Abstract
Type 2 diabetes mellitus (T2D) is a metabolic disease reaching pandemic levels worldwide. In parallel, Alzheimer’s disease (AD) and vascular dementia (VaD) are the two leading causes of dementia in an increasingly long-living Western society. Numerous epidemiological studies support the role of T2D as a risk factor for the development of dementia. However, few basic science studies have focused on the possible mechanisms involved in this relationship. On the other hand, this review of the literature also aims to explore the relationship between T2D, AD and VaD. The data found show that there are several alterations in the central nervous system that may be promoting the development of T2D. In addition, there are some mechanisms by which T2D may contribute to the development of neurodegenerative diseases such as AD or VaD.
1. Introduction
Diabetes mellitus (DM) is a chronic disease characterized by elevated blood glucose levels due to the body’s inability to produce sufficient insulin or use it efficiently. Although there are different types of DM, the most common are type 1 diabetes (T1D) and type 2 diabetes (T2D) [1].
According to the International Diabetes Federation (IDF, 2021), diabetes is the fifth leading cause of death in the world. In 2021, 537 million people were suffering from the disease, and it is estimated that by 2030, 783 million people will be affected (see evolution and prevalence forecasts in Figure 1). In 2021 alone, 6.7 million people died from diabetes, 1 every 5 s [2].
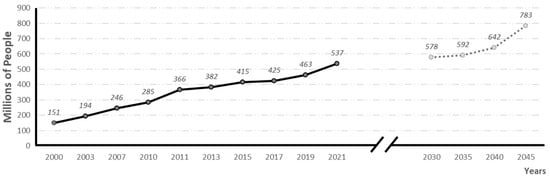
Figure 1.
Global prevalence of diabetes and global prevalence predictions (adapted from 10th edition of IFD diabetes Atlas) [2].
On the other hand, dementia is a syndrome that encompasses a heterogeneous group of diseases of the central nervous system (CNS), characterized by cognitive impairment [3]. In terms of incidence, the main types of dementia are Alzheimer’s disease (AD), vascular dementia (VaD), dementia with Lewy bodies and frontotemporal lobar dementia [4]. According to a WHO report, dementia currently affects around 50 million people, mostly from low- and middle-income countries, and about 10 million cases are registered every year [5]. It is estimated that by 2030, 82 million people will have dementia [5], and this number will increase to 130 million by 2050 [6].
AD is a neurodegenerative disease of the central nervous system and is characterized by a progressive deterioration of higher brain functions, affecting the ability to make decisions and execute them [7,8]. AD patients survive an average of 7 years, and slightly less than 3% of those affected live more than 14 years after diagnosis [9]. In terms of epidemiology, AD is the leading cause of dementia and occurs most frequently in people over 65 years of age (with a 10% prevalence), representing between 60% and 75% of all dementia cases [10,11]. The major risk factor for AD is age, and as life expectancy progressively increases, so does the number of people affected by this disease. Currently, more than 50 million people worldwide suffer from AD, with an associated economic cost of 30,000 EUR/year/patient in developed countries [12]. Projections for 2050 suggest that the number of people affected could exceed 107 million [10]. These rates represent an unbearable economic and social drain.
After AD, VaD is the second most common cause of cognitive impairment (20–30%) affecting 1 in 20 people over 65 years of age [3,13,14]. However, both diseases can coexist [15]. VaD incidence does not follow a homogeneous pattern geographically. Thus, in North America and Europe, it accounts for 15–20% of all dementias, while in Asia and developing countries, it is 30% [14]. As with all other dementias, the main risk factor is age, with the probability of developing dementia doubling every 5.3 years [16]. VaD occurs due to reduced blood flow to the brain, causing damage to different brain structures. This decrease may be caused by cerebrovascular accidents or diseases affecting the blood vessels of the brain, such as arteriosclerosis. Impairment of cognitive functions may vary depending on the location and extent of brain damage caused by reduced blood flow. Although impairment of cognitive functions may vary depending on the location and extent of brain damage, the most common include processing speed, attention, memory, language and communication, as well as executive functioning [17]. Prevention and control of vascular risk factors, such as high blood pressure, DM and high cholesterol, can help reduce the risk of VaD [15].
1.1. Historical Development of Diabetes Mellitus
Society has been affected by DM since ancient times. The first record of its existence dates back to the 15th century AC when the disease was described in Ebers papyrus, found in Egypt. In the 2nd century AC, Aretaeus gave the name diabetes (Greek for “to pass through”) to this ailment because of the polyuria suffered by the patients. But it was not until 1679 that Thomas Willis made a masterly description of the disease, attributing the second name of mellitus (honey flavor) to the disease, given the sweet taste of the urine of sufferers. In the early 19th century, the French clinician Bouchardat associated DM with obesity and a sedentary lifestyle. Paul Langerhans gave a major impetus to basic diabetes research in 1869, when he published his doctoral thesis on the histology of the pancreas where he described the pancreatic beta cells, which formed isolated islets in the pancreas, to which he gave his name, and attributed the ability to synthesize the hormone responsible for the regulation of blood sugar levels. Shortly afterward, in 1889, the surgeons Von Mering and Minkowsky observed that, after removing the pancreas from animals, they became diabetic. All the evidence suggested that the pancreas produced a substance that was released into the blood, the absence of which was responsible for diabetes. This substance was not found or described until 1921 by Frederick Grant Banting and John James Richard Macleod, who succeeded in isolating insulin. They discovered that insulin is produced in the pancreas, in beta cells located in the islets of Langerhans, and demonstrated its hypoglycemic effect. A year later, they described the beneficial effect of treating diabetic patients with pancreatic extracts [18] and received the Novel Prize in Medicine in 1923 for their discoveries. Pig pancreatic extracts were used to treat diabetes until recombinant human insulin was commercialized in 1982.
1.2. Pathophysiological Characteristics of Diabetes Mellitus
As mentioned above, DM is broadly subdivided into two main types: T1D and T2D, with the latter accounting for 90% of all diabetes cases [19]. Although there is a deficiency of insulin production by pancreatic beta cells in both forms, the two types have different histopathological characteristics.
Thus, T1D is an autoimmune disease in which up to 99% of pancreatic islets are eliminated [20], with a consequent lack of insulin leading to a metabolic decompensation called ketoacidosis. T1D usually develops at an early age, while T2D usually occurs from the age of 40 years onward. However, due to the increasing prevalence of obesity, T2D has also been observed in younger people [21,22,23]. The last form of the disease also has insufficient insulin production, similar to T1D, although to a lower magnitude. Thus, the loss of beta mass in T2D is estimated to be approximately 40% (25–60%) [24], whereas in T1D, this loss may affect 90% of beta cells.
T2D is a chronic disease characterized by defects in insulin action (insulin resistance) and secretion [25]. As a result, glucose accumulates in the bloodstream instead of entering the cells, leading to elevated blood sugar levels [2]. The sequence of events that occur in the development of T2D can be observed in Figure 2, which depicts the normal insulin and glucose levels one hour after a meal at the different stages of this disease. The first event in the sequence of processes leading to T2D is insulin resistance, which leads to increased insulin synthesis and secretion and compensatory hyperinsulinemia. This allows metabolic homeostasis to be maintained in the early stages of the disease; this compensatory insulin phase is called prediabetes and can last for years [26]. Once the balance between insulin resistance and secretion is disrupted, biochemical expression (glucose intolerance) and subsequent clinical diabetes begin. Patients with impaired glucose tolerance and short-standing diabetics have hyperinsulinemia, which is a common component of insulin resistance or metabolic syndrome. The loss of β-cells in DM implies that insulin secretion could be restored and hyperglycemia (but not hyperinsulinemia) normalized through the replacement or regeneration of the islets of Langerhans [27]. In fact, it has long been known that hyperglycemia in T1D and T2D can be reversed by pancreas transplantation [28] and the transplantation of isolated islets [29]. However, the number of transplantable pancreases is insufficient for the ever-increasing number of patients with DM, which impedes the widespread application of this intervention and promotes the continued search for other possible therapies for the treatment of this disease.
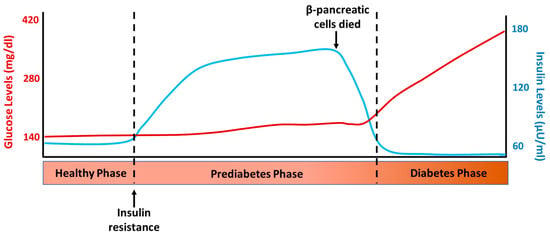
Figure 2.
Representative levels of glucose and insulin after 1 h intake in the different phases of type 2 diabetes development.
Other components of this syndrome that are related to insulin resistance and/or hyperinsulinemia are hypertension, dyslipidemia, obesity, gout, increased prothrombotic factors, fibrinolysis defects, atherosclerosis and dementia. Thus, individuals with T2D are at an increased risk of developing cardiovascular disease. These risks have been extensively studied in clinical and epidemiological research [30,31,32,33,34,35] as well as in basic research [36,37,38,39,40].
1.3. Historical Development of Dementias: Alzheimer’s Disease
The first historical allusion to the term dementia is found in the poem “De rerum natura” by Titus Lucretius in the 1st century BC and in the essay “De Senectute” by Cicero in which dementia is presented as a memory loss contracted with age. In this era, the average life expectancy was around 30 years, so it can be inferred that the term dementia encompasses other pathologies that affect psychiatric disorders. Etiologically, the word dementia means “absent mind” [41]. For centuries, the word dementia has encompassed a heterogeneous set of pathologies that present with memory loss and/or cognitive impairment. It was not until the 17th century that the definitions of dementia and cognitive decline became more precise, distinguishing between mental retardation and age-related cognitive impairment syndromes [41]. By the 19th century, the concept of dementia had acquired the current meaning found in medical literature. The Diagnostic and Statistical Manual of Mental Disorders defines dementia as a syndrome that includes memory loss and the loss of cognitive functions, which incapacitate those who suffer from it to lead a normal work, social and family life [42].
