
Soil Prokaryotic Diversity Responds to Seasonality in Dehesas, Modulated by Tree Identity and Canopy Effect

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Location
2.2. Sampling Design
2.3. Library Preparation and Sequencing
2.4. Statistical Analysis
3. Results
3.1. Sequencing
3.2. Alpha-Diversity
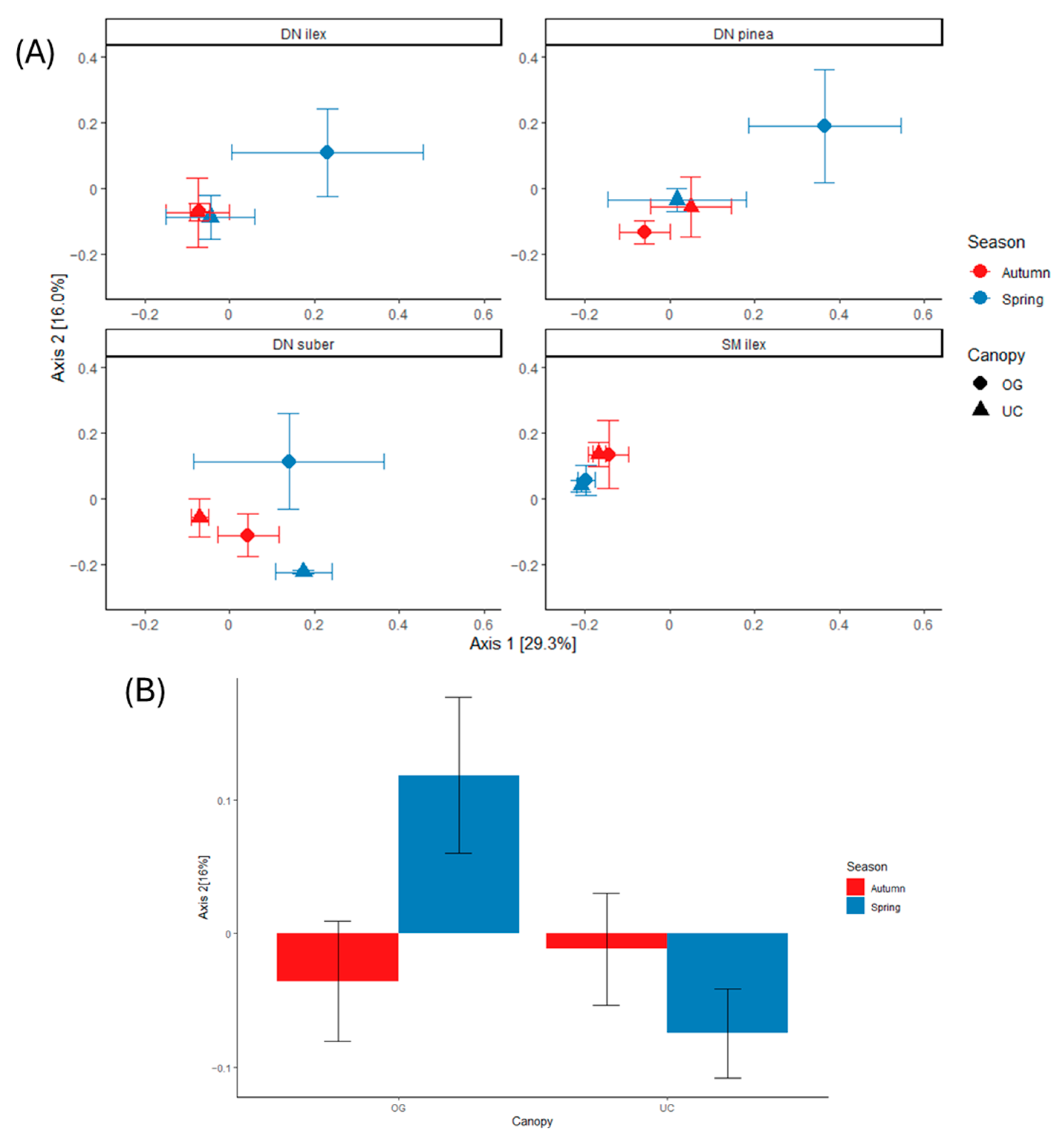
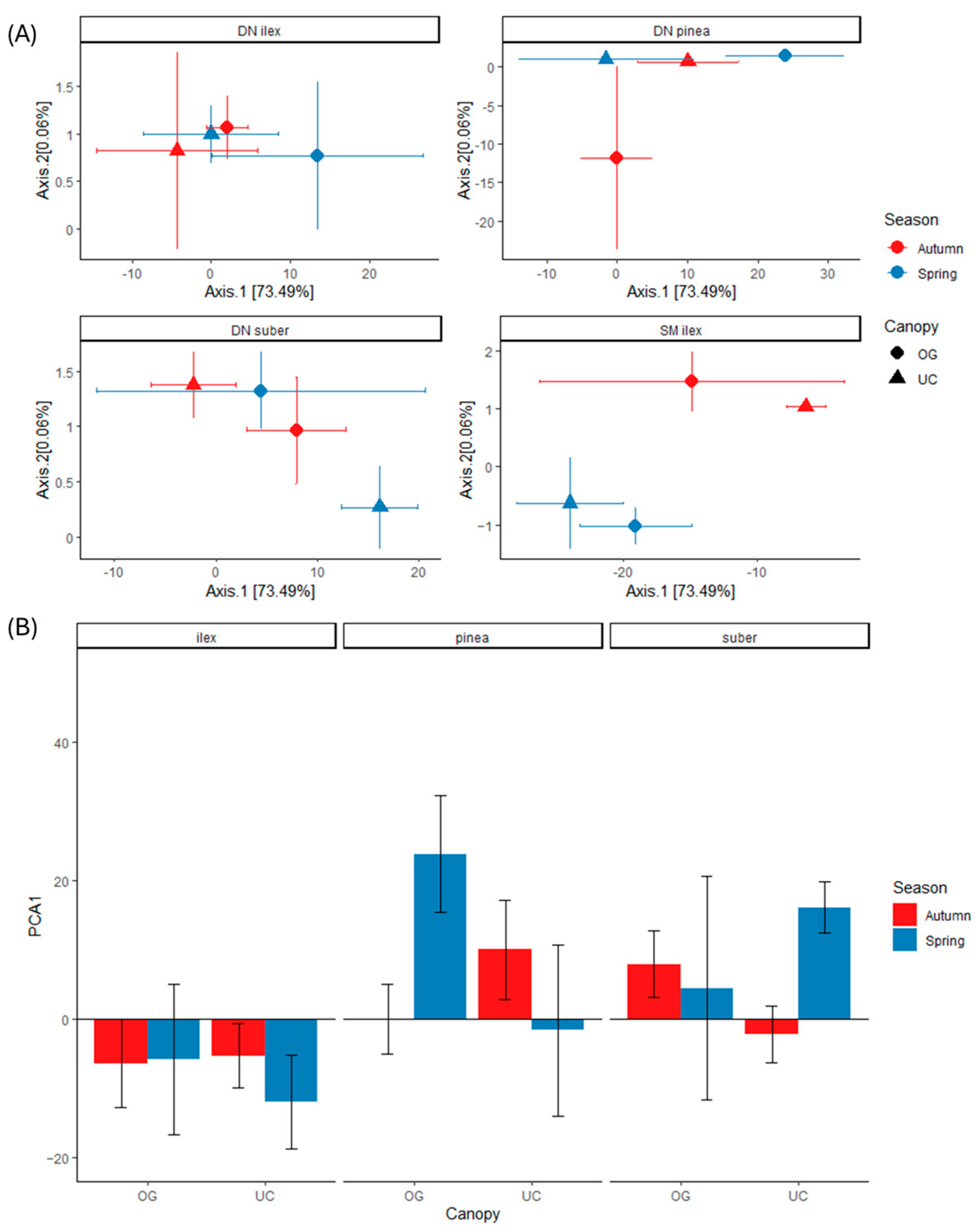
3.3. Community Structure and Inferred Functionality
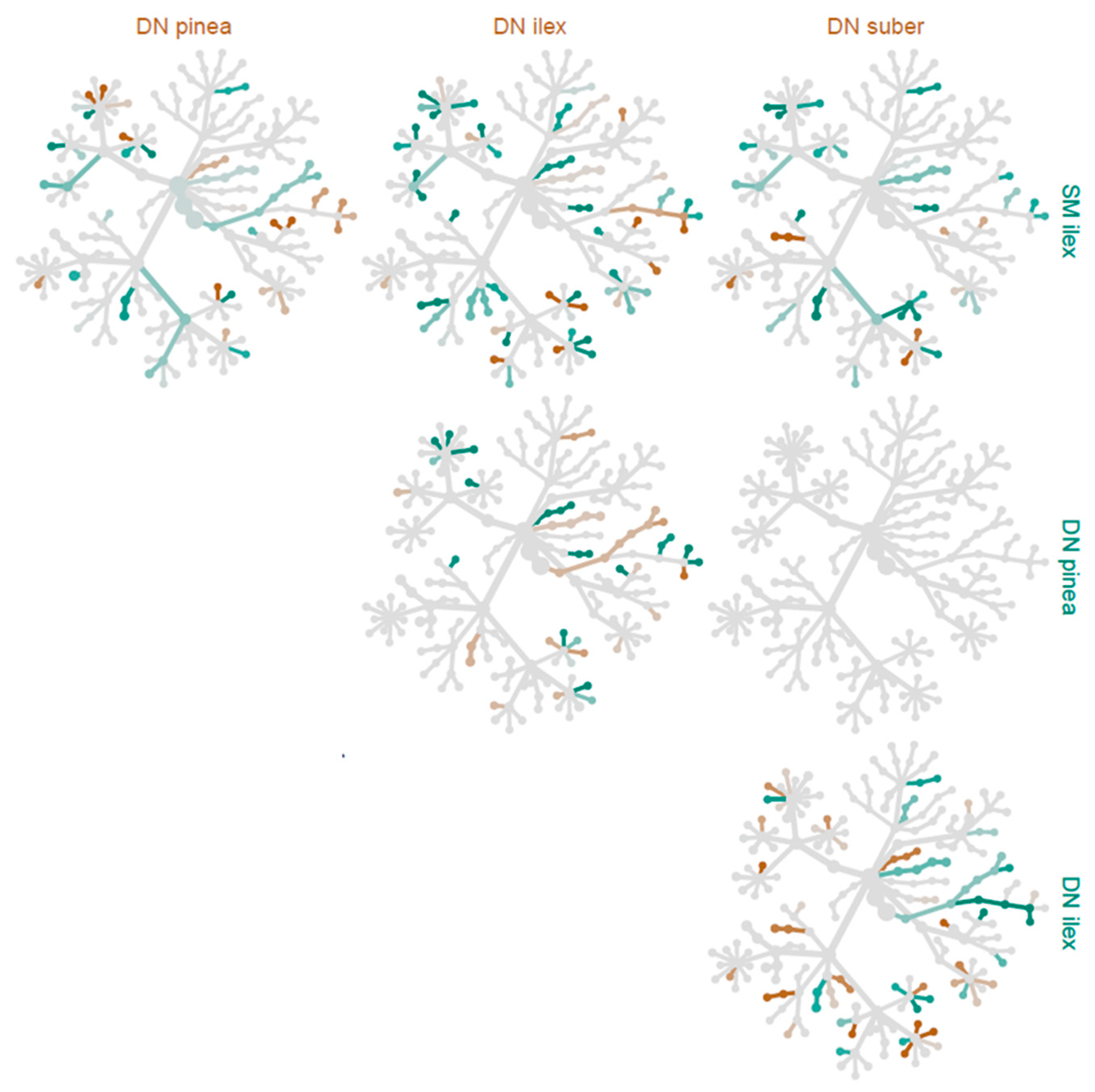
3.4. Differences in Taxonomical Groupings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SM | Sierra Morena |
| DN | Doñana National Park |
| OG | open grassland |
| UC | under canopy |
| OTU | Operational Taxonomic Unit |
References
- Ibañez, M.; Leiva, M.; Chocarro, C.; Aljazairi, S.; Ribas, A.; Sebastia, M.-T. Tree—Open Grassland Structure and Composition Drive Greenhouse Gas Exchange in Holm Oak Meadows of the Iberian Peninsula. Agronomy 2020, 11, 50. [Google Scholar] [CrossRef]
- Tárrega, R.; Calvo, L.; Taboada, Á.; García-Tejero, S.; Marcos, E. Abandonment and Management in Spanish Dehesa Systems: Effects on Soil Features and Plant Species Richness and Composition. For. Ecol. Manag. 2009, 257, 731–738. [Google Scholar] [CrossRef]
- Rodríguez-Rojo, M.P.; Roig, S.; López-Carrasco, C.; Redondo García, M.M.; Sánchez-Mata, D. Which Factors Favour Biodiversity in Iberian Dehesas? Sustainability 2022, 14, 2345. [Google Scholar] [CrossRef]
- Costa, A.; Madeira, M.; Lima Santos, J.; Oliveira, Â. Change and Dynamics in Mediterranean Evergreen Oak Woodlands Landscapes of Southwestern Iberian Peninsula. Landsc. Urban. Plan. 2011, 102, 164–176. [Google Scholar] [CrossRef]
- Liu, K.; Ding, X.; Tang, X.; Wang, J.; Li, W.; Yan, Q.; Liu, Z. Macro and Microelements Drive Diversity and Composition of Prokaryotic and Fungal Communities in Hypersaline Sediments and Saline–Alkaline Soils. Front. Microbiol. 2018, 9, 352. [Google Scholar] [CrossRef]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil Bacterial and Fungal Communities across a pH Gradient in an Arable Soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, D.; Delgado-Baquerizo, M.; Liu, S.; Wang, B.; Wu, J.; Hu, S.; Bai, Y. Long-Term Regional Evidence of the Effects of Livestock Grazing on Soil Microbial Community Structure and Functions in Surface and Deep Soil Layers. Soil. Biol. Biochem. 2022, 168, 108629. [Google Scholar] [CrossRef]
