
Role of pH in Regulating Cancer Pyrimidine Synthesis

 , , , ,
, , , ,
Abstract
:1. Introduction
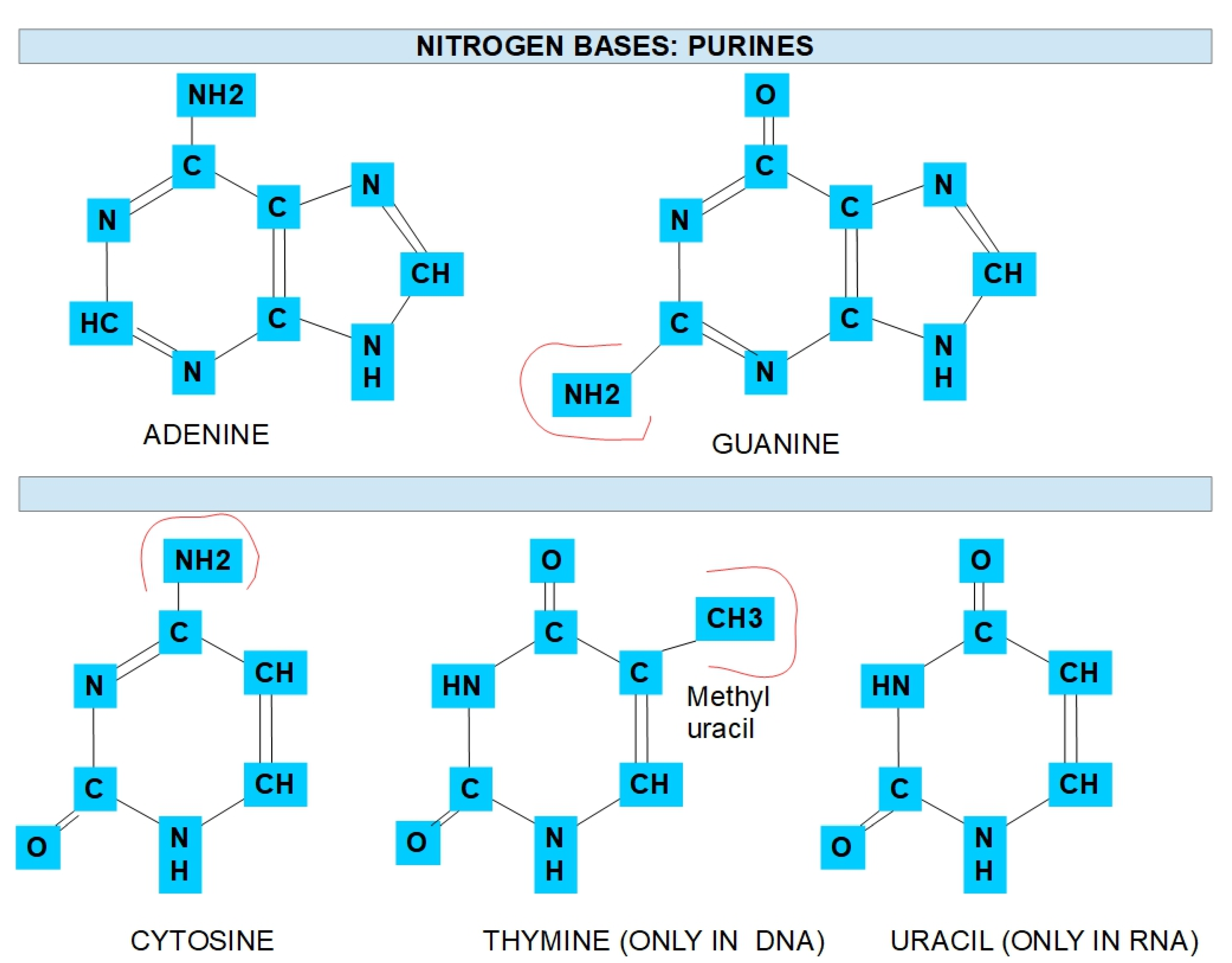
1.1. Nitrogen Bases Nucleotide Synthesis
- The salvage pathway is a process of recycling existing nucleotides and bases that originated in the breakdown of existing nucleic acids.
