Abstract
The pathogenesis of inflammatory bowel disease (IBD) implicates several interconnecting factors. Immunity and external factors interact, and most aspects are still under investigation. Autophagy and apoptosis are two critical pathways that decide the fate of the individual cells of the intestinal mucosa. Experimental and clinical data indicate that the two are closely interconnected and usually mutually exclusive. However, despite the abundant information on their role, very limited translation into therapeutic application has been seen during recent years. In this review, research on these two pathways is presented. After a general overview of autophagy and apoptosis, their association with IBD, including the important mitophagy and ferroptosis, is discussed. The influence of autophagy- and apoptosis-related genes is also discussed. Finally, the interplay of autophagy and apoptosis in IBD is presented and the implications for treatment applications are examined. It is shown that dysregulated autophagy leads to increased apoptosis of enterocytes and impairs the tight junction proteins of the protective intestinal barrier. Dysregulated autophagy also induces the downregulation of lysozyme and the other antimicrobial proteins’ production. Mucus production by the goblet cells is also reduced due to defective autophagy and increased apoptosis.
1. Introduction
Inflammatory bowel disease (IBD) is an intestinal inflammatory disease that includes Crohn’s disease (CD) and ulcerative colitis (UC). CD is a chronic granulomatous lesion that can affect the entire gastrointestinal tract but usually involves the colon and small intestine. UC is a chronic, non-specific inflammation that affects the colonic mucosa and submucosa [1,2]. The small intestinal epithelium is comprised of villi and the crypts of Lieberkühn, while the colonic epithelium consists only of crypts. Inflammation of the intestinal mucosa in association with villus atrophy and a reduction in crypts are histological characteristics of IBD [3].
Globally, more than 2.5 million people of European origin have the disease [4]. However, increasing numbers of IBD cases are observed in Asia and Africa, where it was practically absent at the beginning of this century [5]. The pathogenesis of IBD is a multi-factorial process implicating immunological, environmental, genetic and microbial factors [6]. The intestinal mucosal barrier is the first defense against harmful invaders from the bowel lumen. This barrier consists of three layers, namely the surface mucus, the epithelial cell layer, and the cells of the immune system of the submucosa [7]. The surface mucus inhibits the direct contact of the intestinal bacteria with the epithelial cells [8]. The intestinal epithelial cells consist of enterocytes, goblet cells, Paneth cells, tuft cells and endocrine cells. They are the site of absorption and production of mucus and several important proteins [9]. The immune cells of the submucosa are neutrophils, lymphocytes and macrophages fighting the pathogens that can penetrate into the submucosa. They are involved in the handling of antigens and the secretion of multiple cytokines [10,11].
Recently, the role of two important cellular pathways has been studied in connection with the pathogenesis of IBD. Autophagy and apoptosis are two mechanisms that actively participate in the integrity of the intestinal mucosa [12].
Autophagy is a key process for cell survival and tissue homeostasis, turning damaged organelles and cellular waste into useful reusable metabolites [13,14]. Starvation and infection may initiate autophagy and increase cell viability. Autophagy has been identified in both the small intestinal and colonic mucosa, and it may be implicated in the pathophysiology of IBD [13,14]. On the contrary, apoptosis eliminates damaged cells [15]. The balance between autophagy and apoptosis is one of the factors that determine the fate of intestinal cells, although other forms of cellular death, such as necroptosis or pyroptosis, can also lead to cellular death in IBD in response to inflammatory signals [13].
The complex nature of autophagy and its interplay with apoptosis make it difficult to delineate their exact role in IBD, although current evidence has indicated the participation of autophagy in mucosal homeostasis [16]. Thus, autophagy is involved in the secretion of pro-inflammatory cytokines and activation of inflammasomes, while reactive oxygen species (ROS) are increased in autophagy-deficient macrophages, impairing submucosal immunity [17]. Autophagy is also involved in several functions of intestinal epithelial cells (IECs) and goblet cells, particularly during infection or ER stress [18]. An example is the Salmonella infection, where a deficiency of autophagy in IECs leads to bacterial dissemination and increased inflammation [18]. The intestinal epithelium may then become vulnerable to the infiltrating microbes and their products, and its fate will depend on the interaction of autophagy with the various forms of cellular death [19]. Autophagy is also important for the function of Paneth cells. Paneth cells secrete antimicrobial peptides (AMPs) and other peptides, including lysozymes, alpha-defensins and phospholipase A1, when invaders stimulate the crypt lumen [20]. Paneth cells are critical in the maintenance of a normal microbiota. A form of autophagy to remove pathogens named xenophagy is operative in Paneth cells, allowing maintenance of the normal metabolic functions of the host [21]. Dysfunction of Paneth cells leads to reduced secretion of AMPs and dysbiosis, which is a dysregulation of the composition of intestinal microbiota. Dysbiosis may be implicated in the pathogenesis of IBD [22].
The purpose of this review, therefore, is to present the participation of autophagy and apoptosis in the pathophysiology of IBD. Recently discovered forms of programmed cell death such as ferroptosis will be discussed, as ferroptosis is connected with autophagy and apoptosis. The implications for the treatment of IBD will be also discussed.
2. An Overview of Autophagy and Apoptosis
2.1. Autophagy
Autophagy is a crucial degradative process that is vital for cellular economy and homeostasis. Proteins, lipids, damaged organelles and pathogens are degraded and the products re-used for the synthesis of cellular constituents [23]. Autophagy research led to two Nobel Prizes, one to Christian De Duve, who described the significance of lysosomes in biology, and the second to Yoshinori Ohsumi, who clarified the mechanisms of autophagy [24].
In reality, the process of autophagy is a series of phosphorylations and de-phosphorylations. Three kinases are the main regulators of autophagy: the mammalian target of rapamycin (mTOR), Unc-51-like autophagy activating kinase (ULK1) and the AMP-dependent protein kinase (AMPK) [25]. Decreased nutrients and ATP levels and increased levels of reactive oxygen species (ROS) and damaged DNA are the initiators of AMPK and mTOR [26]. Phosphorylation of ULK1 by mTOR reduces its activity and decreases autophagy, while phosphorylation by AMPK at a different site activates ULK1 and autophagy. AMPK inhibits mTOR, leading to the highest activation of ULK1 [27].
Activated ULK1 phosphorylates Beclin1 and induces autophagy [28,29]. At the same time, JNK-1 phosphorylates Bcl-2, leading to its dissociation from Beclin1. Free Bcl-2 inhibits apoptosis through binding to BAX and BAK proteins. Free Beclin1 subsequently binds to Vps34–Vps15 to promote autophagy through the formation of the class III PI3K complex consisting of Vps34–Vps15–Beclin1 [30,31].
The next step is the autophagosome formation, which is the direct result of ULK1 phosphorylation. Here, the gatekeeper is the phosphorylation of autophagy-related protein 13 (Atg13), leading to a complex formation with Atg5–Atg12, Atg16L1 and LC3-II. Additional proteins that are often studied in autophagy research and are associated with autophagosomes include LC3 (Atg8) and protein sequestosome 1 (p62 SQSTM1) [32,33]. Finally, autophagosomes fuse with lysosomes, forming autolysosomes during the so-called autophagic flux. When autophagosomes are produced faster than autophagic flux, or when the flux is repressed, the levels of LC3 and p62 will increase [34]. Constituents that are destined for lysosomal degradation are either labeled by ubiquitin or attached to cargo receptors such as SQSTM1, and calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2). The lysosomal contents recirculate and useful elements are used again [23].
Another important regulator of autophagy is transcription factor EB (TFEB), which is a controller of lysosomal biogenesis genes [35] in association with TFE3 (transcription factor binding to IGHM enhancer 3) [36]. Reduced expression of TFEB leads to the exacerbation of inflammation [37]. mTOR activation decreases TFEB activity, and the autophagic machinery is suppressed [38].
2.2. Mitophagy
Mitophagy is a specialized form of protective autophagy that selectively degrades damaged mitochondria irrespective of the cause of the damage [39,40]. Two signal pathways initiate mitophagy, either through the PINK1 (PTEN-induced putative kinase 1)–PARKIN (parkin RBR E3 ubiquitin protein ligase) system or through a PINK1/PARKIN-independent pathway [41,42]. Normal mitochondria accumulate PINK1 in the inner mitochondrial membrane (IMM) assisted by the TOMM (translocase of the outer mitochondrial membrane) and TIMM23 (translocase of inner mitochondrial membrane 23) proteins. PINK1 is further cleaved by PARL (presenilin-associated rhomboid like). Damaged mitochondria, on the other hand, accumulate PINK1 in the outer mitochondrial membrane (OMM) instead of the IMM [43,44], leading to the recruitment of ubiquitin and PARKIN from the cytoplasm to the OMM [45]. PARKIN links phosphoubiquitin molecules to several proteins of the mitochondrial surface, such as MFN1 (mitofusin 1) and MFN2 [42,46,47]. Cargo receptor proteins such as SQSTM1/p62 (sequestosome 1), OPTN (optineurin) and CALCOCO2, among others, bind to these OMM proteins to start autophagosome formation [47,48]. Mitophagy may be upregulated by phosphorylation of OPTN via the activation of TBK1 (TANK-binding kinase 1) [49,50]. Details of the mitophagy mechanisms have been recently published [51,52].
2.3. Apoptosis
Apoptosis is a form of programmed cell death. During apoptosis, cellular elements are not spilled into the surrounding environment and do not induce inflammation. Apoptosis, in fact, is a downstream activation of a series of caspases. In the caspase chain, each inactive enzyme pro-form is cleaved and activated by the previous active caspase [53]. There are two apoptotic pathways, namely the intrinsic and the extrinsic pathways [54]. The intrinsic pathway starts with the activation of the pro-apoptotic BH3 domain proteins such as BAX and BAK, which drill pores in the OMM, leading to the reduction of ATP and the liberation of cytochrome c and apoptosis-inducing factor (AIF). Cytosolic AIF is translocated into the nucleus and causes DNA fragmentation. Cytochrome c with Apaf-1 (apoptotic protease-activating factor-1) induces the activation of pro-caspase 9, leading to the activation of pro-caspases 3 and 7, which execute the cell [55]. The extrinsic apoptosis pathway is induced by activation of death receptors such as FAS, DR4/DR5 and TNFR1 by their ligands in the cell surface (FAS ligand, TNF-related apoptosis-inducing ligand (TRAIL) and TNFα, respectively). After death receptor activation, the protein FADD associates with the receptors and causes the activation of pro-caspase 8. The death receptor—FADD—pro-caspase 8 complex is called the death-inducing signaling complex (DISC). The anti-apoptotic protein FLICE-like inhibitor protein (FLIP) inhibits the activation of caspase 8 [56]. Caspase 8 may lead to cell death in one of two ways The first is through the activation of the pro-apoptotic BH3 domain protein BID, which inhibits anti-apoptotic BH3 domain proteins such as BCL-2, BCL-XL and MCL1, allowing the lethal BAX and BAK proteins to aggregate in the OMM and form pores [57] in a similar way to the activation of the intrinsic pathway. The second way for caspase 8 is to directly activate pro-caspase 3, omitting the mitochondrial step [57].
One of the most important inducers of apoptosis through the intrinsic pathway is endoplasmic reticulum (ER) stress. Under normal conditions, the three main ER stress transducers (activating transcription factor 6 (ATF6), the inositol-requiring enzyme 1 (IRE1) and the PRKR-like endoplasmic reticulum kinase (PERK) are inactive and attached to BiP (immunoglobulin heavy-chain binding protein, also referred to as glucose-regulated protein GRP 78). When unfolded proteins accumulate into the ER lumen, BiP dissociates from the ER stress transducers. IRE1 and PERK are involved in apoptosis. IRE1 activates JNK and stimulates the phosphorylation of Jun. Active PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), which results in the successive downregulation of protein translation. eIF2α also induces preferential translation of transcription factor ATF4. Thereby, ATF4 induces expression of C/EBP-homologous protein (CHOP) and ATF3. These proteins trigger an apoptotic program [58].
The most important inducer of apoptosis through the extrinsic pathway is tumor necrosis factor α (TNFα). Under normal circumstances, complex I is formed when RIPK1 (receptor-interacting protein kinase 1) leads to the activation of the pro-inflammatory and protective pathway NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells). In pathologic conditions, either complex IIa or complex IIb can be formed, leading to the activation of caspase-8, which in complex IIa cleaves receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and downstream pro-caspases 3 and 7 to initiate apoptosis. Complex IIb, on the other hand, depends on the active intact RIPK1 kinase to activate caspase-8 and apoptosis [59].
2.4. Ferroptosis
Ferroptosis is a recently described regulated cell death induced by iron accumulation, lipid peroxidation, and the production of ROS, depending on the activity of NADPH oxidases [60].
Cells defend themselves from the lethal lipid peroxidation and ROS formation through different anti-oxidative systems. The most important is the Xc-antiporter system consisting of the SLC7A11 and SLC3A2 subunits, which imports cysteine turning into cysteine and exports glutamine turning into glutamate. γ-glutamylcysteine is formed, followed by the production of glutathione (GSH) and glutathione peroxidase (GPX4) to neutralize the ROS and lipid peroxides produced from either polyunsaturated fatty acids (PUFAs) or phospholipids of the cell membrane through the action of lipoxygenases (LOXs). GSH formation requires nicotinamide adenine dinucleotide phosphate (NADPH). Long-chain-fatty-acid-CoA ligase 4 (ACSL4) facilitates the formation of phosphatidylethanolamine (PE)-PUFAs, finally turning into PE-PUFA hydroperoxides (OOH). Downregulation of GPX4 production favors lipid peroxidation and damage to lipid membranes and mitochondria [61,62]. In addition to the enzymatic oxidation of PUFAs by LOXs, a nonenzymatic reaction, such as a Fenton-type reaction, can also oxidize PUFAs [63,64]. P53 enhances the activity of LOX and promotes lipid peroxidation [62].
However, p53 has a dual role in the regulation of ferroptosis. p53 may induce ferroptosis through inhibition of SLC7A11 or upregulation of arachidonate lipoxygenase (ALOX). On the other hand, p53 may inhibit ferroptosis through either inhibiting dipep-tidyl-peptidase-4, (DPP4) activity or activating the cyclin-dependent kinase inhibitor 1A (CDKN1A)/p21 pathway [65].
Ferrous iron (Fe2+), through the Fenton reaction, promotes the conversion of PE-PUFA into PE-PUFA-OOH, leading to ferroptosis [66].
Besides the Xc system, there are two more antioxidant systems counteracting ferroptosis, namely the ferroptosis suppressor protein 1–coenzyme Q10-NADPH (FSP1–CoQ10-NADPH) axis and the dihydroorotate dehydrogenase (DHODH)–CoQ-NADPH axis acting in parallel to the GPX4-GSH axis [67]. The three main antioxidant systems have discrete cellular localization: the GSH/GPX4 pathway is cytoplasmic, the FSP1–CoQ-NADPH system is located in the cell membrane, and the DHODH–CoQ-NADPH pathway is related to mitochondria. Mitochondria have a critical role in lipid peroxidation and ferroptosis [68,69]. Deprivation of glucose impairs the tricarboxylic acid cycle (TCA) cycle in mitochondria and inhibits ferroptosis via AMPK kinase signaling [70]. On the other hand, free iron activates the mitochondrial membrane protein mitoferrin 2, which in turn increases iron influx into the mitochondria, promoting ROS production and ferroptosis.
Specific inhibitors of ferroptosis, such as deferoxamine (DFO), ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), and potent inducers, such as erastin, RAS-selective lethal 3 (RSL3), FIN56, FINO2, sulfasalazine and sorafenib, have been described [71,72]. Erastin and FIN 56 deplete GSH and allow for increased levels of lipid peroxides and ferroptosis, while RSL3 and FINO2 directly inhibit GPX4. The ferroptosis inhibitors also antagonize ferroptosis through various mechanisms. Thus, iron chelators reduce the labile free iron, downregulating lipid peroxidation. Fer-1 and Lip-1 protect lipids from autoxidation. Lipoxygenase (LOX) inhibitors, such as zileuton, baicalein and NDGA, could counteract LOXs-induced lipid peroxidation. Thiazolidinedione, an ACSL4 (acyl-CoA synthetase long-chain family member 4) inhibitor, represses the esterification of PUFAs, interfering in the step before the production of PE-PUFA-OOHs [67].
2.5. Interaction of Autophagy and Apoptosis
The interplay between apoptosis and autophagy is complex since autophagy is usually an adaptation leading to cell survival and, in most instances, it represses apoptosis. Similar stimuli can initiate either apoptosis or autophagy. However, under certain circumstances, autophagy may turn into an alternative pathway to cellular death, the so-called autophagic cell death. Usually, autophagy and apoptosis move in a mutually exclusive manner. Autophagy and apoptosis may also move in the same direction [73].
Earlier studies demonstrated that TNFα-induced apoptosis was reduced by autophagy [74]. Similar findings were reported in immortal epithelial cells, where autophagy de-graded the death receptor associated protein FADD [75]. The first observation of an inter-action of autophagy and apoptosis was the effect of the anti-apoptotic protein Bcl-2 on the autophagic protein Beclin1, as mentioned before. Beclin1 interacts with Bcl-2 family proteins and dissociation of the complex frees Beclin1 to further promote autophagosome formation [76].
It should be noted that two distinct cellular pools of Bcl-2 exist in each cell. The ER pool is bound to Beclin1, where it inhibits autophagy, and the mitochondrial pool, which is bound to BAX and BAK, where it inhibits apoptosis [77,78]. A BH3-only protein, either BIK, BAD, or NOXA, can bind to Bcl-2, liberating Beclin1, a process augmented through the phosphorylation of Bcl-2 by c-Jun N-terminal kinase (JNK1) during nutrient deprivation [79,80,81]. Contrary to expectations that the binding of Beclin1 to Bcl-2 would induce apoptosis through neutralization of the anti-apoptotic function of Bcl-2, this is not the case and apoptosis is not induced [82].
The caspase 8 inhibitor FLIP also inhibits autophagy by interfering with Atg3 binding in LC3 lipidation [83]. Different regions within the FLIP protein control its anti-autophagic and anti-apoptotic activities [83], and different pools of FLIP may provide separate regulation of FLIP-mediated autophagy and apoptosis. The autophagy-associated Atg5 protein is also implicated in the modulation of autophagy and apoptosis. During apoptosis, Atg5 is cleaved by active caspase 3 and an amino-terminal molecule is produced [84]. This molecule induces apoptotic cell death, as it inhibits the anti-apoptotic Bcl-2 molecules, allowing BAX and BAK to initiate mitochondrial-induced apoptosis. At the same time, the cleaved Atg5 is not able to promote autophagy [84].
During protracted exposure to apoptotic stimuli, cleavage of Beclin1 by caspase 3 produces two molecules: an N-terminal and a C-terminal that are unable to induce autophagy any more. Moreover, the C-terminal molecule enters the mitochondria and in-creases the sensitivity of cells to apoptotic signals [85]. In contrast to the downregulation of autophagy caused by the cleavage of Atg5 or the similar cleavage of Beclin1, the cleavage of Atg4D by caspase 3 generates a molecule that increases autophagy. Moreover, autophagy inhibits apoptosis via either lysosomal degradation of active caspase-8 or via Beclin1 inhibition of the BH3-only protein BID that activates BAX and BAK [30].
Finally, autophagy and apoptosis may cause cell death when acting in concert. The exact mechanisms are still under investigation. One such mechanism may be the liberation of cathepsins from autolysosomes, which may kill the cell during autophagy and, at the same time, cleave BID, increasing apoptosis via activation of BAX and BAK [86,87]. Figure 1 shows the complicated interactions of autophagy and apoptosis in connection to ferroptosis.
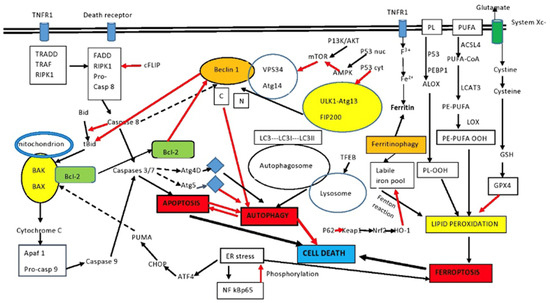
Figure 1.
