Res. 1995, 1, 1165–1170. [Google Scholar] [PubMed]
- Anderson, P.; Tomaras, M.; McConnell, K. Mifamurtide in osteosarcoma—A practical review. Drugs Today 2010, 46, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Meyers, P.; Kleinerman, E.; Oliva, C.; Liu, Y. Mifamurtide (L-MTP-PE) for metastatic and recurrent osteosarcoma (OS): Survival and safety profile from a patient access study. In Annals of Oncology; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival—A report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.; Harasym, T.; Masin, D.; Bally, M.; Mayer, L. Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and therapeutic properties of vincristine in murine and human tumour models. Br. J. Cancer 1995, 72, 896. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.J.; Semple, S.C.; Klimuk, S.K.; Edwards, K.; Eisenhardt, M.L.; Leng, E.C.; Karlsson, G.; Yanko, D.; Cullis, P.R. Therapeutically optimized rates of drug release can be achieved by varying the drug-to-lipid ratio in liposomal vincristine formulations. Biochim. Biophys.Acta (BBA) Biomembr. 2006, 1758, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Webb, M.S.; Onge, G.S.; Mayer, L.D. Liposomal and nonliposomal drug pharmacokinetics after administration of liposome-encapsulated vincristine and their contribution to drug tissue distribution properties. J. Pharmacol. Exp. Ther. 2001, 298, 1206–1212. [Google Scholar] [PubMed]
- Rodriguez, M.; Pytlik, R.; Kozak, T.; Chhanabhai, M.; Gascoyne, R.; Lu, B.; Deitcher, S.R.; Winter, J.N. Vincristine sulfate liposomes injection (Marqibo) in heavily pretreated patients with refractory aggressive non-Hodgkin lymphoma. Cancer 2009, 115, 3475–3482. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Drummond, D.C.; Kirpotin, D. Liposomes Useful for Drug Delivery. U.S. Patent No. US20160030341 A1, 4 February 2016. [Google Scholar]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Lister, J. Amphotericin B lipid complex (Abelcet®) in the treatment of invasive mycoses: The North American experience. Eur. J. Haematol. 1996, 56, 18–23. [Google Scholar] [CrossRef]
- Madden, T.; Janoff, A.; Cullis, P. Incorporation of amphotericin B into large unilamellar vesicles composed of phosphatidylcholine and phosphatidylglycerol. Chem. Phys. Lipids 1990, 52, 189–198. [Google Scholar] [CrossRef]
- Janoff, A.; Boni, L.; Popescu, M.; Minchey, S.; Cullis, P.R.; Madden, T.; Taraschi, T.; Gruner, S.; Shyamsunder, E.; Tate, M. Unusual lipid structures selectively reduce the toxicity of amphotericin B. Proc. Natl. Acad. Sci. USA 1988, 85, 6122–6126. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.J.; Swerdel, M.R.; Blue, B.; Clark, J.M.; Bonner, D.P. Tissue distribution of amphotericin B lipid complex in laboratory animals. J. Pharm. Pharmacol. 1991, 43, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Janoff, A.; Perkins, W.; Saletan, S.; Swenson, C. Amphotericin B lipid complex (ABLC™): A molecular rationale for the attenuation of amphotericin B related toxicities. J. Liposome Res. 1993, 3, 451–471. [Google Scholar] [CrossRef]
- Adedoyin, A.; Bernardo, J.F.; Swenson, C.E.; Bolsack, L.E.; Horwith, G.; DeWit, S.; Kelly, E.; Klasterksy, J.; Sculier, J.-P.; DeValeriola, D. Pharmacokinetic profile of ABELCET (amphotericin B lipid complex injection): Combined experience from phase I and phase II studies. Antimicrob. Agents Chemother. 1997, 41, 2201–2208. [Google Scholar] [PubMed]
- Adler-Moore, J.P.; Proffitt, R.T. Development, characterization, efficacy and mode of action of AmBisome, a unilamellar liposomal formulation of amphotericin B. J. Liposome Res. 1993, 3, 429–450. [Google Scholar] [CrossRef]
- Woodle, M.C.; Storm, G. Long Circulating Liposomes: Old Drugs, New Therapeutics; Springer Science & Business Media: Berlin, Germany, 1998. [Google Scholar]
- Walsh, T.J.; Yeldandi, V.; McEvoy, M.; Gonzalez, C.; Chanock, S.; Freifeld, A.; Seibel, N.I.; Whitcomb, P.O.; Jarosinski, P.; Boswell, G. Safety, tolerance, and pharmacokinetics of a small unilamellar liposomal formulation of amphotericin B (AmBisome) in neutropenic patients. Antimicrob. Agents Chemother. 1998, 42, 2391–2398. [Google Scholar] [PubMed]
