Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release



Abstract
:
1. Introduction
1.1. Overview of Self-Emulsifying Drug Delivery Systems (SEDDS)
1.2. Applications of SEDDS
1.3. Components and Formulation of Liquid/Semisolid SEDDS
1.3.1. Oils
1.3.2. Surfactants
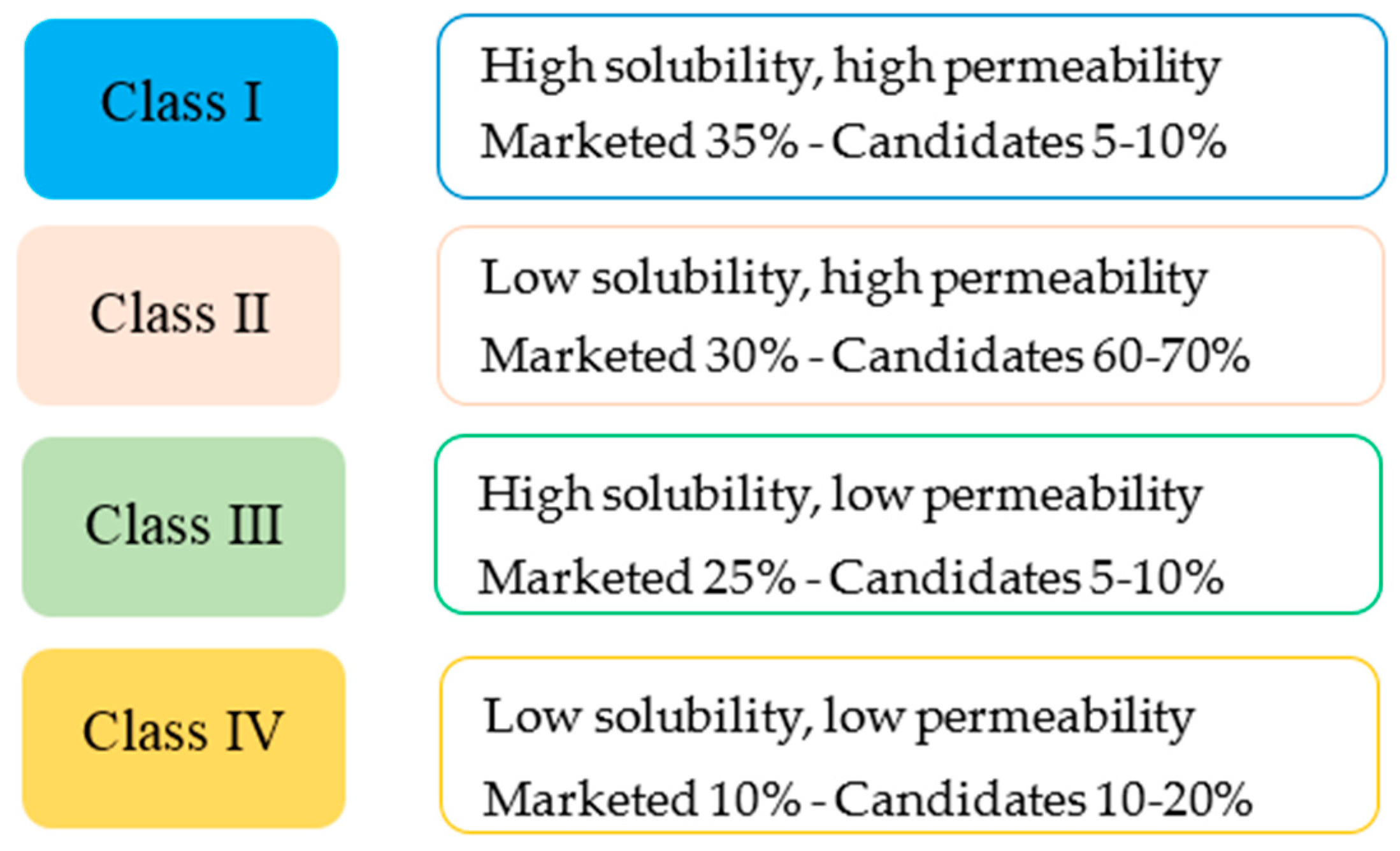
1.3.3. Formulation of SEDDS—Lipid Based Formulation Classification System
2. Solid Self-Emulsifying Drug Delivery Systems (S-SEDDS)
- They can be administered as immediate or controlled release formulations depending on the choice of the powder excipient, with which the SEDDS liquid is formulated.
- They avoid stringent processing requirements since it is a solid dosage form.
- The dose is presented in precise weight of S-SEDDS powder, granules or pellets filled into a capsule or processed into tablet.
- They are easily transferred and stored, thus improving patient compliance.
- Production cost is considerably less compared to liquid capsule filling since self-emulsifying coarse powders, granules and pellets have excellent flowability, allowing fast and reproducible capsule or die-filling, enabling high production rates.
- Self-emulsifying granules or pellets, in particular, being multiple-unit dosage forms provide therapeutic advantages that are characteristic of these dosage forms. They promote reduction of the variation of the gastric emptying time, smooth passage in the gut and low risk of dose dumping. All these conduce to the minimization of the variability in plasma levels [28].
- More importantly, studies have shown that the release of progesterone in dogs from self-emulsifying pellets was equivalent to administration of the microemulsion liquid [3].