Intersection between apoptosis and autophagy. Autophagy and apoptosis have similar signaling pathways and usually but not always exhibit mutual inhibition. Beclin1 and Bcl-2 are important elements in the interplay between the two. Sustained apoptotic stimuli lead to caspase cleavage of Beclin1 and the C-terminal fragment inhibits autophagy. Although apoptosis-associated cleavage of Beclin1 and Atg5 inhibits autophagy, the cleavage of Atg4D by caspase 3 generates a fragment that increases autophagy. Moreover, autophagy inhibits apoptosis by degrading active caspase 8 or preventing activation of Bid by Beclin1. The effect of P53 on autophagy depends on localization. Cytoplasmic P53 inhibits autophagy, while nuclear P53 activates AMPK and increases autophagy. PUFA and PL of the membrane are the sources of lipid peroxides in ferroptosis. p53 and PEBP1 enhance the activity of LOX to promote lipid peroxidation, resulting in ferroptosis. In addition, LOX promotes the conversion of PE-PUFA into PE-PUFA-OOH, leading to ferroptosis, while the free iron pool promotes lipid peroxidation and ferroptosis through the Fenton reaction. The p62/keap1/Nrf2/HO-1 system decreases ferroptosis, along with the main antioxidant system Xc-. Ferritinophagy is a major source of the labile iron pool. Certain pathways have been omitted for clarity. See the text for details. ACSL4: acyl-CoA synthetase long-chain family member 4; ALOX: arachidonate lipoxygenase; Apaf1: apoptosis signal-regulating kinase; ATF4: activating transcription factor 4; GSH: glutathione; BAK: Bcl-2 homologous antagonist killer; BAX: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma-2; Bid: BH3-interacting-domain death agonist 2; CHOP: CCAAT-enhancer-binding protein homologous protein; FADD: Fas-associating protein with death domain; GPX4: glutathione peroxidase 4; LOX: lipoxygenase; LPCAT3: lysophosphatidylcholine acyltransferase 3; mTOR: mammalian target of rapamycin; PEBP1: phosphatidylethanolamine-binding protein-1; PL: phospholipid; PLOOH: phospholipid hydroperoxides; PE: phosphatidylethanolamine; PUFAs: polyunsaturated fatty acids; PUFA-OOH: polyunsaturated fatty acid containing-phospholipid hydroperoxides; PUMA: p53 upregulated modulator of apoptosis; RIPK1: receptor interacting serine/threonine kinase 1; black arrows: activation; red arrows: inhibition; intermittent arrows: cleavage.
Most of these mechanisms and pathways are operative in IBD, as indicated by genetic, experimental and clinical studies [36,37,61,62].
3. Genetics and Autophagy in IBD
Most studies dissecting different genetic or cellular mechanisms of IBD pathophysiology were feasible due to the development of various animal models. The most widely used are those that are chemically induced, such as the trinitrobenzene sulfonic acid (TNBS) model and the oxazolone and dextran sulphate sodium (DSS) models. Additional models are the gene-deficient I-kappa-B kinase gamma (Iκκ-γ) and interleukin (IL)-10 models of spontaneous colitis and the immune cell-induced models such as the CD4+ T-cell transfer model [88]. Comparisons of the different models have shown that the DSS- or TNBS-induced models correspond to human IBD and are reproducible with characteristics indicative of acute inflammation [89]. In addition, use of knock-out models allowed for the identification of discrete pathways in the progression of IBD. Thus, the caspase 1 KO mice TNBS model revealed significant differences between the histopathologies of the wild-type and the KO mouse, indicating the importance of caspase 1 in the inflammasome activation and the pathogenesis of IBD [90]. Models of chronic colitis have also been created [91]. Although murine experiments have contributed immensely to IBD research, the translation to human applications should be performed with caution. Mice and humans differ in their immune responses and their genetic diversity. Moreover, therapeutic interventions are not always consistent between murine models and human trials. Therefore, the limitations of each model should be carefully considered before conclusions can be extrapolated in relation to human IBD [88,92].
The first indications that autophagy is important in the pathogenesis of IBD came when genome-wide association studies (GWAS) identified the autophagy gene ATG16L and the immunity-related GTPase M (IRGM) gene as CD susceptibility genes [93,94,95,96,97,98]. Later, polymorphisms in other autophagy genes were found to be IBD-associated [99,100]. More than 200 loci have been associated with IBD so far [101,102].
Recently, the significance of other autophagy-related genes, such as optineurin (OPTN), TFEB and leucine-rich repeat kinase (LRRK), has been studied in vivo and found to be associated with enhanced susceptibility to bowel inflammation, indicating their involvement in colonic immunity [103,104,105].
Regulation of the intestinal barrier is another function where autophagy is critically involved. A deletion of an autophagy gene may result in the impairment of proteins of the intestinal junctions [106,107,108]. Initiation of autophagy downregulates epithelial barrier permeability because claudin-2 is degraded in lysosomes [106]. An additional factor is the fact that several autophagy proteins have multiple functions not dependent on autophagy. Accordingly, Beclin1 is associated with the junctional protein occludin, leading to endocytosis and modulation of gut permeability [107]. Deficiency of ATG14 leads to intestinal atrophy with the loss of villi and increased TNFα-dependent apoptosis [109,110].
The significance of the main genes involved in the regulation of autophagy in IBD will be presented in more detail.
3.1. ATG16L1
This is the gene most extensively studied. ATG16L1 variations are associated with the IBD risk in different ethnic populations [111,112,113]. An IEC-specific deletion of ATG16L1 led to a reduction in Paneth cell numbers and abnormal morphology and secretory granules in a murine model [114,115]. ATG16L1 T300A/T300A mice have impaired intestinal microbiota compared with the wild type. Bacteria associated with IBD, such as Tyzzerella, Mucispirillum, Ruminococcaceae, and Cyanobacteria, were increased, while Akkermansia, a bacterium associated with mucin production, was reduced. Goblet cells secreted less mucin and autophagy was defective. Not surprisingly, experimental colitis was more severe compared with the wild type [116]. ATG16L1 deletion led to significantly increased susceptibility to dextran sulfate sodium (DSS)-induced colitis [117]. In humans, blood mononuclear cells of CD patients with the ATG16L1 T300A risk variant produced higher amounts of the pro-inflammatory cytokines IL-1β and IL-6 in response to NOD2 ligands [118] and their macrophages overproduced IFN-β and IL-1β [119]. ATG16L1 T300A was also associated with the response to treatment of CD with adalimumab [120]. Interestingly, the risk allele T300A is related to upregulated degradation of the Atg16L1 protein due to caspase 3 activation in both human and mice macrophages and decreased autophagy with enhanced inflammation [121]. Atg16L1 works in tight association with the NOD2 sensor that detects bacterial muramyl-dipeptide (MDP) and recruits Atg16L1. Autophagy is subsequently initiated, resulting in bacterial killing and the presentation of bacterial antigens to MHC class II. Mutations in NOD2 or ATG16L1 impair the whole system [37]. Another pathway that is under the control of ATG16L1 is the IL-22 signaling in IECs. There are conditions where IL-22 may cause tissue damage [122] and drive intestinal inflammation in an autophagy-dependent manner [123]. ATG16L1 regulation of autophagy is not similar in all cell types. Its effect on macrophages was mentioned before. ATG16L1 deficiency in myeloid cells has limited effects in the course of IBD, in contrast to ATG16L1 deficiency in IECs, where apoptosis is increased and inflammation is exacerbated [13].
The interactions of Atg16L with other proteins involved in autophagy or apoptosis are still under investigation. The WD40 domain of Atg16L interacts with the anti-inflammatory protein A20 in IECs [124]. Atg16L upregulates the lysosomal degradation of A20 and A20 upregulates the ubiquitin-mediated degradation of Atg16L. Deletion of these two proteins in a murine model led to spontaneous intestinal inflammation accompanied by high levels of IL-1β and TNF and increased thickness of the jejunal wall, features resembling CD.
3.2. IRGM
This is the second most extensively studied gene. It encodes the only known functional immunity-related GTPase [125]. There is no yeast ATG equivalent and its study in autophagy is therefore difficult. IRGM promotes the initiation complex interacting with ULK1 and BECN1. It is also associated with the NOD2 and Atg16L1 proteins as a protective factor against bacteria and inflammation [125,126,127]. IRGM polymorphisms (rs13361189, rs4958847, and rs11741861) are associated with both CD and UC in many ethnic populations [128,129]. However, this was not the case in a Korean population, where an association was confirmed for CD but not for UC [130]. IRGM1 deletion in mice increased the susceptibility to DSS-induced colitis only when associated with changes in the intestinal microbiota [131]. Paneth cells are also defective in IRGM1 deficiency and mice are susceptible to acute intestinal inflammation [132], while the IRGM protein limited the amount of the intracellular adherent-invasive Escherichia coli (AIEC) in epithelial cells through autophagy activation and phagosomal maturation [133]. In the intestinal mucosa of CD patients, a higher number of AIECs invade IECs and induce TNF-α production compared to healthy controls [134]. miR-196, overexpression in the inflamed epithelium of Crohn’s disease patients reduces an IRGM protective variant but not the risk-associated allele, leading to decreased autophagy and increased replication of intracellular AIECs [135]. The expression of the autophagy-related IRGM gene variants rs4958847 and rs13361189 is associated with increased visceral adipose tissue and increased morbidity in CD patients with non-alcoholic fatty liver disease [136].
However, the importance of IRGM-related autophagy in the pathogenesis of CD is questioned, as activated autophagy in the Paneth cells of CD patients seems to be independent of IRGM variants [137]. Similarly, appendectomy, which protects against UC, was associated with downregulation of autophagy genes and suppression of autophagy may be related to decreased microbial antigen presentation and attenuation of colitis [138].
3.3. LRRK2
Gene studies in leprosy identified the links between the LRRK2 gene and CD [139,140]. LRRK2 is mainly expressed in myeloid cells and B cells and is involved in the secretion of inflammatory cytokines and antimicrobial peptides in macrophages [139,140]. LRRK2 is implicated in autophagic flux, although the regulatory mechanisms of autophagy control have not been clarified [141]. LRRK2 deficiency in murine models increased susceptibility to DSS colitis by decreasing activation of the transcription factor NFAT (Nuclear factor of activated T cells) [142]. Recently, dendritic cells from CD patients showed elevated LRRK2 expression and caused the induction of severe colitis with NF-κB activation and pro-inflammatory cytokine responses in a murine model [105].
3.4. ATG7 and ATG5
Deletion of ATG7 in IECs resulted in impairment of Paneth cells with a reduced size of granules and reduced lysozyme and antimicrobial peptide levels. IECs produced more TNF and IL-1β after lipopolysaccharide challenge compared to normal IECs [143,144,145]. Similar findings in Paneth cells were also found in mice with IEC-specific ATG5 deletion [146,147,148]. Increased colonic inflammation was also demonstrated after ATG7-specific deletion in myeloid cells [149]. In a murine model of IEC-specific X-box-binding protein 1 (XBP1) deficiency with protracted ER stress, the addition of ATG7 deletion exacerbated transmural inflammation, simulating human CD. This finding indicates that autophagy protects against increased apoptosis generated by protracted ER stress [114]. Reduced lysozyme staining was also observed in the Paneth cells of generalized ATG4B-deleted mice [150]. ATG5 deletion in IECs was also associated with increased Salmonella typhimurium presence in IECs and favored the extraintestinal dissemination of this pathogen [151]. Intestinal stem cells (ISCs) are also influenced by the specific deletion of ATG5. The numbers of ISCs are reduced and those that survive have high ROS levels [152]. Increased levels of ROS associated with a reduction in the DNA repair capacity and p53-mediated apoptosis of ISCs were also demonstrated after the loss of ATG7 [153].
Taken together, these findings indicate that autophagy is vital for the maintenance of intestinal homeostasis.
3.5. Other Autophagy-Associated Genes
There are additional genes implicated in IBD’s pathophysiology. Beclin1F121A knock-in mice overexpress a mutant Beclin1 with decreased Bcl-2 affinity and promotion of its ability to increase autophagic flux. These mice have a thicker mucosal layer and decreased ER stress. On the contrary, BCL2AAA knock-in mice overexpress a mutant Bcl-2 with increased affinity to bind Beclin1, with a reduction in autophagic flux associated with a thinner mucosal layer in the colon [154].
The involvement of the autophagy proteins p62 and TFEB and the single nucleotide polymorphisms (SNPs) of NDP52 and optineurin observed in individuals with IBD have been implicated in the pathogenesis of IBD [103,104,155,156].
Accumulation of cytoplasmic p62 is a sign of impaired autophagy because upregulated autophagic flux degrades p62 [157]. However, increased turnover of p62 was observed in human CD and DSS colitis after hypoxia stimulation associated with reduced inflammatory markers and markers indicating increased autophagy [158,159]. On the contrary, the intracellular survival of AIEC LF82 bacteria was higher in cells silenced for p62 [160]. In the inflamed ileal pouch created after colectomy for UC, defective autophagic flux with elevated p62 was demonstrated in IBD tissues [159]. Additionally, the expression of p62 was higher in the epithelial cells of damaged mucosa compared with normal mucosa [161].
Optineurin (OPTN) is an autophagy protein involved in mitophagy and xenophagy [162]. OPTN deficiency has been reported in CD patients and murine models [104,163], leading to an accumulation of IRE1α and exacerbation of colitis during ER stress [115]. OPTN deficiency was associated with reduced production of TNFα and IL-6 and in-creased susceptibility to bacterial colitis [163]. Taken together, these findings indicate that OPTN is involved in macrophage-driven inflammation and bactericidal function related to increased CD susceptibility [163].
The implication of TFEB in IBD is very limited [164]. Mice with a deletion of TFEB in IECs have defective Paneth cell granules, low expression of lipoprotein ApoA1 and exacerbated DSS colitis [103].
The association of IBD with ER stress and the unfolded protein response (UPR) genes such as the XBP1 have long been detected by gene studies [165]. The XBP1 gene was sequenced in a large number of IBD patients and several rare variants of XBP1 were associated with IBD during ER stress [165]. Deletion of XBP1 in IECs led to spontaneous small intestinal inflammation and increased susceptibility to DSS colitis in parallel with abnormal Paneth cells [165].
3.6. NOD2
This is the first gene to be linked to CD susceptibility back in 2001 [166,167,168]. The three most common SNPs, R702W, G908R and L1007f/s, were identified in approximately 30% of patients with CD [169]. Homozygosity increases the risk of CD by 20- to 40-fold [169]. The L1007f/s variant cannot recognize the MDP dipeptide of bacteria and the activation of the host NF-kB is inadequate to mount a proper defense [170]. As mentioned before, the NOD2 protein is directly associated with the autophagy protein Atg16L1 and induces bacterial elimination via autophagy at the entry site [170,171,172]. The mutant NOD2 fails to aggregate Atg16L1 molecules, restricting the autophagic response due to the defective formation of autophagosomes [172]. NOD2 mutations result in two more defensive problems. NOD2-associated intact autophagy of the dendritic cells is necessary for efficient antigen presentation to T lymphocytes and induction of a proper specific immunological reaction against bacterial antigens [170]. Dendritic cells bearing the NOD2 mutations exhibit reduced bacterial killing of AIECs due to inadequate localization of AEICs to lysosomes. This is reversed after administration of rapamycin, which activates autophagy [170].
It should be noted, however, that most IBD genetic studies revealed stronger associations of autophagy-related genes with CD in European white populations rather than in descendants from East Asia [173]. Moreover, two genome-wide scans of African-American CD and UC patients failed to demonstrate differences in the SNPs of autophagy genes between controls and patients with an increased CD risk [174]. The anthropogeography of autophagy in IBD is obviously not over.
The association between autophagy, microbial defense and inflammation in IBD has been recently reviewed [175].
4. Autophagy and Apoptosis in IBD
4.1. Autophagy
Impaired autophagy is observed in several cells of the damaged mucosa of IBD patients [23,161]. Emerging evidence indicates that there is an interplay among three important cellular pathways in the pathogenesis of IBD. These include apoptosis, autophagy and the unfolded protein response induced by endoplasmic reticulum stress [176,177]. Autophagy is the central regulator in IBD pathophysiology, as it is connected to all the defensive mechanisms of the intestinal mucosa. Dysfunctional autophagy affects several processes, such as intracellular microbial clearance, antimicrobial peptide production by Paneth cells, production of mucus by goblet cells, antigen handling by dendritic cells and pro-inflammatory cytokine secretion by macrophages [178,179,180,181]. Some research data on these defense parameters have been presented in the previous subsection but additional collective details are given below.
4.2. Intestinal Functions Affected by Autophagy Dysregulation in IBD
4.2.1. Paneth Cell Autophagy
Paneth Cell Autophagy or, as it is known, crinophagy. Paneth cells produce and secrete via crinophagy antimicrobial peptides such as lysozyme and defensin 5 and 6, which shape, in part, the microbiota of the gut [182]. As mentioned before, CD patients carrying the risk variants of ATG16L1 or NOD2 have abnormal Paneth cells [183], and the same happens in mice with repressed expression of Atg5, Atg7 Atg4B or leucine-rich repeat kinase 2 (LRK2) [184].
4.2.2. Inflammatory Cytokine Regulation
Intestinal mucosa macrophages initially respond to bacterial pathogens with antimicrobial peptide production [185]. This is not the case in IBD patients, where macrophages overproduce pro-inflammatory cytokines and the bacterial clearance is not effective. In murine models, this is associated with specific macro-phage deficiencies of autophagy-related proteins leading to overproduction of IL1β and IL-18 [117,186]. In contrast, inflammatory cytokines are not secreted by IECs despite the presence of the same mutations in autophagy genes [187].
4.2.3. Antigen Presentation by Dendritic Cells
Dysfunctional autophagy interferes with the presentation by dendritic cells of bacterial and other unknown antigens to CD4+ T lymphocytes, as proved in animal models with the deletion of ATG5 or ATG7 [188,189]. NOD2-associated autophagy in dendritic cells is also necessary for the generation of MHC-II-restricted CD4+ cells. Defective antigen presentation has been reported in human IBD. Dendritic cells from CD patients with risk variants in NOD2 or ATG16L1 have impaired MHC II antigen presentation [170] and, possibly, a restricted number of peptides presented to T-cells.
4.2.4. Goblet Cell Mucus Secretion
In IBD patients, the protective mucus layer is defective and microbes of the lumen are in close contact with the epithelium [190]. Reduction in the autophagy proteins Atg5, Atg7 or LC3 results in abnormal goblet cells and decreased mucus production [147,191]. The same findings were reported in a knock-in mice expressing the ATG16L1 T300A variant [18].
4.2.5. ER Stress Response
Increased levels of multiple proteins leading to ER stress and induction of the unfolded protein response were demonstrated in the intestinal mucosa of IBD patients, even in areas without inflammation, and also in the mucosa of healthy people expressing the ATG16L1 T300A allele [192]. CD patients harboring this allele have de-creased stability of the intestinal epithelial proteins due to increased accessibility to caspase cleavage, and an interconnection between ER stress and autophagy leads to epithelial barrier abnormalities [121]. In murine models, the absence of the proteins Atg16L1 and Atg7 is associated with overexpression of ER stress markers such as DDIT3 (DNA dam-age inducible transcript 3) and phospho-EIF2A (eukaryotic translation initiation factor 2A) [193]. ER stress initiates ATF6 and induces autophagy in IECs through the upregulation of death-associated protein kinase 1 (DAPK1) and mATG9 translocation [194,195]. ATF6 is also involved in the interaction between ERS and IECs autophagy in IBD, regulating the downstream molecules XBP1 and Atg16L1 [196]. Importantly, ER stress, UPR, autophagy and apoptosis are all interconnected in the PERK-Elf2a-ATF4 pathway [197,198]. ATF4 induces the expression of the pro-apoptotic protein CHOP and increases the expression of the autophagy gene ATG5 [199]. ATF4 also promotes the expression of the DNA damage response 1 (REDD1) protein [200] that activates autophagy in the neutrophils of the intestinal mucosa inhibiting mTOR. REDD1 expression is closely associated with the severity of UC [201]. ER stress also stimulates the IRE1/JNK and IRE1/XBP1 pathways to release Beclin1 and promote autophagy [202,203]. IKKα kinase is also a mediator capable of repressing ER stress, promoting Atg16L1 stabilization via phosphorylation of Atg16L1 at Ser278. It improves therefore acute intestinal inflammation [204]. ER stress affects Paneth cells as well, where autophagy is increased to counteract the increased ER stress when UPR is not adequate [114]. In agreement with this, the Paneth cells of CD patients expressing the ATG16L1 T300A risk allele and impaired autophagy have increased levels of the ER stress marker pEIF2α, even in the quiescent state [193]. However, they do not show signs of increased apoptosis. Mice with a deletion of ATG16L1 develop ileitis, and a similar ileitis develops earlier in life when a deletion of the unfolded protein response transcription factor XBP1 is added to this model due to increased ER stress [114,115].