- Boswell, G.; Buell, D.; Bekersky, I. AmBisome (liposomal amphotericin B): A comparative review. J. Clin. Pharmacol. 1998, 38, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.S.; Fielding, R.M.; Lasic, D.D.; Hamilton, R.L.; Mufson, D. Novel antifungal drug delivery: Stable amphotericin B-cholesteryl sulfate discs. Int. J. Pharm. 1991, 75, 45–54. [Google Scholar] [CrossRef]
- Fielding, R.; Singer, A.; Wang, L.; Babbar, S.; Guo, L. Relationship of pharmacokinetics and drug distribution in tissue to increased safety of amphotericin B colloidal dispersion in dogs. Antimicrob. Agents Chemother. 1992, 36, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Fielding, R.M.; Smith, P.C.; Guo, L.S. Comparative tissue distribution and elimination of amphotericin B colloidal dispersion (Amphocil®) and Fungizone® after repeated dosing in rats. Pharm. Res. 1995, 12, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.W.; Buchi, K.; Goddard, M.; Lang, J.; Tolman, K. Single-dose pharmacokinetics and tolerance of a cholesteryl sulfate complex of amphotericin B administered to healthy volunteers. Antimicrob. Agents Chemother. 1991, 35, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- White, M.H.; Anaissie, E.J.; Kusne, S.; Wingard, J.R.; Hiemenz, J.W.; Cantor, A.; Gurwith, M.; Mond, C.D.; Mamelok, R.D.; Bowden, R.A. Amphotericin B colloidal dispersion vs. amphotericin B as therapy for invasive aspergillosis. Clin. Infect. Dis. 1997, 24, 635–642. [Google Scholar] [PubMed]
- Bowden, R.A.; Cays, M.; Gooley, T.; Mamelok, R.D.; van Burik, J.-A. Phase I study of amphotericin B colloidal dispersion for the treatment of invasive fungal infections after marrow transplant. J. Infect. Dis. 1996, 173, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Strong, H.A.; Levy, J.; Huber, G.; Fsadni, M. Vision through Photodynamic Therapy of the Eye. U.S. Patent No. US5910510 A, 8 June 1999. [Google Scholar]
- Richter, A.M.; Waterfield, E.; Jain, A.K.; Canaan, A.J.; Allison, B.A.; Levy, J.G. Liposomal delivery of a photosensitizer, benzoporphyrin derivative monoacid ring A (BPD), to tumor tissue in a mouse tumor model. Photochem. Photobiol. 1993, 57, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Bressler, N.M. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: One-year results of 2 randomized clinical trials—TAP report 1. Arch. Ophthalmol. 1999, 117, 1329–1345. [Google Scholar]
- Alam, M.; Hartrick, C.T. Extended-Release Epidural Morphine (DepoDur™): An Old Drug with a New Profile. Pain Pract. 2005, 5, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Hartrick, C.; Manvelian, G. Sustained-Release Epidural Morphine (DepoDur™): A Review. Todays Ther. Trends 2004, 22, 167–180. [Google Scholar]
- Kim, T.; Murdande, S.; Gruber, A.; Kim, S. Sustained-release morphine for epidural analgesia in rats. J. Am. Soc. Anesthesiol. 1996, 85, 331–338. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Kopacz, D.; Hartrick, C.; Martin, G.; Manvelian, G. Single-dose extended-release epidural morphine for pain following hip arthroplasty. Am. J. Ther. 2006, 13, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Angst, M.S.; Drover, D.R. Pharmacology of drugs formulated with DepoFoam™. Clin. Pharmacokinet. 2006, 45, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Richard, B.M.; Rickert, D.E.; Newton, P.E.; Ott, L.R.; Haan, D.; Brubaker, A.N.; Cole, P.I.; Ross, P.E.; Rebelatto, M.C.; Nelson, K.G. Safety evaluation of EXPAREL (DepoFoam bupivacaine) administered by repeated subcutaneous injection in rabbits and dogs: Species comparison. J. Drug Deliv. 2011. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.M.; Barenholz, Y.; Cohen, R.; Haroutiunian, S.; Kagan, L.; Ginosar, Y. High-dose bupivacaine remotely loaded into multivesicular liposomes demonstrates slow drug release without systemic toxic plasma concentrations after subcutaneous administration in humans. Anesth. Analg. 2010, 110, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.; Terem, T.; Kheladze, K.; Mosidze, B. A single administration of depobupivacaine (TM) intraoperatively provides three-day analgesia and reduction in use of rescue opioids in patients undergoing hemorrhoidectomy. In Diseases of the Colon & Rectum; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Clarke, P.D.; Adams, P.; Ibáñez, R.; Herzog, C. Rate, intensity, and duration of local reactions to a virosome-adjuvanted vs. an aluminium-adsorbed hepatitis A vaccine in UK travellers. Travel Med. Infect. Dis. 2006, 4, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bungener, L.; Serre, K.; Bijl, L.; Leserman, L.; Wilschut, J.; Daemen, T.; Machy, P. Virosome-mediated delivery of protein antigens to dendritic cells. Vaccine 2002, 20, 2287–2295. [Google Scholar] [CrossRef]