3. Components of S-SEDDS
3.1. Pellet and Granule Forming Powders
3.1.1. Microcrystalline Cellulose (MCC)
3.1.2. Adsorbents—Potential Alternatives to MCC
3.2. Controlled Release Agents
3.3. Crystallization Inhibitors and Other Additives
4. Formation Mechanisms
4.1. Effect of Drug Incorporation on the Characteristics of SEDDS in Water Emulsions
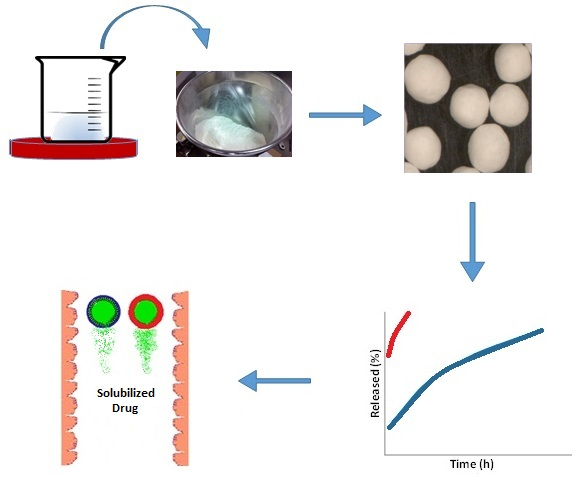
4.2. Self-Emulsifying Powders and Granules
4.3. Instant Release Self-Emulsifying Pellets
4.4. Controlled Release Self-Emulsifying Pellets
5. Relationships between the Characteristics of the Starting Massing Emulsions and the Properties of the S-SEDDS Pellets
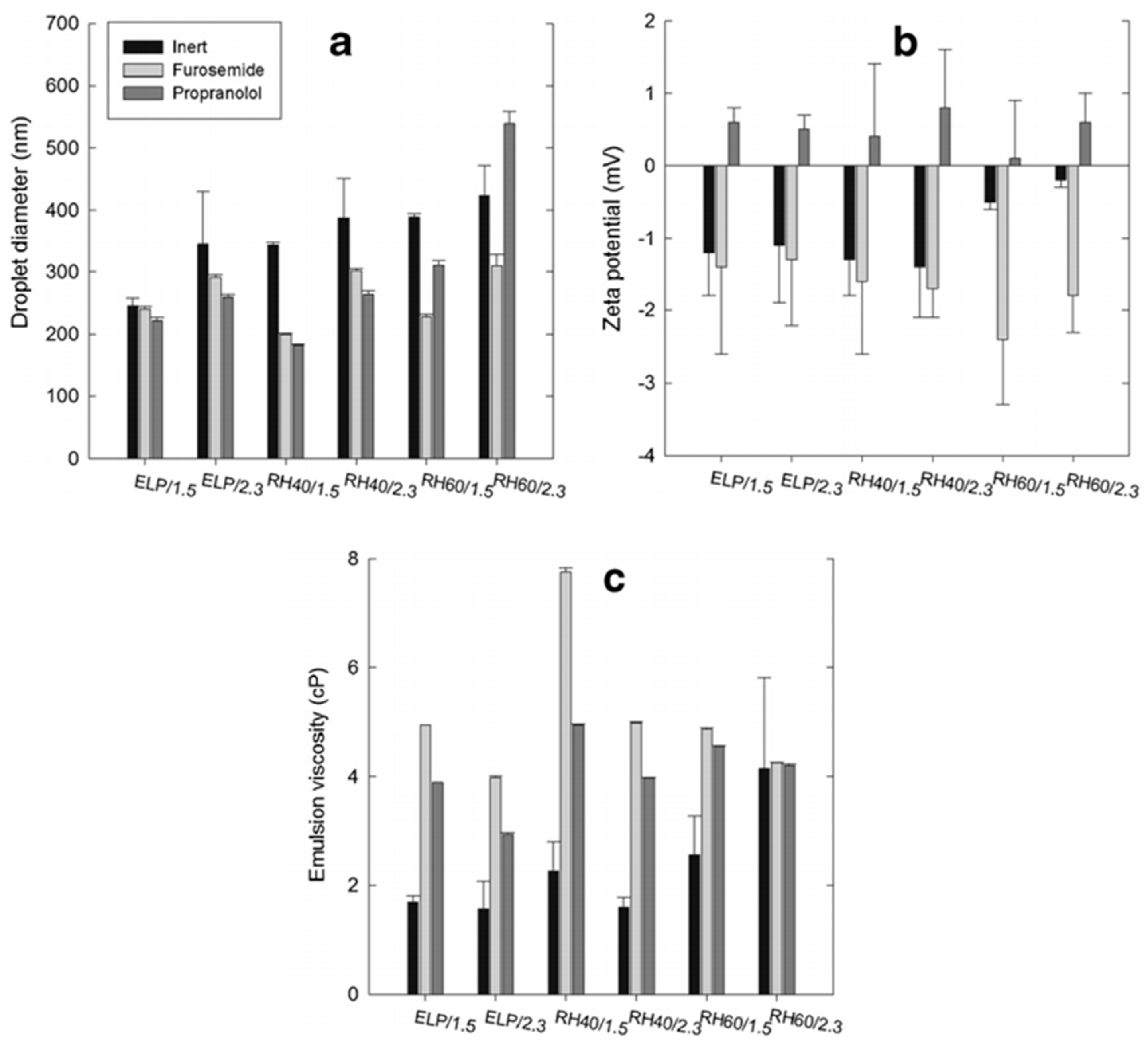
5.1. Droplet Size, Zeta Potential and Viscosity
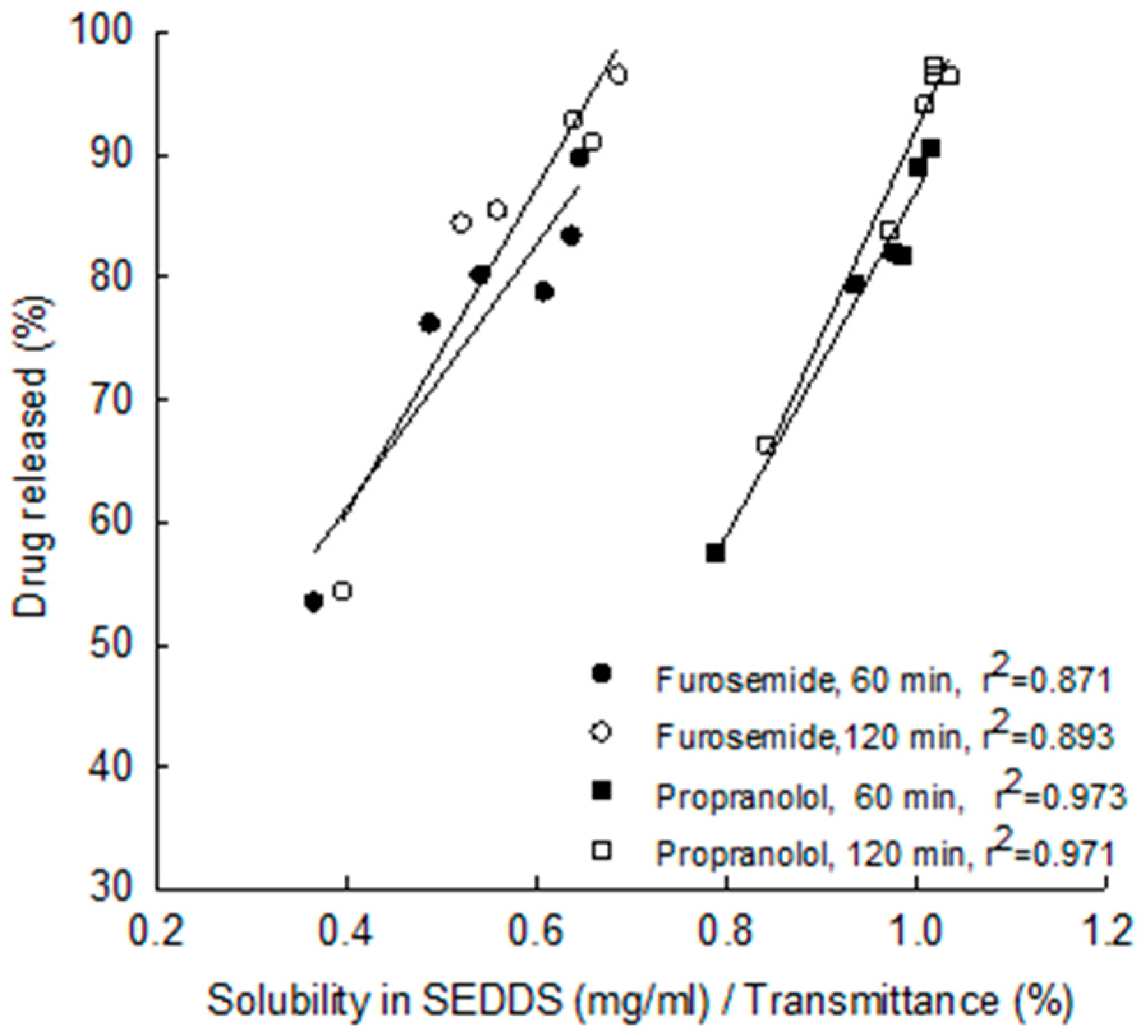
5.2. Rate of Re-Emulsification of Emulsion from the S-SEDDS and Drug Release
5.3. Mechanical Strength
6. Examples of S-SEDDS Formulations—Instant Release and Controlled Release
6.1. Examples of Instant Release S-SEDDS
6.2. Examples of Controlled Release S-SEDDS
7. Conclusions
Conflicts of Interest
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and “self-microemulsifying” drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Tuleu, C.; Newton, M.; Rose, J.; Euler, D.; Saklatvala, R.; Clarke, A.; Booth, S. Comparative Bioavailability study in dogs of a Self-Emulsifying formulation of Progesterone presented in a Pellet and liquid form compared with an aqueous suspension of Progesterone. J. Pharm. Sci. 2004, 93, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, P.; Lee, B.-J.; Oh, H.D.; Kim, J.O.; Hong, J.M.; Jee, J.-P.; Kim, J.A.; Yoo, B.K.; Woo, S.J.; Yong, S.C.; et al. Enhanced oral bioavailability of dexibuprofen by a novel solid Self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Biopharm. 2009, 72, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.A.; Anand, O.; Chun, N.; Conner, P.D.; Mehta, U.M.; Nhu, D.T.; Polli, E.J.; Yu, X.L.; Davit, M.B. Statistics on BCS Classification of Generic Drug Products Approved between 2000 and 2011 in the USA. AAPS J. 2012, 14, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, M.L.; Holm, R.; Abrahamsson, B.; Andersen, J.R.; Müllertz, A. Effect of food intake and co-administration of placebo self-nanoemulsifying drug delivery systems on the absorption of cinnarizine in healthy human volunteers. Eur. J. Pharm. Sci. 2016, 84, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Document Self-nano-emulsifying drug delivery systems: An update of the biopharmaceutical aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Kang, J.-H.; Ngo, L.; Tran, P.; Lee, Y.-B. Preparation and Evaluation of Solid-Self-Emulsifying Drug Delivery System Containing Paclitaxel for Lymphatic Delivery. J. Nanomater. 2016, 2016, 3642418. [Google Scholar] [CrossRef]