In 1906, German scientist Alois Alzheimer published the results of a study on Auguste Deter, a 52-year-old patient whose husband had brought her to the hospital after detecting changes in her behavior consistent with dementia. This study describes neurofibrillary tangles and senile plaques as markers of AD. Over the next 100 years, the disease has become increasingly frequent owing to increased life expectancy. Its neuropathological markers include the accumulation of beta-amyloid plaques and tau pathology. Currently, the amyloid cascade hypothesis is accepted, in which the first event of the disease is the deposition of amyloid-beta (Aβ), followed by synaptic loss, formation of neurofibrillary tangles and neuronal death (for a review, see [7,8]).
1.4. Alzheimer’s Disease Physiopathology
The pathophysiology of AD involves changes at the brain level, including the accumulation of two proteins: Aβ and hyperphosphorylated tau protein [43]. AD currently has no unequivocal premortem diagnosis and can only be diagnosed histologically postmortem in the presence of (1) the deposition of senile plaques, (2) neurofibrillary tangles and (3) neuronal death [7].
Senile plaques are characteristic alterations of AD, quite common in the brains of patients with dementia, and are possibly the origin of the denervation of the disease. Senile plaques result from progressive accumulation of parenchymal Aβ [7]. Aβ is a 39–43 amino acid peptide derived from progressive processing of the beta-amyloid precursor protein (APP) by β- and γ-secretase complexes, where presenilin (PS) is the catalytic component [44]. Aβ is derived from the proteolytic cleavage of the APP protein, which, when processed by β- and γ-secretases, results in three products, including Aβ, and favors the formation of senile plaques. In contrast, when α-secretase acts, APP is cleaved such that its products do not enter the amyloidogenic pathway [7]. Among the possible Aβ isoforms, Aβ40 and Aβ42 are the most common, with Aβ42 being the most fibrillogenic. Accumulation of Aβ aggregates as soluble oligomers and senile plaques plays a relevant role in the pathogenesis of AD [45]. The accumulation of senile plaques can cause interference with neuronal communication, triggering an inflammatory response in the brain, generating oxidative stress at the brain level, activating toxic processes that interfere with normal cellular processes, altering synaptic function and causing neuronal degeneration [43,46]. The pathology and dynamics of senile plaque formation and remodeling, as well as the precise involvement of amyloid deposition, are not fully understood, although senile plaques remain the main therapeutic targets in the development of new alternatives to prevent or reverse the disease [7,8,47]. Aβ peptides in their different aggregation states and compact senile plaques are neurotoxic both in AD and in experimental models [48] and have been associated with synaptic loss and the development of neuritic dystrophies [49,50,51]. Compact senile plaques have also been associated with abnormal curvature of nearby neurites [52,53,54] and may alter cortical synaptic integration [55,56]. In addition, senile plaques tend to accumulate in regions such as the cortex, hippocampus and cortical associative areas. This accumulation of plaques has a significant impact on cognitive functioning, including impairments in attention and concentration, language and communication difficulties, memory impairment and deficits in executive function [46].
1.5. Vascular Dementia Physiopathology
After AD, which is responsible for about 50–75% of all dementia cases [4], VaD is the second cause of dementia. VaD is a clinical cognitive disorder of cerebral vascular origin caused by stroke. The main cause of VaD is cerebrovascular disease, especially cerebrovascular accidents or strokes [57]. This leads to the degeneration of the affected area, usually the cortex, white matter or both, due to hypoxia. Oxygen deprivation preceded by hypoperfusion can be caused by different vascular aetiologies [58,59]. Broadly speaking, there are two types of strokes: (a) ischemic and (b) hemorrhagic. The main cause of VaD is ischemic stroke, which is caused by vascular collapse that impedes blood circulation, resulting in cerebral hypoperfusion. This deprivation of blood supply can be acute or progressive, depending on the degree of hypoperfusion [60]. VaD from hemorrhagic stroke is caused by vascular rupture, which results in blood contents spilling into the extracellular space (see Figure 3) [61]. Hemorrhagic strokes account for approximately 15% of all strokes, increasing the probability of dementia [60].
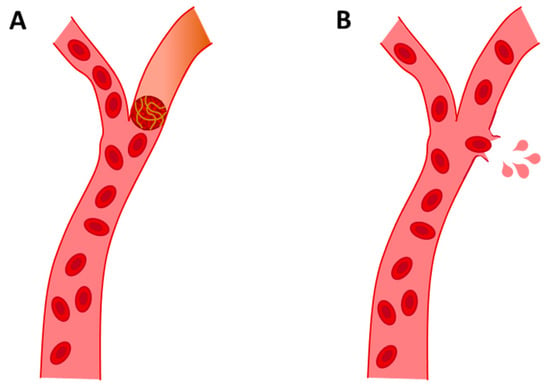
Figure 3.
Representative images of ischemic stroke (A) and hemorrhagic stroke (B).
VaD is composed of a cumulative stroke process that encompasses a heterogeneous pathology of multiple microinfarcts, ischemic small vessel disease, microvascular damage [62] or even the deposition of Aβ in the form of amyloid angiopathy around the cerebral vessels [63]. This heterogeneity of VaD contributes to the presence of numerous clinical phenotypes causing difficulties and controversies in its diagnosis and classification [64,65,66,67].
The pattern of cognitive impairment observed in VaD is variable and can be difficult to distinguish from progressive cognitive decline, especially in the early stages when episodic memory is affected, and it can also be found in the early stages of AD [65]. However, whereas age-related cognitive decline is usually slower, in VaD, it occurs more abruptly and fluctuatingly [68]. Executive function is affected early in VaD to a greater degree than in other dementias, possibly due to the disruption of frontal connections [69]. Similar to AD, mood swings and personality alterations are very pronounced in VaD [67,70]. Another characteristic is decreased serotonin metabolism and a deficit of cholinergic markers, like that described in AD [71,72]. Lesions in white substances can directly affect cholinergic projections. Preclinical and clinical evidence suggests that the cholinergic system may also be involved in VaD [73,74,75]. On the other hand, cognitive impairment associated with subcortical vascular damage may be the result of cortical atrophy of the hippocampus, and although the cause of diffuse cortical atrophy is not well understood, it can be partially correlated with the severity of white matter lesions [75,76].
VaD and AD coexist, and it is possible that vascular pathology contributes to cognitive decline. Thus, it has been speculated that vascular damage may promote AD development. In this sense, blood–brain barrier (BBB) dysfunction could affect the Aβ brain–periphery balance and thus contribute to parenchymal and vascular Aβ deposition [77,78,79]. Furthermore, the pathology of AD can cause vascular damage, such as when Aβ deposition induces inflammation and endothelial damage. The pathological process resulting from vascular damage is associated with alterations in functional markers, such as increased oxidative stress, elevated proinflammatory cytokines or increased activity of matrix metalloproteinases in vessel walls [80,81]. These processes have been linked to neuronal death [82,83] responsible for dementia.
The ultimate triggering cause of VaD is not fully known, although a multitude of studies have been conducted on the relationship between metabolic disorders, lifestyle and vascular dementia [84,85]. In the 1990s, the link between DM and some dementias, such as AD and DaV, was already discussed [86]. The literature suggests that insulin resistance associated with T2D promotes cerebrovascular dysfunction, which would provide a favorable environment for the development of VaD and AD [84,85,87,88]. This review aimed to explore the underlying relationship between T2D, AD and VaD.
2. Link between Diabetes Mellitus, Alzheimer’s Disease and Vascular Dementia
Over the last 15 years, the relationship between T2D, AD and VaD has been extensively studied. Based on published studies, the data suggest that T2D, among other metabolic pathologies, could contribute to the development of a neurodegenerative process that would be a precursor to the development of dementia. Following this idea, DM seems to play a role at this level. The bibliography shows that the incidence of both pathologies increases with age, and it is common to find them coexisting. In fact, some authors claim that 45% of people with T2D suffer from mild cognitive impairment [89], raising the chance of developing dementia by 1.5–2.5 times [90]. In contrast, people with established dementia show insulin disturbances, with altered fasting glucose levels [91].
It also appears that the risk of dementia increases as patients develop other risk factors, including heart disease, hyperlipidemia, hypercholesterolemia or smoking, although T2D is important for its synergistic capacity [34,84,90]. In this relationship, insulin levels and insulin resistance are best correlated with the severity and progression of VaD [92,93]. Furthermore, it seems that DM promotes vascular damage caused by high levels of glucose in the blood, which is accentuated if it coexists with other pathologies that affect blood vessels, such as arteriosclerosis and hypertension [32,94].
The relationship between T2D and VaD can be based on processes that converge between the two pathologies, described below.
2.1. Insulin Receptors in Central Nervous System
Insulin receptors at the central level are located in astrocytes and neuronal synapses and are very abundant in regions of the basal forebrain, such as the septum (origin of cortical and hippocampal cholinergic innervation) [95], and areas particularly relevant for learning and memory processes, such as the cortex and the hippocampus [96]. This wide distribution of insulin receptors explains the involvement of insulin in cognitive processes [97], probably mediated by relevant neurotransmitters in AD, such as norepinephrine and acetylcholine [98,99]. Insulin also contributes to synaptic plasticity mediated by insulin receptors [100]. Similarly, insulin regulates glucose metabolism as the main nutrient of the CNS, directly involved in learning and memory processes [101,102]. In fact, the acute administration of insulin to humans and rodents produces an improvement in cognitive processes and memory [103,104,105,106,107] as well as an increase in the expression of insulin receptors in the dentate gyrus, leading to better performance in spatial memory tests [106,108]. Indeed, some authors have postulated that insulin could counterbalance AD pathology [109].
