
Inherited Developmental and Epileptic Encephalopathies


Abstract
:1. Introduction
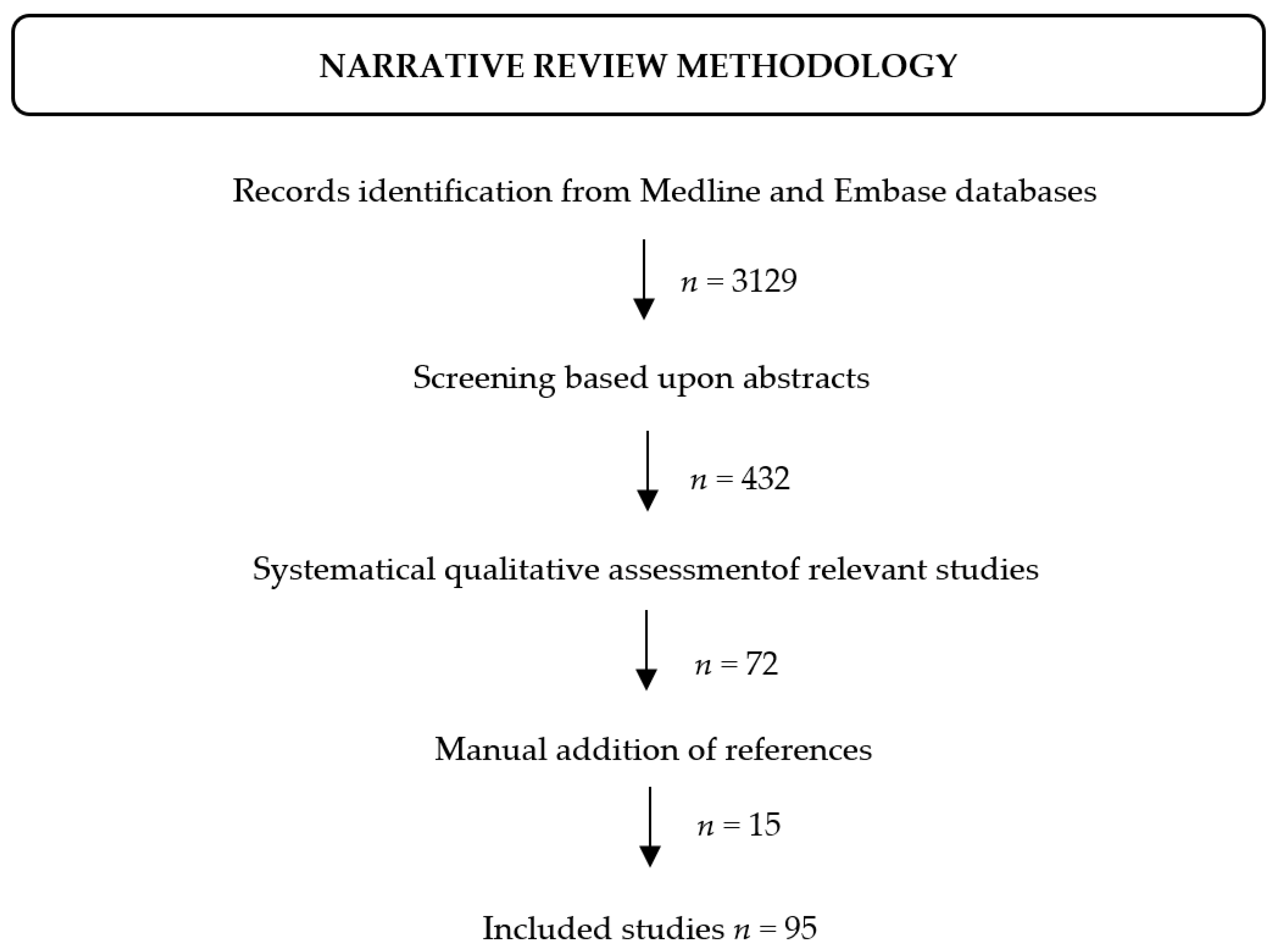
2. Methods
3. Channelopathies
3.1. Voltage-Gated Channels
3.2. Ligand Gated Channels
4. Metabolic Disorders
5. Membrane Trafficking and Exocytosis
6. Cell Adhesion
7. Cell Growth and Proliferation
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; Boas, W.V.E.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.; Scheffer, I. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Møller, R.S.; Liebmann, N.; Larsen, L.H.G.; Stiller, M.; Hentschel, J.; Kako, N.; Abdin, D.; Di Donato, N.; Pal, D.K.; Zacher, P.; et al. Parental mosaicism in epilepsies due to alleged de novo variants. Epilepsia 2019, 60, e63–e66. [Google Scholar] [CrossRef]
- Stosser, M.; Lindy, A.; Butler, E.; Retterer, K.; Piccirillo-Stosser, C.M.; Richard, G.; McKnight, D.A. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 2018, 20, 403–410. [Google Scholar] [CrossRef]
- Symonds, J.; McTague, A. Epilepsy and developmental disorders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 2020, 24, 15–23. [Google Scholar] [CrossRef]
- Barros, J.; Damásio, J.; Tuna, A.; Alves, I.; Silveira, I.; Pereira-Monteiro, J.; Sequeiros, J.; Alonso, I.; Sousa, A.; Coutinho, P. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. 2013, 70, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, E.; Campostrini, R.; Kiferle, L.; Pradella, S.; Rosati, E.; Chinthapalli, K.; Palumbo, P. Epilepsy and brain channelopathies from infancy to adulthood. Neurol. Sci. 2019, 41, 749–761. [Google Scholar] [CrossRef]
- Ryan, D.P.; Ptáček, L.J. Episodic Neurological Channelopathies. Neuron 2010, 68, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution: Voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Escayg, A.; Goldin, A.L. Sodium channel SCN1A and epilepsy: Mutations and mechanisms: Sodium Channel SCN1A and Epilepsy. Epilepsia 2010, 51, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.S.; Westenbroek, R.E.; Roden, W.H.; Kalume, F.; Oakley, J.C.; Jansen, L.A.; Catterall, W.A. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels 2013, 7, 468–472. [Google Scholar] [CrossRef] [Green Version]
- Claes, L.; Del Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The genetics of Dravet syndrome: Genetics of Dravet Syndrome. Epilepsia 2011, 52, 24–29. [Google Scholar] [CrossRef]
- Depienne, C.; Trouillard, O.; Gourfinkel-An, I.; Saint-Martin, C.; Bouteiller, D.; Graber, D.; Barthez-Carpentier, M.-A.; Gautier, A.; Villeneuve, N.; Dravet, C.; et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 2010, 47, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 85, 958–966. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, A.; McMahon, J.; Dibbens, L.M.; Iona, X.; Mulley, J.C.; Scheffer, I.; Berkovic, S.F. Effects of vaccination on onset and outcome of Dravet syndrome: A retrospective study. Lancet Neurol. 2010, 9, 592–598. [Google Scholar] [CrossRef]
- Reyes, I.S.; Hsieh, D.T.; Laux, L.C.; Wilfong, A.A. Alleged Cases of Vaccine Encephalopathy Rediagnosed Years Later as Dravet Syndrome: TABLE 1. Pediatrics 2011, 128, e699–e702. [Google Scholar] [CrossRef]
- Dravet, C. The core Dravet syndrome phenotype: Core Dravet Syndrome. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef]