UPR attempts to counteract ER stress by inducing autophagy to degrade misfolded proteins [205,206]. ER stress activates the ATF6, PERK, and IRE1 pathways. ATF6 activates autophagy either by upregulating the pro-apoptotic cytokine CHOP that mediates mAtg9 translocation or by upregulating DAPK1 [194,195]. PERK activates autophagy through the ATF4 pathway, as mentioned above [197,198,199,207]. IRE1 is associated with both IBD and autophagy through XBP1, which is a vital component of the IRE1 pathway. Decreased genetic expression of XBP1 is associated with IBD. Impairment of the IRE1 pathway in IECs led to reduced numbers of goblet cells and impairment of the intestinal epithelial barrier [208].
Taken together, these data indicate that autophagy and ER stress are mutually regulated [202,203]. ER stress can promote autophagy, and activated autophagy can inhibit overactive ER stress, protecting the integrity of the mucosal barrier and IECs from apoptosis [209].
ER-stress-induced inflammation initiates a direct link between NOD1/2 and the IRE1 pathway facilitated by TRAF2 (TNF receptor-associated factor 2) [210]. Active IRE1 stimulates the JNK pathway to trigger NF-κB signaling [211,212] and autophagy induction [213]. ER stress initiated by infection with Brucella abortus increased inflammation and IL-6 production [210] in human and murine cells. This response was dependent on NOD1/2 and receptor-interacting serine/threonine306 protein kinase 2 (RIPK2), but also on IRE1 kinase activity and TRAF2-induced NF-κB signaling [210].
Interestingly, ER stress effects is also modulated by an innate immune sensor called the stimulator of interferon genes (STING) in response to PAMPs (pathogen-associated molecular patterns) present in live Gram-positive bacteria [214]. This process induces autophagy via inhibition of mTORC1 and localization of STING to autophagosomes. Abnormalities in the above responses in IBD have not been described so far.
4.3. Autophagy Signal Pathways Implicated in IBD
4.3.1. mTOR
Pharmacological inhibition of mTOR and administration of autophagy stimulators improved experimental colitis and oxidative stress [215]. mTOR is activated in the intestines of mice with acute colitis [216,217], and mTORC1-dependent STAT3 (signal transducer and activator of transcription 3) signaling attenuates pathology [216].
4.3.2. AMPK/mTOR
Sodium hydrosulfide (NaHS), which is a donor of H 2 S, reduced the histological injury and pro-inflammatory cytokine expression in a rat model of UC by activating autophagy through the AMPK/mTOR pathway [218]. Sinensetin administration inhibited apoptosis and restored intestinal barrier dysfunction in experimental colitis by activating autophagy of IECs. AMPK knockdown reversed the effects of sinensetin [14]. Similar effects on experimental colitis were reported for dapagliflozin and metformin [219]. Nicotine also attenuated DSS colitis by promoting autophagy and reducing apoptosis through the AMPK/mTOR axis [220].
4.3.3. mTOR/NLRP3
SNPs in the NLRP3 gene confer susceptibility to CD [221]. There is a reciprocal regulatory relationship between NLRP3 expression and autophagy, where activation of autophagy limits NLRP3 activity, leading to a reduction in inflammation [222]. Hypoxia attenuates the colonic inflammation by downregulating the binding of mTOR and NLRP3 and activating autophagy [158].
4.3.4. AKT/mTOR
The activation of the AKT/mTOR signaling pathway negatively regulates autophagy and participates in the pathogenesis of IBD [223]. Thus, the heat shock transcription factor 2, the ring finger protein 8 and the Xianglian pill increase autophagy in IECs by inhibiting the AKT/mTOR pathway and seem to be protective in UC [224,225,226].
4.3.5. NF-kB Pathways
The implication of NF-kB in autophagy and apoptosis is variable. Stimulation of NF-kB can either upregulate [227] or inhibit autophagy [228]. At the same time, autophagy upregulation may inhibit NF-kB activation by interfering with the IkB kinase (IKK), which is an upstream regulator of NF-kB [229,230]. Moreover, the autophagy protein Atg16L1 restricts the pro-inflammatory cytokine response by downregulating NF-kB expression [231]. As a consequence, expression of the Atg16L1T300A polymorphism in macrophages increases the NF-kB-dependent cytokine production and exacerbates the risk of CD [232]. In addition, ginseng polysaccharides improved DSS colitis by inhibiting the TLR4-NF-kB pathway and activating autophagy [233].
4.3.6. Nrf2/HO-1 Pathways
Nuclear transcription factor E2-related factor 2 (Nrf2) is fundamental t the antioxidant response, and the Keap1/Nrf2 pathway is connected to autophagy through the autophagy protein p62 SQSTM1 [234,235]. The activation of the Nrf2/HO-1 pathway attenuated TNBS- and DSS-induced colitis [236,237], while impaired Nrf2 signal is implicated in the pathogenesis of these two colitis models [237,238].
4.3.7. Additional Pathways
Wnt signaling pathway activation through the hypoxia-inducible factor-l (HIF-1) downregulates autophagy in the IECs of patients with IBD via the mTOR pathway [161]. Abnormal regulation of STAT3 in the intestine inhibits autophagy, while inhibition of STAT3 restores autophagy in bacteria-infected cells [239,240]. In models of neonatal necrotizing small-bowel colitis, TNF-a may induce autophagy and regulate the apoptosis of IECs, promoting disease progression [241]. Loss of ATG5 leads to in-creased inflammatory cytokines through activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK kinase) pathway [242].
4.4. Role of Autophagy in CD
The implication of autophagy in CD is multifaceted and cell-specific [36,243]. As mentioned before, dysfunctional autophagy in IECs increases the expression of claudin-2 due to reduced lysosomal degradation and the permeability of the intestinal barrier is in-creased [106,244]. Dysfunctional autophagy allows for the accumulation of damaged mitochondria and ROS production, leading to inflammasome activation and ROS-induced cell death [245,246,247]. The implication of ATG16L in NOD2 and IRGM expression and their importance in CD increased susceptibility have already been analyzed. Dysfunction of NOD2 or LRRK2 results in the degradation of lysozyme in lysosomes and decreased defense [248,249,250].
ERN1 (endoplasmic reticulum to nucleus signaling 1) is an important cellular protein that is degraded in normal autophagy after association with optineurin. Defective autophagy during ER stress reduces the clearance of ERN1, leading to a CD-like transmural ileitis in mice [115]. In intestinal stem cells, defective autophagy increases ROS accumulation and disrupts the regeneration of IECs [152]. In macrophages, dysregulated autophagy impairs the degradation of pathogens. The secretion of pro-inflammatory cytokines by macrophages is increased when autophagy is dysfunctional [119,251]. Defective autophagy leads to impaired antigen presentation and T-cell activation [252], as mentioned before. T-cell activation is supported by the autophagic receptor TAX1BP1 (Tax1 binding protein 1) that binds to LC3 and induces autophagy [253]. Dysfunctional autophagy impairs T-cell proliferation, leading to reduced numbers of CD4+ and CD8+ T cells, and Treg cell survival, while the Th2 and Th17 responses are increased [253,254,255,256,257,258,259,260]. Bacteroides fragilis from the gut microbiota produce molecules that are recognized by DCs and induce IL-10-producing Treg cells, limiting thus the CD4+ cell-mediated inflammatory response. DCs harboring the autophagy-related risk variants fail to induce IL10 production by Treg cells in response to B. fragilis aggravating inflammation in CD experimental models [261,262].
4.5. Role of Autophagy in UC
Data from patients and experimental models indicate the association between dysregulated autophagy impairment and the occurrence of UC [263]. The transcription factor 4 (ATF4), an autophagy-related protein, was reduced in the inflamed intestinal mucosa of patients with UC, suggesting the presence of impaired autophagy [264].
Vitamin D and its receptor (VDR) have an important role in IBD pathogenesis, as they are implicated in the regulation of innate and adaptive immunity and the control of inflammation [265]. VitD/VDR also modulate the intestinal microbiome in IBD. Low levels of intestinal epithelial VDR were associated with reduced autophagy and damage to the barrier integrity, allowing the translocation of pathogens and initiation of inflammation. On the other hand, increased intestinal VDR expression repressed inflammation in a colitis model [266,267].
VDR controls the autophagy and apoptosis in IECs by maintaining the expressions of Beclin1 and Atg16L1. In fact, VDR deficiency leads to decreased AtgL1 and Beclin1, leading to decreased autophagy and increased apoptosis [268,269].
Vitamin D/VDR attenuates the development of TNBS-induced colitis by inhibiting the apoptosis of IECs and protects intestinal cells from apoptosis induced by radiation injury [270,271].
On the other hand, activation of the intestinal nuclear vitamin D receptor (VDR) induces autophagy and ameliorates intestinal inflammation in experimental colitis [272,273]. Low VDR expression was also observed in patients with UC [272]. Moreover, mTOR-dependent autophagy flux deficiency was described in human IECs from active UC patients [215].
In DSS-induced UC models, the nuclear receptor binding factor 2 (NRBF2), a protein of the Beclin1-Atg14-VPS15-VSP34 complex, was crucial for the clearance of apoptotic cells and the repression of inflammation [274]. In addition, it was reported that specific deletion of (meteorin-like) Metrnl, a protein highly expressed in IECs, exacerbated UC severity through the reduction of autophagy via the AMPK-mTOR pathway [275]. Induction of AMPK-mTOR-mediated autophagy, on the other hand, alleviated the intestinal inflammation [276].
It should be stressed, however, that excessive induction of autophagy might be detrimental, leading to autophagic cell death [277]. Deficiency of erbin (Erbb2-interacting protein), a protein required for the polarity of epithelial cells, induced autophagy and exacerbated cell death in DSS colitis [278].
4.6. Lysosomes in IBD
The activation of the lysosomal enzyme cathepsin D is implicated in intestinal inflammation, both in human IECs and a murine model of IBD. Immunohistochemistry identified increased cathepsin D expression in the inflamed intestinal mucosa of IBD patients compared to non-inflamed mucosa. Cathepsin D originates from macrophages, as the mRNA of cathepsin D was only found in isolated macrophages from the inflamed mucosa [279]. Cathepsin D was also identified in the inflamed intestinal mucosa of acute and chronic DSS colitis, but not in normal mucosa. In the same model, apoptosis of IECs was initiated by dietary sphingomyelin through cathepsin D activation [280]. These findings suggest that increased autophagy in macrophages and apoptosis of IECs are interconnected, possibly through the autophagy-associated lysosomal cathepsin D. Interestingly, inhibition of cathepsin D in mice attenuated DSS colitis. Simultaneous inhibition of both cathepsin B and cathepsin L also ameliorated colitis [281].
4.7. Autophagy and Fibrosis in Experimental Colitis
In addition to the ability of autophagy stimulators to improve acute murine colitis [282], data suggest a role of autophagy in modulating fibrosis in the intestinal wall. The TNBS chronic colitis model is associated with intestinal fibrosis. In this model, it has been shown that autophagy inhibition deteriorates and autophagy stimulation prevents fibrosis [283]. Mice treated with an autophagy inhibitor showed the increased expression of profibrogenic CD16+ -M2 macrophage infiltration, while rapamycin activated autophagy and increased the infiltration of macrophages with a regulatory/anti-inflammatory profile [284]. This was confirmed by the demonstration that the initiation of autophagy via mTOR inhibition in Cx3cr1+ mononuclear cells restricted IL-23/IL-22 axis-mediated intestinal fibrosis [285]. In the same TNBS model, the CD147 protein was increased in the colon of fibrotic animals compared to controls. Inhibition of CD147 reduced intestinal fibrosis and collagen deposition through induction of autophagy. Interestingly, there was no effect of the activated TGF-β1 in the fibrotic tissue [286].
Multiple review articles have provided an overview of the general role of autophagy in intestinal function and IBD pathogenesis [37,175,287,288].
4.8. Mitophagy in IBD
Abnormalities in mitophagy are important in IBD as they are related to ROS accumulation and ER stress in IECs, intestinal macrophages and Paneth cells. Mitochondrial stress-induced mitophagy is initiated by increased intracellular ROS accumulation in IBD patients [51,289,290,291]. Deletion of Parkin led to attenuation of colonic inflammation in DSS colitis. This is possibly due to the interaction of Parkin with VDRs, which are highly expressed in IECs [292,293]. Very recent evidence connected VDR to mitophagy. Parkin interacted with VDR and promoted VDR lysosomal degradation. Comparison of colitis in Parkin−/− and Parkin−/−Vdr−/− mice showed that the protective effect of Parkin deletion against colitis was reversed by VDR deletion. Moreover, deletion of Parkin was shown to inhibit apoptosis [294].
Diminished expression of prohibitin1, a mitochondrial chaperone, impairs mitochondrial homeostasis and activates mitophagy, increasing the intracellular ROS in IECs of IBD patients. Reduction in both prohibitin1 and Atg16L1, as frequently found in IBD, exacerbates mitochondrial depolarization and reduces intestinal cell survival [290]. Mitophagy is important for the viability and homeostasis of intestinal stem cells (ISCs) as well. ISCs use oxidative phosphorylation (OXPHOS) as a vital source of ATP supply [295,296,297].
Other intestinal mucosal cells are also influenced by abnormalities in mitophagy. Macrophages are influenced by dysregulated mitophagy in IBD. The CD-associated ATG16L1T300A variant leads to pro-inflammatory transformation of macrophages with impaired mitophagy, ROS accumulation and enhanced IL1B production [17,18]. Moreover, andrographolide administration in mice was found to ameliorate colitis through mitophagy-mediated inhibition of NLRP3 inflammasome activation [246]. Ginsenoside Rd was also reported to inactivate the NLRP3 inflammasome and improve DSS colitis by inducing mitophagy [298]. Palmatine also attenuated intestinal inflammation by inhibiting NLRP3 inflammatory vesicles through mitophagy induction [299]. Macrophages from mice colitis or IBD patients showed reduced mitophagy due to a genetic reduction in IL10 or its receptor and activation of the mTOR pathway resulting in increased inflammasome activation and colonic inflammation [300]. Paneth cell abnormalities were also observed with defective mitophagy in mice after deletion of IRGM [132], which is a mitochondrial protein that triggers mitophagy [301].
4.9. Apoptosis
It has been shown that during inflammation in IBD patients or murine models of colitis, different cell death pathways are operational, including apoptosis, necroptosis and pyroptosis [302,303]. Thus, enhancement of IEC death by apoptosis is demonstrated in histological specimens from patients with IBD [304,305]. Elevated apoptosis of IECs is closely associated with the severity of inflammation in IBD [306].
The extrinsic pathway of apoptosis in enterocytes depends on the activation of any of the death receptors, such as CD95, TRAIL or TNFR1, as mentioned before. The expression of other apoptosis-associated proteins such as the Fas death receptor (CD95), Fas ligand, BAX, and p53 is also increased at the inflamed sites [307]. CD95 in IECs is localized on the basolateral surface [308]. The expression of the CD95 ligand (CD95L) is restricted only to lamina propria immune cells and Paneth cells in normal individuals. Induction of CD95L expression in IECs, however, can be identified in patients with UC [309,310]. Apoptosis of IECs in IBD patients is mainly mediated by p53 and p53-upregulated modulator of apoptosis (PUMA), which inhibits Bcl-2, and controls mitochondria-driven apoptosis. The levels of p53 and PUMA were increased in the mucosa of murine DSS colitis and in patients with UC. Apoptosis of IECs was observed in the lower mucosal crypts in colitis of both humans and mice. Deletion of p53 and PUMA reduced apoptosis of IECs, although the degree of inflammation did not change. Induction of PUMA in mice with p53 deletion indicated that PUMA-mediated apoptosis was independent of p53 [311]. In IECs, the checkpoint kinase Chk1 regulates p53-dependent transcription of PUMA [312]. Thus, increased Chk1 was expected to increase the cell death of IECs and compromise the gut barrier. However, contrary to this expectation, Chk1 activation in mouse models was shown to enhance the epithelial barrier via the transcriptional upregulation of claudins [313,314].
The DNA damage-regulated autophagy modulator 1 (DRAM1) encodes a protein that is localized in the membrane of the lysosomes. It is involved in p53-mediated apoptosis and the progression of autophagy. DRAM1 may act downstream of PUMA to regulate IEC apoptosis by positively regulating JNK. Deletion of DRAM1 abolished the cytotoxic effect of TNF-α on SW480 and HT29 cell viability. However, this may be not true for HT29 cells, as demonstrated by annexin V. The DRAM1 levels were higher in the mucosa and serum of IBD patients compared to healthy individuals. Moreover, DRAM1 expression was lower in CD inflammatory cells compared with UC patients. Importantly, DRAM1 was correlated with the simple endoscopic score for CD and the Mayo endoscopic score for UC [315].
4.9.1. The Role of TNFα
Neutralization of TNFα is the main treatment for IBD, leading to reduced IEC apoptosis and enhanced mucosal repair [316]. In fact, in most cell types, IEC included, transient TNF signaling inhibits apoptosis because of IKKb-dependent NF-kB activation [317]. SNPs in widely expressed genes encoding NF-kB-regulated molecules show a strong association with IBD [318], and activated nuclear NF-kB is present in both IEC and lamina propria macrophages of active disease areas [319]. The administration of the NF-κB inhibitor BAY 11-7082 inhibited IECs apoptosis in experimental colitis [320].
However, the reason for TNF-induced death of IECs is not well understood, because TNF does not damage healthy IECs. In mice, deletion of the NF-κB-mediated survival responses via the ablation of NEMO (NF-κB essential modulator) induced IEC apoptosis in a Raf kinase inhibitor protein (RIPK1)-dependent manner. NEMO-deficient IECs cannot regulate RIPK1 activation [321,322]. It was recently demonstrated that it is the chronic NF-kB activation that induces intestinal crypt apoptosis and severe mucosal erosion after TNF stimulation. This was mediated by the RIPK1 kinase, while RIPK1 inhibitors abolished the TNF-induced apoptosis but preserved its survival properties, which are not dependent on RIPK1 kinase [323,324]. The TNF-induced protein 3 (A20) may play a central role in patients with IBD, as it is a negative regulator of NF-kB [325,326,327]. Mice overexpressing A20 in IECs are prone to TNF-induced IEC death, which can be abolished by administering RIPK1 inhibitors. A20 is also overexpressed in the IECs of patients with IBD, indicating that A20 is the mechanism behind the IEC susceptibility to TNF–RIPK1-mediated cell death [325]. Another critical regulator of NF-κB activation and IECs apoptosis downstream of the TNFR is the TGFβ-activated kinase 1 (TAK1) [328]. Inhibition of TAK1 alone is sufficient to induce TNF-mediated apoptosis [329,330]. TAK1 favors apoptosis over necroptosis of IECs by increasing FLIP degradation. The formation of the RIPK1–FADD–caspase 8 complex is enhanced and apoptosis is initiated [329].
Infiltrating macrophages may also induce IEC apoptosis and further disrupt the integrity of the intestinal barrier due to increased ER stress with the overproduction of TNF-α [331].
4.9.2. Other Factors Affect Apoptosis in IBD
MicroRNAs are important mediators of the pathophysiology of IBD. miR-665 was markedly increased in samples from active colitis patients and promoted apoptosis in DSS colitis. Administration of an miR-665 mimic aggravated DSS colitis, while injection of an antagonist rescued cells from apoptosis [332].