2.2. Type 2 Diabetes Progression Correlated with Pancreatic Amilin Deposition as Brain Aβ Deposition Correlated with Alzheimer’s Disease Progression
The progression of T2D is correlated with pancreatic amylin deposition, which is similar to that of brain Aβ. In addition, insulin, amylin and Aβ are degraded peripherally by the insulin-degrading enzyme (IDE) [110]. This suggests that these substrates may compete at this level [111]. It has been postulated that substrate imbalance may influence the pathogenesis of AD and T2D [112].
From another point of view, insulin resistance detected in AD patients could be mediated by a decrease in the activity of the enzymes responsible for its degradation. Both the insulin-degrading enzyme (IDE) and neprelisine (NEP) are involved in insulin degradation as well as Aβ and amylin degradation [113]. Accordingly, it has been postulated that an imbalance of substrates can affect the degradation rate of other substrates and possibly influence the pathogenesis of T2D and AD [112,114,115,116]. A decrease in IDE expression may result in reduced insulin and Aβ degradation in the brain [115,117]. Consequently, it is conceivable that actions promoted by elevated insulin levels, or its deficiency, may also be a link between T2D and AD.
2.3. Insulin-Like Growth Factor
Insulin and the insulin-like growth factor (IGF) have similar structures and play a significant role in the regulation of aging. IGF acts as a cellular growth factor but also plays a hormonal role in regulating growth and metabolism at the systemic level [118]. IGF is the main prenatal and postnatal growth factor. This is why low levels of insulin during gestation can lead to slower growth and low height and weight of the offspring [107,119]. In contrast, an increase in insulin levels at an early stage, as occurs in maternal T2D, leads to large and overweight children, among other complications [120].
In this context, insulin plays a crucial role in the development of neuronal complexity, as well as in neurogenesis in early neonatal development. In this sense, insulin deprivation during early life may result in reduced neural network development, whereas exogenous administration of insulin may promote increased neural development and complexity [107,121]. A more complex neural network is a protective factor against the development of dementia [122]. Based on the aforementioned, a gestational environment with altered insulin levels could condition the possible development of dementia in the later stages of life [123,124].
Additionally, plasma inulin can cross the BBB in a soluble form and gain access to neurons, microvasculature and even immature neuronal bodies [125]. Insulin plays a key role as a growth-regulating hormone during the early stages of life. In animal models, it has been shown that an imbalance in insulin levels also impairs neurogenesis in early life [107], as well as in more mature and long-lived phases [37]. In addition, the coexistence of T2D with AD pathology reduces neurogenesis and thus neuronal replacement from stages prior to cognitive decline [126]. Insulin is also involved in the regulation of neuronal and synaptic functions in the hippocampus, cortex and cerebellum, protecting neurons from neurodegeneration and cell death [127,128,129,130].
2.4. Insulin Promotes Typical Features of Alzheimer’s Disease
Tau pathology is one of the main features of AD, and many studies have reported the impact of insulin dysfunction and diabetes on it [131,132,133]. Indeed, high insulin levels in the brain, induced by prediabetes or T2D, may increase the hyperphosphorylation of tau protein [134]. It is feasible that higher levels of phosphorylated tau observed in hyperinsulinemia and T2D states could be mediated by insulin receptors at the central level [132]. Additionally, T1D hypoinsulinemia may increase tau hyperphosphorylation [135]. In this regard, a clinical phase I study with the drug SCR-1693 showed a reduction in tau protein phosphorylation levels associated with an improvement in cognitive and central insulin resistance [136].
Inflammation of the CNS is increasingly regarded to play a role in cognitive disorders such as dementia [137]. Insulin promotes the expression of proinflammatory cytokines such as α-TNF and IL6 [138], which are the most important proinflammatory cytokines. Moreover, these cytokines negatively affect the metabolism of Aβ oligomers [139]. Similarly, a proinflammatory state, in concomitance with amyloid pathology, leads to the activation of microglia and astrocytes in the SNC [140]. A recent study in several models of diabetes combined with a classical AD model, such as the APP/PS1 mouse, concludes from the cytokine profiles exhibited in each pathology that neuroinflammation may be the mechanism by which diabetes affects the pathology of AD [80].
On the other hand, one of the mechanisms proposed to link insulin to cognitive impairment is its role in Aβ metabolism [111,115]. In this regard, insulin promotes the amyloidogenic pathway by modulating β and γ-secretases [114,141,142]. Additionally, insulin inhibits or hinders the passage of Aβ through the BBB [143]. Following this idea, insulin would prevent the clearance of Aβ and promote its accumulation in the brain [144]. In return, Aβ interferes with insulin signaling in the CNS; in fact, soluble Aβ oligomers may disrupt insulin signaling in hippocampus neuronal cultures [145]. Furthermore, insulin promotes Aβ deposition in the brain in a similar way to amylin deposition in the pancreas of T2D patients. Amylin, like Aβ, is a toxic species involved in the apoptosis of pancreatic cells and neurons [146,147]. Moreover, amylin and Aβ aggregates alter cellular function by similar mechanisms: mitochondrial dysfunction and the formation of reactive oxygen species [148].
2.5. Role of Prediabetes and Diabetes Mechanism in the Neurodegenerative Process of Alzheimer’s Disease and Vascular Dementia
Previous epidemiological and clinical studies support a close relationship between T2D and AD [148,149]; however, the underlying linking mechanisms are not yet fully understood. It also remains unclear whether hyperinsulinemia and insulin resistance, indicative of a prediabetic state prior to T2D, may induce or accelerate central pathology in AD, in a similar manner to that induced by T2D. Indeed, glucose and insulin play a crucial role in maintaining normal brain activity, and alterations of insulin-dependent functions could be associated with central pathological conditions observed in AD [139,148,150].
The available scientific evidence on this relationship supports that the first pathological event in the DM disorder, as a promoter of dementia, is insulin resistance in the CNS [92,125,151]. Insulin plays an important role in cell growth and differentiation, as well as in protein synthesis [125,152]. Additionally, insulin inhibits catabolic processes such as glycolysis, lipolysis and proteolysis [152]. The wide distribution of insulin receptors throughout the CNS underscores its importance in central glucose homeostasis processes and its role in cognitive processes and neuronal development [125]. In this sense, it has been described that alterations in insulin balance in the CNS accelerate the brain aging process, increasing vascular damage and primarily affecting synaptic plasticity [50,152,153]. Vascular damage is often one of the first central events in diabetes [154], even in the prediabetic stages, and compensatory high insulin levels are sufficient to cause this damage [39]. Simultaneously with vascular damage, brain aging occurs, which is translated into reduced neuronal arborization and synaptic density. The loss of neuronal and synaptic density has been proposed as one of the best pathological markers for the assessment of AD. There is ample scientific evidence that different states of Aβ aggregation, from the most soluble forms to the formation of senile plaques, are neurotoxic [155,156,157]. It has been reported that synaptic loss promotes cell dedifferentiation and ultimately neuronal death [158]. Studies in animal models have shown that T2D conditions decrease synaptic density at the cortical level [38]. This decline is exacerbated when prediabetes and T2D coexist with AD, with greater involvement observed in regions close to senile plaques than in areas away from them [50]. One plausible explanation for this increased involvement is that diabetes, by some yet unknown mechanism, may be promoting soluble and more toxic forms of Aβ [78,79], which would allow for a more extensive distribution and involvement of the brain [159]. However, this effect has been observed in both T1D and T2D animal models [78,79]. The overproduction and absence of insulin produced in T2D and T1D, respectively, lead to vascular damage and a chronic proinflammatory state [38,80]. T1D promotes vascular changes, such as impaired regulation of vascular tone and dysfunction in neovascularization and vasoregression, leading to a blood–brain barrier disruption (for a review, see [160]). T2D can also promote vascular dysfunction and inflammation (for a review, see [161]). Both factors may contribute to a lower capacity to eliminate excess Aβ vascularly or through microglia. This situation promotes its accumulation in a soluble form. In fact, in our murine models of AD-T2D and AD-T1D, the levels of serum Aβ were reduced, indicating the inability to expel peptides from the CNS across the blood–brain barrier, while intracerebral soluble Aβ levels were increased [78,79]. In fact, the higher levels of soluble Aβ40 and 42 in the CNS observed in T1D and T2D are also in agreement with large synaptic loss and neuronal death observations [79,81,162,163,164,165,166]. It is feasible that the shift toward more toxic soluble species might contribute to the observed progressive synapse reduction in prediabetic and diabetic AD mice [50,167] and an in silico study [168]. In addition, prediabetes and T2D promote an increased accumulation of Aβ in the form of amyloid angiopathy (AAC) around blood vessels [81], which is associated with an increased risk of vascular rupture [169]. On the other hand, Aβ clearance might also be affected. In this regard, some studies support that Aβ pathology, in combination with hyperinsulinemia, promotes higher levels of Aβ because IDE is much more selective for insulin than for Aβ [115,170]. In addition, the lack of insulin in T1D seems to be associated with lower levels of enzymatic activity (for a review, see [171]). Additionally, previous studies have shown that the activity of IDE and NEP could be intrinsically altered in AD or DM [163,172,173]. Likewise, vascular damage observed in prediabetic-AD and T2D-AD mice could affect Aβ clearance by altering the BBB, in accordance with previous models showing reduced amyloid clearance along interstitial fluid drainage pathways [174,175], by damage of the artery feeding a particular brain territory.