- Ambrosch, F.; Wiedermann, G.; Jonas, S.; Althaus, B.; Finkel, B.; Glück, R.; Herzog, C. Immunogenicity and protectivity of a new liposomal hepatitis A vaccine. Vaccine 1997, 15, 1209–1213. [Google Scholar] [CrossRef]
- Perez, O.M.; Herzog, C.; Zellmeyer, M.; Loáisiga, A.; Frösner, G.; Egger, M. Efficacy of virosome hepatitis A vaccine in young children in Nicaragua: Randomized placebo-controlled trial. J. Infect. Dis. 2003, 188, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Bovier, P.; Bock, J.; Loutan, L.; Farinelli, T.; Glueck, R.; Herzog, C. Long-term immunogenicity of an inactivated virosome hepatitis A vaccine. J. Med. Virol. 2002, 68, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Gluck, R.; Metcalfe, I. New technology platforms in the development of vaccines for the future. Vaccine 2002, 20, 10–16. [Google Scholar] [CrossRef]
- Gluck, R.; Mischler, R.; Finkel, B.; Que, J.; Cryz, S.; Scarpa, B. Immunogenicity of new virosome influenza vaccine in elderly people. Lancet 1994, 344, 160–163. [Google Scholar] [CrossRef]
- Conne, P.; Gauthey, L.; Vernet, P.; Althaus, B.; Que, J.U.; Finkel, B.; Glück, R.; Cryz, S.J. Immunogenicity of trivalent subunit versus virosome-formulated influenza vaccines in geriatric patients. Vaccine 1997, 15, 1675–1679. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Wurtz, W.; Lee, J.K.; Malinin, V.S.; Durwas-Krishnan, S.; Meers, P.; Perkins, W.R. Characterization of nebulized liposomal amikacin (Arikace™) as a function of droplet size. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P. Clinical trials of lipid-associated aerosolized amikacin: The arikace™ story. In Pediatric Pulmonology; Wiley-Liss Div John Wiley & Sons Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Bilton, D.; Pressler, T.; Fajac, I.; Clancy, J.P.; Sands, D.; Minic, P.; Cipolli, M.; LaRosa, M.; Galeva, I.; Sole, A.A. Phase 3 efficacy and safety data from randomized, multicenter study of liposomal amikacin for inhalation (ARIKACE) Compared with TOBI in cystic fibrosis patients with chronic infection due to Pseudomonas Aeruginosa. Pediatr. Pulmonol. 2013, 48, 207–453. [Google Scholar]
- Clancy, J.; Dupont, L.; Konstan, M.W.; Billings, J.; Fustik, S.; Goss, C.H.; Lymp, J.; Minic, P.; Quittner, A.; Rubenstein, R. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013, 68, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Daley, C.L.; Winthrop, K.L.; Ruoss, S.; Addrizzo-Harris, D.J.; Flume, P.; Dorgan, D.; Salathe, M.A.; Olivier, B.; Brown-Elliott, A. A randomized, double-blind, placebo-controlled study of liposomal amikacin for inhalation (Arikace®) in patients with recalcitrant nontuberculous mycobacterial lung disease. In C27. Diagnosis and Treatment of Nontuberculous Mycobacteria Infections; American Thoracic Society: New York, NY, USA, 2014; p. 4126. [Google Scholar]
- Timmerman, L. Oncothyreon Marches on with ‘Son of Stimuvax’ Cancer Vaccine. Available online: http://www.xconomy.com/seattle/2012/04/10/oncothyreon-marches-on-with-son-of-stimuvax-cancer-vaccine/ (accessed on 27 March 2017).
- Kroemer, G.; Zitvogel, L.; Galluzzi, L. Victories and deceptions in tumor immunology: Stimuvax®. Oncoimmunology 2013, 2, e23687. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.A.; Shepherd, F.A. Immunotherapy for lung cancer. J. Thorac. Oncol. 2008, 3, S164–S170. [Google Scholar] [CrossRef] [PubMed]
- Vergati, M.; Intrivici, C.; Huen, N.-Y.; Schlom, J.; Tsang, K.Y. Strategies for cancer vaccine development. BioMed Res. Int. 2010, 2010, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cart, S.; Alert, N.P.R. Merck KGaA Starts Stimuvax Phase III Study INSPIRE in Asian Patients with Advanced NSCLC. Available online: http://www.businesswire.com/news/home/20091210005488/en/Merck-KGaA-Starts-Stimuvax-Phase-III-Study (accessed on 27 March 2017).