Reduced protein levels of BAX were found in the intestinal mucosa of CD patients. Reduced BAX was localized in the lamina propria, not in the IECs. High levels of Bcl-2 and caspase 3 were seen in the mesenteric adipose tissue (MAT) of these patients, indicating defective apoptosis in MAT [333]. These findings verify previous observations of mucosal T lymphocyte resistance to apoptosis, mostly in steroid-resistant CD [334,335], and indicate that the impairment of apoptosis is BAX-dependent in the lamina propria and Bcl-2-dependent in MAT. The clinical significance of these findings is difficult to explain.
Sam68 (Src-associated substrate during mitosis of 68 KDa), is implicated in various cellular functions, including apoptosis. Sam68 was upregulated in the IECs of patients with UC. In DSS colitis, the overexpression of Sam68 was associated with increased levels of IEC apoptotic markers and NF-κB activation indicators in IECs, indicating that Sam68 regulates apoptosis of IECs via activation of NF-kB in UC [336].
NRBF2 is a positive regulator of autophagy. A recent study demonstrated that it is involved in the clearance of apoptotic bodies by macrophages in DSS colitis. It was also upregulated in biopsies from patients with UC, where NRBF2-positive macrophages engulfed apoptotic bodies. Interestingly, TUNEL-positive apoptotic cells were correlated with UC severity quantified by the Mayo score [274].
4.10. Ferroptosis in IBD
The recently described ferroptotic cell death has been associated with the pathophysiology of IBD. As in other diseases, there is substantial evidence that ferroptosis is an important pathway for cellular death in IBD. Available data refer mostly to murine colitis or human UC. It is, however, likely that ferroptosis will be a central mechanism in CD as well. Ferroptosis in IECs destructs the protective barrier function and bacterial antigens enter into the submucosa and activate the immune cells aggravating IBD in humans or in DSS colitis [62,67,337,338].
The fundamental characteristics of ferroptosis, including enhanced lipid peroxidation, depletion of the antioxidant systems and increased iron deposition, have been re-ported in patients with IBD and murine models [339,340]. Dysregulation of ferroptosis-associated genes affects the severity and morbidity of murine colitis [341,342]. Similarly, ferroptosis genes such as ACSL4, GPX4, and glucose 6 phosphate dehydrogenase are either up- or downregulated in specimens of patients with UC [343]. Moreover, five ferroptosis-related hub genes were identified that can discriminate UC patients from healthy individuals [344].
Specimens from human UC and from mice with DSS colitis have shown that ferroptosis is implicated in UC through IECs death induced by ER stress. NF-kB/p65 phosphorylation repressed ER stress-mediated IEC ferroptosis to attenuate UC [343,345]. This is in agreement with the reported suppression of ER stress by GSK2606414, which led to inhibition of ferroptosis in IECs [343]. Several specific inhibitors of ferroptosis could stop the detrimental effects of ferroptosis in models of UC [337,346,347]. Activation of GPX4 can also inhibit ferroptosis and improve UC symptoms [346,347].
Ferroptosis regulates colitis through the Nrf2/HO-1 signaling pathway, which can inhibit the NF-κB pathway and subsequently suppress secretion of the pro-inflammatory cytokines [339,348]. Astragalus polysaccharide in particular prevented ferroptosis in DSS colitis by inhibiting the Nrf2/HO-1 pathway [349,350]. The Nrf2-GPX4 pathway is inhibited in UC [346]. Deletion of Nrf2 increased the severity of DSS colitis [351,352]. Heme oxygenase-1 (HO-1) has excessive anti-inflammatory and antioxidative properties and protects mice from colitis-associated inflammation injury [353]. Moreover, other ferroptosis mediators, such as NADPH oxidases [354] and CD44 [355], are also associated with the pathogenesis of IBD in patients and colitis models. The exact role of these molecules in mediating ferroptotic regulation in colitis requires additional research [67].
Arachidonic acid, a preferential substrate of lipid peroxidation, is elevated in the phospholipids of the mucosa in patients with UC [356]. This is consistent with the report that selenium supplementation is protective in IBD patients [357], although it is still debatable if the protective action is due to the activation of GPX4 [358]. IECs in Crohn’s disease were also shown to have reduced GPX4 activity and increased markers of lipid peroxidation. PUFAs and specifically arachidonic acid initiate pro-inflammatory cytokines’ production of IECs, which is limited by GPX4, although GPX4 does not directly control arachidonate metabolism. Most importantly, a PUFA-enriched diet administered in mice lacking one GPX4 allele leads to a focal granuloma-like neutrophilic enteritis resembling CD [63].
Lipoxygenase isoforms that participate in the ferroptotic pathway are implicated in the pathogenesis of IBD. Alox15 deletion shows suppression of inflammation and im-proved intestinal barrier integrity, while Alox15 overexpression leads to severe colitis [359]. Alox5 is upregulated in the mucosa of patients with IBD [360]. Phosphatidylethanolamine-binding protein 1 (PEBP1) is a master regulator of Alox15, inducing the production of lipid peroxides that act as ferroptotic death signals [361]. Ferrostatin-1 (Fer-1), a ferroptosis inhibitor, is a poor Alox15 inhibitor, suggesting that this lipoxygenase isoform has a limited role in ferroptosis in IBD. However, Fer-1 effectively inhibits lipid peroxides produced by the Alox15/PEBP1 complex [362]. Moreover, administration of zileuton, which is a strong inhibitor of 5-LOX, prevents a decrease in the tight junctional impairment induced in TNBS colitis [66]. Ferroptosis inhibitors, including Fer-1, Lip-1, and DFO, decreased disease activity scores and attenuated colon length shortening in DSS colitis, proving the beneficial effect of ferroptosis inhibition [363].
Other Ferroptosis Regulators in Intestinal Diseases
Despite evidence that p53 facilitates ferroptosis as it suppresses cystine uptake by decreasing the expression of the cystine/glutamate subunit SLC7A11 [364], it is still debatable if this relevant in IBD. On the other hand, it seems that the reduced expression of the aryl hydrocarbon receptor repressor (Ahrr) in IBD is important in the pathogenesis of the disease. Deletion of Ahrr results in the loss of intestinal intraepithelial lymphocytes (IELs) and increased susceptibility to DSS colitis. Ahrr deficiency leads to increased ferroptosis through increased ROS production and lipid peroxidation [365]. Bioinformatics analysis has shown the significance of the ferroptosis-associated gene ACSF2 (acyl CoA synthetase family member 2) in the pathogenesis of UC. The expression of ACSF2 was downregulated in UC animals and cell models, and the ferroptosis inhibitor Fer-1 reversed the reduced expression of ACSF2, suggesting that ACSF2 plays an important role in ulcerative colitis through the modulation of ferroptosis [342]. A recent finding connected ferroptosis with fibrosis observed in CD but not in UC. Ferroptosis genes such as ACSL4 were upregulated in fibroblasts via single-cell transcriptome analysis. CD fibroblasts also expressed ferroptosis-resistant genes such as GPX4, indicating fibroblast heterogeneity. IECs from patients with CD expressed reduced levels of anti-ferroptotic genes and increased pro-ferroptosis gene levels compared to control cells [366].
4.11. Interaction of Autophagy–Apoptosis in IBD
The common denominator of intersection is ER stress and TNFα. In most cases, autophagy and apoptosis are mutually opposite. Decreased autophagy is associated with increased apoptosis in IBD, and substantial evidence points to the central role of TNFα as a regulator of both increased apoptosis and decreased autophagy. This is important as a possible mechanism of action of anti-TNFα treatment in IBD, in addition to the commonly accepted effect on inflammation. It should be stressed, however, that most studies of inter-actions are referring to the pathogenesis of UC rather than to CD.
Intersection of ERS, Autophagy and Apoptosis
Autophagic proteins regulate epithelial apoptosis during intestinal injury and inflammation [146,247,367]. The effects of ER stress in autophagy and apoptosis have been presented before. The UPR is the critical arm and GRP78 dissociation activates the IRE1, PERK, and ATF6 pathways. ER stress regulates autophagy susceptibility genes and induces IEC apoptosis through the CHOP signaling pathway, leading to UC [263,368]. UPR also interferes with autophagy through the PERK-Elf2a-ATF4 pathway [197,198,199], where decreased ATF4 downregulated the expression of the pro-apoptotic protein CHOP in UC patients [207].
Autophagy protects against cell death induced by inflammation. The autophagy-associated proteins Beclin1 and Atg5 also serve as initiators of apoptosis [367]. This is achieved when Beclin1 interacts with HMGB1 (high mobility group box 1), a nuclear protein that also serves as an extracellular DAMP. HMGB1 inhibits the enzymatic cleavage of Beclin1 and Atg5 and the balance between autophagy and apoptosis is shifted toward autophagy, limiting apoptosis. Decreased intracellular HMGB1 and increased pro-apoptotic Beclin1 and Atg5 were found in the intestinal tissue of patients with IBD, favoring IEC apoptosis [367]. Other autophagy-related proteins are related to the induction of apoptosis in IBD. Mice with a deletion of ATG14 develop villus atrophy and TNF-induced apoptosis of the IECs [109].
In addition, loss of ATG7 leads to p53-induced apoptosis of the intestinal stem cells [153] due to high oxidative stress and impairment of DNA repair.
Dapagliflozin, a widely used anti-diabetic drug, was recently shown to attenuate TNBS-induced rat colitis. The drug increased colonic autophagy by upregulating Beclin1 and downregulating p62 SQSTM1 expression. At the same time, dapagliflozin reduced colonic apoptosis by reducing caspase 3 and the Bax/Bcl-2 ratio. The effect of dapagliflozin against colitis was achieved through activation of the AMPK/mTOR and Nrf2/HO-1 pathways [369].
TNFα is also a central regulator of the interaction between autophagy and apoptosis in IBD, as TNF-induced apoptosis of IECs has been implicated in IBD pathogenesis [370]. TNF, which is released during intestinal damage, was reported to drive apoptosis in different colitis models [13,146]. Lack of functional autophagy in Paneth cells increases sensitivity to TNF apoptosis, along with increased intestinal permeability, in toxoplasma gondii-infected mice [146]. Similarly, autophagy protects against TNF-induced apoptosis in a Helicobacter hepaticus infection model, along with IL10 blockade [13].
Interestingly, the administration of anti-TNFα led to the induction of macrophages with upregulated autophagy. Induction of these macrophages was defective in healthy subjects harboring the ATG16L1T300A variant compared with those with the wild-type allele [371]. The viability of anti-TNF-induced macrophages is additionally dependent on the autophagy-related lysosomal enzyme cathepsin S [371].
It should be noted that augmentation of apoptosis in the IECs was only observed after inflammatory cytokine stimulation or during chronic inflammation. IECs without an inflammatory challenge can counteract the lack of autophagy and exhibit normal proliferative responses [13]. Atg16L1 in IECs is also involved in the protection of Paneth cells in a model of virus-induced IBD [247], suggesting again that IEC autophagy can control inflammation-induced apoptosis. Moreover, alpinetin administration enhanced the integrity of the intestinal barrier via augmentation of autophagy and inhibition of IEC apoptosis [372].
TNFα also regulates the interaction of autophagy and apoptosis by interfering with claudin-2 expression. Claudin-2 is fine-tuned when autophagy is intact. TNF-a inhibits autophagy and induces claudin-2 overexpression, initiating apoptosis and impairment of the intestinal barrier via a leak-flux mechanism [373].
Another protein implicated in apoptosis intersects with autophagy. The X-linked inhibitor of apoptosis (XIAP) is the better studied among the many inhibitor proteins of apoptosis [374]. XIAP is the most potent anti-apoptotic protein characterized so far [375], which binds and inhibits both the initiator and executioner caspases [376]. XIAP is a physiological inhibitor of autophagy as well [377], although its significance in IBD is not certain as only 4% of male patients with early onset CD expressed an XIAP variant [378].
5. Autophagy and Treatment of IBD
All therapy efforts so far, such as anti-TNFα, are directed against the final pathogenetic stage of inflammation. An additional way to counteract inflammation in IBD is through the modulation of intestinal macrophages.
Experimental evidence has shown that several agents seem to be therapeutically effective in IBD through the induction of autophagy-mediated suppression of inflammasomes in macrophages. GL-V9 is a small molecule that activates AMPK induction of macrophage autophagy and protects against experimental colitis through the repression of the NLRP3 inflammasome [379]. Andrographolide exhibited similar protective effects on colitis, downregulating the PIK3CA-Akt1-mTOR pathway [246]. However, kynurenic acid, despite the induction of autophagy degradation of NLRP3 in macrophages, exacerbated experimental social defeat stress colitis. This was possibly due to the repression of the defensive functions of inflammasomes [380]. On the other hand, evodiamine, extracted from Evodiae Fructus, repressed the NLRP3 inflammasome via autophagic regulation of NF-κB in macrophages and alleviated DSS-induced colitis [381].
It is therefore evident that the deficiency of autophagy in macrophages is an important factor in IBD, increasing the macrophage-mediated inflammatory reaction [274,382]. AMPK-mediated induction of autophagy in intestinal macrophages alleviates DSS-induced colitis [383]. Administration of cannabis, a cannabinoid receptor 2 (CB2R) agonist, was reported to reduce the severity of IBD in patients by inducing autophagy and suppression of macrophage-mediated inflammation [384]. HU308, another CB2R agonist, attenuated the DSS-induced colitis in mice via the induction of intestinal macrophage autophagy. The favorable effects of CB2R agonists in IBD are mediated via the AMPK-mTOR pathway [385]. The induction of PINK1)/Parkin-dependent mitophagy also protects against DSS-induced colitis through inhibition of the NLRP3 inflammasome in macrophages [299]. In TNBS-induced colitis, the anti-inflammatory interleukin-33 attenuated colitis by increasing intestinal macrophage autophagy via regulation of the TLR4 signaling pathway [386]. In addition, induction of intestinal autophagy via rapamycin reduced macrophage-induced inflammation and alleviated murine colitis [282].
Several macrophage receptors and receptor regulators attenuate intestinal inflammation in IBD. Optineurin, a selective autophagy receptor, is an important regulator of cytokine secretion by macrophages [51,62,387]. It was shown that the activation of optineurin attenuated IBD via the induction of autophagy and repression of the macrophage-mediated inflammation [388]. Furthermore, the alpha7 nicotinic acetylcholine receptor (α7nAChR) protects against inflammation triggering the cholinergic anti-inflammatory pathway [389,390,391]. Nicotine, an α7nAChR agonist, suppressed the secretion of inflammatory cytokines by macrophages via the STAT system in IBD [389]. This may explain the clinical observation that smoking may protect against UC. The administration of PNU282987, another α7nAChR agonist, protected against DSS colitis through the induction of the AMPK-mTOR-autophagy pathway in intestinal macrophages [391]. On the contrary, the impairment of autophagy via the microRNA-143 inhibition of ATG2B led to increased production of macrophage-induced inflammatory cytokines and a detrimental effect on CD surgical specimens [392].
Ferroptosis is also a promising target in the treatment of IBD. The ferroptotic cells liberate damage-associated molecular patterns (DAMPs) and products of lipid peroxidation, which may induce inflammation activating the NF-kB pathway [363]. Phagocytosis of ferroptotic cells through oxidized phosphatidylethanolamine on the surface of ferroptotic cells attracts macrophages by targeting the TLR2 on macrophages [393]. Ferroptotic cells also secrete macrophage-attracting chemokines that induce macrophage-mediated inflammation [394]. The implication of NF-κB and STAT3 in ferroptosis of IECs is controversial. Activation of NF-κB inhibits ferroptosis in ovary cells upregulating the expression of GPX4, although it induces ferroptosis in glioblastoma cells via suppression of SLC7A11 [395,396]. Phosphorylated STAT3 either inhibits ferroptosis in acute lung injury by increasing SLC7A11 or induces ferroptosis in hypertensive mice by inactivating SLC7A11 [397,398]. Overactivation of NF-κB and STAT3 seems to facilitate ferroptosis in IECs through repression of the SLC7A11 and GPX4, respectively.
Interestingly, traditional medicines may alleviate IBD through modulation of ferroptosis in addition to the classic ferroptosis inhibitors. Several Chinese herbal extracts, such as astragalus polysaccharide, curculigoside, and Shaoyao decoction, suppress ferroptosis and attenuate experimental colitis [347,349,399]. A recent study has revealed that loureirin C extracted from dragon’s blood is also a ferroptotic inhibitor and may be protective in experimental murine colitis [400]. TNBS colitis may be alleviated by Xue-Jie-San (XJS) through a reduction in lipid peroxidation [401]. XJS also ameliorated clinical symptoms and histopathological damages in a rat model of colitis. It also decreased the production of the pro-inflammatory cytokines IL6, IL-17 and TNF-α and increased the anti-inflammatory cytokine IL-10. Additionally, XJS administration inhibited ferroptosis by decreasing iron overload and lipid peroxidation. XJS upregulated the SLC7A11/GSH/GPX4 antioxidant system [402].
Despite the well-proven implication of dysregulated autophagy in IBD and data from in vitro and in vivo experiments, the autophagy pathways have not been directly targeted and no molecules have been approved for treatment of UC or CD [110]. Hydroxychloroquine, an inhibitor of lysosomal degradation, was not effective when tested earlier in a small number of patients [403]. This was not surprising, as hydroxychloroquine further suppresses the already defective autophagy in IBD. On the contrary, rapamycin (sirolimus), an inhibitor of mTOR and inducer of autophagy, was tested in CD patients and was effective in certain cases [404,405], although it is unclear whether the beneficial effect is due to the induction of autophagy [406]. Few specific inducers of autophagy have been produced. The Tat-Beclin peptide produced from the Beclin1 protein is one of the few [407]. Other molecules, such as the BH3-mimetics targeting the Bcl-2 proteins that inhibit Beclin1, could be tried in the treatment of IBD but they may also activate apoptosis [408]. BH3 mimetics that either selectively target the Bcl-2–Beclin1 interaction without affecting apoptosis [409] or are cell-type selective inducers of autophagy [410] seem a promising prospect in future trials. In that respect, it should be remembered that an old drug may act, at least in part, through stimulation of autophagy. Azathioprine induces autophagy through modulation of mTORC1 and stimulation of the UPR sensor PERK [411].
A schematic summary of the interconnections of autophagy and apoptosis in the pathogenesis of IBD is presented in Figure 2.
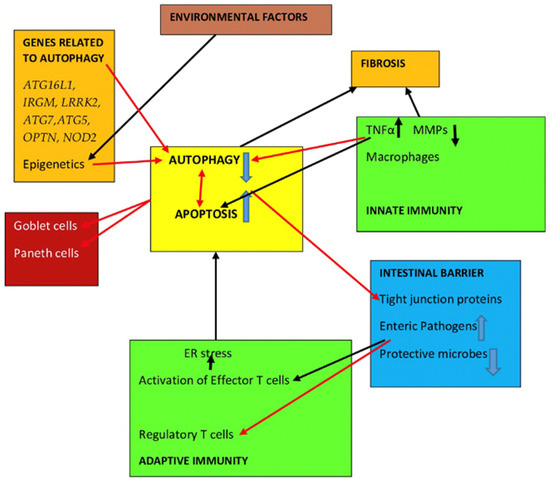
Figure 2.
A schematic overview of the effects of autophagy and apoptosis in the various pathogenetic mechanisms of IBD. Black arrows: activation; Red arrows: inhibition.