- Li, L.; Yi, T.; Lam, C.W. Interactions between human multidrug resistance related protein (MRP2; ABCC2) and excipients commonly used in self-emulsifying drug delivery systems (SEDDS). Int. J. Pharm. 2013, 447, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Elgart, A.; Cherniakov, I.; Aldouby, Y.; Domb, J.A.; Hoffman, A. Improved Oral Bioavailability of BCS Class 2 Compounds by Self Nano-Emulsifying Drug Delivery Systems (SNEDDS): The Underlying Mechanisms for Amiodarone and Talinolol. Pharm. Res. 2013, 30, 3029–3044. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, A.; Mäder, K. Preparation and characterization of a self-emulsifying pellet formulation. Eur. J. Pharm. Biopharm. 2007, 66, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kusawake, T.; Ishida, M.; Tawa, R.; Shibata, N.; Takada, K. Oral solid gentamicin preparation using emulsifier and adsorbent. J. Control Release 2005, 105, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Marketed Drug Molecules According to the BCS Classification System. Available online: http://www.capsugel.com/knowledge-center/webinars/archived-webinars/paginate/P15 (accessed on 2 April 2017).
- Tran, M.; Wang, C. Semi-solid materials for controlled release drug formulation: Current status and future prospects. Front. Chem. Sci. Eng. 2014, 8, 225–232. [Google Scholar] [CrossRef]
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, N.H.; Dalrymple, M.D.; Serajuddin, T.M.A. A Comparative Evaluation of Mono-, Di- and Triglyceride of Medium Chain Fatty Acids by Lipid/Surfactant/Water Phase Diagram, Solubility Determination and Dispersion Testing for Application in Pharmaceutical Dosage Form Development. Pharm. Res. 2012, 292, 285–305. [Google Scholar] [CrossRef] [PubMed]
- Gattefosse Product Information. Lipid Excipients for Oral Dosage Forms; Gattefossé: Lyon, France, 2016. [Google Scholar]
- Buyukozturk, F.; Benneyan, J.C.; Carrier, R.L. Impact of emulsion-based drug delivery systems on intestinal permeability and drug release kinetics. J. Control Release 2010, 142, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Cremophor RH40 and Cremophor RH60; BASF Technical Information: Ludwigshafen, Germany, May 2010.
