
Unveiling the Kadaknath Gut Microbiome: Early Growth Phase Spatiotemporal Diversity
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Maintenance and Sample Collection
2.2. The 16S rRNA Gene Sequencing
2.3. Analysis of the 16S rRNA Sequencing Data
2.4. Statistical Analysis
3. Results
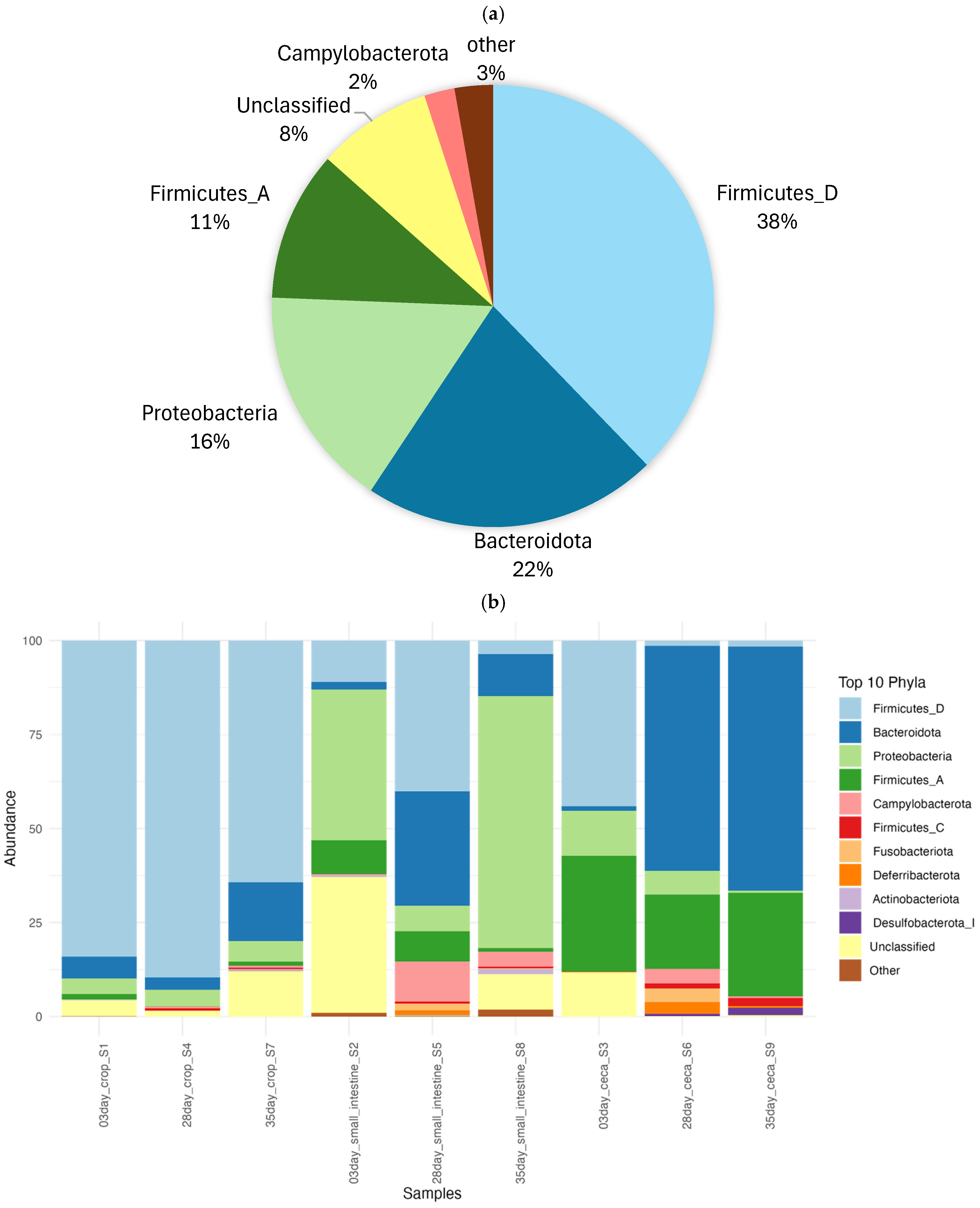
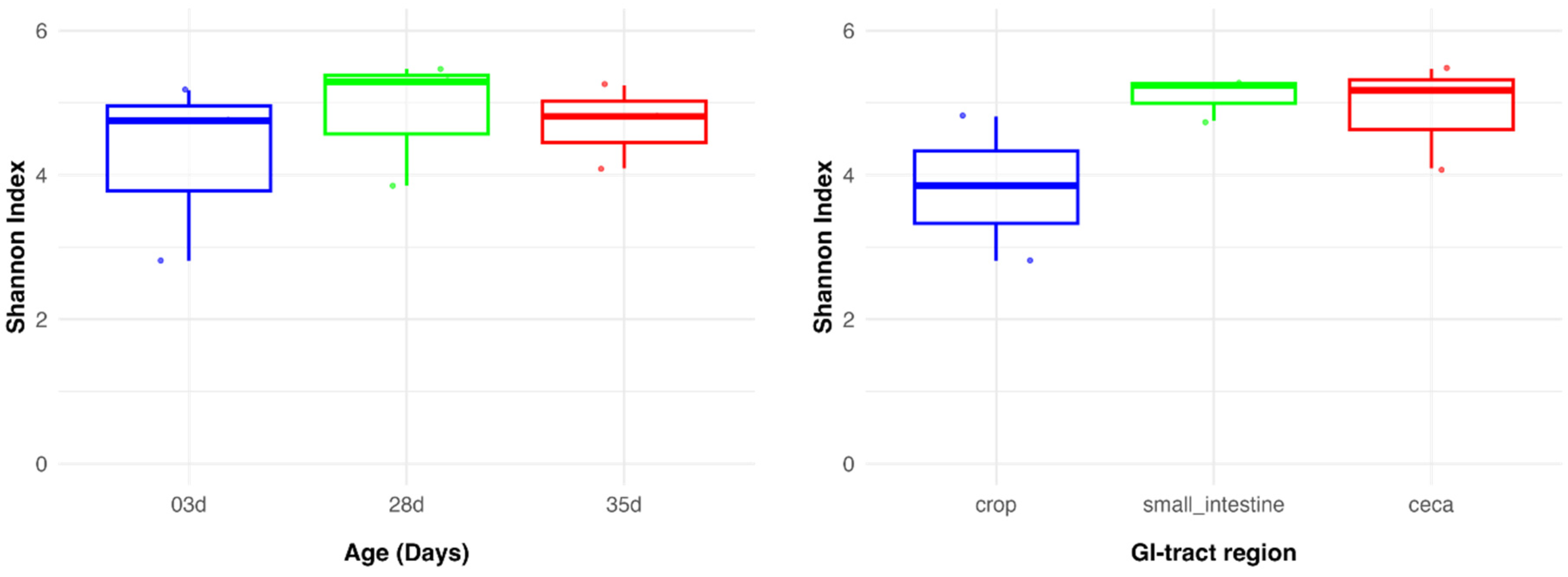
3.1. The Overall Diversity of Kadaknath Gut Microbiome
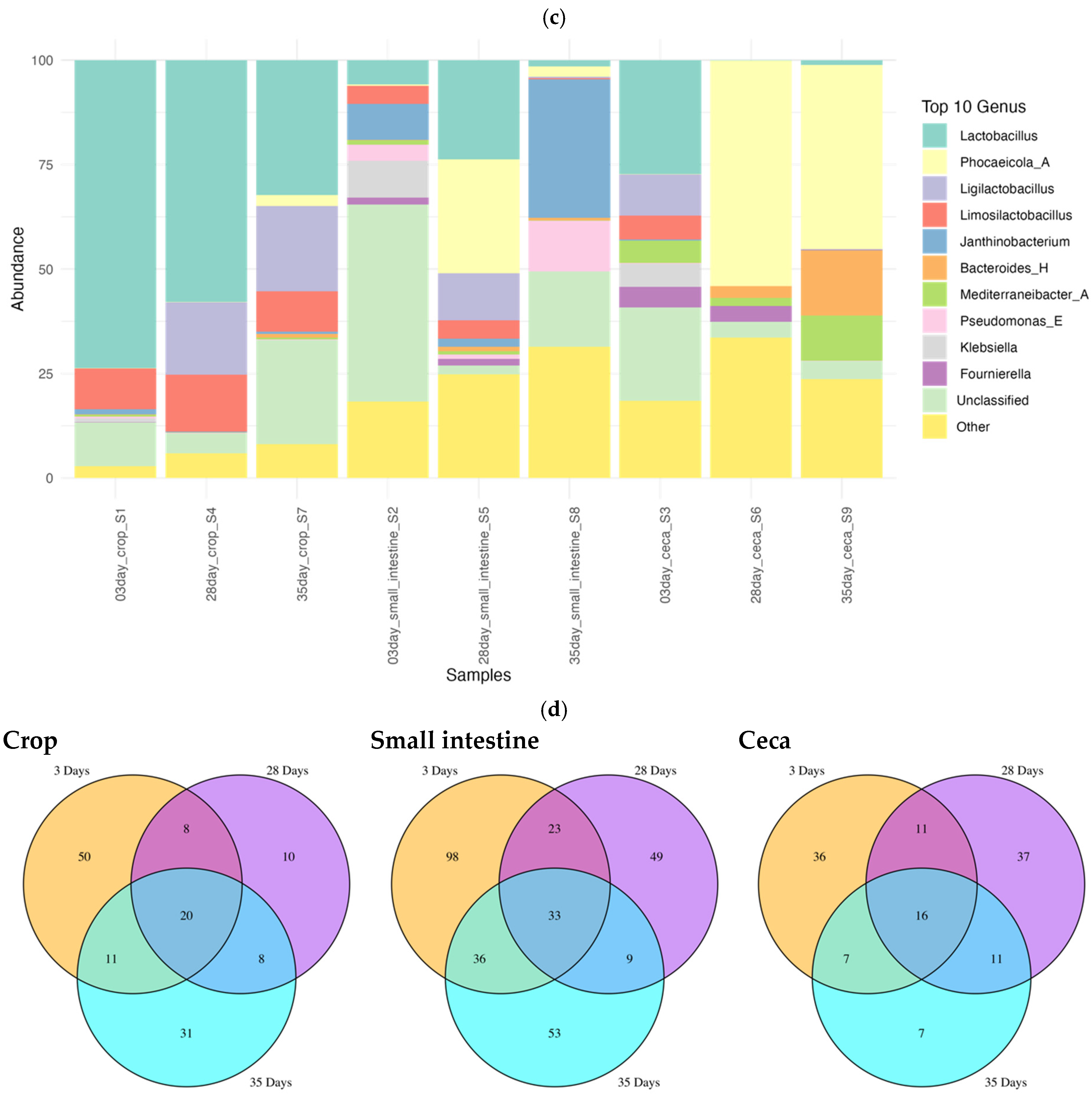
3.2. The Gut Microbiome of Kadaknath Exhibited Diversification and Maturation with Age
3.3. Pathogen Diversity in Kadakanath Chickens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mottet, A.; Tempio, G. Global poultry production: Current state and future outlook and challenges. World’s Poult. Sci. J. 2017, 73, 245–256. [Google Scholar] [CrossRef]
- Economic Survey 2023–2024. Press Information Bureau Government of India. Available online: https://static.pib.gov.in/WriteReadData/specificdocs/documents/2024/jul/doc2024722351601.pdf (accessed on 11 December 2024).
- FAO. Gateway to Poultry Production and Products. Available online: https://www.fao.org/poultry-production-products/products-processing/en/ (accessed on 11 December 2024).
- Haunshi, S.; Prince, L.L. Kadaknath: A popular native chicken breed of India with unique black colour characteristics. World’s Poult. Sci. J. 2021, 77, 427–440. [Google Scholar] [CrossRef]
- Sharma, R.; Sehrawat, R.; Ahlawat, S.; Sharma, V.; Parmar, A.; Thakur, M.S.; Mishra, A.K.; Tantia, M.S. An attempt to valorize the only black meat chicken breed of India by delineating superior functional attributes of its meat. Sci. Rep. 2022, 12, 3555. [Google Scholar] [CrossRef] [PubMed]
- Haunshi, S.; Rajkumar, U.; Padhi, M. Improvement of PD-4 (Aseel), an indigenous chicken, for growth and production traits. Indian J. Anim. Sci. 2019, 89, 419–423. [Google Scholar] [CrossRef]