6. Conclusions
Autophagy is a dominant mechanism in the pathogenesis of inflammatory bowel disease (IBD). Although several pathogenetic aspects of both Crohn’s disease and ulcerative colitis have been extensively studied, a unifying link is still missing. Autophagy and apoptosis abnormalities and their associations could serve this purpose. In this review, we analyzed the intersections of autophagy with most other pathophysiological mechanisms implicated in IBD. In general, human and experimental evidence indicates that autophagy is dysregulated in the intestinal mucosa of patients and murine models with IBD. Autophagy impairment is strongly connected to the genetic control of several proteins implicated in autophagy and/or apoptosis. The presence of certain variants increases the susceptibility of humans to IBD. Dysregulated autophagy leads to increased apoptosis of enterocytes and impairment of the tight junction proteins of the protective intestinal barrier. This allows for the enteric pathogen microbes to have access to the immunocytes of the lamina propria and induce the secretion of pro-inflammatory cytokines and initiation of the ER stress and the unfolded protein response, damaging thus the epithelial cells. ER stress and the secretion of TNFα are the two main mediators that regulate autophagy and apoptosis in IBD. Dysregulated autophagy is the inducer of either loss of Paneth cells or the downregulated production of lysozyme and the other antimicrobial proteins. Mucus production by the goblet cells is also reduced due to defective autophagy and increased apoptosis. Mitophagy, a special form of autophagy, is also involved in IBD. The role of ferroptosis, a recently described discrete form of programmed cell death, is investigated in IBD. Taken together, the current evidence points to the central role of autophagy and apoptosis in the pathogenesis of IBD. Therapeutic exploitation of these new aspects will possibly be useful in the near future.
Author Contributions
Writing—original draft preparation, I.T. and A.V.; Writing—review and editing, I.T., A.V. and E.K.; Supervision, E.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Adams, S.M.; Close, E.D.; Shreenath, A.P. Ulcerative Colitis: Rapid Evidence Review. Am. Fam. Physician 2022, 105, 406–411. [Google Scholar]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Ray, K. IBD: The changing epidemiology of IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 690. [Google Scholar] [CrossRef]
- Ng, S.C.; Leung, W.K.; Shi, H.Y.; Li, M.K.; Leung, C.M.; Ng, C.K.; Lo, F.H.; Hui, Y.T.; Tsang, S.W.; Chan, Y.K.; et al. Epidemiology of Inflammatory Bowel Disease from 1981 to 2014: Results from a Territory-Wide Population-Based Registry in Hong Kong. Inflamm. Bowel Dis. 2016, 22, 1954–1960. [Google Scholar] [CrossRef]
- Jarmakiewicz-Czaja, S.; Zielińska, M.; Sokal, A.; Filip, R. Genetic and Epigenetic Etiology of Inflammatory Bowel Disease: An Update. Genes 2022, 13, 2388. [Google Scholar] [CrossRef]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Hansson, G.C. Membrane mucins of the intestine at a glance. J. Cell Sci. 2020, 133, jcs240929. [Google Scholar] [CrossRef]
- Yin, Y.B.; de Jonge, H.R.; Wu, X.; Yin, Y.L. Enteroids for Nutritional Studies. Mol. Nutr. Food Res. 2019, 63, e1801143. [Google Scholar] [CrossRef]
- Ho, J.; Chan, H.; Liang, Y.; Liu, X.; Zhang, L.; Li, Q.; Zhang, Y.; Zeng, J.; Ugwu, F.N.; Ho, I.H.T.; et al. Cathelicidin preserves intestinal barrier function in polymicrobial sepsis. Crit. Care. 2020, 24, 47. [Google Scholar] [CrossRef]
- Luissint, A.C.; Williams, H.C.; Kim, W.; Flemming, S.; Azcutia, V.; Hilgarth, R.S.; Leary, M.N.O.; Denning, T.L.; Nusrat, A.; Parkos, C.A. Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal. Immunol. 2019, 12, 668–678. [Google Scholar] [CrossRef]
- Gardet, A.; Xavier, R.J. Common alleles that influence autophagy and the risk for inflammatory bowel disease. Curr. Opin. Immunol. 2012, 24, 522–529. [Google Scholar] [CrossRef]
- Pott, J.; Kabat, A.M.; Maloy, K.J. Intestinal Epithelial Cell Autophagy Is Required to Protect against TNF-Induced Apoptosis during Chronic Colitis in Mice. Cell Host Microbe. 2018, 23, 191–202. [Google Scholar] [CrossRef]
- Xiong, Y.J.; Deng, Z.B.; Liu, J.N.; Qiu, J.J.; Guo, L.; Feng, P.P.; Sui, J.R.; Chen, D.P.; Guo, H.S. Enhancement of epithelial cell autophagy induced by sinensetin alleviates epithelial barrier dysfunction in colitis. Pharmacol. Res. 2019, 148, 104461. [Google Scholar] [CrossRef]
- Petrović, A.; Bogojević, D.; Korać, A.; Golić, I.; Jovanović-Stojanov, S.; Martinović, V.; Ivanović-Matić, S.; Stevanović, J.; Poznanović, G.; Grigorov, I. Oxidative stress-dependent contribution of HMGB1 to the interplay between apoptosis and autophagy in diabetic rat liver. J. Physiol. Biochem. 2017, 73, 511–521. [Google Scholar] [CrossRef]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; McGovern, D.P.; Hamill, A.M.; Ichikawa, R.; Kanazawa, Y.; Luu, J.; Kumagai, K.; Cilluffo, M.; Fukata, M.; et al. Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis. J. Immunol. 2017, 198, 2133–2146. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Ouellette, A.J. Paneth cells and innate mucosal immunity. Curr. Opin. Gastroenterol. 2010, 26, 547–553. [Google Scholar] [CrossRef]
- Paulus, G.L.; Xavier, R.J. Autophagy and checkpoints for intracellular pathogen defense. Curr. Opin. Gastroenterol. 2015, 31, 14–23. [Google Scholar] [CrossRef]
- Salzman, N.H.; Bevins, C.L. Dysbiosis—A consequence of Paneth cell dysfunction. Semin. Immunol. 2013, 25, 334–341. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Harnett, M.M.; Pineda, M.A.; Latré de Laté, P.; Eason, R.J.; Besteiro, S.; Harnett, W.; Langsley, G. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed. J. 2017, 40, 9–22. [Google Scholar] [CrossRef]
- Corona Velazquez, A.F.; Jackson, W.T. So Many Roads: The Multifaceted Regulation of Autophagy Induction. Mol. Cell Biol. 2018, 38, e00303-18. [Google Scholar] [CrossRef]
- Liang, N.; He, Q.; Liu, X.; Sun, H. Multifaceted roles of ATM in autophagy: From nonselective autophagy to selective autophagy. Cell Biochem. Funct. 2019, 37, 177–184. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Shahrabi, S.; Paridar, M.; Zeinvand-Lorestani, M.; Jalili, A.; Zibara, K.; Abdollahi, M.; Khosravi, A. Autophagy regulation and its role in normal and malignant hematopoiesis. J. Cell Physiol. 2019, 234, 21746–21757. [Google Scholar] [CrossRef]
- Thorburn, A. Autophagy and disease. J. Biol. Chem. 2018, 293, 5425–5430. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef]
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A Lysosome-Dependent Process with Implications in Cellular Redox Homeostasis and Human Disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Patel, T.; Gores, G.J.; Kaufmann, S.H. The role of proteases during apoptosis. FASEB J. 1996, 10, 587–597. [Google Scholar] [CrossRef]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak Pores: Are We Closing the Circle? Trends Cell Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef]
- Woo, S.M.; Kwon, T.K. E3 ubiquitin ligases and deubiquitinases as modulators of TRAIL-mediated extrinsic apoptotic signaling pathway. BMB Rep. 2019, 52, 119–126. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Kanai, T.; Mikami, Y.; Yoneno, K.; Matsuoka, K.; Hibi, T. Immune aspects of the pathogenesis of inflammatory bowel disease. Pharmacol. Ther. 2013, 137, 283–297. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Yao, S.J.; Das, S.; Guma, M. Dysregulation of Intestinal Epithelial Cell RIPK Pathways Promotes Chronic Inflammation in the IBD Gut. Front. Immunol. 2019, 10, 1094. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef]
- Ocansey, D.K.W.; Yuan, J.; Wei, Z.; Mao, F.; Zhang, Z. Role of ferroptosis in the pathogenesis and as a therapeutic target of inflammatory bowel disease (Review). Int. J. Mol. Med. 2023, 51, 53. [Google Scholar] [CrossRef]
- Mayr, L.; Grabherr, F.; Schwärzler, J.; Reitmeier, I.; Sommer, F.; Gehmacher, T.; Niederreiter, L.; He, G.W.; Ruder, B.; Kunz, K.T.R.; et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat. Commun. 2020, 11, 1775. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Xu, S.; He, Y.; Lin, L.; Chen, P.; Chen, M.; Zhang, S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021, 12, 289. [Google Scholar] [CrossRef]
- Bano, I.; Horky, P.; Abbas, S.Q.; Majid, M.; Bilal, A.H.M.; Ali, F.; Behl, T.; Hassan, S.S.U.; Bungau, S. Ferroptosis: A New Road towards Cancer Management. Molecules 2022, 27, 2129. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, T.; Wu, H. Emerging Pathological Engagement of Ferroptosis in Gut Diseases. Oxid. Med. Cell Longev. 2021, 2021, 4246255. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021, 28, 2843–2856. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Huo, C.; Li, G.; Hu, Y.; Sun, H. The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. Int. J. Mol. Sci. 2022, 23, 14195. [Google Scholar] [CrossRef]
- Lai, B.; Wu, C.H.; Wu, C.Y.; Luo, S.F.; Lai, J.H. Ferroptosis and Autoimmune Diseases. Front. Immunol. 2022, 13, 916664. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Jia, L.; Dourmashkin, R.R.; Allen, P.D.; Gray, A.B.; Newland, A.C.; Kelsey, S.M. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 1997, 98, 673–685. [Google Scholar] [CrossRef]
- Thorburn, J.; Moore, F.; Rao, A.; Barclay, W.W.; Thomas, L.R.; Grant, K.W.; Cramer, S.D.; Thorburn, A. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol. Biol. Cell. 2005, 16, 1189–1199. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Booth, L.A.; Roberts, J.L.; Dent, P. The role of cell signaling in the crosstalk between autophagy and apoptosis in the regulation of tumor cell survival in response to sorafenib and neratinib. Semin. Cancer Biol. 2020, 66, 129–139. [Google Scholar] [CrossRef]
- Hooper, K.M.; Barlow, P.G.; Stevens, C.; Henderson, P. Inflammatory Bowel Disease Drugs: A Focus on Autophagy. J. Crohns Colitis. 2017, 11, 118–127. [Google Scholar] [CrossRef]
- Katsandegwaza, B.; Horsnell, W.; Smith, K. Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. Int. J. Mol. Sci. 2022, 23, 9344. [Google Scholar] [CrossRef]
- Catana, C.S.; Magdas, C.; Tabaran, F.A.; Crăciun, E.C.; Deak, G.; Magdaş, V.A.; Cozma, V.; Gherman, C.M.; Berindan-Neagoe, I.; Dumitraşcu, D.L. Comparison of two models of inflammatory bowel disease in rats. Adv. Clin. Exp. Med. 2018, 27, 599–607. [Google Scholar] [CrossRef]
- Oh, S.Y.; Cho, K.A.; Kang, J.L.; Kim, K.H.; Woo, S.Y. Comparison of experimental mouse models of inflammatory bowel disease. Int. J. Mol. Med. 2014, 33, 333–340. [Google Scholar] [CrossRef]
- Silva, I.; Solas, J.; Pinto, R.; Mateus, V. Chronic Experimental Model of TNBS-Induced Colitis to Study Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 4739. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef]
- Taylor, G.A. IRG proteins: Key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007, 9, 1099–1107. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Lassen, K.G.; McKenzie, C.I.; Mari, M.; Murano, T.; Begun, J.; Baxt, L.A.; Goel, G.; Villablanca, E.J.; Kuo, S.Y.; Huang, H.; et al. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity 2016, 44, 1392–1405. [Google Scholar] [CrossRef]
- Morgan, A.R.; Lam, W.J.; Han, D.Y.; Fraser, A.G.; Ferguson, L.R. Association Analysis of ULK1 with Crohn’s Disease in a New Zealand Population. Gastroenterol. Res. Pract. 2012, 2012, 715309. [Google Scholar] [CrossRef]
- de Lange, K.M.; Barrett, J.C. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 2015, 64, 91–100. [Google Scholar] [CrossRef]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Murano, T.; Najibi, M.; Paulus, G.L.C.; Adiliaghdam, F.; Valencia-Guerrero, A.; Selig, M.; Wang, X.; Jeffrey, K.; Xavier, R.J.; Lassen, K.G.; et al. Transcription factor TFEB cell-autonomously modulates susceptibility to intestinal epithelial cell injury in vivo. Sci. Rep. 2017, 7, 13938. [Google Scholar] [CrossRef]
- Smith, A.M.; Sewell, G.W.; Levine, A.P.; Chew, T.S.; Dunne, J.; O’Shea, N.R.; Smith, P.J.; Harrison, P.J.; Macdonald, C.M.; Bloom, S.L.; et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology 2015, 144, 45–55. [Google Scholar] [CrossRef]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Peter, I.; Tajima, M.; Nakamura, S.; Strober, W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10, eaan8162. [Google Scholar] [CrossRef]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef]
- Saha, K.; Subramenium Ganapathy, A.; Wang, A.; Michael Morris, N.; Suchanec, E.; Ding, W.; Yochum, G.; Koltun, W.; Nighot, M.; Ma, T.; et al. Autophagy Reduces the Degradation and Promotes Membrane Localization of Occludin to Enhance the Intestinal Epithelial Tight Junction Barrier against Paracellular Macromolecule Flux. J. Crohns Colitis 2023, 17, 433–449. [Google Scholar] [CrossRef]
- Wong, M.; Ganapathy, A.S.; Suchanec, E.; Laidler, L.; Ma, T.; Nighot, P. Intestinal epithelial tight junction barrier regulation by autophagy-related protein ATG6/beclin 1. Am. J. Physiol. Cell Physiol. 2019, 316, 753–765. [Google Scholar] [CrossRef]
- Jung, H.; Leal-Ekman, J.S.; Lu, Q.; Stappenbeck, T.S. Atg14 protects the intestinal epithelium from TNF-triggered villus atrophy. Autophagy 2019, 15, 1990–2001. [Google Scholar] [CrossRef]
- Tran, S.; Juliani, J.; Fairlie, W.D.; Lee, E.F. The emerging roles of autophagy in intestinal epithelial cells and its links to inflammatory bowel disease. Biochem. Soc. Trans. 2023, 51, 811–826. [Google Scholar] [CrossRef]
- Girardelli, M.; Basaldella, F.; Paolera, S.D.; Vuch, J.; Tommasini, A.; Martelossi, S.; Crovella, S.; Bianco, A.M. Genetic profile of patients with early onset inflammatory bowel disease. Gene 2018, 645, 18–29. [Google Scholar] [CrossRef]
- Roberts, R.L.; Gearry, R.B.; Hollis-Moffatt, J.E.; Miller, A.L.; Reid, J.; Abkevich, V.; Timms, K.M.; Gutin, A.; Lanchbury, J.S.; Merriman, T.R.; et al. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn’s disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am. J. Gastroenterol. 2007, 102, 2754–2761. [Google Scholar] [CrossRef]
- Yamazaki, K.; Onouchi, Y.; Takazoe, M.; Kubo, M.; Nakamura, Y.; Hata, A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with Crohn’s disease in Japanese patients. J. Hum. Genet. 2007, 52, 575–583. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Liu, H.; Gao, P.; Jia, B.; Lu, N.; Zhu, B.; Zhang, F. IBD-Associated Atg16L1T300A Polymorphism Regulates Commensal Microbiota of the Intestine. Front. Immunol. 2022, 12, 772189. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; van de Veerdonk, F.L.; de Jong, D.J.; Philpott, D.J.; van der Meer, J.W.; Girardin, S.E.; Joosten, L.A.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–12235. [Google Scholar] [CrossRef]
- Samie, M.; Lim, J.; Verschueren, E.; Baughman, J.M.; Peng, I.; Wong, A.; Kwon, Y.; Senbabaoglu, Y.; Hackney, J.A.; Keir, M.; et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 2018, 19, 246–254. [Google Scholar] [CrossRef]
- Nuij, V.J.A.A.; Peppelenbosch, M.P.; van der Woude, C.J.; Fuhler, G.M. Genetic polymorphism in ATG16L1 gene is associated with adalimumab use in inflammatory bowel disease. J. Transl. Med. 2017, 15, 248. [Google Scholar] [CrossRef]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef]
- Wang, C.; Gong, G.; Sheh, A.; Muthupalani, S.; Bryant, E.M.; Puglisi, D.A.; Holcombe, H.; Conaway, E.A.; Parry, N.A.P.; Bakthavatchalu, V.; et al. Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol. 2017, 10, 1504–1517. [Google Scholar] [CrossRef]
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschenthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Häsler, R.; et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp. Med. 2018, 215, 2868–2886. [Google Scholar] [CrossRef]
- Slowicka, K.; Serramito-Gómez, I.; Boada-Romero, E.; Martens, A.; Sze, M.; Petta, I.; Vikkula, H.K.; De Rycke, R.; Parthoens, E.; Lippens, S.; et al. Physical and functional interaction between A20 and ATG16L1-WD40 domain in the control of intestinal homeostasis. Nat. Commun. 2019, 10, 1834. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in infection. Curr. Opin. Cell Biol. 2010, 22, 252–262. [Google Scholar] [CrossRef]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef]
- Palomino-Morales, R.J.; Oliver, J.; Gómez-García, M.; López-Nevot, M.A.; Rodrigo, L.; Nieto, A.; Alizadeh, B.Z.; Martín, J. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: A meta-analysis approach. Genes Immun. 2009, 10, 356–364. [Google Scholar] [CrossRef]
- Rufini, S.; Ciccacci, C.; Di Fusco, D.; Ruffa, A.; Pallone, F.; Novelli, G.; Biancone, L.; Borgiani, P. Autophagy and inflammatory bowel disease: Association between variants of the autophagy-related IRGM gene and susceptibility to Crohn’s disease. Dig. Liver Dis. 2015, 47, 744–750. [Google Scholar] [CrossRef]
- Moon, C.M.; Shin, D.J.; Kim, S.W.; Son, N.H.; Park, A.; Park, B.; Jung, E.S.; Kim, E.S.; Hong, S.P.; Kim, T.I.; et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm. Bowel Dis. 2013, 19, 106–114. [Google Scholar] [CrossRef]
- Rogala, A.R.; Schoenborn, A.A.; Fee, B.E.; Cantillana, V.A.; Joyce, M.J.; Gharaibeh, R.Z.; Roy, S.; Fodor, A.A.; Sartor, R.B.; Taylor, G.A.; et al. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis. Model. Mech. 2018, 11, dmm031070. [Google Scholar] [CrossRef]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef]