- ▶
- In purine and pyrimidine biosynthetic pathways, an amino acid is a precursor of each path:
- ■
- Glycine for purines.
- ■
- Aspartate for pyrimidines.
- ▶
- Glutamine is the source of amino groups.
- ▶
- In purine and pyrimidine de novo pathways, many enzymes are organized as large multi-enzyme complexes.
- ▶
- In both purine and pyrimidine de novo synthesis, a negative feedback loop regulates the number of molecules to be synthesized.
- ▶
- The cellular pools of nucleotides are generally very small compared to the amount needed for DNA or RNA synthesis. Thus, nucleotide synthesis is an essential process for cell replication and growth. This becomes even more evident in highly proliferating cells, such as those found in tumors.
- ▶
- Limiting nucleotide synthesis decreases proliferation and growth.
- ▶
- Drugs that can inhibit nucleotide synthesis can impede, delay, or decrease malignant proliferation.
- ▶
- Pyrimidines are mainly produced by tumor cells, but stromal cells, such as macrophages and cancer-associated fibroblasts, can also produce them. In pancreatic cancer, it has been found that pyrimidines produced by macrophages were able to create resistance to gemcitabine treatment [17].
- ▶
- Glutamine transporters that provide glutamine to the cell (glutamine is a nitrogen donor for pyrimidines) alkalinize the intracellular milieu by simultaneously exporting protons [18].
1.2. Pyrimidine De Novo Synthesis
2. The Pyrimidine Synthesis Pathway
2.1. The Steps in De Novo Pyrimidine Synthesis
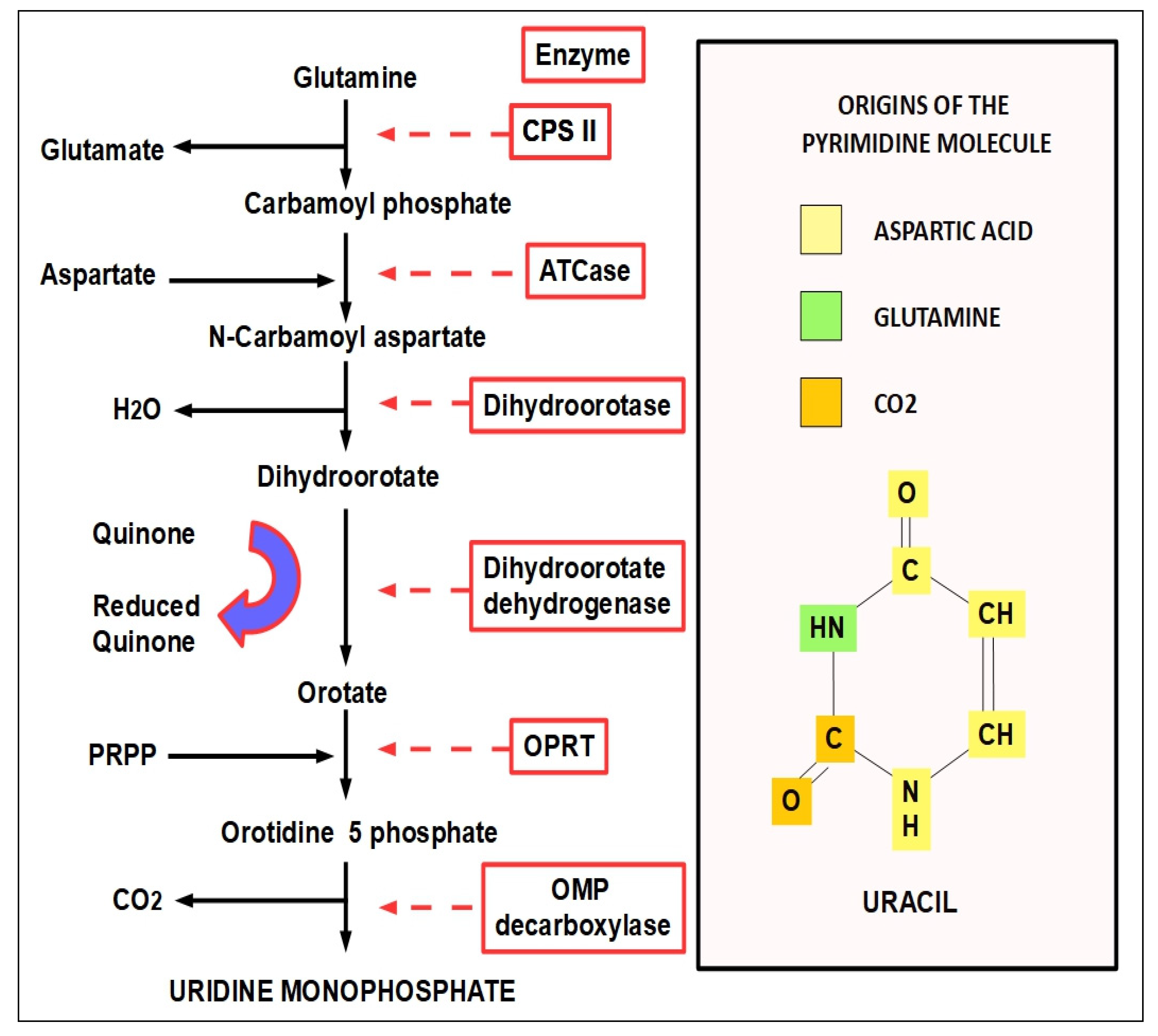
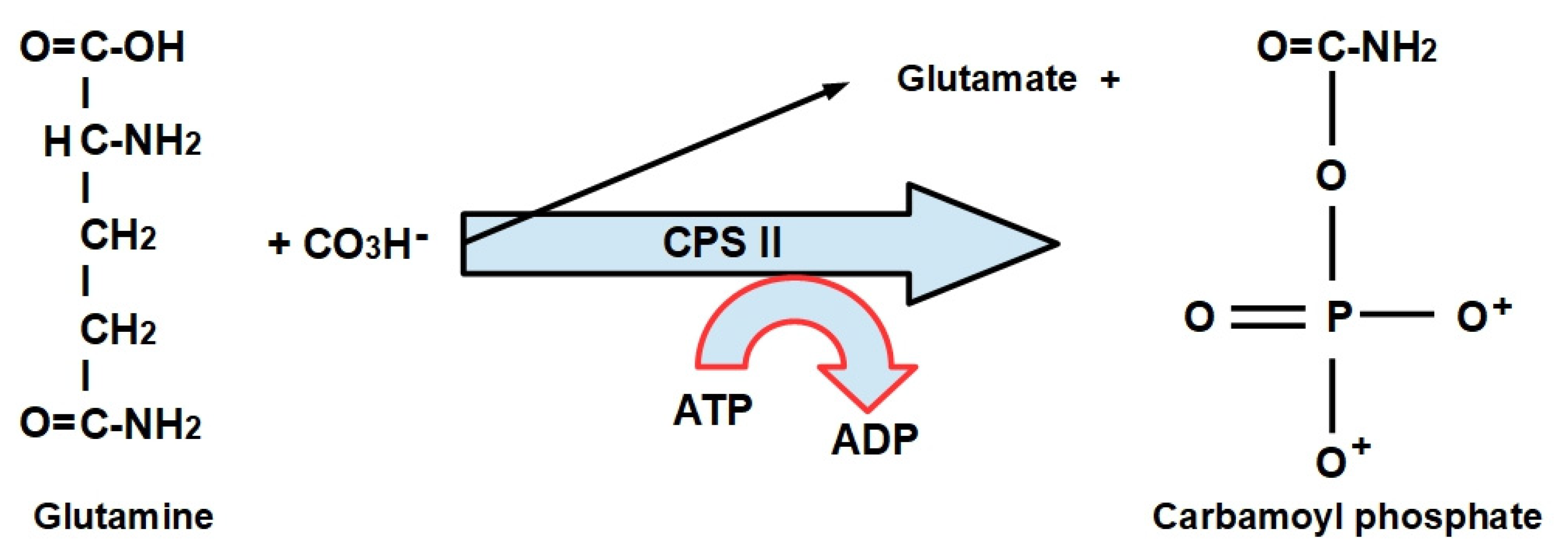
- Step 1:
- (1)
- CPS-I is an intra-mitochondrial enzyme;
- (2)
- CPS-II is cytosolic and is the enzyme that participates in de novo pyrimidine biosynthesis. This is the rate-limiting enzyme in pyrimidine biosynthesis.