- Losito, E.; Kuchenbuch, M.; Chemaly, N.; Laschet, J.; Chiron, C.; Kaminska, A.; Nabbout, R. Age-related “Sleep/nocturnal” tonic and tonic clonic seizure clusters are underdiagnosed in patients with Dravet Syndrome. Epilepsy Behav. 2017, 74, 33–40. [Google Scholar] [CrossRef]
- Scheffer, I.E. Diagnosis and long-term course of Dravet syndrome. Eur. J. Paediatr. Neurol. 2012, 16, S5–S8. [Google Scholar] [CrossRef]
- Verbeek, N.E.; Wassenaar, M.; Van Campen, J.S.; Sonsma, A.C.M.; Gunning, B.; Knoers, N.V.A.M.; Lindhout, D.; Jansen, F.E.; Leijten, F.S.S.; Brilstra, E.H.; et al. Seizure precipitants in Dravet syndrome: What events and activities are specifically provocative compared with other epilepsies? Epilepsy Behav. 2015, 47, 39–44. [Google Scholar] [CrossRef]
- Guerrini, R. Dravet syndrome: The main issues. Eur. J. Paediatr. Neurol. 2012, 16, S1–S4. [Google Scholar] [CrossRef]
- Akiyama, M.; Kobayashi, K.; Yoshinaga, H.; Ohtsuka, Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 2009, 51, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Genton, P.; Velizarova, R.; Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 2011, 52, 44–49. [Google Scholar] [CrossRef]
- Fasano, A.; Borlot, F.; Lang, A.E.; Andrade, D.M. Antecollis and levodopa-responsive parkinsonism are late features of Dravet syndrome. Neurology 2014, 82, 2250–2251. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Deprez, L.; Maljevic, S.; Pitsch, J.; Claes, L.; Hristova, D.; Jordanova, A.; Ala-Mello, S.; Bellan-Koch, A.; Blazevic, D.; et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010, 133, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Kato, M.; Osaka, H.; Yamashita, S.; Nakagawa, E.; Haginoya, K.; Tohyama, J.; Okuda, M.; Wada, T.; Shimakawa, S.; et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 2013, 81, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Gardella, E.; Marini, C.; Trivisano, M.; Fitzgerald, M.P.; Alber, M.; Howell, K.B.; Darra, F.; Siliquini, S.; Bölsterli, B.K.; Masnada, S.; et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018, 91, e1112–e1124. [Google Scholar] [CrossRef] [Green Version]
- Johannesen, K.M.; Gardella, E.; Encinas, A.C.; Lehesjoki, A.; Linnankivi, T.; Petersen, M.B.; Lund, I.C.B.; Blichfeldt, S.; Miranda, M.J.; Pal, D.K.; et al. The spectrum of intermediate SCN8A-related epilepsy. Epilepsia 2019, 60, 830–844. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.; Carvill, G.L.; Gardella, E.; Kluger, G.; Schmiedel, G.; Barisic, N.; Depienne, C.; Brilstra, E.; Mang, Y.; Nielsen, J.E.K.; et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015, 84, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Denis, J.; Villeneuve, N.; Cacciagli, P.; Mignon-Ravix, C.; Lacoste, C.; Lefranc, J.; Napuri, S.; Damaj, L.; Villega, F.; Pedespan, J.; et al. Clinical study of 19 patients with SCN8A-related epilepsy: Two modes of onset regarding EEG and seizures. Epilepsia 2019, 60, 845–856. [Google Scholar] [CrossRef]
- Trivisano, M.; Pavia, G.C.; Ferretti, A.; Fusco, L.; Vigevano, F.; Specchio, N. Generalized tonic seizures with autonomic signs are the hallmark of SCN8A developmental and epileptic encephalopathy. Epilepsy Behav. 2019, 96, 219–223. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [Green Version]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A Potassium Channel Mutation in Neonatal Human Epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [Google Scholar] [CrossRef]