- Rolhion, N.; Darfeuille-Michaud, A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 1277–1283. [Google Scholar] [CrossRef]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hébuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Simon, T.G.; Van Der Sloot, K.W.J.; Chin, S.B.; Joshi, A.D.; Lochhead, P.; Ananthakrishnan, A.N.; Xavier, R.; Chung, R.T.; Khalili, H. IRGM Gene Variants Modify the Relationship Between Visceral Adipose Tissue and NAFLD in Patients With Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 2247–2257. [Google Scholar] [CrossRef]
- Thachil, E.; Hugot, J.P.; Arbeille, B.; Paris, R.; Grodet, A.; Peuchmaur, M.; Codogno, P.; Barreau, F.; Ogier-Denis, E.; Berrebi, D.; et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology 2012, 142, 1097–1099.e4. [Google Scholar] [CrossRef]
- Cheluvappa, R.; Luo, A.S.; Grimm, M.C. Autophagy suppression by appendicitis and appendectomy protects against colitis. Inflamm. Bowel Dis. 2014, 20, 847–855. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef]
- Zhang, F.R.; Huang, W.; Chen, S.M.; Sun, L.D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.X.; Yang, H.T.; Yang, R.D.; et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef]
- Toledo Pinto, T.G.; Batista-Silva, L.R.; Medeiros, R.C.A.; Lara, F.A.; Moraes, M.O. Type I Interferons, Autophagy and Host Metabolism in Leprosy. Front. Immunol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef]
- Fujishima, Y.; Nishiumi, S.; Masuda, A.; Inoue, J.; Nguyen, N.M.; Irino, Y.; Komatsu, M.; Tanaka, K.; Kutsumi, H.; Azuma, T.; et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-κB activation. Arch. Biochem. Biophys. 2011, 506, 223–235. [Google Scholar] [CrossRef]
- Tsuboi, K.; Nishitani, M.; Takakura, A.; Imai, Y.; Komatsu, M.; Kawashima, H. Autophagy Protects against Colitis by the Maintenance of Normal Gut Microflora and Secretion of Mucus. J. Biol. Chem. 2015, 290, 20511–20526. [Google Scholar] [CrossRef]
- Wittkopf, N.; Günther, C.; Martini, E.; Waldner, M.; Amann, K.U.; Neurath, M.F.; Becker, C. Lack of intestinal epithelial atg7 affects paneth cell granule formation but does not compromise immune homeostasis in the gut. Clin. Dev. Immunol. 2012, 2012, 278059. [Google Scholar] [CrossRef]
- Burger, E.; Araujo, A.; López-Yglesias, A.; Rajala, M.W.; Geng, L.; Levine, B.; Hooper, L.V.; Burstein, E.; Yarovinsky, F. Loss of Paneth Cell Autophagy Causes Acute Susceptibility to Toxoplasma gondii-Mediated Inflammation. Cell Host Microbe 2018, 23, 177–190.e4. [Google Scholar] [CrossRef]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.L.; Saitoh, T.; Akira, S.; Seglen, P.O.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.; Zhao, W.; He, C.; Ding, J.; Dai, R.; Xu, K.; Xiao, L.; Luo, L.; Liu, S.; et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl. Environ. Microbiol. 2018, 84, e00880-18. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, J.; Quan, W.; Lee, J.C.; Kim, M.S.; Kim, S.H.; Bae, J.W.; Hur, K.Y.; Lee, M.S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef]
- Cabrera, S.; Fernández, A.F.; Mariño, G.; Aguirre, A.; Suárez, M.F.; Español, Y.; Vega, J.A.; Laurà, R.; Fueyo, A.; Fernández-García, M.S.; et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013, 9, 1188–1200. [Google Scholar] [CrossRef]
- Benjamin, J.L.; Sumpter, R., Jr.; Levine, B.; Hooper, L.V. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 2013, 13, 723–734. [Google Scholar] [CrossRef]
- Asano, J.; Sato, T.; Ichinose, S.; Kajita, M.; Onai, N.; Shimizu, S.; Ohteki, T. Intrinsic Autophagy Is Required for the Maintenance of Intestinal Stem Cells and for Irradiation-Induced Intestinal Regeneration. Cell Rep. 2017, 20, 1050–1060. [Google Scholar] [CrossRef]
- Trentesaux, C.; Fraudeau, M.; Pitasi, C.L.; Lemarchand, J.; Jacques, S.; Duche, A.; Letourneur, F.; Naser, E.; Bailly, K.; Schmitt, A.; et al. Essential role for autophagy protein ATG7 in the maintenance of intestinal stem cell integrity. Proc. Natl. Acad. Sci. USA 2020, 117, 11136–11146. [Google Scholar] [CrossRef]
- Naama, M.; Telpaz, S.; Awad, A.; Ben-Simon, S.; Harshuk-Shabso, S.; Modilevsky, S.; Rubin, E.; Sawaed, J.; Zelik, L.; Zigdon, M.; et al. Autophagy controls mucus secretion from intestinal goblet cells by alleviating ER stress. Cell Host Microbe 2023, 31, 433–446.e4. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Zhang, H.; Zeissig, S.; Lipinski, S.; Till, A.; Jiang, T.; Stade, B.; Bromberg, Y.; Ellinghaus, E.; Keller, A.; et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 2013, 145, 339–347. [Google Scholar] [CrossRef]
- Till, A.; Lipinski, S.; Ellinghaus, D.; Mayr, G.; Subramani, S.; Rosenstiel, P.; Franke, A. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy 2013, 9, 1256–1257. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.; Adams, P.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef]
- Paiva, N.M.; Pascoal, L.B.; Negreiros, L.M.V.; Portovedo, M.; Coope, A.; Ayrizono, M.L.S.; Coy, C.S.R.; Milanski, M.; Leal, R.F. Ileal pouch of ulcerative colitis and familial adenomatous polyposis patients exhibit modulation of autophagy markers. Sci. Rep. 2018, 8, 2619. [Google Scholar] [CrossRef]
- Mimouna, S.; Bazin, M.; Mograbi, B.; Darfeuille-Michaud, A.; Brest, P.; Hofman, P.; Vouret-Craviari, V. HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC). Autophagy 2014, 10, 2333–2345. [Google Scholar] [CrossRef]
- Ortiz-Masiá, D.; Cosín-Roger, J.; Calatayud, S.; Hernández, C.; Alós, R.; Hinojosa, J.; Apostolova, N.; Alvarez, A.; Barrachina, M.D. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: Relevance in IBD. Mucosal Immunol. 2014, 7, 929–938. [Google Scholar] [CrossRef]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Chew, T.S.; O’Shea, N.R.; Sewell, G.W.; Oehlers, S.H.; Mulvey, C.M.; Crosier, P.S.; Godovac-Zimmermann, J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis. Model. Mech. 2015, 8, 817–829. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]
- Hampe, J.; Cuthbert, A.; Croucher, P.J.; Mirza, M.M.; Mascheretti, S.; Fisher, S.; Frenzel, H.; King, K.; Hasselmeyer, A.; MacPherson, A.J.; et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 2001, 357, 1925–1928. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Niess, J.H.; Klaus, J.; Stephani, J.; Pflüger, C.; Degenkolb, N.; Spaniol, U.; Mayer, B.; Lahr, G.; von Boyen, G.B. NOD2 polymorphism predicts response to treatment in Crohn’s disease--first steps to a personalized therapy. Dig. Dis. Sci. 2012, 57, 879–886. [Google Scholar] [CrossRef]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef]
- Mak, W.Y.; Zhao, M.; Ng, S.C.; Burisch, J. The epidemiology of inflammatory bowel disease: East meets west. J. Gastroenterol. Hepatol. 2020, 35, 380–389. [Google Scholar] [CrossRef]
- Brant, S.R.; Okou, D.T.; Simpson, C.L.; Cutler, D.J.; Haritunians, T.; Bradfield, J.P.; Chopra, P.; Prince, J.; Begum, F.; Kumar, A.; et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans with Inflammatory Bowel Disease. Gastroenterology 2017, 152, 206–217.e2. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Kaser, A.; Blumberg, R.S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011, 140, 1738–1747. [Google Scholar] [CrossRef]
- Xavier, R.J.; Huett, A.; Rioux, J.D. Autophagy as an important process in gut homeostasis and Crohn’s disease pathogenesis. Gut 2008, 57, 717–7120. [Google Scholar] [CrossRef]
- El-Khider, F.; McDonald, C. Links of Autophagy Dysfunction to Inflammatory Bowel Disease Onset. Dig. Dis. 2016, 34, 27–34. [Google Scholar] [CrossRef]
- Elliott, T.R.; Hudspith, B.N.; Rayment, N.B.; Prescott, N.J.; Petrovska, L.; Hermon-Taylor, J.; Brostoff, J.; Boussioutas, A.; Mathew, C.G.; Sanderson, J.D. Defective macrophage handling of Escherichia coli in Crohn’s disease. J. Gastroenterol. Hepatol. 2015, 30, 1265–1274. [Google Scholar] [CrossRef]
- Kernbauer, E.; Cadwell, K. Autophagy, viruses, and intestinal immunity. Curr. Opin. Gastroenterol. 2014, 30, 539–546. [Google Scholar] [CrossRef]
- Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Rocha, J.D.; Schlossmacher, M.G.; Philpott, D.J. LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat. Immunol. 2015, 16, 898–900. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W., 4th; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef]
- Smythies, L.E.; Shen, R.; Bimczok, D.; Novak, L.; Clements, R.H.; Eckhoff, D.E.; Bouchard, P.; George, M.D.; Hu, W.K.; Dandekar, S.; et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J. Biol. Chem. 2010, 285, 19593–19604. [Google Scholar] [CrossRef]
- Randall-Demllo, S.; Chieppa, M.; Eri, R. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef]
- Scharl, M.; Wojtal, K.A.; Becker, H.M.; Fischbeck, A.; Frei, P.; Arikkat, J.; Pesch, T.; Kellermeier, S.; Boone, D.L.; Weber, A.; et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm. Bowel Dis. 2012, 18, 1287–1302. [Google Scholar] [CrossRef]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef]
- Liu, E.; Van Grol, J.; Subauste, C.S. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-γ production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015, 17, 275–284. [Google Scholar] [CrossRef]
- Swidsinski, A.; Loening-Baucke, V.; Herber, A. Mucosal flora in Crohn’s disease and ulcerative colitis—An overview. J. Physiol. Pharmacol. 2009, 60, 61–71. [Google Scholar]
- Chen, G.Y.; Stappenbeck, T.S. Mucus, it is not just a static barrier. Sci. Signal. 2014, 7, 11. [Google Scholar] [CrossRef]
- Baxt, L.A.; Xavier, R.J. Role of Autophagy in the Maintenance of Intestinal Homeostasis. Gastroenterology 2015, 149, 553–562. [Google Scholar] [CrossRef]
- Deuring, J.J.; Fuhler, G.M.; Konstantinov, S.R.; Peppelenbosch, M.P.; Kuipers, E.J.; de Haar, C.; van der Woude, C.J. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut 2014, 63, 1081–1091. [Google Scholar] [CrossRef]
- Lopes, F.; Keita, Å.V.; Saxena, A.; Reyes, J.L.; Mancini, N.L.; Al Rajabi, A.; Wang, A.; Baggio, C.H.; Dicay, M.; van Dalen, R.; et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J. Biol. Chem. 2018, 293, 3073–3087. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Dai, C.; Tang, S.; Yang, X.; Li, D.; Zhao, K.; Xiao, X. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol. Toxicol. 2016, 32, 141–152. [Google Scholar] [CrossRef]
- Stengel, S.T.; Fazio, A.; Lipinski, S.; Jahn, M.T.; Aden, K.; Ito, G.; Wottawa, F.; Kuiper, J.W.P.; Coleman, O.I.; Tran, F.; et al. Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells Upon Endoplasmic Reticulum Stress. Gastroenterology 2020, 159, 1357–1374. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef]
- Moon, H.S.; Kim, B.; Gwak, H.; Suh, D.H.; Song, Y.S. Autophagy and protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 alpha kinase (eIF2α) pathway protect ovarian cancer cells from metformin-induced apoptosis. Mol. Carcinog. 2016, 55, 346–356. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Kimball, S.R.; Jefferson, L.S. Induction of REDD1 gene expression in the liver in response to endoplasmic reticulum stress is mediated through a PERK, eIF2α phosphorylation, ATF4-dependent cascade. Biochem. Biophys. Res. Commun. 2012, 427, 485–489. [Google Scholar] [CrossRef]
- Angelidou, I.; Chrysanthopoulou, A.; Mitsios, A.; Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Ritis, D.; Tsironidou, V.; Moschos, I.; Dalla, V.; et al. REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1β Inflammatory Response in Active Ulcerative Colitis. J. Immunol. 2018, 200, 3950–3961. [Google Scholar] [CrossRef]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef]
- Rather, R.A.; Bhagat, M.; Singh, S.K. Oncogenic BRAF, endoplasmic reticulum stress, and autophagy: Crosstalk and therapeutic targets in cutaneous melanoma. Mutat. Res. Rev. Mutat. Res. 2020, 785, 108321. [Google Scholar] [CrossRef]
- Diamanti, M.A.; Gupta, J.; Bennecke, M.; De Oliveira, T.; Ramakrishnan, M.; Braczynski, A.K.; Richter, B.; Beli, P.; Hu, Y.; Saleh, M.; et al. IKKα controls ATG16L1 degradation to prevent ER stress during inflammation. J. Exp. Med. 2017, 214, 423–437. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Tréton, X.; Pédruzzi, E.; Cazals-Hatem, D.; Grodet, A.; Panis, Y.; Groyer, A.; Moreau, R.; Bouhnik, Y.; Daniel, F.; Ogier-Denis, E. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology 2011, 141, 1024–1035. [Google Scholar] [CrossRef]
- Zhang, H.S.; Chen, Y.; Fan, L.; Xi, Q.L.; Wu, G.H.; Li, X.X.; Yuan, T.L.; He, S.Q.; Yu, Y.; Shao, M.L.; et al. The Endoplasmic Reticulum Stress Sensor IRE1α in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J. Biol. Chem. 2015, 290, 15327–15336. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol. Pharm. Bull. 2003, 26, 931–935. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef]
- Moretti, J.; Roy, S.; Bozec, D.; Martinez, J.; Chapman, J.R.; Ueberheide, B.; Lamming, D.W.; Chen, Z.J.; Horng, T.; Yeretssian, G.; et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 2017, 171, 809–823. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, L.; Li, X.; Zhang, X.; Liu, S.; Gao, N.; Li, L.; Gao, G.; Wei, G.; Chen, Z.; et al. Repression of Mammalian Target of Rapamycin Complex 1 Inhibits Intestinal Regeneration in Acute Inflammatory Bowel Disease Models. J. Immunol. 2015, 195, 339–346. [Google Scholar] [CrossRef]
- Gutiérrez-Martínez, I.Z.; Rubio, J.F.; Piedra-Quintero, Z.L.; Lopez-Mendez, O.; Serrano, C.; Reyes-Maldonado, E.; Salinas-Lara, C.; Betanzos, A.; Shibayama, M.; Silva-Olivares, A.; et al. mTORC1 Prevents Epithelial Damage During Inflammation and Inhibits Colitis-Associated Colorectal Cancer Development. Transl. Oncol. 2019, 12, 24–35. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, R.; Qiang, Z.; Yang, W.; Cao, J.; Zeng, H. Exogenous H2S Protects Colon Cells in Ulcerative Colitis by Inhibiting NLRP3 and Activating Autophagy. DNA Cell Biol. 2021, 40, 748–756. [Google Scholar] [CrossRef]
- Deng, J.; Zeng, L.; Lai, X.; Li, J.; Liu, L.; Lin, Q.; Chen, Y. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J. Cell Mol. Med. 2018, 22, 546–557. [Google Scholar] [CrossRef]
- Gao, Q.; Bi, P.; Luo, D.; Guan, Y.; Zeng, W.; Xiang, H.; Mi, Q.; Yang, G.; Li, X.; Yang, B. Nicotine-induced autophagy via AMPK/mTOR pathway exerts protective effect in colitis mouse model. Chem. Biol. Interact. 2020, 317, 108943. [Google Scholar] [CrossRef]
- Villani, A.C.; Lemire, M.; Fortin, G.; Louis, E.; Silverberg, M.S.; Collette, C.; Baba, N.; Libioulle, C.; Belaiche, J.; Bitton, A.; et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 2009, 41, 71–76. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Jiang, W.; Han, Y.P.; Hu, M.; Bao, X.Q.; Yan, Y.; Chen, G. A study on regulatory mechanism of miR-223 in ulcerative colitis through PI3K/Akt-mTOR signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4865–4872. [Google Scholar] [CrossRef]
- Wang, B.; Gong, Z.; Zhan, J.; Yang, L.; Zhou, Q.; Yuan, X. Xianglian Pill Suppresses Inflammation and Protects Intestinal Epithelial Barrier by Promoting Autophagy in DSS Induced Ulcerative Colitis Mice. Front. Pharmacol. 2021, 11, 594847. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, W.; Niu, J.; Yang, G.; Luo, J.; Lan, D.; Wu, J.; Li, M.; Sun, Y.; Wang, K.; et al. Heat-shock transcription factor 2 promotes sodium butyrate-induced autophagy by inhibiting mTOR in ulcerative colitis. Exp. Cell Res. 2020, 388, 111820. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, Y.; Ke, X.; Xuan, L.; Ma, Z. RNF8 induces autophagy and reduces inflammation by promoting AKT degradation via ubiquitination in ulcerative colitis mice. J. Biochem. 2020, 168, 445–453. [Google Scholar] [CrossRef]
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.P.; Hetz, C.; Atkin, J.D.; et al. NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol. Biol. Cell. 2016, 27, 1712–1727. [Google Scholar] [CrossRef]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef]
- Gerster, R.; Eloranta, J.J.; Hausmann, M.; Ruiz, P.A.; Cosin-Roger, J.; Terhalle, A.; Ziegler, U.; Kullak-Ublick, G.A.; von Eckardstein, A.; Rogler, G. Anti-inflammatory Function of High-Density Lipoproteins via Autophagy of IκB Kinase. Cell Mol. Gastroenterol. Hepatol. 2014, 1, 171–187. [Google Scholar] [CrossRef]
- Shibata, Y.; Oyama, M.; Kozuka-Hata, H.; Han, X.; Tanaka, Y.; Gohda, J.; Inoue, J. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat. Commun. 2012, 3, 1061. [Google Scholar] [CrossRef]
- Honjo, H.; Watanabe, T.; Arai, Y.; Kamata, K.; Minaga, K.; Komeda, Y.; Yamashita, K.; Kudo, M. ATG16L1 negatively regulates RICK/RIP2-mediated innate immune responses. Int. Immunol. 2021, 33, 91–105. [Google Scholar] [CrossRef]
- Gao, P.; Liu, H.; Huang, H.; Sun, Y.; Jia, B.; Hou, B.; Zhou, X.; Strober, W.; Zhang, F. The Crohn Disease-associated ATG16L1T300A polymorphism regulates inflammatory responses by modulating TLR- and NLR-mediated signaling. Autophagy 2022, 18, 2561–2575. [Google Scholar] [CrossRef]
- Wang, D.; Shao, S.; Zhang, Y.; Zhao, D.; Wang, M. Insight Into Polysaccharides From Panax ginseng C. A. Meyer in Improving Intestinal Inflammation: Modulating Intestinal Microbiota and Autophagy. Front. Immunol. 2021, 12, 683911. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A. Molecular Interactions Between Reactive Oxygen Species and Autophagy in Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3791. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Zheng, Z.; Luo, L.; Wang, P.; Liu, K.; Namani, A.; Jiang, Z.; Wang, X.J.; Tang, X. Mkp-1 cross-talks with Nrf2/Ho-1 pathway protecting against intestinal inflammation. Free Radic. Biol. Med. 2018, 124, 541–549. [Google Scholar] [CrossRef]
- Pompili, S.; Sferra, R.; Gaudio, E.; Viscido, A.; Frieri, G.; Vetuschi, A.; Latella, G. Can Nrf2 Modulate the Development of Intestinal Fibrosis and Cancer in Inflammatory Bowel Disease? Int. J. Mol. Sci. 2019, 20, 4061. [Google Scholar] [CrossRef]