In the second stage, we propose that the sequence of events involves the inflammatory process and glial activation. It has been suggested that the insulin resistance typical of prediabetes and T2D may exacerbate the inflammatory process when it interacts with the presence of Aβ [139]. Previous studies report that the presence of Aβ is sufficient to promote microglial activation and the inflammatory process [176,177]. Vascular damage and the T2D-induced imbalance of Aβ pathology toward more soluble forms have been shown to induce an increase in cytotoxic and pro-inflammatory cytokines and increase microglial activity [80,178]. This inflammatory process would increase oxidative molecular species, generating a harmful environment that would promote neurodegenerative processes [179]. Microglial cells, faced with vascular damage and the accumulation of toxic substances such as Aβ, begin to produce proinflammatory cytokines (IL-1β, IL-6, IL-18 and tumor necrosis factor-α (TNF-α)), chemokines (CCL1, CCL5, CXCL1), small messenger molecules (prostaglandins, NO) and ROS [180]. Although most cells involved in this process of neuroinflammation are microglia and astrocytes, capillary endothelial cells are also involved, as well as some infiltrating blood cells, which are more frequent when there is tissue damage in the BBB [180,181]. This pro-inflammatory ecosystem could lead to synaptic dysfunction, neuronal death and inhibition of neurogenesis [80,126,182]. Similarly, it has been shown that the increase in prostaglandin 2 by the damaged vascular endothelium increases the production of IL-1β, which is associated with synaptic loss through the activation of the postsynaptic N-methyl-D-aspartate receptor [182]. Under these circumstances, activation of the complement system, which promotes phagocytosis by microglia, may also be involved in the process of synaptic loss [183]. The described mechanism involves neuronal death by TNF-α-mediated caspase-8 recruitment and TNF receptor type 1 [184].
The following event would affect one of the classic AD pathologies: the hyperphosphorylation of the tau protein [7], which can also be found in other neurodegenerative diseases such as VaD. Previous studies have shown early tau phosphorylation in diabetic mouse models [185], whereas other studies support the idea that this pathology only appears in the latest phases of diabetes [38]. This apparent discrepancy may be explained by the fact that tau phosphorylation is highly dependent on the specific phosphorylated residues studied [185,186]. Altogether, these data support the step-through role of early prediabetes and consolidated T1D and T2D at the central level, which seem to preferentially affect the cortex and spread to the hippocampus as the disease progresses [39,78,79,187,188,189].
Described central alterations ultimately lead to learning and memory dysfunction. Prediabetes or early diabetes does not affect learning or memory [38,39]. However, late diabetes is related to poor cognitive preservation, and episodic and spatial memories are affected in animal models [38]. A similar outcome was observed in patients with T2D [190,191] and T1D [192], with lower levels of global cognition and episodic or working memory. In fact, a recent study supports that low levels of cognition in aging people are associated with body fat and higher body max index, which are risk factors for suffering AD, VaD and T2D [193]. Metabolic parameters are correlated with CNS alterations in animal models; a study supports that glucose levels, insulin levels and body weight are good predictors of cortical atrophy and impaired cortical and hippocampal cell proliferation, suggesting the role of the diabetic process at the central level [37]. In accordance with this, human studies have also shown significant associations between insulin levels in T2D patients and brain alterations detected by magnetic resonance imaging (MRI) [189,194,195]. Curiously, whereas worse metabolic conditions correlate with lower rates of central cell proliferation, affected metabolism also predicts increased neurogenesis rates, suggesting that as the pathology progresses, the overall cellular production is impaired, and the system tries to compensate for this by generating new neurons [37].
3. Future Perspectives
Growing evidence suggests that T2D may increase the risk of developing AD. Several studies have found that insulin resistance and metabolic dysfunction associated with T2D negatively affect the brain and increase the accumulation of Aβ. Also, T2D promotes chronic inflammation, oxidative stress and vascular dysfunction in the brain which links T2D with AD and VaD.
Understanding the relationship between DM and AD is essential to prevent the development of AD through metabolic control. Likewise, knowledge of the involvement of DM in the progress and development of AD can help us address new therapeutic strategies for its treatment. In this sense, treatment to control T2D, such as metformin, has shown a beneficial effect, like neuroprotective, anti-inflammatory and antioxidant action in animal models [196,197,198]. However, limited studies in humans have shown a more discrete positive relationship with the use of metformin as a treatment for AD [199]. The possible beneficial effects of metformin as a treatment for Alzheimer’s disease are still being studied and are a hot topic (for a review, see [200]).
Another emerging candidate for a therapeutic approach to AD an VaD is the application of intranasal insulin. This treatment has been shown to have beneficial effects in reducing inflammation and improving immune function. In addition, intranasal insulin improves cognitive status and helps to maintain cognitive abilities [201,202]. Other studies have shown that intranasal insulin as a treatment for AD has neuroprotective effects, and its use can maintain the insulin brain signaling to improve cognitive health [203] by reducing the P-tau/Aβ42 ratio [204] and preserving the white matter volume in deep and frontal regions, which correlates with AD progression [205]. However, recent long-term studies in humans have shown limited efficacy of intranasal insulin as a treatment for AD [204,206]. The current disagreement in the literature shows the need to carry out studies that demonstrate the usefulness of this treatment in the control of AD.
Finally, as the understanding of the relationship between T2D, AD and VaD increases, there are new opportunities for prevention and treatment. Improving glycemic control and promoting healthy lifestyles should be explored further. Thus, early attention to risk factors associated with dementia, such as T2D, may play a crucial role in preventing or delaying dementia.
4. Conclusions
The findings of this review clarify the underlying relationship between AD, T2D and VaD. Thus, early hyperinsulinemia, without clinically established T2D, is sufficient to worsen AD pathology. T2D promotes vascular damage and increases inflammatory processes at the central level. Moreover, both T2D and T1D accelerate AD pathological features, increasing more toxic Aβ species and resulting in a more severe version of AD (Figure 4). Increased levels of soluble Aβ and phosphotau may contribute to synaptic loss, neuronal death, brain atrophy and final cognitive impairment. Altogether, T2D promotes an early and more severe version of AD and VaD pathology. Published evidence provides a relevant tool to further explore the relationship between T2D, AD and vascular implications, offering the possibility to assess therapeutic approaches that can delay or prevent AD and VaD pathology by improving metabolic control.
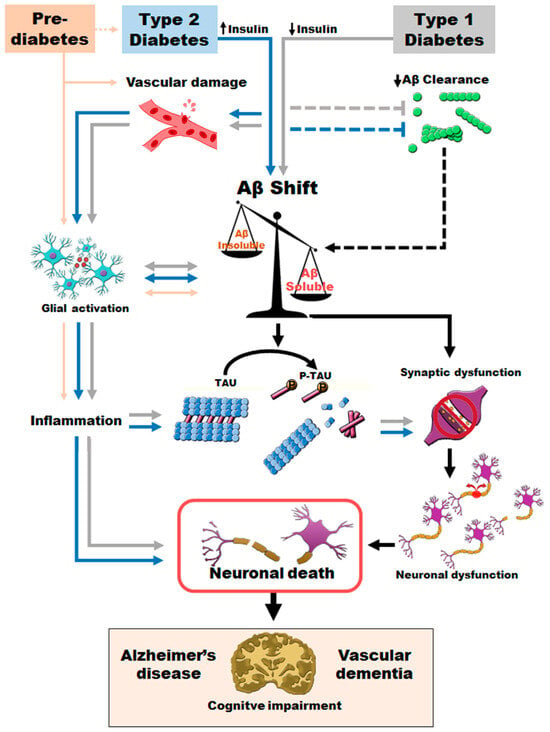
Figure 4.
Proposed sequence of pathogenic events promoted when T2D or T1D coexist with AD and VaD.
Author Contributions
Conceptualization and supervision: H.M.-M. and J.J.R.-R.; methodology: T.P.-M., A.G.-A., M.I.T.-G., L.M.-R. and H.M.-M.; resources: T.P.-M., E.M.S.L. and J.J.R.-R.; writing—original draft preparation: H.M.-M., V.G.-M., L.M.-R. and A.G.-A.; writing—review and editing: M.I.T.-G., E.M.S.L., V.G.-M. and J.J.R.-R. All authors have read and agreed to the published version of the manuscript.
Funding
L.M.-R. and J.J.R.-R. are funded by P20-01293 from Junta de Andalucía, Spain. J.J.R.-R. and V.G.-M. are additionally funded by P20-01061 from Junta de Andalucía, Spain, and PID2019-110960GB-I00 from the Ministry of Science and Innovation, Spain.
Conflicts of Interest
The authors declare no conflict of interest.