- Zahid, S.; Brownell, I. Repairing DNA damage in xeroderma pigmentosum: T4N5 lotion and gene therapy. J. Drugs Dermatol. 2008, 7, 405–408. [Google Scholar] [PubMed]
- Yarosh, D.B.; Kibitel, J.T.; Green, L.A.; Spinowitz, A. Enhanced unscheduled DNA synthesis in UV-irradiated human skin explants treated with T4N5 liposomes. J. Investig. Dermatol. 1991, 97, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Müllegger, R.R.; Soyer, H.P.; Hofer, A.; Smolle, J.; Horn, M.; Cerroni, L.; Hofmann-Wellenhof, R.; Kerl, H.; Maier, H. Topical treatment with liposomes containing T4 endonuclease V protects human skin in vivo from ultraviolet-induced upregulation of interleukin-10 and tumor necrosis factor-α. J. Investig. Dermatol. 2000, 114, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Cafardi, J.A.; Elmets, C.A. T4 endonuclease V: Review and application to dermatology. Expert Opin. Biol. Ther. 2008, 8, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-I.; Yeh, M.-K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar]
- Summers, D.; Ruff, D.; Smalling, R.; Cardoza, D.; Dottavio, D.; Lasic, D. Administration of liprostin (TM) for the treatment of critical limb ischemia (CLI) and peripheral arterial disease (PAD). In Journal of Liposome Research; Marcel Dekker Inc.: New York, NY, USA, 2003. [Google Scholar]
- Li, J.; Wang, B.; Wang, Y.; Wu, F.; Li, P.; Li, Y.; Zhao, L.; Cui, W.; Ding, Y.; An, Q. Therapeutic effect of liposomal prostaglandin E1 in acute lower limb ischemia as an adjuvant to hybrid procedures. Exp. Ther. Med. 2013, 5, 1760–1764. [Google Scholar] [PubMed]
- Chen, J.; He, C.-Q.; Lin, A.-H.; Gu, W.; Chen, Z.-P.; Li, W.; Cai, B.-C. Thermosensitive liposomes with higher phase transition temperature for targeted drug delivery to tumor. Int. J. Pharm. 2014, 475, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Mylonopouloua, E.; Arvanitisa, C.D.; Bazan-Peregrinoa, M.; Arora, M.; Coussios, C.C. Ultrasonic activation of thermally sensitive liposomes. In Proceedings of the 9th International Symposium on Therapeutic Ultrasound: Istu—2009, Aix-en-Provence, France, 24–26 September 2009; AIP Publishing: New York, NY, USA, 2010. [Google Scholar]
- Puri, A. Phototriggerable liposomes: Current research and future perspectives. Pharmaceutics 2013, 6, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.; Poon, R.; Neeman, Z.; Eugeni, M.; Locklin, J.; Dromi, S.; Kachala, S.; Probhakar, R.; Hahne, W.; Libutti, S. Phase I study of thermally sensitive liposomes containing doxorubicin (ThermoDox) given during radiofrequency ablation (RFA) in patients with unresectable hepatic malignancies. In Proceedings of the Gastrointestinal Cancers Symposium, Orlando, FL, USA, 19–21 January 2007; The American Society of Clinical Oncology: Alexandria, VA, USA, 2007. [Google Scholar]
- Dromi, S.; Quijano, J.; Xie, J.; Frenkel, V.; Wood, B.; Li, K. Pulsed-high intensity focused ultrasound (HIFU) enhanced delivery of Doxorubicin using heat sensitive liposome (Thermodox TM). In Proceedings of the 91st Annual Meeting of the Radiological Society of North America, Chicago, IL, USA, 27 November–2 December 2005. [Google Scholar]
- Palazzi, M.; Maluta, S.; Dall’Oglio, S.; Romano, M. The role of hyperthermia in the battle against cancer. Tumori 2010, 96, 902. [Google Scholar] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Egusquiaguirre, S.P.; Igartua, M.; Hernández, R.M.; Pedraz, J.L. Nanoparticle delivery systems for cancer therapy: Advances in clinical and preclinical research. Clin. Transl. Onco. 2012, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Boulikas, T. Clinical overview on Lipoplatin™: A successful liposomal formulation of cisplatin. Expert Opin. Investig. Drugs 2009, 18, 1197–1218. [Google Scholar] [CrossRef] [PubMed]
- Sotiriosrigatos, E.D.; Viliotou, V.; Stathopoulos, J.G. Pharmacokinetics and adverse reactions of a new liposomal cisplatin (Lipoplatin): Phase I study. Oncol. Rep. 2005, 13, 589–595. [Google Scholar]
- Stathopoulos, G.P.; Boulikas, T.; Vougiouka, M.; Rigatos, S.K.; Stathopoulos, J.G. Liposomal cisplatin combined with gemcitabine in pretreated advanced pancreatic cancer patients: A phase I-II study. Oncol. Rep. 2006, 15, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, N.; Athanasiou, A.; Ziras, N.; Angel, J.; Rapti, A.; Lampaki, S.; Politis, N.; Karanikas, C.; Kosmas, C. Phase II study of liposomal cisplatin (Lipoplatin™) plus gemcitabine versus cisplatin plus gemcitabine as first line treatment in inoperable (stage IIIB/IV) non-small cell lung cancer. Lung Cancer 2010, 68, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Boulikas, T.; Stathopoulos, G.P.; Volakakis, N.; Vougiouka, M. Systemic Lipoplatin infusion results in preferential tumor uptake in human studies. Anticancer Res. 2005, 25, 3031–3039. [Google Scholar] [PubMed]