- Costa, D.; Freitas, H.; Sousa, J.P. Influence of Seasons and Land-Use Practices on Soil Microbial Activity and Metabolic Diversity in the “Montado Ecosystem”. Eur. J. Soil Biol. 2013, 59, 22–30. [Google Scholar] [CrossRef]
- Araya, Y.N.; Bartelheimer, M.; Valle, C.J.; Crujeiras, R.M.; García-Baquero, G. Does Functional Soil Microbial Diversity Contribute to Explain Within-Site Plant β-Diversity in an Alpine Grassland and a Dehesa Meadow in Spain? J. Veg. Sci. 2017, 28, 1018–1027. [Google Scholar] [CrossRef]
- Ibañez, M.; Chocarro, C.; Leiva, M.; Sebastia, M.-T. Tree—Open Grassland Mosaics Drive the Herbaceous Structure and Diversity in Mediterranean Wood Pastures. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Andreu, A.; Kustas, W.P.; Polo, M.J.; Carrara, A.; González-Dugo, M.P. Modeling Surface Energy Fluxes over a Dehesa (Oak Savanna) Ecosystem Using a Thermal Based Two-Source Energy Balance Model (TSEB) I. Remote Sens. 2018, 10, 567. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Wang, F.; Jiang, Y.; Zhang, X.-X. Assessing the Relative Effects of Geographic Location and Soil Type on Microbial Communities Associated with Straw Decomposition. Appl. Environ. Microbiol. 2013, 79, 3327–3335. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A Comprehensive Benchmarking Study of Protocols and Sequencing Platforms for 16S rRNA Community Profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into Biases and Sequencing Errors for Amplicon Sequencing with the Illumina MiSeq Platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef] [PubMed]
- Ovreås, L.; Forney, L.; Daae, F.L.; Torsvik, V. Distribution of Bacterioplankton in Meromictic Lake Saelenvannet, as Determined by Denaturing Gradient Gel Electrophoresis of PCR-Amplified Gene Fragments Coding for 16S rRNA. Appl. Environ. Microbiol. 1997, 63, 3367–3373. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S rRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm v2: Highly-Scalable and High-Resolution Amplicon Clustering. PeerJ 2015, 3, e1420. [Google Scholar] [CrossRef]