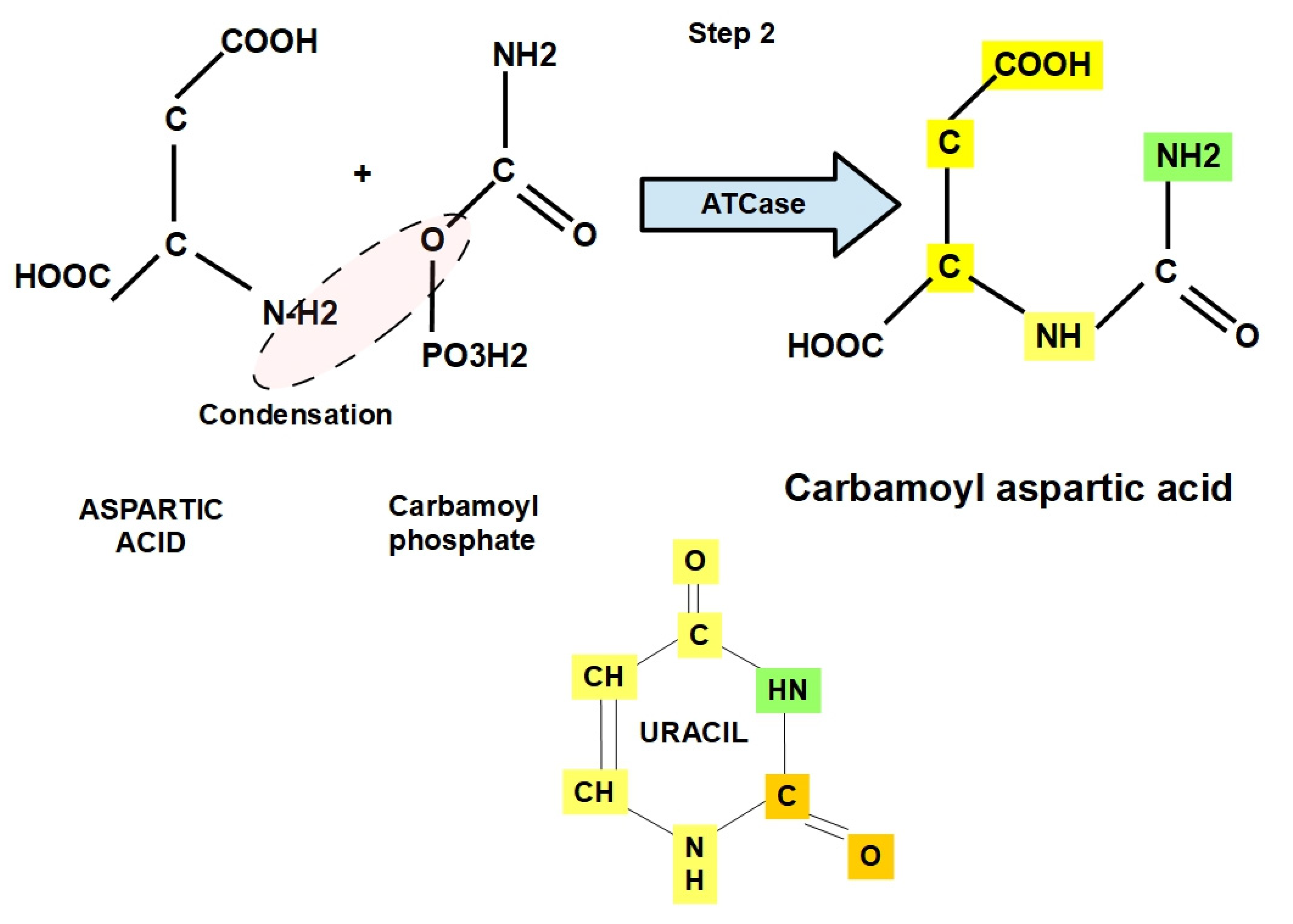
- Step 2:
- Step 3:
2.2. Dihydroorotase Inhibitors
- Step 4:
- ▶
- Solid tumors have a higher level of pyrimidines that require toxic doses of brequinar, while leukemias respond to lower doses due to a decreased pool of uridine;
- ▶
- The pyrimidine synthetic pathway is somehow related to the inhibition of myeloid differentiation because the inhibition of dihydroorotate dehydrogenase overcame a differentiation blockade in acute myeloid leukemia in vivo [78].