- Harper, B.W.; Krause-Heuer, A.M.; Grant, M.P.; Manohar, M.; Garbutcheon-Singh, K.B.; Aldrich-Wright, J.R. Advances in platinum chemotherapeutics. Chem. Eur. J. 2010, 16, 7064–7077. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297. [Google Scholar]
- Farrell, N.P. Platinum formulations as anticancer drugs clinical and pre-clinical studies. Curr. Top. Med. Chem. 2011, 11, 2623–2631. [Google Scholar] [CrossRef]
- Dragovich, T.; Mendelson, D.; Hoos, A.; Lewis, J.; Kurtin, S.; Richardson, K.; von Hoff, D. 268 A phase II trial of aroplatin (L-NDDP), a liposomal DACH platinum, in patients with metastatic colorectal cancer (CRC)-a preliminary report. Eur. J. Cancer Suppl. 2003, 1, S82–S83. [Google Scholar] [CrossRef]
- Jakupec, M.; Galanski, M.; Keppler, B. Tumour-inhibiting platinum complexes—State of the art and future perspectives. In Reviews of physiology, Biochemistry and Pharmacology; Springer: New York, NY, USA, 2003; pp. 1–53. [Google Scholar]
- Zou, Y.; Priebe, W.; Perez-Soler, R. Lyophilized preliposomal formulation of the non-cross-resistant anthracycline annamycin: Effect of surfactant on liposome formation, stability and size. Cancer Chemother. Pharmacol. 1996, 39, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Wasan, K.M.; Perez-Soler, R. Distribution of free and liposomal annamycin within human plasma is regulated by plasma triglyceride concentrations but not by lipid transfer protein. J. Pharm. Sci. 1995, 84, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ling, Y.H.; Van, N.T.; Priebe, W.; Perez-Soler, R. Antitumor activity of free and liposome-entrapped annamycin, a lipophilic anthracycline antibiotic with non-cross-resistance properties. Cancer Res. 1994, 54, 1479–1484. [Google Scholar] [PubMed]
- Pui, C.-H.; Jeha, S. New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat. Rev. Drug Discov. 2007, 6, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Wetzler, M.; Thomas, D.A.; Wang, E.S.; Shepard, R.; Ford, L.A.; Heffner, T.L.; Parekh, S.; Andreeff, M.; O’Brien, S.; Kantarjian, H.M. Phase I/II trial of nanomolecular liposomal annamycin in adult patients with relapsed/refractory acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2013, 13, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Rowlinson-Busza, G.; Syrigos, K.N.; Vile, R.G.; Uster, P.S.; Peters, A.M.; Stewart, J.S.W. Pegylated liposome-encapsulated doxorubicin and cisplatin enhance the effect of radiotherapy in a tumor xenograft model. Clin. Cancer Res. 2000, 6, 4939–4949. [Google Scholar] [PubMed]
- Zamboni, W.C.; Gervais, A.C.; Egorin, M.J.; Schellens, J.H.; Zuhowski, E.G.; Pluim, D.; Joseph, E.; Hamburger, D.R.; Working, P.K.; Colbern, G. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother. Pharmacol. 2004, 53, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Bendele, R.; Giles, F.J.; Brown, E.; Gray, A.; Hart, K.; LeRay, J.D.; Meyer, D.; Pelanne, M.; Emerson, D.L. OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk. Res. 2003, 27, 1039–1050. [Google Scholar] [CrossRef]
- Yu, N.Y.; Conway, C.; Pena, R.L.; Chen, J.Y. STEALTH® liposomal CKD-602, a topoisomerase I inhibitor, improves the therapeutic index in human tumor xenograft models. Anticancer Res. 2007, 27, 2541–2545. [Google Scholar] [PubMed]
- Zamboni, W.C.; Strychor, S.; Joseph, E.; Walsh, D.R.; Zamboni, B.A.; Parise, R.A.; Tonda, M.E.; Ning, Y.Y.; Engbers, C.; Eiseman, J.L. Plasma, tumor, and tissue disposition of STEALTH liposomal CKD-602 (S-CKD602) and nonliposomal CKD-602 in mice bearing A375 human melanoma xenografts. Clin. Cancer Res. 2007, 13, 7217–7223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.A.; Xuan, T.; Parmar, M.; Ma, L.; Ugwu, S.; Ali, S.; Ahmad, I. Development and characterization of a novel liposome-based formulation of SN-38. Int. J. Pharm. 2004, 270, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Su, Y.S.; Samuelson, C.G.; Liebes, L.F.; Chamberlain, M.C.; Hofman, F.M.; Schönthal, A.H.; Chen, T.C. Irinotecan: A potential new chemotherapeutic agent for atypical or malignant meningiomas. J. Neurosurg. 