- Abdalla, A.; Mäder, K. ESR studies on the influence of physiological dissolution and digestion media on the lipid phase characteristics of SEDDS and SEDDS pellets. Int. J. Pharm. 2009, 367, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Vithani, K.; Hawley, A.; Jannin, V.; Pouton, C.; Boyd, B.J. Inclusion of Digestible Surfactants in Solid SMEDDS Formulation Removes Lag Time and Influences the Formation of Structured Particles During Digestion. AAPS J. 2017, 19, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Pouton, W.C. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Thi, T.D.; Van Speybroeck, M.; Barillaro, V.; Martens, J.; Annaert, P.; Augustijns, P.; Van Humbeeck, J.; Vermant, J.; Van den Mooter, G. Formulate-ability of ten compounds with different physicochemical profiles in SMEDDS. Eur. J. Pharm. Sci. 2009, 38, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Matsaridou, I.; Barmpalexis, P.; Salis, A.; Nikolakakis, I. The influence of surfactant HLB and oil/surfactant ratio on the formation and properties of Self-emulsifying pellets and microemulsion reconstitution. AAPS PharmSciTech. 2012, 13, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Jannin, V.; Chevrier, S.; Michenaud, M.; Camille Dumont, C.; Belotti, S.; Chavant, Y.; Demarne, F. Development of self emulsifying lipid formulations of BCS class II drugs with low to medium lipophilicity. Int. J. Pharm. 2015, 495, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.T.; Cadé, D.; Benameur, H. Challenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administration. Adv. Drug Deliv. Rev. 2008, 60, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Chu, M.; Itagaki, K.; Xin, P.; Zhou, X.; Zhang, D.; Wang, Y.; Fu, J.; Sun, S. Formulation and In Vitro Characterization of a Novel Solid Lipid-Based Drug Delivery System. Chem. Pharm. Bull. 2014, 62, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Abuhelwa, Y.A.; Foster, J.R.D.; Upton, N.R. A Quantitative Review and Meta-models of the Variability and Factors Affecting Oral Drug Absorption—Part II: Gastrointestinal Transit Time. AAPS J. 2016, 18, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.; Petersson, J.; Podczeck, F.; Clarke, A.; Booth, S. The influence of formulation variables on the properties of pellets containing a self-emulsifying mixture. J. Pharm. Sci. 2001, 90, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Nikolakakis, I. Influence of SEDDS on the shape of pellets prepared by extrusion/spheronization. Unpublished data. 2014.
- Nikolakakis, I.; Panagopoulou, A.; Salis, A.; Malamataris, S. Relationships between the properties of Self-Emulsifying pellets and of the Emulsions used as massing liquids for their preparation. AAPS PharmSciTech. 2015, 16, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lin, C.; Chen, D.; Zhang, J.; Liu, Z.; Wu, W.; Song, H. Sirolimus solid self-microemulsifying pellets: Formulation development, characterization and bioavailability evaluation. Int. J. Pharm. 2012, 438, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kallakunta, V.R.; Eedara, B.B.; Jukanti, R.; Ajmeera, R.K.; Bandari, S. A Gelucire 44/14 and labrasol based solid self-emulsifying drug delivery system: Formulation and evaluation. J. Pharm. Investig. 2013, 43, 185–196. [Google Scholar] [CrossRef]
- Patel, J.; Dhingani, A.; Tilala, J.; Raval, M.; Sheth, N. Formulation and development of Self-Nanoemulsifying granules of Olmesartan Medoxomil for Bioavailability enhancement. Part. Sci. Technol. 2014, 32, 274–290. [Google Scholar] [CrossRef]
- Krupa, A.; Jachowicz, R.; Kurek, M.; Figiel, W.; Kwiecień, M. Preparation of solid self-emulsifying drug delivery systems using magnesium aluminometasilicates and fluid-bed coating process. Powder Technol. 2014, 266, 329–339. [Google Scholar] [CrossRef]
- Cho, W.; Kim, M.S.; Kim, J.S.; Park, J.; Park, H.J.; Cha, K.H.; Park, J.S.; Hwang, S.J. Optimized formulation of solid self-microemulsifying sirolimus delivery systems. Int. J. Nanomed. 2013, 8, 1673–1682. [Google Scholar]
- Desai, N.S.; Nagarsenker, M.S. Design and evaluation of self-nanoemulsifying pellets of Repaglinide. AAPS PharmSciTech. 2013, 14, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Chavan, B.R.; Modi, R.S.; Bansal, K.A. Role of solid carriers in pharmaceutical performance of solid supersaturable SEDDS of celecoxib. Int. J. Pharm. 2015, 495, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.M. Extrusion and Extruders. In Encyclopaedia of Pharmaceutical Technology, 3rd ed.; Swarbrick, J., Ed.; CRC Press: Boca Raton, Fl, USA, 2007; Volume 3, p. 1717. [Google Scholar]
- Basit, A.W.; Newton, J.M.; Lacey, L.F. Formulation of ranitidine pellets by extrusion-spheronization with little or no microcrystalline cellulose. Pharm. Dev. Technol. 1999, 4, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Steele, D.F.; Edge, S.; Tobyn, M.J.; Moreton, R.C.; Staniforth, J.N. Adsorption of an amine drug onto microcrystalline cellulose and silicified microcrystalline cellulose samples. Drug Dev. Ind. Pharm. 2003, 29, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Al-Nimry, S.S.; Assaf, S.M.; Jalal, I.M.; Najib, N.M. Adsorption of ketotifen onto some pharmaceutical excipients. Int. J. Pharm. 1997, 149, 115–121. [Google Scholar] [CrossRef]
- Okada, S.; Nakahara, H. Adsorption of drugs on microcrystalline cellulose suspended in aqueous solutions. Chem. Pharm. Bull. 1987, 35, 761–768. [Google Scholar] [CrossRef]
- Rivera, S.L.; Ghodbane, S. In vitro adsorption-desorption of famotidine on microcrystalline cellulose. Int. J. Pharm. 1994, 108, 31–38. [Google Scholar] [CrossRef]
- Balaxi, M.; Nikolakakis, I.; Kachrimanis, K.; Malamataris, S. Combined Effects of wetting, drying, and microcrystalline cellulose type on the mechanical strength and disintegration of pellets. J. Pharm. Sci. 2009, 98, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Serratoni, M.; Newton, M.; Booth, S.; Clarke, A. Controlled drug release from pellets containing water-insoluble drugs dissolved in a self-emulsifying system. Eur. J. Pharm. Biopharm. 2007, 65, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, S.; Chen, Y.; Ping, Q. Self-emulsifying bifendate pellets: Preparation, characterization and oral bioavailability in rats. Drug Dev. Ind. Pharm. 2013, 39, 724–732. [Google Scholar] [PubMed]
- Fuji Chemical Industries. Product Information. Available online: http://www.neusilin.com/product/index.php (accessed on 2 April 2017).