- Lanzén, A.; Jørgensen, S.L.; Huson, D.H.; Gorfer, M.; Grindhaug, S.H.; Jonassen, I.; Øvreås, L.; Urich, T. CREST—Classification Resources for Environmental Sequence Tags. PLoS ONE 2012, 7, e49334. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-Based Accuracy of Metagenome Prediction Tools Varies across Sample Types and Functional Categories. Microbiome 2020, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package; R Foundation: Vienna, Austria, 2022. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R Package for Visualization and Manipulation of Community Taxonomic Diversity Data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef]
- Ma, L.; Liu, L.; Lu, Y.; Chen, L.; Zhang, Z.; Zhang, H.; Wang, X.; Shu, L.; Yang, Q.; Song, Q.; et al. When Microclimates Meet Soil Microbes: Temperature Controls Soil Microbial Diversity along an Elevational Gradient in Subtropical Forests. Soil. Biol. Biochem. 2022, 166, 108566. [Google Scholar] [CrossRef]
- Gallardo, A. Effect of Tree Canopy on the Spatial Distribution of Soil Nutrients in a Mediterranean Dehesa. Pedobiologia 2003, 47, 117–125. [Google Scholar] [CrossRef]
- García del Barrio, J.M.; Alonso Ponce, R.; Benavides, R.; Roig, S. Species Richness of Vascular Plants along the Climatic Range of the Spanish Dehesas at Two Spatial Scales. For. Syst. 2014, 23, 111. [Google Scholar] [CrossRef]
- López-Sánchez, A.; San Miguel, A.; López-Carrasco, C.; Huntsinger, L.; Roig, S. The Important Role of Scattered Trees on the Herbaceous Diversity of a Grazed Mediterranean Dehesa. Acta Oecologica 2016, 76, 31–38. [Google Scholar] [CrossRef]
- Iovieno, P.; Alfani, A.; Bååth, E. Soil Microbial Community Structure and Biomass as Affected by Pinus Pinea Plantation in Two Mediterranean Areas. Appl. Soil. Ecol. 2010, 45, 56–63. [Google Scholar] [CrossRef]
- Iovieno, P.; Alfani, A. Influence of Pinus pinea plantation on physico-chemical and biological soil properties in Quercus ilex climax areas in Campania (Southern Italy). In Proceedings of the International Workshop MEDPINE 3: Conservation, Regeneration and Restoration of Mediterranean Pines and Their Ecosystems; Bari, Italy, 26–30 September 2005, Leone, V., Lovreglio, R., Eds.; CIHEAM: Bari, Italy, 2007; pp. 143–148. [Google Scholar]