- de Almeida, A.M.; Zuber, U. The effect of the Naked Neck genotype (Nana), feeding and outdoor rearing on growth and carcass characteristics of free-range broilers in a hot climate. Trop. Anim. Health Prod. 2010, 42, 99–107. [Google Scholar] [CrossRef]
- Ramasamy, K.T.; Reddy, M.R.; Raveendranathan, D.N.; Murugesan, S.; Chatterjee, R.N.; Ullengala, R.; Haunshi, S. Differential expression of toll-like receptor mRNA in white leghorn and indigenous chicken of India. Vet. Res. Commun. 2010, 34, 633–639. [Google Scholar] [CrossRef]
- Jones, P.J.; Niemi, J.; Christensen, J.P.; Tranter, R.B.; Bennett, R.M. A review of the financial impact of production diseases in poultry production systems. Anim. Prod. Sci. 2019, 59, 1585. [Google Scholar] [CrossRef]
- Naundrup Thofner, I.C.; Poulsen, L.L.; Bisgaard, M.; Christensen, H.; Olsen, R.H.; Christensen, J.P. Longitudinal study on causes of mortality in Danish broiler breeders. Avian Dis. 2019, 63, 400–410. [Google Scholar] [CrossRef]
- Maki, J.J.; Klima, C.L.; Sylte, M.J.; Looft, T. The microbial pecking order: Utilization of intestinal microbiota for poultry health. Microorganisms 2019, 7, 376. [Google Scholar] [CrossRef]
- Crhanova, M.; Hradecka, H.; Faldynova, M.; Matulova, M.; Havlickova, H.; Sisak, F.; Rychlik, I. Immune response of chicken gut to natural colonization by gut microflora and to Salmonella enterica serovar enteritidis infection. Infect. Immun. 2011, 79, 2755–2763. [Google Scholar] [CrossRef]
- Wigley, P. Blurred lines: Pathogens, commensals, and the healthy gut. Front. Vet. Sci. 2015, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Waters, J.L.; Clark, A.G.; Ley, R.E. Cross-species comparisons of host genetic associations with the microbiome. Science 2016, 352, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lan, F.; Li, X.; Yan, W.; Sun, C.; Li, J.; Yang, N.; Wen, C. The spatial and temporal characterization of gut microbiota in broilers. Front. Vet. Sci. 2021, 8, 712226. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Qi, G.H.; Wang, J.; Zhang, H.J.; Qiu, K.; Wu, S.G. Intestinal microbiota of layer hens and its association with egg quality and safety. Poult. Sci. 2022, 101, 102008. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, I. Composition and function of chicken gut microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef]