- Grace Davison Discovery Sciences. Silica Excipient for Pharmaceutical Applications. Available online: https://grace.com/pharma-and-biotech/en-us/Documents/Syloid/M298_Syloid244FPSilica_TechInfo.pdf (accessed on 2 April 2017).
- Agarwal, V.; Siddiqui, A.; Ali, H.; Nazzal, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm. 2009, 366, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kanuganti, S.; Jukanti, R.; Veerareddy, P.R.; Bandari, S. Paliperidone-Loaded Self-Emulsifying drug delivery systems (SEDDS) for improved oral delivery. J. Dispers. Sci. Technol. 2012, 33, 506–515. [Google Scholar] [CrossRef]
- Milović, M.; Djuriš, J.; Djekić, L.; Vasiljević, D.; Ibrić, S. Characterization and evaluation of solid self-microemulsifying drug delivery systems with porous carriers as systems for improved carbamazepine release. Int. J. Pharm. 2012, 436, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Jena, S.S.; Patra, C.N.; Rizwan, M.; Swain, S.; Sruti, J.; Rao, M.E.B.; Singh, B. Development of solid self-nanoemulsifying granules (SSNEGs) of ondansetron hydrochloride with enhanced bioavailability potential. Colloid Surf. B 2013, 101, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Suthar, V.; Butani, S.; Gohel, M. Solid self-emulsified nanostructures of Lercanidipine hydrochloride: A potential approach to improve the fraction of the dose absorbed. J. Drug Del. Sci. Technol. 2016, 31, 11–21. [Google Scholar] [CrossRef]
- Patil, P.; Joshi, P.; Paradkar, A. Effect of Formulation Variables on Preparation and Evaluation of Gelled Self-emulsifying Drug Delivery System (SEDDS) of Ketoprofen. AAPS PharmSciTech. 2004, 5, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Setthacheewakul, S.; Kedjinda, W.; Maneenuan, D.; Wiwattanapatapee, R. Controlled release of oral Tetrahydrocurcumin from a novel Self-Emulsifying floating drug delivery system (SEFDDS). AAPS PharmSciTech. 2011, 12, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Nikolakakis, I. Ibuprofen self-emulsifying pellets prepared with colloidal silicon dioxide/microcrystalline cellulose and triglycerides. Unpublished work. 2014. [Google Scholar]
- Alderman, D.A. A review of cellulose ethers in hydrophilic matrices for oral controlled—Release dosage forms. Int. J. Pharm. Technol. Prod. Manuf. 1984, 5, 1–9. [Google Scholar]
- Li, L.C.; Martini, G.L.; Ford, L.J.; Roberts, M. The use of hypromellose in oral drug delivery. J. Pharm. Pharmacol. 2005, 57, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, R.; Wu, J.; Shen, Q. Characterization and evaluation of self-microemulsifying sustained-release pellet formulation of puerarin for oral delivery. Int. J. Pharm. 2012, 427, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; Chen, J.; Huang, A.; Zhang, J.; Lin, B.; Liu, Z.; Zhang, M.; Chen, X.; Zeng, L.; Zhang, L.; et al. Development of solidified self-microemulsifying delivery systems with enhanced stability of sirolimus and extended release. Int. J. Pharm. 2016, 513, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Rush, B.D.; Pfund, W.P.; Huang, T.; Bauer, J.M.; Morozowich, W.; Kuo, M.S.; Hageman, M.J. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 2003, 92, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Ye, X.; Shang, X.; Peng, X.; Bao, Q.; Liu, M.; Guo, M.; Li, F. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf. A Physicochem. Eng. Asp. 2012, 396, 22–28. [Google Scholar] [CrossRef]
- Song, W.H.; Park, J.H.; Yeom, D.W.; Ahn, B.K.; Lee, K.M.; Lee, S.G.; Woo, H.S.; Choi, Y.W. Enhanced dissolution of celecoxib by supersaturating self-emulsifying drug delivery system (S-SEDDS) formulation. Arch. Pharm. Res. 2013, 36, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Franceschinis, E.; Voinovich, D.; Grassi, M.; Perissutti, B.; Filipovic-Grcic, J.; Martinac, A.; Meriani-Merlo, F. Self-emulsifying pellets prepared by wet granulation in high-shear mixer: Influence of formulation variables and preliminary study on the in vitro absorption. Int. J. Pharm. 2005, 291, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.P.; Nagarsenker, M.S. Self-nanoemulsifying granules of ezetimibe: Design, optimization and evaluation. Eur. J. Pharm. Sci. 2008, 35, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Nekkanti, V.; Karatgi, P.; Prabhu, R.; Pillai, R. Solid Self-Microemulsifying formulation for Candesartan Cilexetil. AAPS PharmSciTech. 2009, 11, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, J.; Wang, Y.; Liu, X.; Liu, Y.; Fu, Q.; Meng, P.; He, Z. Solid self-emulsifying nitrendipine pellets: Preparation and in vitro/in vivo evaluation. Int. J. Pharm. 2010, 383, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Franceschinis, E.; Bortoletto, C.; Perissutti, B.; Dal Zotto, M.; Voinovich, D.; Realdon, N. Self-emulsifying pellets in a lab-scale high shear mixer: Formulation and production design. Powder Technol. 2011, 207, 113–118. [Google Scholar] [CrossRef]