- Dedek, K.; Fusco, L.; Teloy, N.; Steinlein, O.K. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003, 54, 21–27. [Google Scholar] [CrossRef]
- Miceli, F.; Striano, P.; Soldovieri, M.V.; Fontana, A.; Nardello, R.; Robbiano, A.; Bellini, G.; Elia, M.; Zara, F.; Taglialatela, M.; et al. A novelKCNQ3mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 2014, 56, e15–e20. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, C.; Barcia, G.; Jennesson, M.; Le Guyader, G.; Schneider, A.; Mignot, C.; Lesca, G.; Breuillard, D.; Montomoli, M.; Keren, B.; et al. Expanding the genetic and phenotypic relevance of KCNB1 variants in developmental and epileptic encephalopathies: 27 new patients and overview of the literature. Hum. Mutat. 2020, 41, 69–80. [Google Scholar] [CrossRef]
- McTague, A.; Appleton, R.; Avula, S.; Cross, H.; King, M.D.; Jacques, T.S.; Bhate, S.; Cronin, A.; Curran, A.; Desurkar, A.; et al. Migrating partial seizures of infancy: Expansion of the electroclinical, radiological and pathological disease spectrum. Brain 2013, 136, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Milligan, C.J.; Li, M.; Gazina, E.V.; Heron, S.E.; Nair, U.; Trager, C.; Reid, C.A.; Venkat, A.; Younkin, D.P.; Dlugos, D.J.; et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine: KCNT1 and Human Epilepsy. Ann. Neurol. 2014, 75, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, C.; Van Gils, J.; Bigourdan, A.; Jouk, P.-S.; Lacombe, D.; Menegon, P.; Moutton, S.; Riant, F.; Sole, G.; Tournier-Lasserve, E.; et al. Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a CACNA1A pathogenic variant. Eur. J. Med. Genet. 2019, 62, 103530. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Rispoli, M.G.; Pellegrino, N.; Graziosi, A.; Rotondo, E.; Napoli, C.; Pietrobon, D.; Brighina, F.; Parisi, P. Diagnostic and therapeutic aspects of hemiplegic migraine. J. Neurol Neurosurg. Psychiatry 2020, 91, 764–771. [Google Scholar] [CrossRef]
- Indelicato, E.; Boesch, S. From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front. Neurol. 2021, 12, 639994. [Google Scholar] [CrossRef]
- Le Roux, M.; Barth, M.; Gueden, S.; de Cepoy, P.D.; Aeby, A.; Vilain, C.; Hirsch, E.; de Saint Martin, A.; des Portes, V.; Lesca, G.; et al. CACNA1A-associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. Eur. J. Paediatr. Neurol. 2021, 33, 75–85. [Google Scholar] [CrossRef]
- Reinson, K.; Õiglane-Shlik, E.; Talvik, I.; Vaher, U.; Õunapuu, A.; Ennok, M.; Teek, R.; Pajusalu, S.; Murumets, Ü.; Tomberg, T.; et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am. J. Med. Genet. A 2016, 170, 2173–2176. [Google Scholar] [CrossRef]
- Johannesen, K.; Marini, C.; Pfeffer, S.; Møller, R.S.; Dorn, T.; Niturad, C.E.; Gardella, E.; Weber, Y.; Søndergård, M.; Hjalgrim, H.; et al. Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology 2016, 87, 1140–1151. [Google Scholar] [CrossRef]
- Carvill, G.L.; Regan, B.; Yendle, S.C.; O’Roak, B.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy: GRIN2B Mutations in Epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; Al Mahmoud, R.A.; Mekki, M.; El-Hattab, A.W. Metabolic Seizures. Front. Neurol. 2021, 12, 985. [Google Scholar] [CrossRef]
- Van Konijnenburg, E.M.H.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D. Metabolic Evaluation of Epilepsy: A Diagnostic Algorithm with Focus on Treatable Conditions. Front. Neurol. 2018, 9, 1016. [Google Scholar]