- Rubio, L.A. Possibilities of early life programming in broiler chickens via intestinal microbiota modulation. Poult. Sci. 2019, 98, 695–706. [Google Scholar] [CrossRef]
- Broom, L.J.; Kogut, M.H. The role of the gut microbiome in shaping the immune system of chickens. Vet. Immunol. Immunopathol. 2018, 204, 44–51. [Google Scholar] [CrossRef]
- Pandit, R.; Hinsu, A.; Patel, N.; Koringa, P.; Jakhesara, S.J.; Thakkar, J.; Joshi, C.G.; Limon, G.; Psifidi, A.; Guitián, J.; et al. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome 2018, 6, 1. [Google Scholar] [CrossRef]
- Hay, M.C.; Hinsu, A.T.; Koringa, P.G.; Pandit, R.J.; Liu, P.Y.; Parekh, M.J.; Jakhesara, S.J.; Dai, X.; Crotta, M.; Fosso, B.; et al. Chicken caecal enterotypes in indigenous Kadaknath and commercial Cobb chicken lines are associated with Campylobacter abundance and influenced by farming practices. Front. Microbiomes 2023, 2, 1301609. [Google Scholar] [CrossRef]
- Yan, W.; Sun, C.; Zheng, J.; Wen, C.; Ji, C.; Zhang, D. Efficacy of fecal sampling as a gut proxy in the study of chicken gut microbiota. Front. Microbiol. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Shang, Y.; Kumar, S.; Oakley, B.; Kim, W.K. Chicken gut microbiota: Importance and detection technology. Front. Vet. Sci. 2018, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Kim, S.B.; Choi, S.H.; Kim, S. Comparison of 16S rRNA Gene Based Microbial Profiling Using Five Next-Generation Sequencers and Various Primers. Front. Microbiol. 2021, 12, 693021. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Bernard, M.; Lecoeur, A.; Coville, J.L.; Bruneau, N.; Jardet, D.; Lagarrigue, S.; Meynadier, A.; Calenge, F.; Pascal, G.; Zerjal, T. Relationship between feed efficiency and gut microbiota in laying chickens under contrasting feeding conditions. Sci. Rep. 2024, 14, 8210. [Google Scholar] [CrossRef]