- Pund, S.; Shete, Y.; Jagadale, S. Multivariate analysis of physicochemical characteristics of lipid based nanoemulsifying cilostazol—Quality by design. Colloid Surf. B 2014, 115, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Nikolakakis, I.; Malamataris, S. Self-Emulsifying pellets: Relations between kinetic parameters of drug release and emulsion Reconstitution—Influence of formulation variables. J. Pharm. Sci. 2014, 103, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Franceschinis, E.; Santomaso, A.C.; Benda, L.; Perissutti, B.; Voinovich, D.; Realdon, N. Influence of process variables on the properties of simvastatin self-emulsifying granules obtained through high shear wet granulation. Powder Technol. 2015, 274, 173–179. [Google Scholar] [CrossRef]
- Mudie, M.D.; Shi, Y.; Ping, H.; Gao, P.; Amidon, L.G.; Amidon, E.G. Mechanistic analysis of solute transport in an in vitro physiological two-phase dissolution apparatus. Biopharm. Drug Dispos. 2012, 33, 378–402. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Benet, B.L. Predicting Drug Disposition via Application of BCS: Transport/Absorption/Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Taupitz, T.; Dressman, B.J.; Klein, S. New formulation approaches to improve solubility and drug release from fixed dose combinations: Case examples pioglitazone/glimepiride and ezetimibe/simvastatin. Eur. J. Pharm. Biopharm. 2013, 84, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of orally administered drugs on the World Health Organization model list of essential medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Takano, R.; Sugano, K.; Higashida, A.; Hayashi, Y.; Machida, M.; Aso, Y.; Yamashita, S. Oral Absorption of Poorly Water-Soluble Drugs: Computer Simulation of Fraction Absorbed in Humans from a Miniscale Dissolution Test. Pharm. Res. 2006, 23, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Pawar, B.Y.; Bansal, K.A. Identification of permeability-related hurdles in oral delivery of curcumin using the Caco-2 cell model. Eur. J. Pharm. Biopharm. 2011, 77, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Shohin, I.E.; Kulinich, I.J.; Ramenskaya, V.G.; Abrahamsson, B.; Kopp, S.; Langguth, P.; Polli, E.J.; Shah, P.V.; Groot, D.W.; Barends, M.D.; et al. Biowaiver Monographs for Immediate Release Solid Oral Dosage Forms: Piroxicam. J. Pharm. Sci. 2014, 103, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Rath, B.; Dwivedi, A.K. Dissolution rate enhancement of BCS class II drug, paliperidone by spray drying. Res. J. Pharm. Biol. Chem. Sci. 2013, 4, 145–155. [Google Scholar]
- Petruševska, M.; Homar, M.; Petek, B.; Resman, A.; Kocjan, D.; Urle, U.; Peternel, L. Hydroxypropyl Methylcellulose Mediated Precipitation Inhibition of Sirolimus: From a Screening Campaign to a Proof-of-Concept Human Study. Mol. Pharm. 2013, 10, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, L.; Liu, Y.; Wang, G.; Wang, G.; Qiao, Y. Biopharmaceutics classification of puerarin and comparison of perfusion approaches in rats. Int. J. Pharm. 2014, 466, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Jinno, J.; Kamada, N.; Miyake, M.; Yamada, K.; Mukai, T.; Odomi, M.; Toguchi, H.; Liversidge, G.G.; Higaki, K.; Kimura, T. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J. Control Release 2006, 111, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liao, J.; Qi, X.; Zhang, J. Coamorphous repaglinide-saccharin with enhanced dissolution. Int. J. Pharm. 2013, 450, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Cristofoletti, R.; Dressman, J.B. Dissolution Methods to Increasing Discriminatory Power of In Vitro Dissolution Testing for Ibuprofen Free Acid and Its Salts. J. Pharm. Sci. 2017, 106, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Vogelpoel, H.; Welink, J.; Amidon, G.L.; Junginger, H.E.; Midha, K.K.; Moller, H.; Olling, M.; Shah, V.P.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: Verapamil hydrochloride, propranolol hydrochloride, and atenolol. J. Pharm. Sci. 2010, 99, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Granero, G.E.; Longhi, M.R.; Mora, M.J.; Junginger, H.E.; Midha, K.K.; Shah, V.P.; Stavchansky, S.; Dressman, J.B.; Barends, D.M. Biowaivermonographs for immediate release solid oral dosage forms: Furosemide. J. Pharm. Sci. 2010, 99, 2544–2556. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.Y.H.; Du, Z.; Wang, N.G.; Chan, H.M.; Chang, Q.; Lai, L.C.; Chow, A.H.L.; Zheng, Y. Spray freeze drying with polyvinylpyrrolidone and sodium caprate for improved dissolution and oral bioavailability of oleanolic acid, a BCS Class IV compound. Int. J. Pharm. 2001, 404, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Zur, M.; Cohen, N.; Agbaria, R.; Dahan, A. The biopharmaceutics of successful controlled release drug product: Segmental-dependent permeability of glipizide vs. metoprolol throughout the intestinal tract. Int. J. Pharm. 2015, 489, 304–310. [Google Scholar] [PubMed]
- Yazdanian, M.; Briggs, K.; Jankovsky, C.; Hawi, A. The “High Solubility” Definition of the Current FDA Guidance on Biopharmaceutical Classification System May Be Too Strict for Acidic Drugs. Pharm. Res. 2004, 2, 293–299. [Google Scholar] [CrossRef]
- Pubchem Open Chemistry Base. Available online: https://pubchem.ncbi.nlm. nih.gov/compound (accessed on 2 April 2017).