References
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42 (Suppl. S1), S13–S28. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2021, 183, 109119. [Google Scholar] [CrossRef]
- Santos, M.A.O.; Bezerra, L.S.; Correia, C.d.C.; Bruscky, I.S. Neuropsychiatric symptoms in vascular dementia: Epidemiologic and clinical aspects. Dement. Neuropsychol. 2018, 12, 40–44. [Google Scholar] [CrossRef]
- Cunningham, E.L.; McGuinness, B.; Herron, B.; Passmore, A.P. Dementia. Ulst. Med. J. 2015, 84, 79–87. [Google Scholar]
- OMS. Dementia; 2019. [Google Scholar]
- Garre-Olmo, J. Epidemiology of Alzheimer’s disease and other dementias. Rev. Neurol. 2018, 66, 377–386. [Google Scholar]
- Garcia-Morales, V.; Gonzalez-Acedo, A.; Melguizo-Rodriguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodriguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef]
- Molsa, P.K.; Marttila, R.J.; Rinne, U.K. Long-term survival and predictors of mortality in Alzheimer’s disease and multi-infarct dementia. Acta Neurol. Scand. 1995, 91, 159–164. [Google Scholar] [CrossRef]
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef]
- Lleo, A. Alzheimer’s disease: An ignored condition. Med. Clin. 2018, 150, 432–433. [Google Scholar] [CrossRef]
- Bjerre, L.M.; Farrell, B.; Hogel, M.; Graham, L.; Lemay, G.; McCarthy, L.; Raman-Wilms, L.; Rojas-Fernandez, C.; Sinha, S.; Thompson, W.; et al. Deprescribing antipsychotics for behavioural and psychological symptoms of dementia and insomnia: Evidence-based clinical practice guideline. Can. Fam. Physician 2018, 64, 17–27. [Google Scholar] [PubMed]
- Wolters, F.J.; Ikram, M.A. Epidemiology of Vascular Dementia. Arter. Thromb. Vasc. Biol. 2019, 39, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Primers 2018, 4, 18003. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Sachdev, P.; Kalaria, R.; O’Brien, J.; Skoog, I.; Alladi, S.; Black, S.E.; Blacker, D.; Blazer, D.G.; Chen, C.; Chui, H.; et al. Diagnostic criteria for vascular cognitive disorders: A VASCOG statement. Alzheimer Dis. Assoc. Disord. 2014, 28, 206–218. [Google Scholar] [CrossRef]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar]
- Cloete, L. Diabetes mellitus: An overview of the types, symptoms, complications and management. Nurs. Stand. 2022, 37, 61–66. [Google Scholar] [CrossRef]
- Syed, F.Z. Type 1 Diabetes Mellitus. Ann. Intern. Med. 2022, 175, itc33–itc48. [Google Scholar] [CrossRef]
- Lascar, N.; Brown, J.; Pattison, H.; Barnett, A.H.; Bailey, C.J.; Bellary, S. Type 2 diabetes in adolescents and young adults. Lancet Diabetes Endocrinol. 2018, 6, 69–80. [Google Scholar] [CrossRef]
- Luo, J.; Hodge, A.; Hendryx, M.; Byles, J.E. Age of obesity onset, cumulative obesity exposure over early adulthood and risk of type 2 diabetes. Diabetologia 2020, 63, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Zucker, I.; Afek, A.; Cukierman-Yaffe, T.; Bendor, C.D.; Derazne, E.; Lutski, M.; Shohat, T.; Mosenzon, O.; Tzur, D.; et al. Adolescent Obesity and Early-Onset Type 2 Diabetes. Diabetes Care 2020, 43, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Pearson, E.R. Type 2 diabetes: A multifaceted disease. Diabetologia 2019, 62, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Rett, K.; Gottwald-Hostalek, U. Understanding prediabetes: Definition, prevalence, burden and treatment options for an emerging disease. Curr. Med. Res. Opin. 2019, 35, 1529–1534. [Google Scholar] [CrossRef]
- De Klerk, E.; Hebrok, M. Stem Cell-Based Clinical Trials for Diabetes Mellitus. Front. Endocrinol. 2021, 12, 631463. [Google Scholar] [CrossRef]
- Robertson, R.P. Successful islet transplantation for patients with diabetes—Fact or fantasy? N. Engl. J. Med. 2000, 343, 289–290. [Google Scholar] [CrossRef]
- Ryan, E.A.; Paty, B.W.; Senior, P.A.; Bigam, D.; Alfadhli, E.; Kneteman, N.M.; Lakey, J.R.; Shapiro, A.M.J. Five-Year Follow-Up After Clinical Islet Transplantation. Diabetes 2005, 54, 2060–2069. [Google Scholar] [CrossRef]
- Becker, M.A.; MacDonald, P.A.; Hunt, B.J.; Jackson, R.L. Diabetes and gout: Efficacy and safety of febuxostat and allopurinol. Diabetes Obes. Metab. 2013, 15, 1049–1055. [Google Scholar] [CrossRef]
- Kaze, A.D.; Santhanam, P.; Erqou, S.; Bertoni, A.G.; Ahima, R.S.; Echouffo-Tcheugui, J.B. Microvascular disease and cardiovascular outcomes among individuals with type 2 diabetes. Diabetes Res. Clin. Pract. 2021, 176, 108859. [Google Scholar] [CrossRef]
- Nagar, S.D.; Qian, J.; Boerwinkle, E.; Cicek, M.; Clark, C.R.; Cohn, E.; Gebo, K.; Loperena, R.; Mayo, K.; Mockrin, S.; et al. Investigation of hypertension and type 2 diabetes as risk factors for dementia in the All of Us cohort. Sci. Rep. 2022, 12, 19797. [Google Scholar] [CrossRef] [PubMed]
- Nazarzadeh, M.; Bidel, Z.; Canoy, D.; Copland, E.; Wamil, M.; Majert, J.; Byrne, K.S.; Sundström, J.; Teo, K.; Davis, B.R.; et al. Blood pressure lowering and risk of new-onset type 2 diabetes: An individual participant data meta-analysis. Lancet 2021, 398, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Xu, W.; Ou, Y.-N.; Cao, X.-P.; Tan, M.-S.; Tan, L.; Yu, J.-T. Diabetes mellitus and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 144 prospective studies. Ageing Res. Rev. 2019, 55, 100944. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Chen, Z.; Baudier, R.L.; Krousel-Wood, M.; Anderson, A.H.; Fonseca, V.A.; Mauvais-Jarvis, F. Early Menopause and Cardiovascular Disease Risk in Women With or Without Type 2 Diabetes: A Pooled Analysis of 9,374 Postmenopausal Women. Diabetes Care 2021, 44, 2564–2572. [Google Scholar] [CrossRef]
- Preguiça, I.; Alves, A.; Nunes, S.; Gomes, P.; Fernandes, R.; Viana, S.D.; Reis, F. Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients 2020, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Molina-Gil, S.; Ortiz-Barajas, O.; Jimenez-Palomares, M.; Perdomo, G.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; Garcia-Alloza, M. Central Proliferation and Neurogenesis Is Impaired in Type 2 Diabetes and Prediabetes Animal Models. PLoS ONE 2014, 9, e89229. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz, O.; Jimenez-Palomares, M.; Kay, K.R.; Berrocoso, E.; Murillo-Carretero, M.I.; Perdomo, G.; Spires-Jones, T.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 2013, 38, 2462–2475. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz-Barajas, O.; Gamero-Carrasco, C.; de la Rosa, P.R.; Infante-Garcia, C.; Zopeque-Garcia, N.; Lechuga-Sancho, A.M.; Garcia-Alloza, M. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology 2014, 48, 123–135. [Google Scholar] [CrossRef]
- Rodríguez, J.E.; Ruiz-Hernández, A.; Hernández-Díazcouder, A.; Huang, F.; Hong, E.; Villafaña, S. Chronic diabetes and hypertension impair the in vivo functional response to phenylephrine independent of α1-adrenoceptor expression. Eur. J. Pharmacol. 2020, 883, 173283. [Google Scholar] [CrossRef]
- Custodio, N.; Montesisnos, R.; Alarcon, J.O. Evolución histórica del concepto y criterios actuales para el diagnóstico de demencia. In Rev. De Neruo-Psiquiatr. 2018, 81, 250. [Google Scholar] [CrossRef]
- Eramudugolla, R.; Mortby, M.E.; Sachdev, P.; Meslin, C.; Kumar, R.; Anstey, K.J. Evaluation of a research diagnostic algorithm for DSM-5 neurocognitive disorders in a population-based cohort of older adults. Alzheimer’s Res. Ther. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef]
- Urbanc, B.; Cruz, L.; Le, R.; Sanders, J.; Ashe, K.H.; Duff, K.; Stanley, H.E.; Irizarry, M.C.; Hyman, B.T. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 13990–13995. [Google Scholar] [CrossRef]
- Grace, E.A.; Rabiner, C.A.; Busciglio, J. Characterization of neuronal dystrophy induced by fibrillar amyloid beta: Implications for Alzheimer’s disease. Neuroscience 2002, 114, 265–273. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Spires-Jones, T.; Pooler, A.M.; Lechuga-Sancho, A.M.; Bacskai, B.J.; Garcia-Alloza, M. Progressive Neuronal Pathology and Synaptic Loss Induced by Prediabetes and Type 2 Diabetes in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2016, 54, 3428–3438. [Google Scholar] [CrossRef]
- Koutarapu, S.; Ge, J.; Jha, D.; Blennow, K.; Zetterberg, H.; Lashley, T.; Michno, W.; Hanrieder, J. Correlative Chemical Imaging Identifies Amyloid Peptide Signatures of Neuritic Plaques and Dystrophy in Human Sporadic Alzheimer’s Disease. Brain Connect. 2023, 13, 297–306. [Google Scholar] [CrossRef]
- D’Amore, J.D.; Kajdasz, S.T.; McLellan, M.E.; Bacskai, B.J.; Stern, E.A.; Hyman, B.T. In Vivo Multiphoton Imaging of a Transgenic Mouse Model of Alzheimer Disease Reveals Marked Thioflavine-S-Associated Alterations in Neurite Trajectories. J. Neuropathol. Exp. Neurol. 2003, 62, 137–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Infante-Garcia, C.; Ramos-Rodriguez, J.J.; Delgado-Olmos, I.; Gamero-Carrasco, C.; Fernandez-Ponce, M.T.; Casas, L.; Mantell, C.; Garcia-Alloza, M. Long-Term Mangiferin Extract Treatment Improves Central Pathology and Cognitive Deficits in APP/PS1 Mice. Mol. Neurobiol. 2017, 54, 4696–4704. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Meyer-Luehmann, M.; Stern, E.A.; McLean, P.J.; Skoch, J.; Nguyen, P.T.; Bacskai, B.J.; Hyman, B.T. Dendritic Spine Abnormalities in Amyloid Precursor Protein Transgenic Mice Demonstrated by Gene Transfer and Intravital Multiphoton Microscopy. J. Neurosci. 2005, 25, 7278–7287. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.A.; Bacskai, B.J.; Hickey, G.A.; Attenello, F.J.; Lombardo, J.A.; Hyman, B.T. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J. Neurosci. 2004, 24, 4535–4540. [Google Scholar] [CrossRef]