- Oladele, P.; Ngo, J.; Chang, T.; Johnson, T.A. Temporal dynamics of fecal microbiota community succession in broiler chickens, calves, and piglets under aerobic exposure. Microbiol. Spectr. 2024, 12, e0408423. [Google Scholar] [CrossRef]
- Deryabin, D.; Lazebnik, C.; Vlasenko, L.; Karimov, I.; Kosyan, D.; Zatevalov, A.; Duskaev, G. Broiler Chicken Cecal Microbiome and Poultry Farming Productivity: A Meta-Analysis. Microorganisms 2024, 12, 747. [Google Scholar] [CrossRef]
- Clausen, U.; Vital, S.T.; Lambertus, P.; Gehler, M.; Scheve, S.; Wohlbrand, L.; Rabus, R. Catabolic Network of the Fermentative Gut Bacterium Phocaeicola vulgatus (Phylum Bacteroidota) from a Physiologic-Proteomic Perspective. Microb. Physiol. 2024, 34, 88–107. [Google Scholar] [CrossRef]
- Bajagai, Y.S.; Van, T.T.H.; Joat, N.; Chousalkar, K.; Moore, R.J.; Stanley, D. Layer chicken microbiota: A comprehensive analysis of spatial and temporal dynamics across all major gut sections. J. Anim. Sci. Biotechnol. 2024, 15, 20. [Google Scholar] [CrossRef]
- Swelum, A.A.; Elbestawy, A.R.; El-Saadony, M.T.; Hussein, E.O.; Alhotan, R.; Suliman, G.M.; Taha, A.E.; Ba-Awadh, H.; El-Tarabily, K.A.; Abd El-Hack, M.E. Ways to minimize bacterial infections, with special reference to Escherichia coli, to cope with the first-week mortality in chicks: An updated overview. Poult. Sci. 2021, 100, 101039. [Google Scholar] [CrossRef]
- Awad, W.A.; Mann, E.; Dzieciol, M.; Hess, C.; Schmitz-Esser, S.; Wagner, M.; Hess, M. Age-related differences in the luminal and mucosa-associated gut microbiome of broiler chickens and shifts associated with Campylobacter jejuni infection. Front. Cell. Infect. Microbiol. 2016, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, M.; Liu, Y.; Yang, X.; Wei, F.; Jin, X.; Liu, D.; Guo, Y.; Hu, Y. Quantitative microbiome profiling reveals the developmental trajectory of the chicken gut microbiota and its connection to host metabolism. iMeta 2023, 2, 105. [Google Scholar] [CrossRef] [PubMed]