2007, 106, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.; Jonker, D.; Seymour, L.; Goel, R.; Vincent, M.; Kocha, W.; Cripps, C.; Fisher, B.; Lister, D.; Malpage, A. A National Cancer Institute of Canada Clinical Trials Group Study–IND. 135: Phase I/II study of irinotecan (camptosar), oxaliplatin and raltitrexed (tomudex)(COT) in patients with advanced colorectal cancer. Eur. J. Cancer 2006, 42, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.A.; Anyarambhatla, G.; Ma, L.; Ugwu, S.; Xuan, T.; Sardone, T.; Ahmad, I. Development and characterization of a novel Cremophor® EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur. J. Pharm. Biopharm. 2005, 59, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Hanauske, A.; Gelderblom, H.; Scheulen, M.; van Warmerdam, L.; Rosing, H.; Fetterly, G.; Shu, V.; Sherman, J.; Rubin, E. Results of a clinical pharmacokinetic (PK) bioequiulence (BE) study of liposomal paclitaxel (LEP-ETU) versus paclitaxel (T) in patients with advanced cancer. In Journal of Clinical Oncology; American Society of Clinical Oncology: Alexandria, VA, USA, 2006. [Google Scholar]
- Slingerland, M.; Guchelaar, H.-J.; Rosing, H.; Scheulen, M.E.; van Warmerdam, L.J.; Beijnen, J.H.; Gelderblom, H. Bioequivalence of Liposome-Entrapped Paclitaxel Easy-To-Use (LEP-ETU) formulation and paclitaxel in polyethoxylated castor oil: A randomized, two-period crossover study in patients with advanced cancer. Clin. Ther. 2013, 35, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, M.E.; Ischenko, I.; Luedemann, S.; Strieth, S.; Papyan, A.; Werner, A.; Bohnenkamp, H.; Guenzi, E.; Preissler, G.; Michaelis, U. Vascular targeting by EndoTAG™-1 enhances therapeutic efficacy of conventional chemotherapy in lung and pancreatic cancer. Int. J. Cancer 2010, 126, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Schuch, G. EndoTAG-1. MediGene. Curr. Opin. Investig. Drugs 2005, 6, 1259. [Google Scholar] [PubMed]
- Lohr, M.; Haas, S.; Bechstein, W.; Bodoky, G.; Maerten, A.; Fischbach, W.; Lilla, C.; Mescheder, A.; Pap, A.; Fölsch, U. A phase II trial of cationic liposomal paclitaxel in combination with gemcitabine in patients with unresectable pancreatic cancer. In Proceedings of the ASCO Gastrointestinal Cancers Symposium, San Francisco, CA, USA, 15–17 January 2009. [Google Scholar]
- Wallace, T.L.; Larson, J.L.; Bazemore, S.A.; Wilson, C.W.; Cossum, P.A. The nonclinical safety evaluation of the anticancer drug ATRAGEN® (Liposomal all-trans-retinoic acid). Int. J. Toxicol. 2000, 19, 33–42. [Google Scholar] [CrossRef]
- Douer, D.; Estey, E.; Santillana, S.; Bennett, J.M.; Lopez-Bernstein, G.; Boehm, K.; Williams, T. Treatment of newly diagnosed and relapsed acute promyelocytic leukemia with intravenous liposomal all-transretinoic acid. Blood 2001, 97, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.C. Drug carriers in pharmaceutical design: Promises and progress. Curr. Pharm. Des. 2007, 13, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Wang, Y.-F.; Ahmad, I. Separation of liposome-entrapped mitoxantrone from nonliposomal mitoxantrone in plasma: Pharmacokinetics in mice. Methods Enzymol. 2005, 391, 176–185. [Google Scholar] [PubMed]
- Tayi, W.; Cheng, K. Advanced drug delivery in cancer therapy. In Advanced Drug Delivery; Mitra, C.H.L.A., Cheng, K., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Tari, A.M.; Gutiérrez-Puente, Y.; Monaco, G.; Stephens, C.; Sun, T.; Rosenblum, M.; Belmont, J.; Arlinghaus, R.; Lopez-Berestein, G. Liposome-incorporated Grb2 antisense oligodeoxynucleotide increases the survival of mice bearing bcr-abl-positive leukemia xenografts. Int. J. Oncol. 2007, 31, 1243–1250. [Google Scholar] [PubMed]
- Ashizawa, A.T.; Cortes, J. Liposomal delivery of nucleic acid-based anticancer therapeutics: BP-100–1.01. Expert Opin. Drug deliv. 2015, 12, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, M.; Kantarjian, H.M.; Ravandi, F.; Borthakur, G.; Garcia-Manero, G.; Andreeff, M.; Jabbour, E.; Konopleva, M.; Lim, M.; Pierce, S. Safety, Pharmacokinetics, and Efficacy of BP-100–1.01 (Liposomal Grb-2 Antisense Oligonucleotide) in Patients with Refractory or Relapsed Acute Myeloid Leukemia (AML), Philadelphia Chromosome Positive Chronic Myelogenous Leukemia (CML), Acute Lymphoblastic Leukemia (ALL), and Myelodysplastic Syndrome (MDS). Blood 2015, 126, 3801–3801. [Google Scholar]