- Domínguez-Álvaro, M.; Montero-Crespo, M.; Blazquez-Llorca, L.; Plaza-Alonso, S.; Cano-Astorga, N.; DeFelipe, J.; Alonso-Nanclares, L. 3D Analysis of the Synaptic Organization in the Entorhinal Cortex in Alzheimer’s Disease. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef]
- Korczyn, A.D. What is new in vascular dementia? BMC Med. 2016, 14, 175. [Google Scholar] [CrossRef]
- Yang, T.; Sun, Y.; Lu, Z.; Leak, R.K.; Zhang, F. The impact of cerebrovascular aging on vascular cognitive impairment and dementia. Ageing Res. Rev. 2016, 34, 15–29. [Google Scholar] [CrossRef]
- Pangprasertkul, S.; Borisoot, W.; Buawangpong, N.; Sirikul, W.; Wiwattanadittakul, N.; Katanyuwong, K.; Sanguansermsri, C. Comparison of Arterial Ischemic and Hemorrhagic Pediatric Stroke in Etiology, Risk Factors, Clinical Manifestations, and Prognosis. Pediatr. Emerg. Care 2022, 38, e1569–e1573. [Google Scholar] [CrossRef]
- Frederiksen, K.S. Vascular dementia. Ugeskr. Laeger 2017, 179, V10160701. [Google Scholar] [PubMed]
- Greenberg, S.M.; Grabowski, T.; Gurol, M.E.; Skehan, M.E.; Nandigam, R.N.; Becker, J.A.; Garcia-Alloza, M.; Prada, C.; Frosch, M.P.; Rosand, J.; et al. Detection of isolated cerebrovascular beta-amyloid with Pittsburgh compound B. Ann. Neurol. 2008, 64, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Lim, W.S.; Sahadevan, S. Stage-Independent and Stage-Specific Phenotypic Differences between Vascular Dementia and Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2008, 26, 513–521. [Google Scholar] [CrossRef]
- Kalaria, R. Similarities between Alzheimer’s disease and vascular dementia. J. Neurol. Sci. 2002, 203–204, 29–34. [Google Scholar] [CrossRef]
- Román, G.; Pascual, B. Contribution of Neuroimaging to the Diagnosis of Alzheimer’s Disease and Vascular Dementia. Arch. Med. Res. 2012, 43, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Groves, W.C.; Brandt, J.; Steinberg, M.; Warren, A.; Rosenblatt, A.; Baker, A.; Lyketsos, C.G. Vascular dementia and Alzheimer’s disease: Is there a difference? A comparison of symptoms by disease duration. J. Neuropsychiatry Clin. Neurosci. 2000, 12, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Luck, T.; Luppa, M.; Briel, S.; Riedel-Heller, S.G. Incidence of Mild Cognitive Impairment: A Systematic Review. Dement. Geriatr. Cogn. Disord. 2010, 29, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Royall, D.R. Executive Control Function: A Rational Basis for the Diagnosis of Vascular Dementia. Alzheimer Dis. Assoc. Disord. 1999, 13 (Suppl. S3), S69–S80. [Google Scholar] [CrossRef]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cattaruzza, T.; Cazzato, G.; Bava, A. Olanzapine as a possible treatment for anxiety due to vascular dementia: An open study. Am. J. Alzheimer’s Dis. Other Dement. 2004, 19, 81–88. [Google Scholar] [CrossRef]
- Ban, Y.; Watanabe, T.; Miyazaki, A.; Nakano, Y.; Tobe, T.; Idei, T.; Iguchi, T.; Ban, Y.; Katagiri, T. Impact of increased plasma serotonin levels and carotid atherosclerosis on vascular dementia. Atherosclerosis 2007, 195, 153–159. [Google Scholar] [CrossRef]
- Gottfries, C.; Blennow, K.; Karlsson, I.; Wallin, A. The Neurochemistry of Vascular Dementia. Dement. Geriatr. Cogn. Disord. 1994, 5, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ruiz, C.; Court, J.; Lee, M.; Piggott, M.; Johnson, M.; Ballard, C.; Kalaria, R.; Perry, R.; Perry, E. Nicotinic receptors in dementia of Alzheimer, Lewy body and vascular types. Acta Neurol. Scand. Suppl. 2000, 102, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Swartz, R.H.; Sahlas, D.J.; Black, S.E. Strategic involvement of cholinergic pathways and executive dysfunction: Does location of white matter signal hyperintensities matter? J. Stroke Cerebrovasc. Dis. 2003, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Kwon, H.; Lee, Y. Effect of cholinergic pathway disruption on cortical and subcortical volumes in subcortical vascular cognitive impairment. Eur. J. Neurol. 2019, 27, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Fein, G.; Di Sclafani, V.; Tanabe, J.; Cardenas, V.; Weiner, M.W.; Jagust, W.J.; Reed, B.R.; Norman, D.; Schuff, N.; Kusdra, L.; et al. Hippocampal and cortical atrophy predict dementia in subcortical ischemic vascular disease. Neurology 2000, 55, 1626–1635. [Google Scholar] [CrossRef]
- Infante-Garcia, C.; Ramos-Rodriguez, J.J.; Galindo-Gonzalez, L.; Garcia-Alloza, M. Long-term central pathology and cognitive impairment are exacerbated in a mixed model of Alzheimer’s disease and type 2 diabetes. Psychoneuroendocrinology 2015, 65, 15–25. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Infante-Garcia, C.; Galindo-Gonzalez, L.; Garcia-Molina, Y.; Lechuga-Sancho, A.; Garcia-Alloza, M. Increased Spontaneous Central Bleeding and Cognition Impairment in APP/PS1 Mice with Poorly Controlled Diabetes Mellitus. Mol. Neurobiol. 2016, 53, 2685–2697. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Jimenez-Palomares, M.; Murillo-Carretero, M.I.; Infante-Garcia, C.; Berrocoso, E.; Hernandez-Pacho, F.; Lechuga-Sancho, A.M.; Cozar-Castellano, I.; Garcia-Alloza, M. Central vascular disease and exacerbated pathology in a mixed model of type 2 diabetes and Alzheimer’s disease. Psychoneuroendocrinology 2015, 62, 69–79. [Google Scholar] [CrossRef]
- Sankar, S.B.; Infante-Garcia, C.; Weinstock, L.D.; Ramos-Rodriguez, J.J.; Hierro-Bujalance, C.; Fernandez-Ponce, C.; Wood, L.B.; Garcia-Alloza, M. Amyloid beta and diabetic pathology cooperatively stimulate cytokine expression in an Alzheimer’s mouse model. J. Neuroinflamm. 2020, 17, 38. [Google Scholar] [CrossRef]
- Vargas-Soria, M.; Ramos-Rodriguez, J.J.; del Marco, A.; Hierro-Bujalance, C.; Carranza-Naval, M.J.; Calvo-Rodriguez, M.; van Veluw, S.J.; Stitt, A.W.; Simó, R.; Bacskai, B.J.; et al. Accelerated amyloid angiopathy and related vascular alterations in a mixed murine model of Alzheimer’s disease and type two diabetes. Fluids Barriers CNS 2022, 19, 88. [Google Scholar] [CrossRef]
- Brown, C.E.; Aminoltejari, K.; Erb, H.; Winship, I.R.; Murphy, T.H. In Vivo Voltage-Sensitive Dye Imaging in Adult Mice Reveals That Somatosensory Maps Lost to Stroke Are Replaced over Weeks by New Structural and Functional Circuits with Prolonged Modes of Activation within Both the Peri-Infarct Zone and Distant Sites. J. Neurosci. 2009, 29, 1719–1734. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Swarnakar, S. Implication of matrix metalloproteinases in regulating neuronal disorder. Mol. Biol. Rep. 2015, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Tahmi, M.; Palta, P.; Luchsinger, J.A. Metabolic Syndrome and Cognitive Function. Curr. Cardiol. Rep. 2021, 23, 180. [Google Scholar] [CrossRef]
- Stewart, R.; Liolitsa, D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet. Med. 1999, 16, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.R. Linking cardiometabolic disorders to sporadic Alzheimer’s disease: A perspective on potential mechanisms and mediators. J. Neurochem. 2010, 115, 551–562. [Google Scholar] [CrossRef]
- Lyu, F.; Wu, D.; Wei, C.; Wu, A. Vascular cognitive impairment and dementia in type 2 diabetes mellitus: An overview. Life Sci. 2020, 254, 117771. [Google Scholar] [CrossRef]
- You, Y.; Liu, Z.; Chen, Y.; Xu, Y.; Qin, J.; Guo, S.; Huang, J.; Tao, J. The prevalence of mild cognitive impairment in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Acta Diabetol. 2021, 58, 671–685. [Google Scholar] [CrossRef]
- Ninomiya, T. Epidemiological Evidence of the Relationship Between Diabetes and Dementia. Adv. Exp. Med. Biol. 2019, 1128, 13–25. [Google Scholar] [CrossRef]
- Lee, D.Y.; Kim, J.; Park, S.; Park, S.Y.; Yu, J.H.; Seo, J.A.; Kim, N.H.; Yoo, H.J.; Kim, S.G.; Choi, K.M.; et al. Fasting Glucose Variability and the Risk of Dementia in Individuals with Diabetes: A Nationwide Cohort Study. Diabetes Metab. J. 2022, 46, 923–935. [Google Scholar] [CrossRef]
- Bhattamisra, S.K.; Shin, L.Y.; Saad, H.I.B.M.; Rao, V.; Candasamy, M.; Pandey, M.; Choudhury, H. Interlink Between Insulin Resistance and Neurodegeneration with an Update on Current Therapeutic Approaches. CNS Neurol. Disord. Drug Targets 2020, 19, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Cetinkalp, S.; Simsir, I.Y.; Ertek, S. Insulin Resistance in Brain and Possible Therapeutic Approaches. Curr. Vasc. Pharmacol. 2014, 12, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Lin, M.Y.; Hwang, S.J.; Liu, C.K.; Lee, H.L.; Wu, M.T. Factors associated with type 2 diabetes in patients with vascular dementia: A population-based cross-sectional study. BMC Endocr. Disord. 2018, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, F.; Lauretta, M.P.; Tewari, A.; Haque, M.; Hara, N.; Uchino, H.; Rosa, G. Insulin and the Brain: A Sweet Relationship With Intensive Care. J. Intensive Care Med. 2017, 32, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Roberson, M.R.; Harrell, L.E. Cholinergic activity and amyloid precursor protein metabolism. Brain Res. Rev. 1997, 25, 50–69. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef]