- Pouton, C.W. Effects of the inclusion of a model drug on the performance of self emulsifying formulations. J. Pharm. Pharmacol. 1985, 37. [Google Scholar] [CrossRef]
- Sznitowska, M.; Janicki, S.; Dabrowska, E.; Zurowska-Pryczkowska, K. Submicron emulsions as drug carriers Studies on destabilization potential of various drugs. Eur. J. Pharm. Sci. 2001, 12, 175–179. [Google Scholar] [CrossRef]
- Patil, S.S.; Venugopal, E.; Bhat, S.; Mahadik, R.K.; Paradkar, R.A. Microstructural Elucidation of Self-Emulsifying System: Effect of Chemical Structure. Pharm. Res. 2012, 29, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Cavinato, M.; Franceschinis, E.; Cavallari, S.; Realdon, N.; Santomaso, A. Relationship between particle shape and some process variables in high shear wet granulation using binders of different viscosity. Chem. Eng. J. 2010, 164, 292–298. [Google Scholar] [CrossRef]
- Newton, M.J.; Bazzigialuppi, M.; Podczeck, F.; Booth, S.; Clarke, A. The rheological properties of self-emulsifying systems, water and microcrystalline cellulose. Eur. J. Pharm. Sci. 2005, 26, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Podczeck, F. A novel aid for the preparation of pellets by extrusion/spheronizationpellets. Pharm. Technol. Eur. 2008, 20, 26–31. [Google Scholar]
- Agrawal, A.G.; Kumar, A.; Gide, P.S. Self emulsifying drug delivery system for enhanced solubility and dissolution of glipizide. Colloid Surf. B 2015, 126, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Podczeck, F.; Newton, J.M. A Shape Factor to Characterize The Quality of Spheroids. J. Pharm. Pharmacol. 1994, 46, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Gershanik, T.; Benzeno, S.; Benita, S. Interaction of a self-emulsifying lipid drug delivery system with the everted rat intestinal mucosa as a function of droplet size and surface charge. Pharm. Res. 1998, 15, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Yap, S.P.; Yuen, K.H. Influence of lipolysis and droplet size on tocotrienol absorption from self-emulsifying formulations. Int. J. Pharm. 2004, 281, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Iosio, T.; Voinovich, D.; Grassi, M.; Pinto, J.F.; Perissutti, B.; Zacchigna, M.; Quintavalle, U.; Serdoz, F. Bi-layered self-emulsifying pellets prepared by co-extrusion and spheronization: Influence of formulation variables and preliminary study on the in vivo absorption. Eur. J. Pharm. Biopharm. 2008, 69, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.G.; Kumar, A.; Gide, P.S. Formulation of solid self-nanoemulsifying drug delivery systems using N-methyl pyrrolidone as cosolvent. Drug Dev. Ind. Pharm. 2014, 41, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Nazzal, S.; Smalyukh, I.I.; Lavrentovich, O.D.; Khan, M.A. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: Mechanism and progress of emulsion formation. Int. J. Pharm. 2002, 235, 247–265. [Google Scholar] [CrossRef]
- Erkoboni, D. Extrusion/Spheronization. In Pharmaceutical Extrusion Technology; Ghebre-Sellassie, I., Martin, C., Eds.; Marcel Dekker Inc: New York, NY, USA, 2003; pp. 288–291. [Google Scholar]
- Cuine, J.F.; Charman, W.N.; Pouton, C.W.; Edwards, G.A.; Porter, C.J. Increasing the proportional content of surfactant (Cremophor EL) relative to lipid in self-emulsifying lipid-based formulations of danazol reduces oral bioavailability in beagle dogs. Pharm. Res. 2007, 24, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Hamed Almurisi, S.; Ahmed Mahdi Dukhan, A.; Mandal, U.K.; Sengupta, P. Controversies with self-emulsifying drug delivery system from pharmacokinetic point of view. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef] [PubMed]
- Carley, D.; Fedde, K.; Koleng, J.; Letendre, P. Sustained Release Cannabinoid Medicaments. U.S. Patent 20120231083 A1, 13 September 2012. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Drug/LogP/BCS Class | Oil | Surfactant/Cosurfactant | Powder Carriers | Presentation | Evaluation | Reference |
|---|---|---|---|---|---|---|---|
| 1 | Progesterone LogP = 3.87 Class IV | C8, C10 mono and di-glycerides (Imwitor 742®) | Tween 80 | MCC | Pellets | In vitro dissolution & bioavailability improvement | Tuleu et al. 2004 [3] |
| 2 | Gentamicin LogP = n.a. Class III | PEG-8 caprylic capric glyceride (Labrasol) | Tween 80 | Mg Aluminosilicate, silicon dioxide, calcium silicate | Powder filled into enteric capsules | In vitro dissolution & absorption enhancement | Ito et al. 2005 [12] |
| 3 | Nimesulide LogP = 2.60 Class II | C8, C10 mono and di-glycerides (Cithrol GMO®) | Tween 80 | MCC, Lactose | Granules | In vitro dissolution & ex vivo permeability improvement | Franceschinis et al. 2005 [65] |
| 4 | Methyl Paraben LogP = 1.96 Propyl Paraben LogP = 3.04 Class n.c. | C8, C10 mono and di-glycerides (Imwitor 742) | Tween 80 | MCC | Controlled release pellets | In vitro release enhancement | Serratoni et al. 2006 [46] |
| 5 | Diazepam LogP = 2.82 Class I | C18 mono and di-glycerides (Cithrol GMS) | Solutol HS 15 | MCC | Pellets | In vitro dissolution & bioavailability improvement | Abdalla & Mader 2007 [11] |
| 6 | Ezetimibe LogP = 4.50 Class II | C8, C10 triglycerides (Miglyol, Labrafac lipophile WL 1349) | Capryol 90, Cremophor EL, Transcutol P | CSD | Coarse powders | In vitro dissolution improvement | Dixit & Nagarsenker 2008 [66] |
| 7 | Grizeofulvin LogP = 2.18 Class II | C8, C10 triglycerides (Captex 355) | Tween 80, Labrasol | Calcium silicate, Mg Aluminosilicate, silicon dioxide | Coarse powders | In vitro , dissolution improvement | Agarwal et al. 2009 [50] |
| 8 | Candesartan Cilexetil LogP = 4.0–5.1 Class II | C8, C10 triglycerides (Miglyol 812) | Tween 80, Labrasol | MCC, CSD, Sodium croscarmellose | Coarse powders | In vitro , dissolution & bioavailability improvement | Nekkanti et al. 2009 [67] |
| 9 | Nitrendipine LogP = 2.9 Class II | C8, C10 triglycerides (Miglyol 812) | Cremophor RH40, Tween 80, Transcutol P | MCC, Lactose, CSD, Crospovidone | Pellets | In vitro dissolution & absorption improvement | Wang et al. 2010 [68] |