- Sharma, S.; Prasad, A.N. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int. J. Mol. Sci. 2017, 18, E1384. [Google Scholar] [CrossRef] [Green Version]
- Van Karnebeek, C.D.M.; Jaggumantri, S. Current Treatment and Management of Pyridoxine-Dependent Epilepsy. Curr. Treat. Options Neurol. 2015, 17, 335. [Google Scholar] [CrossRef]
- Khayat, M.; Korman, S.H.; Frankel, P.; Weintraub, Z.; Hershckowitz, S.; Sheffer, V.F.; Ben Elisha, M.; Wevers, R.A.; Falik-Zaccai, T.C. PNPO deficiency: An under diagnosed inborn error of pyridoxine metabolism. Mol. Genet. Metab. 2008, 94, 431–434. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Trifiletti, R.R.; Jacobson, R.I.; Ronen, G.M.; Behmand, R.A.; Harik, S.I. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N. Engl. J. Med. 1991, 325, 703–709. [Google Scholar] [CrossRef]
- Ardicli, D.; Haliloglu, G.; Gocmen, R.; Gunbey, C.; Topcu, M. Unraveling neuronal ceroid lipofuscinosis type 2 (CLN2) disease: A tertiary center experience for determinants of diagnostic delay. Eur. J. Paediatr. Neurol. 2021, 33, 94–98. [Google Scholar] [CrossRef]
- Beltrán, L.; Valenzuela, G.R.; Loos, M.; Vargas, R.; Lizama, R.; Spinsanti, P.; Caraballo, R. Late-onset childhood neuronal ceroid lipofuscinosis: Early clinical and electroencephalographic markers. Epilepsy Res. 2018, 144, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; de Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S. Mitochondrial diseases and status epilepticus. Epilepsia 2018, 59, 70–77. [Google Scholar] [CrossRef]
- Ciara, E.; Rokicki, D.; Halat, P.; Karkucińska-Więckowska, A.; Piekutowska-Abramczuk, D.; Mayr, J.; Trubicka, J.; Szymańska-Dębińska, T.; Pronicki, M.; Pajdowska, M.; et al. Difficulties in recognition of pyruvate dehydrogenase complex deficiency on the basis of clinical and biochemical features. The role of next-generation sequencing. Mol. Genet. Metab. Rep. 2016, 7, 70–76. [Google Scholar] [CrossRef]
- Wesół-Kucharska, D.; Rokicki, D.; Jezela-Stanek, A. Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment. Children 2021, 8, 532. [Google Scholar] [CrossRef]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine 2020, 99, e18634. [Google Scholar] [CrossRef]
- Lamperti, C.; Zeviani, M. Myoclonus epilepsy in mitochondrial disorders. Epileptic Disord. 2016, 18, 94–102. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Smirnova, T.; Miniou, P.; Viegas-Péquignot, E.; Mallet, J. Assignment of the Human Syntaxin 1B Gene (STX) to Chromosome 16p11.2 by Fluorescence in Situ Hybridization. Genomics 1996, 36, 551–553. [Google Scholar] [CrossRef]
- Schubert, J.; EuroEPINOMICS RES Consortium; Siekierska, A.; Langlois, M.; May, P.; Huneau, C.; Becker, F.; Muhle, H.; Suls, A.; Lemke, J.R.; et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 2014, 46, 1327–1332. [Google Scholar] [CrossRef]
- Vlaskamp, D.R.; Rump, P.; Callenbach, P.M.; Vos, Y.J.; Sikkema-Raddatz, B.; van Ravenswaaij-Arts, C.M.; Brouwer, O.F. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 489–492. [Google Scholar] [CrossRef]
- Wolking, S.; May, P.; Mei, D.; Møller, R.S.; Balestrini, S.; Helbig, K.L.; Altuzarra, C.D.; Chatron, N.; Kaiwar, C.; Stöhr, K.; et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology 2019, 92, e1238–e1249. [Google Scholar] [CrossRef] [Green Version]