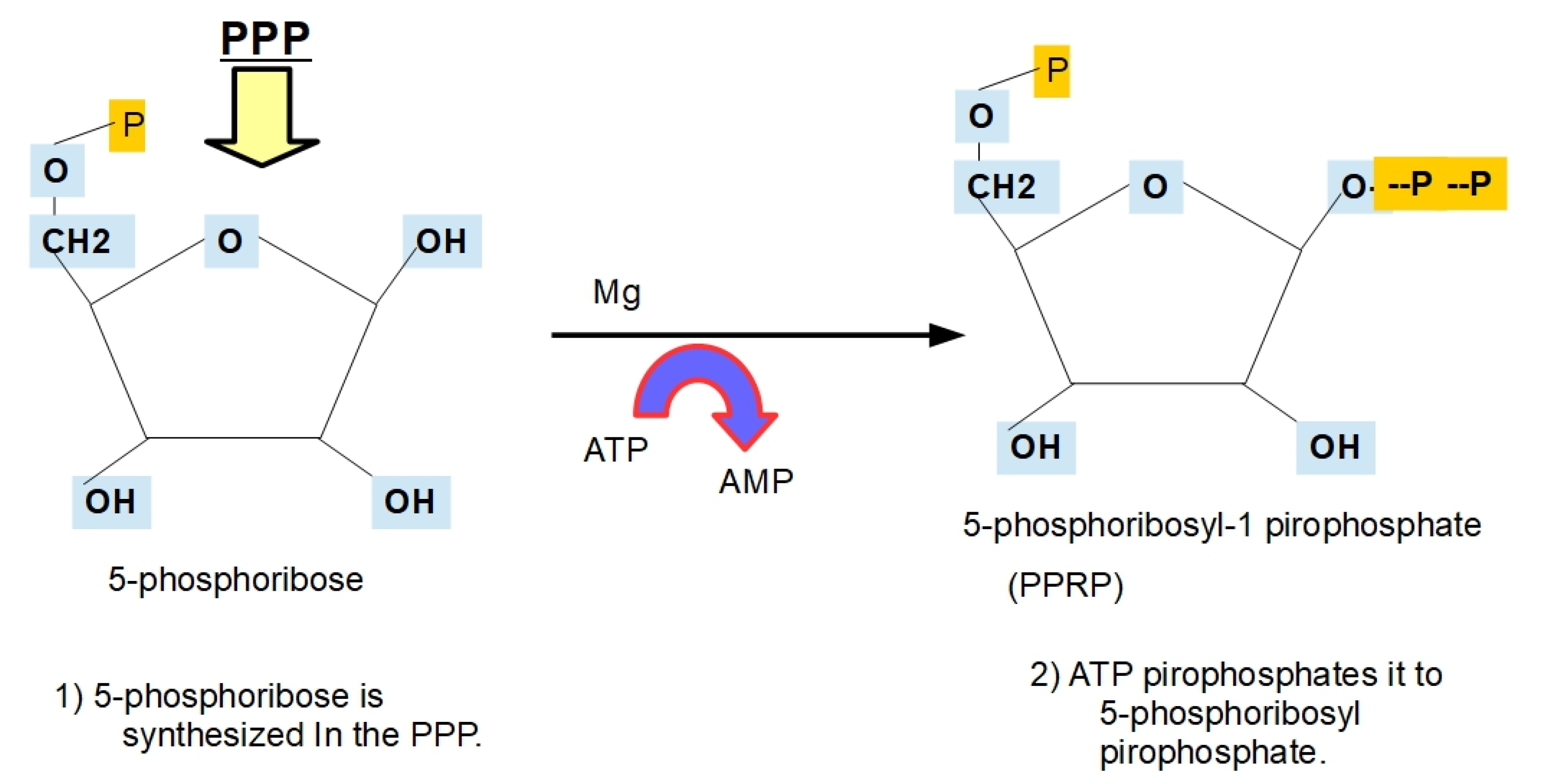
