
Clinical and Genetic Analysis of a Patient with CMT4J

Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical
2.2. Genetic Analysis
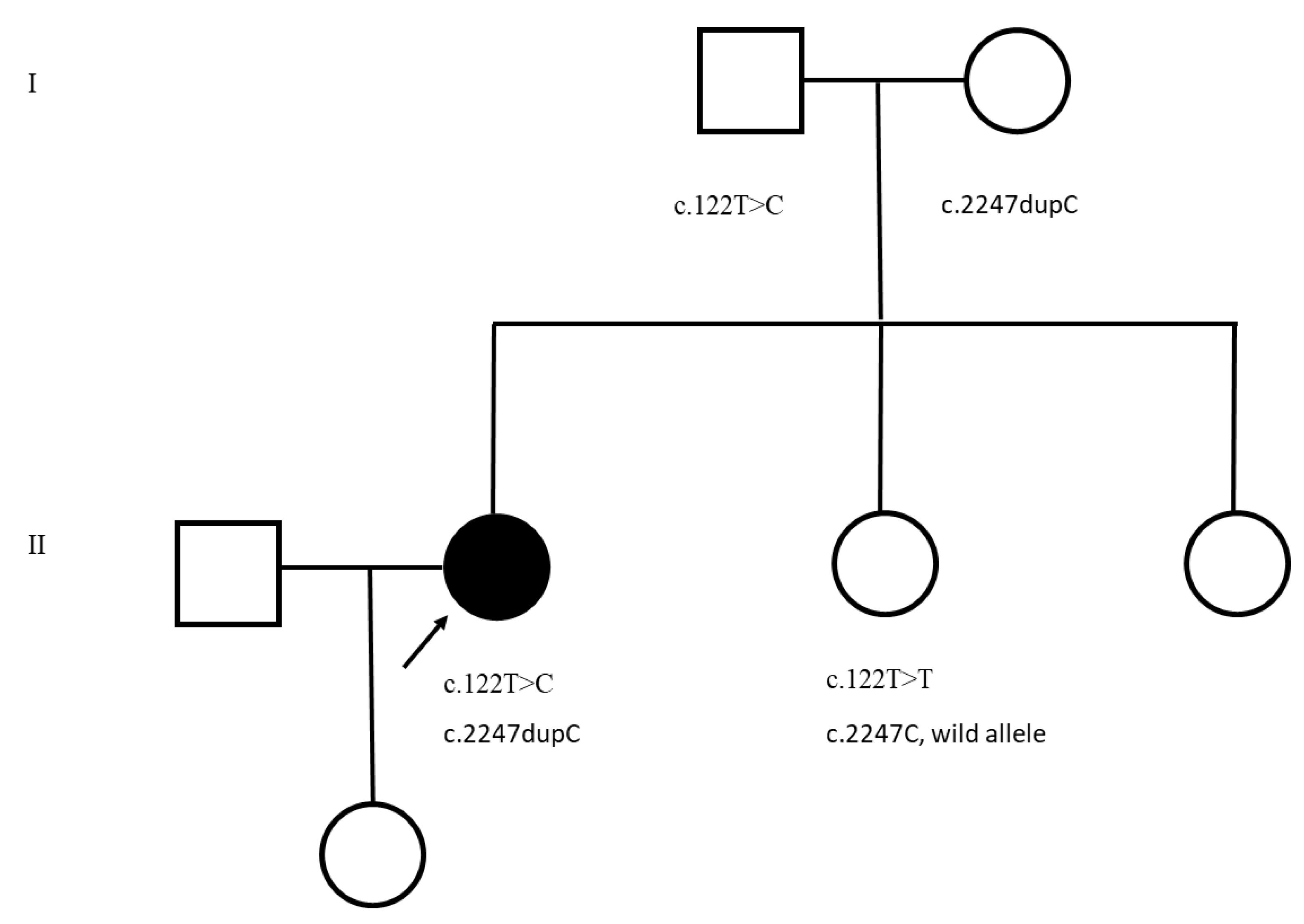
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.; Li, J.; Zhang, X.; et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Gentil, B.J.; O’Ferrall, E.; Chalk, C.; Santana, L.F.; Durham, H.D.; Massie, R. A New Mutation in FIG4 Causes a Severe Form of CMT4J Involving TRPV4 in the Pathogenic Cascade. J. Neuropathol. Exp. Neurol. 2017, 76, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, G.; Lenk, G.M.; Reddel, S.W.; Grant, A.E.; Towne, C.F.; Ferguson, C.J.; Simpson, E.; Scheuerle, A.; Yasick, M.; Hoffman, S.; et al. Distinctive genetic and clinical features of CMT4J: A severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain 2011, 134, 1959–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- DiVincenzo, C.; Elzinga, C.D.; Medeiros, A.C.; Karbassi, I.; Jones, J.R.; Evans, M.C.; Braastad, C.D.; Bishop, C.M.; Jaremko, M.; Wang, Z.; et al. The allelic spectrum of Charcot–Marie–Tooth disease in over 17,000 individuals with neuropathy. Mol. Genet. Genom. Med. 2014, 2, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Cottenie, E.; Menezes, M.P.; Rossor, A.; Morrow, J.M.; Yousry, T.A.; Dick, D.J.; Anderson, J.R.; Jaunmuktane, Z.; Brandner, S.; Blake, J.C.; et al. Rapidly progressive asymmetrical weakness in Charcot–Marie–Tooth disease type 4J resembles chronic inflammatory demyelinating polyneuropathy. Neuromuscul. Disord. 2013, 23, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Michaelidou, K.; Tsiverdis, I.; Erimaki, S.; Papadimitriou, D.; Amoiridis, G.; Papadimitriou, A.; Mitsias, P.; Zaganas, I. Whole exome sequencing establishes diagnosis of Charcot–Marie–Tooth 4J, 1C, and X1 subtypes. Mol. Genet. Genom. Med. 2020, 8, e1141. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.; Jin, Y.; Zhang, H.; Qiang, W.; Shekhtman, E.; Shao, D.; Revoe, D.; Villamarin, R.; Ivanchenko, E.; Kimura, M.; et al. ALFA: Allele Frequency Aggregator; National Center for Biotechnology Information, U.S. National Library of Medicine: Bethesda, MD, USA, 2020. Available online: www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/ (accessed on 2 January 2022).