- Myers, C.T.; Hollingsworth, G.; Muir, A.M.; Schneider, A.L.; Thuesmunn, Z.; Knupp, A.; King, C.; Lacroix, A.; Mehaffey, M.G.; Berkovic, S.F.; et al. Parental Mosaicism in “De Novo” Epileptic Encephalopathies. N. Engl. J. Med. 2018, 378, 1646–1648. [Google Scholar] [CrossRef]
- Depienne, C.; LeGuern, E. PCDH19-related infantile epileptic encephalopathy: An unusual X-linked inheritance disorder. Hum. Mutat. 2012, 33, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Gecz, J.; Thomas, P.Q. Disentangling the paradox of the PCDH19 clustering epilepsy, a disorder of cellular mosaics. Curr. Opin. Genet. Dev. 2020, 65, 169–175. [Google Scholar] [CrossRef]
- Pederick, D.T.; Richards, K.L.; Piltz, S.G.; Kumar, R.; Mincheva-Tasheva, S.; Mandelstam, S.A.; Dale, R.C.; Scheffer, I.E.; Gecz, J.; Petrou, S.; et al. Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 2018, 97, 59–66.e5. [Google Scholar] [CrossRef] [Green Version]
- Depienne, C.; Bouteiller, D.; Keren, B.; Cheuret, E.; Poirier, K.; Trouillard, O.; Benyahia, B.; Quelin, C.; Carpentier, W.; Julia, S.; et al. Sporadic Infantile Epileptic Encephalopathy Caused by Mutations in PCDH19 Resembles Dravet Syndrome but Mainly Affects Females. PLoS Genet. 2009, 5, e1000381. [Google Scholar] [CrossRef]
- Friocourt, G.; Parnavelas, J. Mutations in ARX result in several defects involving GABAergic neurons. Front. Cell. Neurosci. 2010, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Poirier, K.; Abriol, J.; Souville, I.; Laroche-Raynaud, C.; Beldjord, C.; Gilbert, B.; Chelly, J.; Bienvenu, T. Maternal mosaicism for mutations in the ARX gene in a family with X linked mental retardation. Qual. Life Res. 2005, 118, 45–48. [Google Scholar] [CrossRef]
- Sherr, E.H. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): One gene leads to many phenotypes. Curr. Opin. Pediatr. 2003, 15, 567–571. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Cavallin, M. Tubulinopathies Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Jauhari, P.; Farmania, R.; Chakrabarty, B.; Kumar, A.; Gulati, S. Electrographic pattern recognition: A simple tool to predict clinical outcome in children with lissencephaly. Seizure 2020, 83, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Kolbjer, S.; Martin, D.A.; Pettersson, M.; Dahlin, M.; Anderlid, B.-M. Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects. Eur. J. Paediatr. Neurol. 2021, 30, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Dobyns, W.B.; Guerrini, R. Malformations of Cortical Development and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022392. [Google Scholar] [CrossRef] [PubMed]
- Brock, S.; Cools, F.; Jansen, A.C. Neuropathology of genetically defined malformations of cortical development-A systematic literature review. Neuropathol. Appl. Neurobiol. 2021, 47, 585–602. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E.; Gross, C.; Calza, L.; Bartesaghi, R.; Ciani, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3β signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131, 2647–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 Deficiency Disorder—A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Cheadle, J.P.; Gill, H.; Fleming, N.; Maynard, J.; Kerr, A.; Leonard, H.; Krawczak, M.; Cooper, D.N.; Lynch, S.; Thomas, N.; et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: Correlation of disease severity with mutation type and location. Hum. Mol. Genet. 2000, 9, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, R.; Parrini, E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia 2012, 53, 2067–2078. [Google Scholar] [CrossRef]