- Street, R.A.; Owen, S.; Duckham, S.C.; Boissard, C.; Hewitt, C.N. Effect of Habitat and Age on Variations in Volatile Organic Compound (VOC) Emissions from Quercus Ilex and Pinus Pinea. Atmos. Environ. 1997, 31, 89–100. [Google Scholar] [CrossRef]
- Kämpfer, P.; Arun, A.B.; Busse, H.-J.; Langer, S.; Young, C.-C.; Chen, W.-M.; Schumann, P.; Syed, A.A.; Rekha, P. D Georgenia Soli Sp. Nov., Isolated from Iron-Ore-Contaminated Soil in India. Int. J. Syst. Evol. Microbiol. 2010, 60, 1027–1030. [Google Scholar] [CrossRef]
- Tang, S.-K.; Wang, Y.; Lee, J.-C.; Lou, K.; Park, D.-J.; Kim, C.-J.; Li, W.-J. 2010 Georgenia Halophila Sp. Nov., a Halophilic Actinobacterium Isolated from a Salt Lake. Int. J. Syst. Evol. Microbiol. 2010, 60, 1317–1421. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-Q.; Li, J.; Qin, S.; Tian, X.-P.; Wang, F.-Z.; Zhang, S. 2013 Georgenia Sediminis Sp. Nov., a Moderately Thermophilic Actinobacterium Isolated from Sediment. Int. J. Syst. Evol. Microbiol. 2010, 63, 4243–4247. [Google Scholar] [CrossRef]
- Kovaleva, O.L.; Merkel, A.Y.; Novikov, A.A.; Baslerov, R.V.; Toshchakov, S.V.; Bonch-Osmolovskaya, E.A. Tepidisphaera Mucosa Gen. Nov., Sp. Nov., a Moderately Thermophilic Member of the Class Phycisphaerae in the Phylum Planctomycetes, and Proposal of a New Family, Tepidisphaeraceae Fam. Nov., and a New Order, Tepidisphaerales Ord. Nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 549–555. [Google Scholar] [CrossRef]
- Li, W.; Yuan, L.; Lan, X.; Shi, R.; Chen, D.; Feng, D.; Zhao, X.; Chen, H. Effects of Long-Term Warming on Soil Prokaryotic Communities in Shrub and Alpine Meadows on the Eastern Edge of the Qinghai-Tibetan Plateau. Appl. Soil. Ecol. 2023, 188, 104871. [Google Scholar] [CrossRef]
- Fuerst, J.A.; Sagulenko, E. Beyond the Bacterium: Planctomycetes Challenge Our Concepts of Microbial Structure and Function. Nat. Rev. Microbiol. 2011, 9, 403–413. [Google Scholar] [CrossRef]
- Jung, J.; Park, W. Acinetobacter Species as Model Microorganisms in Environmental Microbiology: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 2533–2548. [Google Scholar] [CrossRef]