- Lafontaine, M.; Lia, A.; Bourthoumieu, S.; Beauvais-Dzugan, H.; Derouault, P.; Arné-Bes, M.; Sarret, C.; Laffargue, F.; Magot, A.; Sturtz, F.; et al. Clinical features of homozygous FIG4-p.Ile41Thr Charcot-Marie-Tooth 4J patients. Ann. Clin. Transl. Neurol. 2021, 8, 471–476. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Nerve | Distal Latencies, ms (nl) | Response Amplitude, mv | Conduction Velocity, m/s | F-Wave Latency, ms | Comments |
|---|---|---|---|---|---|
| Motor | |||||
| L. Median | 5.1 (<4.2) 5.0 | 4.0 (>4.0) 2.3 | 31.0 (>50) | 41.0 (<30) | |
| L. Ulnar | 4.3 (<3.3) | 3.6 (>3.5) 1.4 (BE) 1.0 (AE) | 30.0 (>50) 21 | 37.58 (<30) 41.4 | |
| Bilateral Fibular | NR (<6.2) | NR (>2.6) | NR (>40) | NR | No response recording the extensor digitorum brevis |
| Bilateral Tibial | NR (<6.0) | NR (>4.0) | NR (>40) | NR | |
| Sensory | |||||
| Bilateral Sural | NR | ||||
| Bilateral Fibular | NR | ||||
| L. Median | 4.8 | 10.2 (>20) | 38.0 (>50) | ||
| L. Ulnar | 3.1 | 2.7 (>17) | 50.0 (<50) | ||
| L. Radial | 3.9 | 5.0(>15) | 44.0 (>50) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peddareddygari, L.R.; Grewal, R.P. Clinical and Genetic Analysis of a Patient with CMT4J. Neurol. Int. 2022, 14, 207-211. https://doi.org/10.3390/neurolint14010017
Peddareddygari LR, Grewal RP. Clinical and Genetic Analysis of a Patient with CMT4J. Neurology International. 2022; 14(1):207-211. https://doi.org/10.3390/neurolint14010017
Chicago/Turabian StylePeddareddygari, Leema Reddy, and Raji P. Grewal. 2022. "Clinical and Genetic Analysis of a Patient with CMT4J" Neurology International 14, no. 1: 207-211. https://doi.org/10.3390/neurolint14010017
APA StylePeddareddygari, L. R., & Grewal, R. P. (2022). Clinical and Genetic Analysis of a Patient with CMT4J. Neurology International, 14(1), 207-211. https://doi.org/10.3390/neurolint14010017