| 10 | Tetrahydro-curcumin LogP = 3.5–4.0 Class IV | Propylene glycol dicaprylocaprate (Labrafac PG) | Capryol 90, Cremophor EL, Labrasol | MCC, CSD, Glyceryl behenate, Pregelatinised starch, Starch glycolate | Floating pellets—controlled release | In vitro solubility and dissolution improvement | Setthacheewakul et al. 2011 [56] |
| 11 | Piroxicam LogP = 3.0 Class II | Propylene glycol-monolaurate (Lauroglycol™ 90) | Cremophor EL, Transcutol HP | MCC, Lactose, PVP | Pellets | In vitro dissolution improvement | Franceschinis et al. 2011 [69] |
| 12 | Paliperidone LogP = 1.8 Class II | Oleic acid, C8, C10 mono and di-glycerides (Capmul MCM) | Tween 80 | Mg Aluminometasilicate | Coarse powders | In vitro dissolution & ex vivo permeability improvement | Kanuganti et al. 2012 [51] |
| 13 | Sirolimus LogP = 4.3 Class II | n.a. | Labrafil 1944CS Cremophor EL, Transcutol P | MCC, Lactose, Na carboxymethyl starch | Pellets | In vitro dissolution & absorption improvement | Hu et al. 2012 [32] |
| 14 | Carbamazepine LogP = 2.45 Class II | C8, C10 triglycerides (Miglyol 812) | Tween 80, Cremophor RH 40 | CSD, Mg Aluminometasilicate | Coarse powders | In vitro dissolution improvement | Milovic et al. 2012 [52] |
| 15 | Puerarin LogP = n.a. Class IV | Castor oil | Cremophor E4, Propylene glycol | MCC, HPMC | Pellets—sustained release | In vitro dissolution & bioavailability improvement | Zhang et al. 2012 [60] |
| 16 | Cilostazol LogP = 2.3 Class II | C8, C10 mono and di-glycerides (Capmul MCM) | Tween 80, Transcutol P | Mg Aluminometasilicate | Coarse powders | In vitro solubility improvement | Pund et al. 2013 [70] |
| 17 | Sirolimus LogP = 4.3 Class II | n.a. | Capryol, PGMC E-T PGS, glycofurol | Mannitol, Sucrose monopalmitate | Granules | In vitro solubility & dissolution improvement | Cho et al. 2013 [36] |
| 18 | Lercanidipine HCl LogP = 6.4 Class n.c. | n.a. | Gelucire 44/14, Labrasol, Transcutol P | Mg Aluminometasilicate | Coarse powders | In vitro , dissolution improvement | Kallakunta et al. 2013 [33] |
| 19 | Repaglinide LogP = 5.3 Class II | n.a. | Capryol 90, Cremophor EL, Solutol HS-15 | MCC, Lactose, Kollidon CL, PVP | Pellets | In vitro , dissolution improvement | Desai & Negarsenker 2013 [37] |
| 20 | Ondasetron HCl LogP=2.40 Class II | Medium Chain Mono- and Diglycerides (Capmul MCM) | Labrasol, Tween 20 | Silica, Mg Aluminometasilicate | Coarse powders | In vitro dissolution & bioavailability improvement | Beg et al. 2013 [53] |
| 21 | Bifendate LogP = 2.80 Class n.c. | Propylene Glycol Dicaprylate/Dicaprate (Miglyol® 840) | Cremorphor® EL, Solutol HS® 15 (1:2, w/w)/Transcutol HP | MCC, lactose, mannitol | Pellets | In vitro dissolution & bioavailability improvement | Xiao et al. 2013 [47] |
| 22 | Atorvastatin calcium LogP = 5.7 Class II | Polyglycerol-3-oleate (Caprol 3GO) | Cremophor EL, Tween 20, Tween 80, N-methylpyrrolidone | MCC, CSD, Mg Aluminometasilicate | Coarse powders | In vitro dissolution & ex vivo permeability improvement | Agrawal et al. 2014 [50] |
| 23 | Olmesartan medoxomil LogP = 5.9 Class II | n.a. | Acconon Sorb-20, Tween 80, Carbitol | MCC, CSD, PVPP XL | Granules | In vitro dissolution & bioavailability improvement | Patel et al. 2014 [34] |
| 24 | Ibuprofen LogP = 3.97 Class II | n.a. | PEG 200 Labrasol | Mg Aluminometasilicate, MCC, Lactose | Pellets coated with SEDDS | In vitro , dissolution improvement | Krupa et al. 2014 [35] |
| 25 | Furosemide LogP = 2.03 Class II | C8, C10 triglycerides (Radia 7104) | Cremophor ELP, Cremophor RH40, Cremophor RH60 | MCC | Pellets | In vitro , dissolution & solubility improvement | Nikolakakis et al. 2014 [71] |
| 26 | Propranolol LogP =3.48 Class II | ||||||
| 27 | Oleanolic acid LogP na Class IV | Ethyl oleate | Labrasol, Transcutol P | Mannitol | Granules | In vitro , dissolution improvement | Ma et al. 2014 [27] |
| 28 | Simvastatin LogP = 4.68 Class II | Lauroglycol | Cremophor EL, Transcutol | MCC, Lactose, PVP | Granules | In vitro dissolution | Franceschinis et al. 2015 [72] |
| 29 | Glipiside LogP = 1.91 Class II | Phosphatidyl choline (Phosal 53 MCT), Capmul MCT | Tween 80, Transcutol | Silica (Syloid 244 FP) | Coarse powders | In vitro dissolution & bioavailability improvement | Agarwal et al. 2015 [50] |
| 30 | Celecoxib LogP = 3.9 Class II | n.a. | Capryol 90, Tween 20, Transcutol HP | CSD, Soluplus | Coarse powders | In vitro dissolution & bioavailability improvement | Chavan et al. 2015 [38] |
| 31 | Ibuprofen LogP = 3.97 Class II | C8, C10 triglycerides | Cremophor EL | MCC, CSD | Pellets | In vitro , dissolution improvement | Panagopoulou et al. 2015 [57] |
| 32 | Lercanidipine HCl Class n.c. | Rice brown oil/Clyceryl monooleate 1/9 | Tween 80, Propionic acid | Mg Aluminometasilicate | Coarse powders | In vitro dissolution & absorption improvement | Suthar et al. 2016 [54] |
| 33 | Sirolimus LogP = 4.3 Class II | n.a. | Labrafil 1944CS Cremophor EL, Transcutol P | MCC, HPMC 100LV | Tablets—extended release | Stability improvement | Tao et al. 2016 [61] |
| CSD/MCC | Consumption (mL) | % in Size Fraction (850–1200 μm) | Median Diameter (μm) | Aspect Ratio | Shape Factor (eR) |
|---|---|---|---|---|---|
| 0/10 | 17 | 56.8 | 1070 | 1.101 | 0.433 |
| 3/7 | 25 | 74.4 | 1240 | 1.093 | 0.453 |
| 7/3 | 46 | 87.7 | 1250 | 1.130 | 0.419 |
| 10/0 | 58 | 79.1 # | 1310 # | 1.181 | 0.342 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolakakis, I.; Partheniadis, I. Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release. Pharmaceutics 2017, 9, 50. https://doi.org/10.3390/pharmaceutics9040050
Nikolakakis I, Partheniadis I. Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release. Pharmaceutics. 2017; 9(4):50. https://doi.org/10.3390/pharmaceutics9040050
Chicago/Turabian StyleNikolakakis, Ioannis, and Ioannis Partheniadis. 2017. "Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release" Pharmaceutics 9, no. 4: 50. https://doi.org/10.3390/pharmaceutics9040050
APA StyleNikolakakis, I., & Partheniadis, I. (2017). Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release. Pharmaceutics, 9(4), 50. https://doi.org/10.3390/pharmaceutics9040050