- Belser, L.W.; Schmidt, E.L. Diversity in the Ammonia-Oxidizing Nitrifier Population of a Soil. Appl. Environ. Microbiol. 1978, 36, 584–588. [Google Scholar] [CrossRef]
- Nierychlo, M.; Andersen, K.S.; Xu, Y.; Green, N.; Jiang, C.; Albertsen, M.; Dueholm, M.S.; Nielsen, P.H. MiDAS 3: An Ecosystem-Specific Reference Database, Taxonomy and Knowledge Platform for Activated Sludge and Anaerobic Digesters Reveals Species-Level Microbiome Composition of Activated Sludge. Water Res. 2020, 182, 115955. [Google Scholar] [CrossRef]
- Dueholm, M.K.D.; Nierychlo, M.; Andersen, K.S.; Rudkjøbing, V.; Knutsson, S.; Albertsen, M.; Nielsen, P.H. MiDAS 4: A Global Catalogue of Full-Length 16S rRNA Gene Sequences and Taxonomy for Studies of Bacterial Communities in Wastewater Treatment Plants. Nat. Commun. 2022, 13, 1908. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, T.; Li, H.; Ren, C.; Chen, J.; Yang, G.; Han, X.; Feng, Y.; Ren, G.; Wang, X. Variations of Soil Nitrogen-Fixing Microorganism Communities and Nitrogen Fractions in a Robinia Pseudoacacia Chronosequence on the Loess Plateau of China. CATENA 2019, 174, 316–323. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Y.; Shang, Y.; Liu, D.; Liesack, W.; Cui, Z.; Peng, J.; Zhang, F. Peat-Vermiculite Alters Microbiota Composition towards Increased Soil Fertility and Crop Productivity. Plant Soil. 2022, 470, 21–34. [Google Scholar] [CrossRef]
- Beauvais, M.; Schatt, P.; Montiel, L.; Logares, R.; Galand, P.E.; Bouget, F.-Y. Functional Redundancy of Seasonal Vitamin B12 Biosynthesis Pathways in Coastal Marine Microbial Communities. Environ. Microbiol. 2023, 25, 3753–3770. [Google Scholar] [CrossRef] [PubMed]
- Ramond, P.; Galand, P.E.; Logares, R. Microbial Functional Diversity and Redundancy: Moving Forward. FEMS Microbiol. Rev. 2025, 49, fuae031. [Google Scholar] [CrossRef]
- Oggioni, S.D.; Ochoa-Hueso, R.; Peco, B. Livestock Grazing Abandonment Reduces Soil Microbial Activity and Carbon Storage in a Mediterranean Dehesa. Appl. Soil. Ecol. 2020, 153, 103588. [Google Scholar] [CrossRef]