- Abbott, M.A.; Wells, D.G.; Fallon, J.R. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci. 1999, 19, 7300–7308. [Google Scholar] [CrossRef]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Mullins, R.J.; Mustapic, M.; Goetzl, E.J.; Kapogiannis, D. Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer’s disease. Hum. Brain Mapp. 2017, 38, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Rai, S.; Tota, S.K.; Kumar, A.; Ahmad, A.S. Streptozotocin Intracerebroventricular-Induced Neurotoxicity and Brain Insulin Resistance: A Therapeutic Intervention for Treatment of Sporadic Alzheimer’s Disease (sAD)-Like Pathology. Mol. Neurobiol. 2016, 53, 4548–4562. [Google Scholar] [CrossRef]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving Influence of Insulin on Cognitive Functions in Humans. Neuroendocrinology 2001, 74, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Rajasekar, N.; Nath, C.; Hanif, K.; Shukla, R. Intranasal insulin improves cerebral blood flow, Nrf-2 expression and BDNF in STZ (ICV)-induced memory impaired rats. Life Sci. 2017, 173, 1–10. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Sanchez-Sotano, D.; Doblas-Marquez, A.; Infante-Garcia, C.; Lubian-Lopez, S.; Garcia-Alloza, M. Intranasal insulin reverts central pathology and cognitive impairment in diabetic mother offspring. Mol. Neurodegener. 2017, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [PubMed]
- Semprini, R.; Koch, G.; Belli, L.; Lorenzo, F.D.; Ragonese, M.; Manenti, G.; Sorice, G.P.; Martorana, A. Insulin and the Future Treatment of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2016, 15, 660–664. [Google Scholar] [CrossRef]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Miranda, H.V. Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.M.; Lim, Y.-A. Common features between diabetes mellitus and Alzheimer’s disease. Cell. Mol. Life Sci. 2009, 66, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowski, K.; Brokos, B.; Leszek, J.; Tarasov, V.; Ashraf, G.; Aliev, G. Insulin Resistance in Alzheimer Disease: p53 and MicroRNAs as Important Players. Curr. Top Med. Chem. 2017, 17, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Eckman, E.A.; Eckman, C.B. Abeta-degrading enzymes: Modulators of Alzheimer’s disease pathogenesis and targets for therapeutic intervention. Biochem. Soc. Trans. 2005, 33 Pt 5, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, H.; Fang, G.G.; Walsh, K.; Mwamburi, M.; Wolozin, B.; Abdul-Hay, S.O.; Ikezu, T.; Leissring, M.A.; Qiu, W.Q. Characterization of insulin degrading enzyme and other amyloid-beta degrading proteases in human serum: A role in Alzheimer’s disease? J. Alzheimers Dis. 2012, 29, 329–340. [Google Scholar] [CrossRef]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Stoeckel, L.E. Brain insulin resistance as a contributing factor to dementia and psychiatric disease. Exp. Neurol. 2020, 326, 113205. [Google Scholar] [CrossRef]
- Harvey, N.C.; Holroyd, C.; Ntani, G.; Javaid, K.; Cooper, P.; Moon, R.; Cole, Z.; Tinati, T.; Godfrey, K.; Dennison, E.; et al. Vitamin D supplementation in pregnancy: A systematic review. Health Technol. Assess. 2014, 18, 1–190. [Google Scholar] [CrossRef]
- Moon, J.H.; Jang, H.C. Gestational Diabetes Mellitus: Diagnostic Approaches and Maternal-Offspring Complications. Diabetes Metab. J. 2022, 46, 3–14. [Google Scholar] [CrossRef]
- Abbasi, Z.; Seno, M.M.G.; Fereidoni, M. A Neonatal Mild Defect in Brain Insulin Signaling Predisposes a Subclinical Model of Sporadic Alzheimer’s to Develop the Disease. J. Mol. Neurosci. 2021, 71, 1473–1484. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.G.; Desai, M.; Khorram, O.; McKnight, R.A.; Lane, R.H.; Torday, J. Gestational programming of offspring obesity: A potential contributor to Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 213–217. [Google Scholar] [CrossRef]
- Cordner, Z.A.; Khambadkone, S.G.; Boersma, G.J.; Song, L.; Summers, T.N.; Moran, T.H.; Tamashiro, K.L. Maternal high-fat diet results in cognitive impairment and hippocampal gene expression changes in rat offspring. Exp. Neurol. 2019, 318, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Sah, S. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef] [PubMed]
- Hierro-Bujalance, C.; del Marco, A.; Ramos-Rodríguez, J.J.; Infante-Garcia, C.; Gomez-Santos, S.B.; Herrera, M.; Garcia-Alloza, M. Cell proliferation and neurogenesis alterations in Alzheimer’s disease and diabetes mellitus mixed murine models. J. Neurochem. 2020, 154, 673–692. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Chiu, S.-L.; Chen, C.-M.; Cline, H.T. Insulin Receptor Signaling Regulates Synapse Number, Dendritic Plasticity, and Circuit Function In Vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef]
- Morrison, C.D. Leptin signaling in brain: A link between nutrition and cognition? Biochim. Biophys. Acta 2009, 1792, 401–408. [Google Scholar] [CrossRef]
- Ferrario, C.R.; Reagan, L. Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology 2018, 136 Pt B, 182–191. [Google Scholar] [CrossRef]
- El Khoury, N.B.; Gratuze, M.; Papon, M.-A.; Bretteville, A.; Planel, E. Insulin dysfunction and Tau pathology. Front. Cell. Neurosci. 2014, 8, 22. [Google Scholar] [CrossRef]
- Mittal, K.; Katare, D. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. 2016, 10 (Suppl. S1), S144–S149. [Google Scholar] [CrossRef] [PubMed]
- Park, C. Cognitive effects of insulin in the central nervous system. Neurosci. Biobehav. Rev. 2001, 25, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Liang, Y.; Yue, L.; Liu, P.; Xu, Y.; Zhu, C. The identities of insulin signaling pathway are affected by overexpression of Tau and its phosphorylation form. Front. Aging Neurosci. 2022, 14, 1057281. [Google Scholar] [CrossRef] [PubMed]
- Schechter, R.; Beju, D.; Miller, K.E. The effect of insulin deficiency on tau and neurofilament in the insulin knockout mouse. Biochem. Biophys. Res. Commun. 2005, 334, 979–986. [Google Scholar] [CrossRef]
- Bi, A.; An, W.; Wang, C.; Hua, Y.; Fang, F.; Dong, X.; Chen, R.; Zhang, Z.; Luo, L. SCR-1693 inhibits tau phosphorylation and improves insulin resistance associated cognitive deficits. Neuropharmacology 2020, 168, 108027. [Google Scholar] [CrossRef]
- Bevan-Jones, W.R.; Surendranathan, A.; Passamonti, L.; Rodríguez, P.V.; Arnold, R.; Mak, E.; Su, L.; Coles, J.P.; Fryer, T.D.; Hong, Y.T.; et al. Neuroimaging of Inflammation in Memory and Related Other Disorders (NIMROD) study protocol: A deep phenotyping cohort study of the role of brain inflammation in dementia, depression and other neurological illnesses. BMJ Open 2017, 7, e013187. [Google Scholar] [CrossRef]
- Mitrou, P.; Boutati, E.; Lambadiari, V.; Tsegka, A.; Raptis, A.E.; Tountas, N.; Economopoulos, T.; Raptis, S.A.; Dimitriadis, G. Insulin resistance in hyperthyroidism: The role of IL6 and TNF alpha. Eur. J. Endocrinol. 2010, 162, 121–126. [Google Scholar] [CrossRef]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Wacławczyk, D.; Silberring, J.; Grasso, G. The insulin-degrading enzyme as a link between insulin and neuropeptides metabolism. J. Enzym. Inhib. Med. Chem. 2021, 36, 183–187. [Google Scholar] [CrossRef]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004, 18, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jaspan, J.B.; Huang, W.; Kastin, A.J. Transport of Insulin Across the Blood-Brain Barrier: Saturability at Euglycemic Doses of Insulin. Peptides 1997, 18, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef]
- Konarkowska, B.; Aitken, J.F.; Kistler, J.; Zhang, S.; Cooper, G.J. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006, 273, 3614–3624. [Google Scholar] [CrossRef]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef]
- Craft, S. The role of metabolic disorders in Alzheimer disease and vascular dementia: Two roads converged. Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, Defective Insulin Signaling, and Mitochondrial Dysfunction as Common Molecular Denominators Connecting Type 2 Diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef]
- Garcia-Alloza, M. Streptozotocin as a tool to induce central pathology and cognitive impairment in rodents. In Streptozotocin: Uses, Mechanism of Action and SideEffects; Gauthier, E.L., Ed.; New Developments in Medical Research; Nova Biomedical: New York, NY, USA, 2014. [Google Scholar]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Insulin and Insulin Resistance in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Chopp, M.; Chen, J. Blood–Brain Barrier Disruption, Vascular Impairment, and Ischemia/Reperfusion Damage in Diabetic Stroke. J. Am. Heart Assoc. 2017, 6, e005819. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Lauwers, E.; Verstreken, P. Presynaptic protein homeostasis and neuronal function. Curr. Opin. Genet. Dev. 2017, 44, 38–46. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Sharma, S.; Brown, C.E. Microvascular basis of cognitive impairment in type 1 diabetes. Pharmacol. Ther. 2022, 229, 107929. [Google Scholar] [CrossRef]