- Hermans, D.; Van Deun, K.; Martel, A.; Van Immerseel, F.; Messens, W.; Heyndrickx, M.; Haesebrouck, F.; Pasmans, F. Colonization factors of Campylobacter jejuni in the chicken gut. Vet. Res. 2011, 42, 82. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Pielsticker, C.; Gerzova, L.; Rychlik, I.; Rautenschlein, S. The influence of age on Campylobacter jejuni infection in chicken. Dev. Comp. Immunol. 2016, 62, 58–71. [Google Scholar] [CrossRef]
- Van Immerseel, F.; De Buck, J.; Pasmans, F.; Huyghebaert, G.; Haesebrouck, F.; Ducatelle, R. Clostridium perfringens in poultry: An emerging threat for animal and public health. Avian Pathol. 2004, 33, 537–549. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.; Doijad, S.P.; Suryavanshi, M.V.; Dey, A.; Singh Malik, S.V.; Dutilh, B.E.; Barbuddhe, S.B. Unveiling the Kadaknath Gut Microbiome: Early Growth Phase Spatiotemporal Diversity. Microbiol. Res. 2025, 16, 54. https://doi.org/10.3390/microbiolres16030054
Nair A, Doijad SP, Suryavanshi MV, Dey A, Singh Malik SV, Dutilh BE, Barbuddhe SB. Unveiling the Kadaknath Gut Microbiome: Early Growth Phase Spatiotemporal Diversity. Microbiology Research. 2025; 16(3):54. https://doi.org/10.3390/microbiolres16030054
Chicago/Turabian StyleNair, Amruta, Swapnil Prakash Doijad, Mangesh Vasant Suryavanshi, Anwesha Dey, Satya Veer Singh Malik, Bas E. Dutilh, and Sukhadeo Baliram Barbuddhe. 2025. "Unveiling the Kadaknath Gut Microbiome: Early Growth Phase Spatiotemporal Diversity" Microbiology Research 16, no. 3: 54. https://doi.org/10.3390/microbiolres16030054
APA StyleNair, A., Doijad, S. P., Suryavanshi, M. V., Dey, A., Singh Malik, S. V., Dutilh, B. E., & Barbuddhe, S. B. (2025). Unveiling the Kadaknath Gut Microbiome: Early Growth Phase Spatiotemporal Diversity. Microbiology Research, 16(3), 54. https://doi.org/10.3390/microbiolres16030054