- Magarzo, A.; Olsson, S.; Sanz-Benito, I.; Mediavilla, O.; Oria-de-Rueda, J.A.; Villafuerte-Jordán, R.; Martínez-Jauregui, M.; Martín-Pinto, P. Wild Ungulate Effects on Soil Fungal Diversity in Mediterranean Mixed Forests. For. Ecol. Manag. 2024, 562, 121928. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | SM–ilex | DN-Mixed | DN–pinea |
|---|---|---|---|
| pH | 6.7 | 7.9 | 7.0 |
| Organic C (%) | 0.8 | 0.32 | 0.51 |
| Total N (%) | 0.85 | 0.15 | 0.11 |
| Clay (%) | 18 | 10 | 16 |
| Silt (%) | 29 | 25 | 21 |
| Sand (%) | 54 | 65 | 63 |
| Degrees of Freedom | Sum of Squares | Mean of Squares | F Statistic | p-Value | |
|---|---|---|---|---|---|
| Site | 2 | 57,819,710 | 28,909,855 | 9.145141 | 0.00066 * |
| Season | 1 | 6,631,683 | 6,631,683 | 2.09782 | 0.15666 |
| Tree sp. | 1 | 2,451,204 | 2,451,204 | 0.77539 | 0.38473 |
| Canopy | 1 | 7,753,639 | 7,753,639 | 2.45273 | 0.12658 |
| Season: Tree sp. | 2 | 9,026,212 | 4,513,106 | 1.42764 | 0.25388 |
| Season: Canopy | 1 | 11,788,968 | 11,788,968 | 3.72924 | 0.06183 |
| Tree sp.:Canopy | 2 | 1,143,875 | 571,938 | 0.18092 | 0.83529 |
| Season: Tree sp.:Canopy | 2 | 8,784,181 | 4,392,091 | 1.38936 | 0.26302 |
| Residuals | 34 | 107,481,631 | 3,161,224 |
| Degrees of Freedom | Sum of Squares | Mean of Squares | F Statistic | p-Value | |
|---|---|---|---|---|---|
| Site | 2 | 0.010124 | 0.0050619 | 2.4421 | 0.10216 |
| Season | 1 | 0.007127 | 0.0077039 | 3.7373 | 0.0724 |
| Tree sp. | 1 | 0.000000 | 0.0000002 | 0.0001 | 0.9331 |
| Canopy | 1 | 0.004347 | 0.0043468 | 2.0971 | 0.1567 |
| Season: Tree sp. | 2 | 0.000453 | 0.0002265 | 0.1093 | 0.8968 |
| Season: Canopy | 1 | 0.000470 | 0.0004701 | 0.2268 | 0.6369 |
| Tree sp.:Canopy | 2 | 0.004416 | 0.0022078 | 1.0651 | 0.3559 |
| Season: Tree sp.:Canopy | 2 | 0.009949 | 0.0049746 | 2.4000 | 0.1059 |
| Residuals | 34 | 0.070475 | 0.0020728 |
| Degrees of Freedom | Sum of Squares | Mean of Squares | F Statistic | p-Value | |
|---|---|---|---|---|---|
| site | 2 | 0.0002593 | 0.00012963 | 1.2836 | 0.2887751 |
| season | 1 | 0.0013157 | 0.00131575 | 13.0282 | 0.0008824 * |
| tree_sp | 1 | 0.0004033 | 0.00040326 | 3.9930 | 0.0528829 |
| canopy | 1 | 0.0000743 | 0.00003717 | 0.3680 | 0.6945274 |
| season: tree_sp | 2 | 0.0000613 | 0.00003064 | 0.3034 | 0.7400813 |
| season: canopy | 1 | 0.0004564 | 0.00022818 | 2.2593 | 0.1182688 |