- Singh, R.; Kumari, P.; Kumar, S. Nanotechnology for enhanced bioactivity of bioactive phytomolecules. In Nutrient Delivery; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Egusquiaguirre, S.P.; Pedraz, J.L.; Hernandez, R.M.; Igartua, M. Nanotherapeutic platforms for cancer treatment: From preclinical development to clinical application. In Nanoarchitectonics for Smart Delivery and Drug Targeting; Grumezescu, A., Holban, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 813–869. [Google Scholar]
- Lee, J.-M.; Yoon, T.-J.; Cho, Y.-S. Recent developments in nanoparticle-based siRNA delivery for cancer therapy. BioMed Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Resnier, P.; Montier, T.; Mathieu, V.; Benoit, J.-P.; Passirani, C. A review of the current status of siRNA nanomedicines in the treatment of cancer. Biomaterials 2013, 34, 6429–6443. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; Appeldoorn, C.C.; Dorland, R.; van Kregten, J.; Manca, F.; Vugts, D.J.; Windhorst, B.; van Dongen, G.A.; de Vries, H.E.; Maussang, D. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione PEGylated liposomal doxorubicin (2B3–101). PLoS ONE 2014, 9, e82331. [Google Scholar] [CrossRef]
- Gaillard, P.J.; Visser, C.C.; Appeldoorn, C.C.; Rip, J. Enhanced brain drug delivery: Safely crossing the blood–brain barrier. Drug Discov. Today Technol. 2012, 9, e155–e160. [Google Scholar] [CrossRef] [PubMed]
- Birngruber, T.; Raml, R.; Gladdines, W.; Gatschelhofer, C.; Gander, E.; Ghosh, A.; Kroath, T.; Gaillard, P.J.; Pieber, T.R.; Sinner, F. Enhanced Doxorubicin Delivery to the Brain Administered Through Glutathione PEGylated Liposomal Doxorubicin (2B3-101) as Compared with Generic Caelyx,®/Doxil®—A Cerebral Open Flow Microperfusion Pilot Study. J. Pharm. Sci. 2014, 103, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SN | Clinical Products (Approval Year) | Administration | Active Agent | Lipid/Lipid:Drug Molar Ratio | Indication | Company |
|---|---|---|---|---|---|---|
| 1. | Doxil® (1995) | i.v. | Doxorubicin | HSPC:Cholesterol:PEG 2000-DSPE (56:39:5 molar ratio) | Ovarian, breast cancer, Kaposi’s sarcoma | Sequus Pharmaceuticals |
| 2. | DaunoXome® (1996) | i.v. | Daunorubicin | DSPC and Cholesterol (2:1 molar ratio) | AIDS-related Kaposi’s sarcoma | NeXstar Pharmaceuticals |
| 3. | Depocyt® (1999) | Spinal | Cytarabine/Ara-C | DOPC, DPPG, Cholesterol and Triolein | Neoplastic meningitis | SkyPharma Inc. |
| 4. | Myocet® (2000) | i.v. | Doxorubicin | EPC:Cholesterol (55:45 molar ratio) | Combination therapy with cyclophosphamide in metastatic breast cancer | Elan Pharmaceuticals |
| 5. | Mepact® (2004) | i.v. | Mifamurtide | DOPS:POPC (3:7 molar ratio) | High-grade, resectable, non-metastatic osteosarcoma | Takeda Pharmaceutical Limited |
| 6. | Marqibo® (2012) | i.v. | Vincristine | SM:Cholesterol (60:40 molar ratio) | Acute lymphoblastic leukaemia | Talon Therapeutics, Inc. |
| 7. | Onivyde™ (2015) | i.v. | Irinotecan | DSPC:MPEG-2000:DSPE (3:2:0.015 molar ratio) | Combination therapy with fluorouracil and leucovorin in metastatic adenocarcinoma of the pancreas | Merrimack Pharmaceuticals Inc. |
| 8. | Abelcet® (1995) | i.v. | Amphotericin B | DMPC:DMPG (7:3 molar ratio) | Invasive severe fungal infections | Sigma-Tau Pharmaceuticals |
| 9. | Ambisome® (1997) | i.v. | Amphotericin B | HSPC:DSPG:Cholesterol:Amphotericin B (2:0.8:1:0.4 molar ratio) | Presumed fungal infections | Astellas Pharma |
| 10. | Amphotec® (1996) | i.v. | Amphotericin B | Cholesteryl sulphate:Amphotericin B (1:1 molar ratio) | Severe fungal infections | Ben Venue Laboratories Inc. |
| 11. | Visudyne® (2000) | i.v. | Verteporphin | Verteporphin:DMPC and EPG (1:8 molar ratio) | Choroidal neovascularisation | Novartis |
| 12. | DepoDur™ (2004) | Epidural | Morphine sulfate | DOPC, DPPG, Cholesterol and Triolein | Pain management | SkyPharma Inc. |
| 13. | Exparel® (2011) | i.v. | Bupivacaine | DEPC, DPPG, Cholesterol and Tricaprylin | Pain management | Pacira Pharmaceuticals, Inc. |
| 14. | Epaxal® (1993) | i.m. | Inactivated hepatitis A virus (strain RGSB) | DOPC:DOPE (75:25 molar ratio) | Hepatitis A | Crucell, Berna Biotech |
| 15. | Inflexal® V (1997) | i.m. | Inactivated hemaglutinine of Influenza virus strains A and B | DOPC:DOPE (75:25 molar ratio) | Influenza | Crucell, Berna Biotech |
| SN | Products | Administration | Active Agent | Lipid Composition | Indication | Company |
|---|---|---|---|---|---|---|
| Phase III | ||||||
| 1. | Arikace | Aerosol delivery | Amikacin | DPPC and cholesterol | Lung infections | Transave Inc. |
| 2. | Stimuvax | s.c. | Tecemotide | Cholesterol, DMPG, DPPC | Non-small cell lung cancer | Oncothyreon Inc. |