- Farzaei, M.H.; El-Senduny, F.F.; Momtaz, S.; Parvizi, F.; Iranpanah, A.; Tewari, D.; Naseri, R.; Abdolghaffari, A.H.; Rezaei, N. An update on dietary consideration in inflammatory bowel disease: Anthocyanins and more. Expert. Rev. Gastroenterol. Hepatol. 2018, 12, 1007–1024. [Google Scholar] [CrossRef]
- Fu, X.Y. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006, 16, 214–219. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Zhu, X.; Lu, R.; Messer, J.S.; Xia, Y.; Chang, E.B.; Sun, J. Intestinal epithelial HMGB1 inhibits bacterial infection via STAT3 regulation of autophagy. Autophagy 2019, 15, 1935–1953. [Google Scholar] [CrossRef]
- Yuan, Y.; Ding, D.; Zhang, N.; Xia, Z.; Wang, J.; Yang, H.; Guo, F.; Li, B. TNF-α induces autophagy through ERK1/2 pathway to regulate apoptosis in neonatal necrotizing enterocolitis model cells IEC-6. Cell Cycle. 2018, 17, 1390–1402. [Google Scholar] [CrossRef]
- Kubota, M.; Kakimoto, K.; Nakagawa, T.; Koubayashi, E.; Nakazawa, K.; Tawa, H.; Hirata, Y.; Okada, T.; Kawakami, K.; Asai, A.; et al. Autophagy deficiency exacerbates colitis through excessive oxidative stress and MAPK signaling pathway activation. PLoS ONE 2019, 14, e0225066. [Google Scholar] [CrossRef]
- Chen, S.L.; Li, C.M.; Li, W.; Liu, Q.S.; Hu, S.Y.; Zhao, M.Y.; Hu, D.S.; Hao, Y.W.; Zeng, J.H.; Zhang, Y. How autophagy, a potential therapeutic target, regulates intestinal inflammation. Front. Immunol. 2023, 14, 1087677. [Google Scholar] [CrossRef]
- Zhang, C.; Yan, J.; Xiao, Y.; Shen, Y.; Wang, J.; Ge, W.; Chen, Y. Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers. Int. J. Mol. Sci. 2017, 18, 157. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Z.; Yang, X.; Li, T.; Liu, M.; Tang, H. miR-346 functions as a pro-survival factor under ER stress by activating mitophagy. Cancer Lett. 2018, 413, 69–81. [Google Scholar] [CrossRef]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Zuo, Z.; Zhang, Q.; Pan, Y.; Zeng, B.; Li, W.; Wei, H.; Liu, Z. Rip2 Is Required for Nod2-Mediated Lysozyme Sorting in Paneth Cells. J. Immunol. 2017, 198, 3729–3736. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, Y.; Yan, R.; Zeng, B.; Wang, H.; Zhang, X.; Li, W.; Wei, H.; Liu, Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015, 16, 918–926. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Liu, C. Intestinal Autophagy and Its Pharmacological Control in Inflammatory Bowel Disease. Front. Immunol. 2017, 7, 695. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Whang, M.I.; Tavares, R.M.; Benjamin, D.I.; Kattah, M.G.; Advincula, R.; Nomura, D.K.; Debnath, J.; Malynn, B.A.; Ma, A. The Ubiquitin Binding Protein TAX1BP1 Mediates Autophagasome Induction and the Metabolic Transition of Activated T Cells. Immunity 2017, 46, 405–420. [Google Scholar] [CrossRef]
- Jia, W.; He, Y.W. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J. Immunol. 2011, 186, 5313–5322. [Google Scholar] [CrossRef]
- Kabat, A.M.; Harrison, O.J.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.F.; Laing, A.; Abeler-Dörner, L.; Forman, S.P.; Grencis, R.K.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife 2016, 5, 12444. [Google Scholar] [CrossRef]
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.G.; Windle, J.J.; Zhang, X.; Lu, B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152. [Google Scholar] [CrossRef]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P.; et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife 2014, 3, 03706. [Google Scholar] [CrossRef]
- Schlie, K.; Westerback, A.; DeVorkin, L.; Hughson, L.R.; Brandon, J.M.; MacPherson, S.; Gadawski, I.; Townsend, K.N.; Poon, V.I.; Elrick, M.A.; et al. Survival of effector CD8+ T cells during influenza infection is dependent on autophagy. J. Immunol. 2015, 194, 4277–4286. [Google Scholar] [CrossRef]
- Wildenberg, M.E.; Vos, A.C.; Wolfkamp, S.C.; Duijvestein, M.; Verhaar, A.P.; Te Velde, A.A.; van den Brink, G.R.; Hommes, D.W. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology 2012, 142, 1493–1503. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef]
- Shen, Y.; Giardino Torchia, M.L.; Lawson, G.W.; Karp, C.L.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef]
- Qiao, D.; Zhang, Z.; Zhang, Y.; Chen, Q.; Chen, Y.; Tang, Y.; Sun, X.; Tang, Z.; Dai, Y. Regulation of Endoplasmic Reticulum Stress-Autophagy: A Potential Therapeutic Target for Ulcerative Colitis. Front. Pharmacol. 2021, 12, 697360. [Google Scholar] [CrossRef]
- Hu, X.; Deng, J.; Yu, T.; Chen, S.; Ge, Y.; Zhou, Z.; Guo, Y.; Ying, H.; Zhai, Q.; Chen, Y.; et al. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology 2019, 156, 1098–1111. [Google Scholar] [CrossRef]
- Vernia, F.; Valvano, M.; Longo, S.; Cesaro, N.; Viscido, A.; Latella, G. Vitamin D in Inflammatory Bowel Diseases. Mechanisms of Action and Therapeutic Implications. Nutrients 2022, 14, 269. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.O.; Claud, E.C.; Chen, D.; Chang, E.B.; Carmeliet, G.; et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2015, 64, 1082–1094. [Google Scholar] [CrossRef]
- Battistini, C.; Ballan, R.; Herkenhoff, M.E.; Saad, S.M.I.; Sun, J. Vitamin D Modulates Intestinal Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2020, 22, 362. [Google Scholar] [CrossRef]
- Sun, J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy 2016, 12, 1057–1058. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, Y.G.; Xia, Y.; Sun, J. Imbalance of autophagy and apoptosis in intestinal epithelium lacking the vitamin D receptor. FASEB J. 2019, 33, 11845–11856. [Google Scholar] [CrossRef]
- Zhu, T.; Liu, T.J.; Shi, Y.Y.; Zhao, Q. Vitamin D/VDR signaling pathway ameliorates 2,4,6-trinitrobenzene sulfonic acid-induced colitis by inhibiting intestinal epithelial apoptosis. Int. J. Mol. Med. 2015, 35, 1213–1218. [Google Scholar] [CrossRef]
- Li, W.; Lin, Y.; Luo, Y.; Wang, Y.; Lu, Y.; Li, Y.; Guo, H. Vitamin D Receptor Protects against Radiation-Induced Intestinal Injury in Mice via Inhibition of Intestinal Crypt Stem/Progenitor Cell Apoptosis. Nutrients 2021, 13, 2910. [Google Scholar] [CrossRef]
- Bakke, D.; Sun, J. Ancient Nuclear Receptor VDR With New Functions: Microbiome and Inflammation. Inflamm. Bowel Dis. 2018, 24, 1149–1154. [Google Scholar] [CrossRef]
- Law, A.D.; Dutta, U.; Kochhar, R.; Vaishnavi, C.; Kumar, S.; Noor, T.; Bhadada, S.; Singh, K. Vitamin D deficiency in adult patients with ulcerative colitis: Prevalence and relationship with disease severity, extent, and duration. Indian. J. Gastroenterol. 2019, 38, 6–14. [Google Scholar] [CrossRef]
- Wu, M.Y.; Liu, L.; Wang, E.J.; Xiao, H.T.; Cai, C.Z.; Wang, J.; Su, H.; Wang, Y.; Tan, J.; Zhang, Z.; et al. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy 2021, 17, 1096–1111. [Google Scholar] [CrossRef]
- Zhang, S.L.; Li, Z.Y.; Wang, D.S.; Xu, T.Y.; Fan, M.B.; Cheng, M.H.; Miao, C.Y. Aggravated ulcerative colitis caused by intestinal Metrnl deficiency is associated with reduced autophagy in epithelial cells. Acta Pharmacol. Sin. 2020, 41, 763–770. [Google Scholar] [CrossRef]
- Shao, B.Z.; Yao, Y.; Zhai, J.S.; Zhu, J.H.; Li, J.P.; Wu, K. The Role of Autophagy in Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 621132. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghosh, S.; Sinha, K.; Chowdhury, S.; Sil, P.C. Sulphur dioxide ameliorates colitis related pathophysiology and inflammation. Toxicology 2019, 412, 63–78. [Google Scholar] [CrossRef]
- Shen, T.; Li, S.; Cai, L.D.; Liu, J.L.; Wang, C.Y.; Gan, W.J.; Li, X.M.; Wang, J.R.; Sun, L.N.; Deng, M.; et al. Erbin exerts a protective effect against inflammatory bowel disease by suppressing autophagic cell death. Oncotarget 2018, 9, 12035–12049. [Google Scholar] [CrossRef]
- Hausmann, M.; Obermeier, F.; Schreiter, K.; Spottl, T.; Falk, W.; Schölmerich, J.; Herfarth, H.; Saftig, P.; Rogler, G. Cathepsin D is up-regulated in inflammatory bowel disease macrophages. Clin. Exp. Immunol. 2004, 136, 157–167. [Google Scholar] [CrossRef]
- Fischbeck, A.; Leucht, K.; Frey-Wagner, I.; Bentz, S.; Pesch, T.; Kellermeier, S.; Krebs, M.; Fried, M.; Rogler, G.; Hausmann, M.; et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut 2011, 60, 55–65. [Google Scholar] [CrossRef]
- Menzel, K.; Hausmann, M.; Obermeier, F.; Schreiter, K.; Dunger, N.; Bataille, F.; Falk, W.; Scholmerich, J.; Herfarth, H.; Rogler, G. Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin. Exp. Immunol. 2006, 146, 169–180. [Google Scholar] [CrossRef]
- Macias-Ceja, D.C.; Cosín-Roger, J.; Ortiz-Masiá, D.; Salvador, P.; Hernández, C.; Esplugues, J.V.; Calatayud, S.; Barrachina, M.D. Stimulation of autophagy prevents intestinal mucosal inflammation and ameliorates murine colitis. Br. J. Pharmacol. 2017, 174, 2501–2511. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Canet, F.; Macias-Ceja, D.C.; Gisbert-Ferrándiz, L.; Ortiz-Masiá, D.; Esplugues, J.V.; Alós, R.; Navarro, F.; Barrachina, M.D.; Calatayud, S. Autophagy Stimulation as a Potential Strategy against Intestinal Fibrosis. Cells 2019, 8, 1078. [Google Scholar] [CrossRef]
- Salvador, P.; Macías-Ceja, D.C.; Gisbert-Ferrándiz, L.; Hernández, C.; Bernardo, D.; Alós, R.; Navarro-Vicente, F.; Esplugues, J.V.; Ortiz-Masiá, D.; Barrachina, M.D.; et al. CD16+ Macrophages Mediate Fibrosis in Inflammatory Bowel Disease. J. Crohns Colitis. 2018, 12, 589–599. [Google Scholar] [CrossRef]
- Mathur, R.; Alam, M.M.; Zhao, X.F.; Liao, Y.; Shen, J.; Morgan, S.; Huang, T.; Lee, H.; Lee, E.; Huang, Y.; et al. Induction of autophagy in Cx3cr1+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol. 2019, 12, 612–623. [Google Scholar] [CrossRef]
- Butera, A.; Quaranta, M.T.; Crippa, L.; Spinello, I.; Saulle, E.; Di Carlo, N.; Campanile, D.; Boirivant, M.; Labbaye, C. CD147 Targeting by AC-73 Induces Autophagy and Reduces Intestinal Fibrosis Associated with TNBS Chronic Colitis. J. Crohns Colitis. 2022, 16, 1751–1761. [Google Scholar] [CrossRef]
- Haq, S.; Grondin, J.; Banskota, S.; Khan, W.I. Autophagy: Roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci. 2019, 26, 19. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, L.; Wang, B.; Sun, Q.; Zhao, P.; Li, W. The role of autophagy in maintaining intestinal mucosal barrier. J. Cell Physiol. 2019, 234, 19406–19419. [Google Scholar] [CrossRef]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating Mitophagy in Mitochondrial Disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef]
- Kathiria, A.S.; Butcher, L.D.; Feagins, L.A.; Souza, R.F.; Boland, C.R.; Theiss, A.L. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS ONE 2012, 7, e31231. [Google Scholar] [CrossRef]
- Vincent, G.; Novak, E.A.; Siow, V.S.; Cunningham, K.E.; Griffith, B.D.; Comerford, T.E.; Mentrup, H.L.; Stolz, D.B.; Loughran, P.; Ranganathan, S.; et al. Nix-Mediated Mitophagy Modulates Mitochondrial Damage During Intestinal Inflammation. Antioxid. Redox Signal. 2020, 33, 1–19. [Google Scholar] [CrossRef]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef]
- Chetcuti Zammit, S.; Ellul, P.; Girardin, G.; Valpiani, D.; Nielsen, K.R.; Olsen, J.; Goldis, A.; Lazar, D.; Shonová, O.; Nováková, M.; et al. Vitamin D deficiency in a European inflammatory bowel disease inception cohort: An Epi-IBD study. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1297–1303. [Google Scholar] [CrossRef]
- Ma, Z.; Wu, J.; Wu, Y.; Sun, X.; Rao, Z.; Sun, N.; Fu, Y.; Zhang, Z.; Li, J.; Xiao, M.; et al. Parkin increases the risk of colitis by downregulation of VDR via autophagy-lysosome degradation. Int. J. Biol. Sci. 2023, 19, 1633–1644. [Google Scholar] [CrossRef]
- Berger, E.; Rath, E.; Yuan, D.; Waldschmitt, N.; Khaloian, S.; Allgäuer, M.; Staszewski, O.; Lobner, E.M.; Schöttl, T.; Giesbertz, P.; et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016, 7, 13171. [Google Scholar] [CrossRef]
- Khaloian, S.; Rath, E.; Hammoudi, N.; Gleisinger, E.; Blutke, A.; Giesbertz, P.; Berger, E.; Metwaly, A.; Waldschmitt, N.; Allez, M.; et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 2020, 69, 1939–1951. [Google Scholar] [CrossRef]
- Rodríguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Yang, Y.; Liu, X.; Zhu, Y.; Zou, J.; Peng, S.; Le, T.H.; Chen, Y.; Zhao, S.; et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem. Pharmacol. 2018, 155, 366–379. [Google Scholar] [CrossRef]
- Mai, C.T.; Wu, M.M.; Wang, C.L.; Su, Z.R.; Cheng, Y.Y.; Zhang, X.J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol. Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef]
- Patel, J. IL-10 reprogramming of metabolism in macrophages through mitophagy. Cardiovasc. Res. 2017, 113, 40–41. [Google Scholar] [CrossRef]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef]
- Eckmann, L.; Nebelsiek, T.; Fingerle, A.A.; Dann, S.M.; Mages, J.; Lang, R.; Robine, S.; Kagnoff, M.F.; Schmid, R.M.; Karin, M.; et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc. Natl. Acad. Sci. USA. 2008, 105, 15058–15063. [Google Scholar] [CrossRef]
- Patankar, J.V.; Becker, C. Cell death in the gut epithelium and implications for chronic inflammation. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 543–556. [Google Scholar] [CrossRef]
- Iwamoto, M.; Koji, T.; Makiyama, K.; Kobayashi, N.; Nakane, P.K. Apoptosis of crypt epithelial cells in ulcerative colitis. J. Pathol. 1996, 180, 152–159. [Google Scholar] [CrossRef]
- Pedersen, J.; LaCasse, E.C.; Seidelin, J.B.; Coskun, M.; Nielsen, O.H. Inhibitors of apoptosis (IAPs) regulate intestinal immunity and inflammatory bowel disease (IBD) inflammation. Trends Mol. Med. 2014, 20, 652–665. [Google Scholar] [CrossRef]
- Parker, A.; Vaux, L.; Patterson, A.M.; Modasia, A.; Muraro, D.; Fletcher, A.G.; Byrne, H.M.; Maini, P.K.; Watson, A.J.M.; Pin, C. Elevated apoptosis impairs epithelial cell turnover and shortens villi in TNF-driven intestinal inflammation. Cell Death Dis. 2019, 10, 108. [Google Scholar] [CrossRef]
- Vetuschi, A.; Latella, G.; Sferra, R.; Caprilli, R.; Gaudio, E. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig. Dis. Sci. 2002, 47, 1447–1457. [Google Scholar] [CrossRef]
- Sträter, J.; Möller, P. Expression and function of death receptors and their natural ligands in the intestine. Ann. N. Y Acad. Sci. 2000, 915, 162–170. [Google Scholar] [CrossRef]
- Möller, P.; Walczak, H.; Reidl, S.; Sträter, J.; Krammer, P.H. Paneth cells express high levels of CD95 ligand transcripts: A unique property among gastrointestinal epithelia. Am. J. Pathol. 1996, 149, 9–13. [Google Scholar]
- Sträter, J.; Wellisch, I.; Riedl, S.; Walczak, H.; Koretz, K.; Tandara, A.; Krammer, P.H.; Möller, P. CD95 (APO-1/Fas)-mediated apoptosis in colon epithelial cells: A possible role in ulcerative colitis. Gastroenterology 1997, 113, 160–167. [Google Scholar] [CrossRef]
- Dirisina, R.; Katzman, R.B.; Goretsky, T.; Managlia, E.; Mittal, N.; Williams, D.B.; Qiu, W.; Yu, J.; Chandel, N.S.; Zhang, L.; et al. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011, 141, 1036–1045. [Google Scholar] [CrossRef]
- Sidi, S.; Sanda, T.; Kennedy, R.D.; Hagen, A.T.; Jette, C.A.; Hoffmans, R.; Pascual, J.; Imamura, S.; Kishi, S.; Amatruda, J.F.; et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008, 133, 864–877. [Google Scholar] [CrossRef]
- Greenow, K.R.; Clarke, A.R.; Jones, R.H. Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene 2009, 28, 1443–1453. [Google Scholar] [CrossRef]
- Watari, A.; Hasegawa, M.; Yagi, K.; Kondoh, M. Checkpoint Kinase 1 Activation Enhances Intestinal Epithelial Barrier Function via Regulation of Claudin-5 Expression. PLoS ONE 2016, 11, e0145631. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Li, Y.; Li, Y.; Wang, Y.; Zhu, L.; Chen, P.; Tian, Z.; Qiu, Y.; Feng, R.; et al. DNA Damage-Regulated Autophagy Modulator 1 (DRAM1) Mediates Autophagy and Apoptosis of Intestinal Epithelial Cells in Inflammatory Bowel Disease. Dig. Dis. Sci. 2021, 66, 3375–3390. [Google Scholar] [CrossRef]
- Zeissig, S.; Bojarski, C.; Buergel, N.; Mankertz, J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut 2004, 53, 1295–1302. [Google Scholar] [CrossRef]
- Lin, A.; Karin, M. NF-kappaB in cancer: A marked target. Semin. Cancer Biol. 2003, 13, 107–114. [Google Scholar] [CrossRef]
- Vereecke, L.; Beyaert, R.; van Loo, G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009, 30, 383–391. [Google Scholar] [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Qiu, W.; Wu, B.; Wang, X.; Buchanan, M.E.; Regueiro, M.D.; Hartman, D.J.; Schoen, R.E.; Yu, J.; Zhang, L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Investig. 2011, 121, 1722–1732. [Google Scholar] [CrossRef]
- Lin, W.; Ma, C.; Su, F.; Jiang, Y.; Lai, R.; Zhang, T.; Sun, K.; Fan, L.; Cai, Z.; Li, Z.; et al. Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut 2017, 66, 597–610. [Google Scholar] [CrossRef]
- Vlantis, K.; Wullaert, A.; Polykratis, A.; Kondylis, V.; Dannappel, M.; Schwarzer, R.; Welz, P.; Corona, T.; Walczak, H.; Weih, F.; et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-κB-Dependent and -Independent Functions. Immunity 2016, 44, 553–567. [Google Scholar] [CrossRef]
- Wong, J.; Garcia-Carbonell, R.; Zelic, M.; Ho, S.B.; Boland, B.S.; Yao, S.J.; Desai, S.A.; Das, S.; Planell, N.; Harris, P.A.; et al. RIPK1 Mediates TNF-Induced Intestinal Crypt Apoptosis During Chronic NF-κB Activation. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 295–312. [Google Scholar] [CrossRef]