| tree_sp: canopy | 2 | 0.0001770 | 0.00008851 | 0.8764 | 0.4245050 |
| season: tree_sp:canopy | 2 | 0.0000516 | 0.00002582 | 0.2557 | 0.7757173 |
| Residuals | 34 | 0.00038377 | 0.00010099 |
| Code | Pathway | T | S | C | T*S | T*C | C*S | T*S*C | Comments |
|---|---|---|---|---|---|---|---|---|---|
| Synthesis | |||||||||
| PWY.6350 | archaetidylinositol biosynthesis | X | Archaea | ||||||
| PWY.6167 | flavin biosynthesis II (archaea) | X | Archaea | ||||||
| PWY.6654 | phosphopantothenate biosynthesis III (archaea) | X | Archaea | ||||||
| PWY.6349 | C20,20 CDP-archaeol biosynthesis | X | Archaea | ||||||
| P381.PWY | adenosylcobalamin biosynthesis II (aerobic) | X | B12 Vitamin | ||||||
| PWY.3081 | adenosylcobalamin biosynthesis II (aerobic) | X | B12 Vitamin | ||||||
| PWY.3941 | β-alanine biosynthesis II | X | X | Bacteria (General) | |||||
| PWY.622 | starch biosynthesis | X | Bacteria (General) | ||||||
| PWY.7316 | dTDP-N-acetylviosamine biosynthesis | X | X | X | Gram-Bacteria | ||||
| KDO.NAGLIPASYN.PWY | superpathway of (Kdo)2-lipid A biosynthesis | X | Gram-Bacteria | ||||||
| PWY.7198 | pyrimidine deoxyribonucleotides de novo biosynthesis IV | X | Methane involved organisms | ||||||
| PWY.5198 | factor 420 biosynthesis II (mycobacteria) | X | Methane involved organisms | ||||||
| PWY.1622 | formaldehyde assimilation I (serine pathway) | X | Methane involved organisms | ||||||
| PWY.7347 | sucrose biosynthesis III | X | Methane involved organisms | ||||||
| SUCSYN.PWY | sucrose biosynthesis I (Photos) | X | Photosynthetic organisms | ||||||
| PWY.1422 | vitamin E biosynthesis (tocopherols) | X | Photosynthetic organisms | ||||||
| PWY.7286 | 7-(3-amino-3-carboxypropyl)-wyosine biosynthesis | X | X | RNA modification | |||||
| AEROBACTINSYN.PWY | aerobactin biosynthesis | X | Siderophore | ||||||
| PWY.6749 | CMP-legionaminate biosynthesis I | X | Signaling molecules | ||||||
| Degradation | |||||||||
| PWY.3801 | Sucrose degradation II | X | Anaerobic | ||||||
| PWY.6572 | chondroitin sulfate degradation I (bacterial) | X | Animal matter degradation | ||||||
| PWY.5532 | nucleoside and nucleotide degradation (archaea) | X | X | Archaea | |||||