- Caban, C.; Khan, N.; Hasbani, D.M.; Crino, P.B. Genetics of tuberous sclerosis complex: Implications for clinical practice. Appl. Clin. Genet. 2016, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Franz, D.N.; Lawson, J.A.; Yapici, Z.; Brandt, C.; Kohrman, M.H.; Wong, M.; Milh, M.; Wiemer-Kruel, A.; Voi, M.; Coello, N.; et al. Everolimus dosing recommendations for tuberous sclerosis complex-associated refractory seizures. Epilepsia 2018, 59, 1188–1197. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Clinical Overview | Typical Age at Onset | |
|---|---|---|
| Early Myoclonic Encephalopathy | Myoclonic seizures are frequent, distinguishing this syndrome from Ohtahara syndrome | 1–3 months |
| Ohtahara syndrome | Tonic seizures predominate, myoclonic seizures are uncommon | 1–3 months |
| West syndrome | Epileptic spasms, hypsarrhythmia and global developmental delay | 3–12 months |
| Dravet syndrome | Normal child with prolonged, febrile and afebrile, focal and tonic-clonic seizures at onset | Around 6 months of age |
| Epilepsy Of Infancy With Migrating Focal Seizures | Focal seizures arise independently in both hemispheres and can migrate from one cortical region to another randomly but consecutively in the same seizure. | First 6 months |
| Epilepsy With Myoclonic-Atonic Seizures | Myoclonic-atonic seizures in an otherwise normal child who may have a history of febrile and/or afebrile seizures. | 6 months–6 years |
| Epileptic Encephalopathy With Continuous Spike-And-Wave During Sleep | Focal seizures, atypical absences, negative myoclonus/atonic seizures and neurocognitive regression | 2–12 years |
| Landau-Kleffner syndrome | Subacute onset of acquired aphasia. Seizures may not occur in all cases and when present are infrequent and self-limiting | 2–8 years |
| Affected Gene | Main Imaging Phenotypes |
|---|---|
| ARX | X-linked lissencephaly with abnormal genitalia |
| DCX | Anteriorly predominant lissencephaly (males) Subcortical band heterotopia (females) |
| LIS1 | Posteriorly predominant lissencephaly |
| TUBA1A | Posteriorly predominant lissencephaly ± absent corpus callous, and cerebellar hypoplasia; polymicrogyria like pattern |
| TUBB2B | Posteriorly predominant lissencephaly ± cerebellar hypoplasia; polymicrogyria like |
| TUBB3 | Absent corpus callosum, polymicrogyria like pattern |
| TUBB [TUBB5] | Absent corpus callosum, polymicrogyria like pattern |
| TUBG1 | Posteriorly predominant lissencephaly |
| DYNC1H1 | Posteriorly predominant lissencephaly |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartolini, E. Inherited Developmental and Epileptic Encephalopathies. Neurol. Int. 2021, 13, 555-568. https://doi.org/10.3390/neurolint13040055
Bartolini E. Inherited Developmental and Epileptic Encephalopathies. Neurology International. 2021; 13(4):555-568. https://doi.org/10.3390/neurolint13040055
Chicago/Turabian StyleBartolini, Emanuele. 2021. "Inherited Developmental and Epileptic Encephalopathies" Neurology International 13, no. 4: 555-568. https://doi.org/10.3390/neurolint13040055
APA StyleBartolini, E. (2021). Inherited Developmental and Epileptic Encephalopathies. Neurology International, 13(4), 555-568. https://doi.org/10.3390/neurolint13040055