- Yao, X.; Cadwell, K. Tumor Necrosis Factor-α-Induced Apoptosis in the Intestinal Epithelium due to Chronic Nuclear Factor Kappa B Signaling Is Mediated by Receptor Interacting Serine/Threonine Kinase 1. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 337–338. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Wong, J.; Kim, J.Y.; Close, L.A.; Boland, B.S.; Wong, T.L.; Harris, P.A.; Ho, S.B.; Das, S.; Ernst, P.B.; et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, E9192–E9200. [Google Scholar] [CrossRef]
- Kattah, M.G.; Shao, L.; Rosli, Y.Y.; Shimizu, H.; Whang, M.I.; Advincula, R.; Achacoso, P.; Shah, S.; Duong, B.H.; Onizawa, M.; et al. A20 and ABIN-1 synergistically preserve intestinal epithelial cell survival. J. Exp. Med. 2018, 215, 1839–1852. [Google Scholar] [CrossRef]
- Zaidi, D.; Huynh, H.Q.; Carroll, M.W.; Baksh, S.; Wine, E. Tumor necrosis factor α-induced protein 3 (A20) is dysregulated in pediatric Crohn disease. Clin. Exp. Gastroenterol. 2018, 11, 217–231. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Chen, Y.; Li, L.; Li, J.; Liu, Q. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 2016, 7, e2381. [Google Scholar] [CrossRef]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029–1039. [Google Scholar] [CrossRef]
- Goretsky, T.; Dirisina, R.; Sinh, P.; Mittal, N.; Managlia, E.; Williams, D.B.; Posca, D.; Ryu, H.; Katzman, R.B.; Barrett, T.A. p53 mediates TNF-induced epithelial cell apoptosis in IBD. Am. J. Pathol. 2012, 181, 1306–1315. [Google Scholar] [CrossRef]
- Li, M.; Zhang, S.; Qiu, Y.; He, Y.; Chen, B.; Mao, R.; Cui, Y.; Zeng, Z.; Chen, M. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis. 2017, 8, e2699. [Google Scholar] [CrossRef]
- Dias, C.B.; Milanski, M.; Portovedo, M.; Horita, V.; Ayrizono Mde, L.; Planell, N.; Coy, C.S.; Velloso, L.A.; Meirelles, L.R.; Leal, R.F. Defective apoptosis in intestinal and mesenteric adipose tissue of Crohn’s disease patients. PLoS ONE 2014, 9, e98547. [Google Scholar] [CrossRef]
- Itoh, J.; de La Motte, C.; Strong, S.A.; Levine, A.D.; Fiocchi, C. Decreased Bax expression by mucosal T cells favours resistance to apoptosis in Crohn’s disease. Gut 2001, 49, 35–41. [Google Scholar] [CrossRef][Green Version]
- Santaolalla, R.; Mañé, J.; Pedrosa, E.; Lorén, V.; Fernández-Bañares, F.; Mallolas, J.; Carrasco, A.; Salas, A.; Rosinach, M.; Forné, M.; et al. Apoptosis resistance of mucosal lymphocytes and IL-10 deficiency in patients with steroid-refractory Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 1490–1500. [Google Scholar] [CrossRef]
- Qian, J.; Zhao, W.; Miao, X.; Li, L.; Zhang, D. Sam68 modulates apoptosis of intestinal epithelial cells via mediating NF-κB activation in ulcerative colitis. Mol. Immunol. 2016, 75, 48–59. [Google Scholar] [CrossRef]
- Tang, B.; Zhu, J.; Fang, S.; Wang, Y.; Vinothkumar, R.; Li, M.; Weng, Q.; Zheng, L.; Yang, Y.; Qiu, R.; et al. Pharmacological inhibition of MELK restricts ferroptosis and the inflammatory response in colitis and colitis-propelled carcinogenesis. Free Radic. Biol. Med. 2021, 172, 312–329. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.; Ji, J.; Zhao, X.; Yuan, J.; Wang, H.; Lv, G. High-fat diet alleviates colitis by inhibiting ferroptosis via solute carrier family seven member 11. J. Nutr. Biochem. 2022, 109, 109106. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, P.; Chen, W.; Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 2020, 225, 9–15. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, S.; Li, X.; Huang, Y.; He, S.; Luo, L. STAT3-mediated ferroptosis is involved in ulcerative colitis. Free Radic. Biol. Med. 2022, 188, 375–385. [Google Scholar] [CrossRef]
- Cui, D.J.; Chen, C.; Yuan, W.Q.; Yang, Y.H.; Han, L. Integrative analysis of ferroptosis-related genes in ulcerative colitis. J. Int. Med. Res. 2021, 49, 3000605211042975. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, S.; Guo, N.; Li, H.; He, S. ACSF2-mediated ferroptosis is involved in ulcerative colitis. Life Sci. 2023, 313, 121272. [Google Scholar] [CrossRef]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef]
- Qian, R.; Tang, M.; Ouyang, Z.; Cheng, H.; Xing, S. Identification of ferroptosis-related genes in ulcerative colitis: A diagnostic model with machine learning. Ann. Transl. Med. 2023, 11, 177. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A Double-Edged Sword in Gastrointestinal Disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef]
- Dong, S.; Lu, Y.; Peng, G.; Li, J.; Li, W.; Li, M.; Wang, H.; Liu, L.; Zhao, Q. Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway. Dig. Liver Dis. 2021, 53, 1276–1285. [Google Scholar] [CrossRef]
- Wang, S.; Liu, W.; Wang, J.; Bai, X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sci. 2020, 259, 118356. [Google Scholar] [CrossRef]
- Rahman, M.S.; Alam, M.B.; Kim, Y.K.; Madina, M.H.; Fliss, I.; Lee, S.H.; Yoo, J.C. Activation of Nrf2/HO-1 by Peptide YD1 Attenuates Inflammatory Symptoms through Suppression of TLR4/MYyD88/NF-κB Signaling Cascade. Int. J. Mol. Sci. 2021, 22, 5161. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Li, J.; Zhu, J.; Wang, R.; Xi, Q.; Wu, H.; Shi, T.; Chen, W. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. Eur. J. Pharmacol. 2021, 911, 174518. [Google Scholar] [CrossRef]
- Mei, Y.; Wang, Z.; Zhang, Y.; Wan, T.; Xue, J.; He, W.; Luo, Y.; Xu, Y.; Bai, X.; Wang, Q.; et al. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Ameliorates DSS-Induced Colitis Against Oxidative Stress by Activating Nrf2/HO-1 Pathway. Front. Immunol. 2020, 10, 2969. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Prawan, A.; Liu, Y.; Hao, X.; Yu, S.; Cheung, W.K.; Chan, J.Y.; Reddy, B.S.; Yang, C.S.; et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. [Google Scholar] [CrossRef]
- Su, D.; Wang, X.; Ma, Y.; Hao, J.; Wang, J.; Lu, Y.; Liu, Y.; Wang, X.; Zhang, L. Nrf2-induced miR-23a-27a-24-2 cluster modulates damage repair of intestinal mucosa by targeting the Bach1/HO-1 axis in inflammatory bowel diseases. Free Radic. Biol. Med. 2021, 163, 1–9. [Google Scholar] [CrossRef]
- Stenke, E.; Aviello, G.; Singh, A.; Martin, S.; Winter, D.; Sweeney, B.; McDermott, M.; Bourke, B.; Hussey, S.; Knaus, U.G. NADPH oxidase 4 is protective and not fibrogenic in intestinal inflammation. Redox Biol. 2020, 37, 101752. [Google Scholar] [CrossRef]
- Wittig, B.M.; Sabat, R.; Holzlöhner, P.; Witte-Händel, E.; Heilmann, K.; Witte, K.; Triebus, J.; Tzankov, A.; Laman, J.D.; Bokemeyer, B.; et al. Absence of specific alternatively spliced exon of CD44 in macrophages prevents colitis. Mucosal Immunol. 2018, 11, 846–860. [Google Scholar] [CrossRef]
- de Silva, P.S.; Olsen, A.; Christensen, J.; Schmidt, E.B.; Overvaad, K.; Tjonneland, A.; Hart, A.R. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology 2010, 139, 1912–1917. [Google Scholar] [CrossRef]
- Vargas-Robles, H.; Castro-Ochoa, K.F.; Citalán-Madrid, A.F.; Schnoor, M. Beneficial effects of nutritional supplements on intestinal epithelial barrier functions in experimental colitis models in vivo. World J. Gastroenterol. 2019, 25, 4181–4198. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279. [Google Scholar] [CrossRef]
- Kroschwald, S.; Chiu, C.Y.; Heydeck, D.; Rohwer, N.; Gehring, T.; Seifert, U.; Lux, A.; Rothe, M.; Weylandt, K.H.; Kuhn, H. Female mice carrying a defective Alox15 gene are protected from experimental colitis via sustained maintenance of the intestinal epithelial barrier function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 866–880. [Google Scholar] [CrossRef]
- Jupp, J.; Hillier, K.; Elliott, D.H.; Fine, D.R.; Bateman, A.C.; Johnson, P.A.; Cazaly, A.M.; Penrose, J.F.; Sampson, A.P. Colonic expression of leukotriene-pathway enzymes in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 537–546. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.; Lv, G.; Wang, H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front. Pharmacol. 2023, 14, 1095366. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, 47563. [Google Scholar] [CrossRef]
- Panda, S.K.; Peng, V.; Sudan, R.; Ulezko Antonova, A.; Di Luccia, B.; Ohara, T.E.; Fachi, J.L.; Grajales-Reyes, G.E.; Jaeger, N.; Trsan, T.; et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity 2023, 56, 797–812. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Du, C.; Sang, L.; Liu, L.; Li, Y.; Wang, F.; Fan, W.; Tang, P.; Zhang, S.; et al. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J. Transl. Med. 2022, 20, 363. [Google Scholar] [CrossRef]
- Zhu, X.; Messer, J.S.; Wang, Y.; Lin, F.; Cham, C.M.; Chang, J.; Billiar, T.R.; Lotze, M.T.; Boone, D.L.; Chang, E.B. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Investig. 2015, 125, 1098–1110. [Google Scholar] [CrossRef]
- Hooper, K.M.; Barlow, P.G.; Henderson, P.; Stevens, C. Interactions Between Autophagy and the Unfolded Protein Response: Implications for Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 661–671. [Google Scholar] [CrossRef]
- Arab, H.H.; Al-Shorbagy, M.Y.; Saad, M.A. Activation of autophagy and suppression of apoptosis by dapagliflozin attenuates experimental inflammatory bowel disease in rats: Targeting AMPK/mTOR, HMGB1/RAGE and Nrf2/HO-1 pathways. Chem. Biol. Interact. 2021, 335, 109368. [Google Scholar] [CrossRef]
- Blander, J.M. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J. 2016, 283, 2720–2730. [Google Scholar] [CrossRef]
- Levin, A.D.; Koelink, P.J.; Bloemendaal, F.M.; Vos, A.C.; D’Haens, G.R.; van den Brink, G.R.; Wildenberg, M.E. Autophagy Contributes to the Induction of Anti-TNF Induced Macrophages. J. Crohns Colitis. 2016, 10, 323–329. [Google Scholar] [CrossRef]
- Miao, Y.; Lv, Q.; Qiao, S.; Yang, L.; Tao, Y.; Yan, W.; Wang, P.; Cao, N.; Dai, Y.; Wei, Z. Alpinetin improves intestinal barrier homeostasis via regulating AhR/suv39h1/TSC2/mTORC1/autophagy pathway. Toxicol. Appl. Pharmacol. 2019, 384, 114772. [Google Scholar] [CrossRef]
- Hu, C.A.; Hou, Y.; Yi, D.; Qiu, Y.; Wu, G.; Kong, X.; Yin, Y. Autophagy and tight junction proteins in the intestine and intestinal diseases. Anim. Nutr. 2015, 1, 123–127. [Google Scholar] [CrossRef]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216. [Google Scholar] [CrossRef]
- Zeissig, Y.; Petersen, B.S.; Milutinovic, S.; Bosse, E.; Mayr, G.; Peuker, K.; Hartwig, J.; Keller, A.; Kohl, M.; Laass, M.W.; et al. XIAP variants in male Crohn’s disease. Gut 2015, 64, 66–76. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Zhao, K.; Zhou, Y.; Li, W.; Pan, C.; Qiang, L.; Li, Z.; Lu, N. Small molecule GL-V9 protects against colitis-associated colorectal cancer by limiting NLRP3 inflammasome through autophagy. Oncoimmunology 2017, 7, e1375640. [Google Scholar] [CrossRef]
- Zheng, X.; Hu, M.; Zang, X.; Fan, Q.; Liu, Y.; Che, Y.; Guan, X.; Hou, Y.; Wang, G.; Hao, H. Kynurenic acid/GPR35 axis restricts NLRP3 inflammasome activation and exacerbates colitis in mice with social stress. Brain Behav. Immun. 2019, 79, 244–255. [Google Scholar] [CrossRef]
- Ding, W.; Ding, Z.; Wang, Y.; Zhu, Y.; Gao, Q.; Cao, W.; Du, R. Evodiamine Attenuates Experimental Colitis Injury Via Activating Autophagy and Inhibiting NLRP3 Inflammasome Assembly. Front. Pharmacol. 2020, 11, 573870. [Google Scholar] [CrossRef]
- Samoilă, I.; Dinescu, S.; Costache, M. Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development-A Focus on Ulcerative Colitis. Cells 2020, 9, 1647. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Ma, H.; Yang, Q.; Shang, Q.; Song, L.; Zheng, Z.; Zhang, S.; Pan, Y.; Huang, P.; et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, Hif-1α upregulation and autophagy. Free Radic. Biol. Med. 2020, 161, 339–350. [Google Scholar] [CrossRef]
- Tartakover Matalon, S.; Ringel, Y.; Konikoff, F.; Drucker, L.; Pery, S.; Naftali, T. Cannabinoid receptor 2 agonist promotes parameters implicated in mucosal healing in patients with inflammatory bowel disease. United Eur. Gastroenterol. J. 2020, 8, 271–283. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Wei, W.; Han, B.Z.; Chen, X.W.; Su, D.F.; Liu, C. Activation of Cannabinoid Receptor 2 Ameliorates DSS-Induced Colitis through Inhibiting NLRP3 Inflammasome in Macrophages. PLoS ONE 2016, 11, e0155076. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, L.; Hua, S.; Qi, C.; Fang, M. IL-33 ameliorates experimental colitis involving regulation of autophagy of macrophages in mice. Cell Biosci. 2019, 9, 10. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, Y.; Chen, X.F.; Yang, S.L.; Tang, X.L.; Xiang, Z.G. Intestinal Macrophage Autophagy and its Pharmacological Application in Inflammatory Bowel Disease. Front. Pharmacol. 2021, 12, 803686. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E. The Selective Autophagy Receptor Optineurin in Crohn’s Disease. Front. Immunol. 2018, 9, 766. [Google Scholar] [CrossRef]
- Qin, Z.; Wan, J.J.; Sun, Y.; Wu, T.; Wang, P.Y.; Du, P.; Su, D.F.; Yang, Y.; Liu, X. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J. Mol. Med. 2017, 95, 221–233. [Google Scholar] [CrossRef]
- Shao, B.Z.; Ke, P.; Xu, Z.Q.; Wei, W.; Cheng, M.H.; Han, B.Z.; Chen, X.W.; Su, D.F.; Liu, C. Autophagy Plays an Important Role in Anti-inflammatory Mechanisms Stimulated by Alpha7 Nicotinic Acetylcholine Receptor. Front. Immunol. 2017, 8, 553. [Google Scholar] [CrossRef]
- Shao, B.Z.; Wang, S.L.; Pan, P.; Yao, J.; Wu, K.; Li, Z.S.; Bai, Y.; Linghu, E.Q. Targeting NLRP3 Inflammasome in Inflammatory Bowel Disease: Putting out the Fire of Inflammation. Inflammation 2019, 42, 1147–1159. [Google Scholar] [CrossRef]
- Lin, X.T.; Zheng, X.B.; Fan, D.J.; Yao, Q.Q.; Hu, J.C.; Lian, L.; Wu, X.J.; Lan, P.; He, X.S. MicroRNA-143 Targets ATG2B to Inhibit Autophagy and Increase Inflammatory Responses in Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 781–791. [Google Scholar] [CrossRef]
- Luo, X.; Gong, H.B.; Gao, H.Y.; Wu, Y.P.; Sun, W.Y.; Li, Z.Q.; Wang, G.; Liu, B.; Liang, L.; Kurihara, H.; et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021, 28, 1971–1989. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, J.; Ma, J.; Ma, J.; Liu, J.; Wang, F.; Tang, X. Inhibiting Ferroptosis: A Novel Approach for Ulcerative Colitis Therapeutics. Oxid. Med. Cell Longev. 2022, 2022, 9678625. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-κB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid. Med. Cell Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Tan, W.; Dai, F.; Yang, D.; Deng, Z.; Gu, R.; Zhao, X.; Cheng, Y. MiR-93-5p promotes granulosa cell apoptosis and ferroptosis by the NF-kB signaling pathway in polycystic ovary syndrome. Front. Immunol. 2022, 13, 967151. [Google Scholar] [CrossRef]
- Qiang, Z.; Dong, H.; Xia, Y.; Chai, D.; Hu, R.; Jiang, H. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxid. Med. Cell. Longev. 2020, 2020, 5146982. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Li, J.; Tian, X.; Liu, J.; Mo, Y.; Guo, X.; Qiu, Y.; Liu, Y.; Ma, X.; Wang, Y.; Xiong, Y. Therapeutic material basis and underling mechanisms of Shaoyao Decoction-exerted alleviation effects of colitis based on GPX4-regulated ferroptosis in epithelial cells. Chin. Med. 2022, 17, 96. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Y.; Wang, Y.; Meng, Q.; Xu, L.; Liu, Y.; Zhou, D.; Wang, Y.; Liang, D.; Li, W.; et al. Loureirin C inhibits ferroptosis after cerebral ischemia reperfusion through regulation of the Nrf2 pathway in mice. Phytomedicine 2023, 113, 154729. [Google Scholar] [CrossRef]
- Hong, Y.; Wu, B.; Xu, Z.; Du, J.; Gao, Y.; Wen, K.; Sun, X.L. Exploration of the effect of Xuejie San on expressions of Notch1, Nrf2, GPX4 and PTGS2 in colon tissues of Crohn’s disease rats based on a bioinformatic analysis. J. Liaoning Univ. TCM 2022, 24, 38–42. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Z.; Du, J.; Yang, X.; Wang, X.; Wen, K.; Sun, X. Xue-Jie-San restricts ferroptosis in Crohn’s disease via inhibiting FGL1/NF-κB/STAT3 positive feedback loop. Front. Pharmacol. 2023, 14, 1148770. [Google Scholar] [CrossRef]
- Louis, E.; Belaïche, J. Hydroxychloroquine (Plaquenil) for recurrence prevention of Crohn’s disease after curative surgery. Gastroenterol. Clin. Biol. 1995, 19, 233–234. [Google Scholar]
- Mutalib, M.; Borrelli, O.; Blackstock, S.; Kiparissi, F.; Elawad, M.; Shah, N.; Lindley, K. The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J. Crohns Colitis. 2014, 8, 1730–1734. [Google Scholar] [CrossRef]
- Zhong, M.; Cui, B.; Xiang, J.; Wu, X.; Wen, Q.; Li, Q.; Zhang, F. Rapamycin is Effective for Upper but not for Lower Gastrointestinal Crohn’s Disease-Related Stricture: A Pilot Study. Front. Pharmacol. 2021, 11, 617535. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Yang, H.; Zhou, Y.; Tang, L. Autophagy induction by rapamycin ameliorates experimental colitis and improves intestinal epithelial barrier function in IL-10 knockout mice. Int. Immunopharmacol. 2020, 81, 105977. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]
- Fairlie, W.D.; Tran, S.; Lee, E.F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 115–158. [Google Scholar] [CrossRef]
- Dong, X.; Liang, Q.; Pan, Y.Z.; Wang, X.; Kuo, Y.C.; Chiang, W.C.; Zhang, X.; Williams, N.S.; Rizo, J.; Levine, B.; et al. Novel Bcl-2 Inhibitors Selectively Disrupt the Autophagy-Specific Bcl-2-Beclin 1 Protein-Protein Interaction. ACS Med. Chem. Lett. 2022, 13, 1510–1516. [Google Scholar] [CrossRef]
- Calis, S.; Dogan, B.; Durdagi, S.; Celebi, A.; Yapicier, O.; Kilic, T.; Turanli, E.T.; Avsar, T. A novel BH3 mimetic Bcl-2 inhibitor promotes autophagic cell death and reduces in vivo Glioblastoma tumor growth. Cell Death Discov. 2022, 8, 433. [Google Scholar] [CrossRef]
- Hooper, K.M.; Casanova, V.; Kemp, S.; Staines, K.A.; Satsangi, J.; Barlow, P.G.; Henderson, P.; Stevens, C. The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 2019, 25, 1481–1496. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).