- Serlin, Y.; Levy, J.; Shalev, H. Vascular Pathology and Blood-Brain Barrier Disruption in Cognitive and Psychiatric Complications of Type 2 Diabetes Mellitus. Cardiovasc. Psychiatry Neurol. 2011, 2011, 609202. [Google Scholar] [CrossRef]
- Maesako, M.; Uemura, K.; Kubota, M.; Kuzuya, A.; Sasaki, K.; Asada, M.; Watanabe, K.; Hayashida, N.; Ihara, M.; Ito, H.; et al. Environmental enrichment ameliorated high-fat diet-induced Abeta deposition and memory deficit in APP transgenic mice. Neurobiol. Aging 2012, 33, 1011.e11–1011.e23. [Google Scholar] [CrossRef]
- Maesako, M.; Uemura, K.; Kubota, M.; Kuzuya, A.; Sasaki, K.; Hayashida, N.; Asada-Utsugi, M.; Watanabe, K.; Uemura, M.; Kihara, T.; et al. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J. Biol. Chem. 2012, 287, 23024–23033. [Google Scholar] [CrossRef] [PubMed]
- Shie, F.S.; Jin, L.W.; Cook, D.G.; Leverenz, J.B.; LeBoeuf, R.C. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport 2002, 13, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Synaptic Plasticity, Dementia and Alzheimer Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 220–233. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef]
- Molina-Fernández, R.; Picón-Pagès, P.; Barranco-Almohalla, A.; Crepin, G.; Herrera-Fernández, V.; García-Elías, A.; Fanlo-Ucar, H.; Fernàndez-Busquets, X.; García-Ojalvo, J.; Oliva, B.; et al. Differential regulation of insulin signalling by monomeric and oligomeric amyloid beta-peptide. Brain Commun. 2022, 4, fcac243. [Google Scholar] [CrossRef]
- Parodi-Rullán, R.M.; Javadov, S.; Fossati, S. Dissecting the Crosstalk between Endothelial Mitochondrial Damage, Vascular Inflammation, and Neurodegeneration in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Cells 2021, 10, 2903. [Google Scholar] [CrossRef]
- Roriz-Filho, J.S.; Sa-Roriz, T.M.; Rosset, I.; Camozzato, A.L.; Santos, A.C.; Chaves, M.L.; Moriguti, J.C.; Roriz-Cruz, M. (Pre)diabetes, brain aging, and cognition. Biochim. Biophys. Acta 2009, 1792, 432–443. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef]
- Gallagher, J.J.; Minogue, A.M.; Lynch, M.A. Impaired performance of female APP/PS1 mice in the Morris water maze is coupled with increased Abeta accumulation and microglial activation. Neurodegener. Dis. 2013, 11, 33–41. [Google Scholar] [CrossRef]
- Stargardt, A.; Gillis, J.; Kamphuis, W.; Wiemhoefer, A.; Kooijman, L.; Raspe, M.; Benckhuijsen, W.; Drijfhout, J.W.; Hol, E.M.; Reits, E. Reduced amyloid-beta degradation in early Alzheimer’s disease but not in the APPswePS1dE9 and 3xTg-AD mouse models. Aging Cell 2013, 12, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Gregory, J.; Kuchibhotla, K.V.; Fine, S.; Wei, Y.; Ayata, C.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Cerebrovascular lesions induce transient beta-amyloid deposition. Brain 2011, 134 Pt 12, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D. Modulation of microglial activation state following passive immunization in amyloid depositing transgenic mice. Neurochem. Int. 2006, 49, 190–194. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Ferrara, B.J.; Dodwell, S.A.; Hickey, G.A.; Hyman, B.T.; Bacskai, B.J. A limited role for microglia in antibody mediated plaque clearance in APP mice. Neurobiol. Dis. 2007, 28, 286–292. [Google Scholar] [CrossRef]
- Dey, A.; Hao, S.; Erion, J.R.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid sensitization of microglia in a genetic mouse model of obesity and diabetes. J. Neuroimmunol. 2014, 269, 20–27. [Google Scholar] [CrossRef][Green Version]
- Onyango, A.N. Cellular Stresses and Stress Responses in the Pathogenesis of Insulin Resistance. Oxidative Med. Cell. Longev. 2018, 2018, 4321714. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Mishra, A.; Kim, H.J.; Shin, A.H.; Thayer, S.A. Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J. Neuroimmune. Pharmacol. 2012, 7, 571–578. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Backus, C.; Oh, S.; Hayes, J.M.; Feldman, E.L. Increased Tau Phosphorylation and Cleavage in Mouse Models of Type 1 and Type 2 Diabetes. Endocrinology 2009, 150, 5294–5301. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Deng, J.; Sheng, W.; Zuo, Z. Metformin attenuates Alzheimer’s disease-like neuropathology in obese, leptin-resistant mice. Pharmacol. Biochem. Behav. 2012, 101, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Brundel, M.; Heuvel, M.v.D.; de Bresser, J.; Kappelle, L.J.; Biessels, G.J. Cerebral cortical thickness in patients with type 2 diabetes. J. Neurol. Sci. 2010, 299, 126–130. [Google Scholar] [CrossRef]
- Heijer, T.D.; Vermeer, S.E.; van Dijk, E.J.; Prins, N.D.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003, 46, 1604–1610. [Google Scholar] [CrossRef]
- Ferris, J.K.; Peters, S.; Brown, K.E.; Tourigny, K.; Boyd, L.A. Type-2 diabetes mellitus reduces cortical thickness and decreases oxidative metabolism in sensorimotor regions after stroke. J. Cereb. Blood Flow Metab. 2017, 38, 823–834. [Google Scholar] [CrossRef]
- Geijselaers, S.L.; Sep, S.J.; Schram, M.T.; van Boxtel, M.P.; Henry, R.M.; Verhey, F.R.; Kroon, A.A.; Schaper, N.C.; Dagnelie, P.C.; van der Kallen, C.J.; et al. Insulin resistance and cognitive performance in type 2 diabetes—The Maastricht study. J. Diabetes Complicat. 2017, 31, 824–830. [Google Scholar] [CrossRef]
- Hassing, L.B.; Grant, M.D.; Hofer, S.M.; Pedersen, N.L.; Nilsson, S.E.; Berg, S.; Mcclearn, G.; Johansson, B. Type 2 diabetes mellitus contributes to cognitive decline in old age: A longitudinal population-based study. J. Int. Neuropsychol. Soc. 2004, 10, 599–607. [Google Scholar] [CrossRef]
- Van Duinkerken, E.; Ijzerman, R.G.; Klein, M.; Moll, A.C.; Snoek, F.J.; Scheltens, P.; Pouwels, P.J.; Barkhof, F.; Diamant, M.; Tijms, B.M. Disrupted subject-specific gray matter network properties and cognitive dysfunction in type 1 diabetes patients with and without proliferative retinopathy. Hum. Brain Mapp. 2016, 37, 1194–1208. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; Mohamed-Mohamed, H.; Rivas-Dominguez, A.; Garcia-Morales, V.; Garcia-Lara, R.A.; Suleiman-Martos, S.; Bermudez-Pulgarin, B.; Ramos-Rodriguez, J.J. Poor Cognitive Agility Conservation in Obese Aging People. Biomedicines 2023, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Manschot, S.M.; Biessels, G.J.; de Valk, H.; Algra, A.; Rutten, G.E.H.M.; van der Grond, J.; Kappelle, L.J.; on behalf of the Utrecht Diabetic Encephalopathy Study Group. Metabolic and vascular determinants of impaired cognitive performance and abnormalities on brain magnetic resonance imaging in patients with type 2 diabetes. Diabetologia 2007, 50, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Raji, C.A.; Ho, A.J.; Parikshak, N.N.; Becker, J.T.; Lopez, O.L.; Kuller, L.H.; Hua, X.; Leow, A.D.; Toga, A.W.; Thompson, P.M. Brain structure and obesity. Hum. Brain Mapp. 2010, 31, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, W.H.; Braga, C.F.; Lós, D.B.; Araújo, S.M.R.; França, M.R.; Duarte-Silva, E.; Rodrigues, G.B.; Rocha, S.W.S.; Peixoto, C.A. Metformin prevents p-tau and amyloid plaque deposition and memory impairment in diabetic mice. Exp. Brain Res. 2021, 239, 2821–2839. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Roesler, E.; Niehoff, M.L.; Roby, D.A.; McKee, A.; Morley, J.E. Metformin Improves Learning and Memory in the SAMP8 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef]
- Zheng, J.; Xu, M.; Walker, V.; Yuan, J.; Korologou-Linden, R.; Robinson, J.; Huang, P.; Burgess, S.; Yeung, S.L.A.; Luo, S.; et al. Evaluating the efficacy and mechanism of metformin targets on reducing Alzheimer’s disease risk in the general population: A Mendelian randomisation study. Diabetologia 2022, 65, 1664–1675. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Saad, H.M.; Batiha, G.E.-S. Long-term use of metformin and Alzheimer’s disease: Beneficial or detrimental effects. Inflammopharmacology 2023, 31, 1107–1115. [Google Scholar] [CrossRef]
- AboEl-Azm, Y.H.; El-Samahy, M.; Hendi, N.I.; Arar, A.; Yasen, N.S.; Ramadan, S.; Zedan, E.M.; Al-Dardery, N.M.; Khaity, A. Safety and efficacy of intranasal insulin in patients with Alzheimer’s disease: A systematic review and meta-analysis. J. Clin. Transl. Res. 2023, 9, 222–235. [Google Scholar]
- Kellar, D.; Register, T.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A randomized trial. Sci. Rep. 2022, 12, 1346. [Google Scholar] [CrossRef]
- Novak, V.; Milberg, W.; Hao, Y.; Munshi, M.; Novak, P.; Galica, A.; Manor, B.; Roberson, P.; Craft, S.; Abduljalil, A. Enhancement of Vasoreactivity and Cognition by Intranasal Insulin in Type 2 Diabetes. Diabetes Care 2014, 37, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2021, 8, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).