| PWY.5519 | D-arabinose degradation III | X | Archaea | ||||||
| PWY.6713 | L-rhamnose degradation II | X | Bacteria (General) | ||||||
| PWY.5499 | vitamin B6 degradation I | X | Bacteria (General) | ||||||
| PWY5F9.12 | biphenyl degradation | X | Bacteria (General) | ||||||
| PWY.7013 | (S)-propane-1,2-diol degradation | X | Bacteria (General) | ||||||
| LACTOSECAT.PWY | lactose degradation I | X | X | Gram+ Bacteria | |||||
| P441.PWY | superpathway of N-acetylneuraminate degradation | X | Gram+ Bacteria | ||||||
| PWY.5677 | succinate fermentation to butanoate | X | Gram+ Bacteria | ||||||
| GLCMANNANAUT.PWY | superpathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation | X | N related metabolisms | ||||||
| PWY.3661 | glycine betaine degradation I | X | Osmoregulation | ||||||
| VALDEG.PWY | L-valine degradation I | X | Bacteria (General) | ||||||
| Nitrogen | |||||||||
| PWY.7084 | nitrifier denitrification | X | N related metabolisms |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manjón-Cabeza, J.; Ibáñez, M.; Leiva, M.J.; Chocarro, C.; Lanzén, A.; Epelde, L.; Sebastià, M.T. Soil Prokaryotic Diversity Responds to Seasonality in Dehesas, Modulated by Tree Identity and Canopy Effect. Microbiol. Res. 2025, 16, 153. https://doi.org/10.3390/microbiolres16070153
Manjón-Cabeza J, Ibáñez M, Leiva MJ, Chocarro C, Lanzén A, Epelde L, Sebastià MT. Soil Prokaryotic Diversity Responds to Seasonality in Dehesas, Modulated by Tree Identity and Canopy Effect. Microbiology Research. 2025; 16(7):153. https://doi.org/10.3390/microbiolres16070153
Chicago/Turabian StyleManjón-Cabeza, José, Mercedes Ibáñez, María José Leiva, Cristina Chocarro, Anders Lanzén, Lur Epelde, and Maria Teresa Sebastià. 2025. "Soil Prokaryotic Diversity Responds to Seasonality in Dehesas, Modulated by Tree Identity and Canopy Effect" Microbiology Research 16, no. 7: 153. https://doi.org/10.3390/microbiolres16070153
APA StyleManjón-Cabeza, J., Ibáñez, M., Leiva, M. J., Chocarro, C., Lanzén, A., Epelde, L., & Sebastià, M. T. (2025). Soil Prokaryotic Diversity Responds to Seasonality in Dehesas, Modulated by Tree Identity and Canopy Effect. Microbiology Research, 16(7), 153. https://doi.org/10.3390/microbiolres16070153