| 3. | T4N5 liposomal lotion | Topical | T4 endonuclease V | Egg lecithin | Xeroderma pigmentosum | AGI Dermatics Inc. |
| 4. | Liprostin | i.v. | Prostaglandin E-1 (PGE-1) | Unknown | Restenosis after angioplasty | Endovasc Inc. |
| 5. | ThermoDox | i.v. | Doxorubicin | DPPC, Myristoyl stearyl phosphatidylcholine and DSPE-N-[amino(polyethylene glycol)-2000] | Hepatocellular carcinoma and also recurring chest wall breast cancer | Celsion |
| 6. | Lipoplatin | i.v. | Cisplatin | DPPG, soy phosphatidyl choline, mPEG-distearoyl phosphatidylethanolamine lipid conjugate and cholesterol | Non-small cell lung cancer | Regulon Inc. |
| Phase II | ||||||
| 7. | Aroplatin | i.v. | Platinum analogue cis-(trans- R,R-1,2-diaminocyclohexane) bis (neodecanoato) platinum (II) | DMPC and DMPG | Metastatic colorectal cancer | Agenus Inc. |
| 8. | Liposomal annamycin | i.v. | Semi-synthetic doxorubicin analogue annamycin | DMPC and DMPG | Relapsed or refractory acute myeloid leukaemia | Aronex Pharmaceuticals |
| 9. | SPI-077 | i.v. | Cisplatin | Soybean phosphatidylcholine, cholesterol | Lung, head and neck cancer | Alza Corporation |
| 10. | OSI-211 | i.v. | Lurtotecan | HSPC and cholesterol | Ovarian, head and neck cancer | OSI Pharmaceuticals |
| 11. | S-CKD602 | i.v. | Potent topoisomerase I inhibitor | Phospholipids covalently bound to mPEG | Cancer | Alza Corporation |
| 12. | LE-SN38 | i.v. | Irinotecan’s active metabolite | DOPC, cholesterol and cardiolipin | Advanced colorectal cancer | NeoPharm Labs Ltd. |
| 13. | LEP-ETU | i.v. | Paclitaxel | DOPC, cholesterol and cardiolipin | Cancer | NeoPharm Labs Ltd. |
| 14. | Endotag-I | i.v. | Paclitaxel | DOTAP: DOPC: Paclitaxel | Breast and pancreatic cancers | Medigene |
| 15. | Atragen | i.v. | All-trans retinoic acid | DMPC and soybean oil | Hormone-resistant prostate cancer, renal cell carcinoma and acute myelogenous leukaemia | Aronex Pharmaceuticals |
| Phase I | ||||||
| 16. | LEM-ETU | i.v. | Mitoxantrone | DOPC, cholesterol and cardiolipin | Various cancers | NeoPharm Labs Ltd. |
| 17. | Liposomal Grb-2 | i.v. | Antisense oligodeoxynucleotide growth factor receptor bound protein 2 (Grb-2) | Unknown | Hematologic malignancies | Bio-Path holdings |
| 18. | INX-0125 | i.v. | Vinorelbine tartrate | Cholesterol and sphingomyelin | Advanced solid tumours | Inex Pharmaceuticals |
| 19. | INX-0076 | i.v. | Topotecan | Cholesterol and sphingomyelin | Advanced solid tumours | Inex Pharmaceuticals |
| 20. | TKM-080301 | Hepatic intra-arterial administration | PLK1 siRNA | Unique LNP technology (formerly referred to as stable nucleic acid-lipid particles or SNALP) | Neuroendocrine tumours | Tekmira Pharmaceuticals |
| 21. | Atu027 | i.v. | PKN3 siRNA | AtuFECT01 | Pancreatic cancer | Silence Therapeutics |
| 22. | 2B3-101 | i.v. | Doxorubicin | Glutathione PEGylated liposomes | Solid tumours | 2-BBB therapeutic |
| 23. | MTL-CEBPA | i.v. | CEBPA siRNA | SMARTICLES® liposomal nanoparticles | Liver cancer | MiNA Therapeutics |
| 24. | ATI-1123 | i.v. | Docetaxel | Protein stabilizing liposomes (PSL™) | Solid tumours | Azaya therapeutic |
| 25. | LiPlaCis | i.v. | Cisplatin | The lipid composition of the LiPlasomes is tailored to be specifically sensitive to degradation by the sPLA2 enzyme | Advanced solid tumours | Oncology Venture |
| 26. | MCC-465 | i.v. | Doxorubicin | DPPC, cholesterol and maleimidated palmitoyl phosphatidyl ethanolamine; immunoliposomes tagged with PEG and the F(ab′)2 fragment of human monoclonal antibody GAH | Metastatic stomach cancer | Mitsubishi Tanabe Pharma Corporation |
| 27. | SGT-53 | i.v. | p53 gene | Cationic lipids complexed with plasmid DNA encoding wild-type p53 tumour suppressor protein | Various solid tumours | SynerGene Therapeutics |
| 28. | Alocrest | i.v. | Vinorelbine | Sphingomyelin/cholesterol (OPTISOME™) | Breast and lung cancers | Spectrum Pharmaceuticals |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. https://doi.org/10.3390/pharmaceutics9020012
Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics. 2017; 9(2):12. https://doi.org/10.3390/pharmaceutics9020012
Chicago/Turabian StyleBulbake, Upendra, Sindhu Doppalapudi, Nagavendra Kommineni, and Wahid Khan. 2017. "Liposomal Formulations in Clinical Use: An Updated Review" Pharmaceutics 9, no. 2: 12. https://doi.org/10.3390/pharmaceutics9020012
APA StyleBulbake, U., Doppalapudi, S., Kommineni, N., & Khan, W. (2017). Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics, 9(2), 12. https://doi.org/10.3390/pharmaceutics9020012