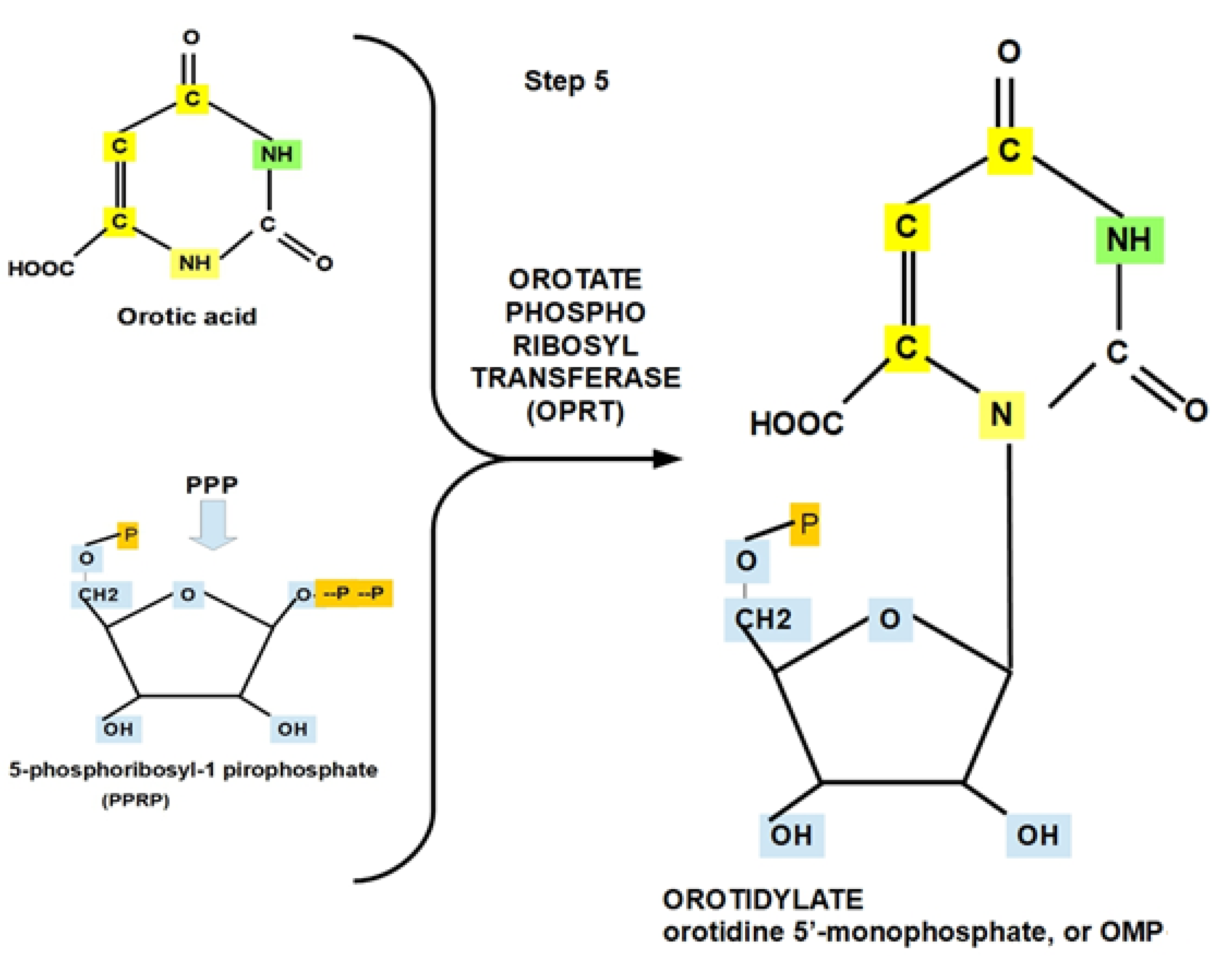
- Step 5:
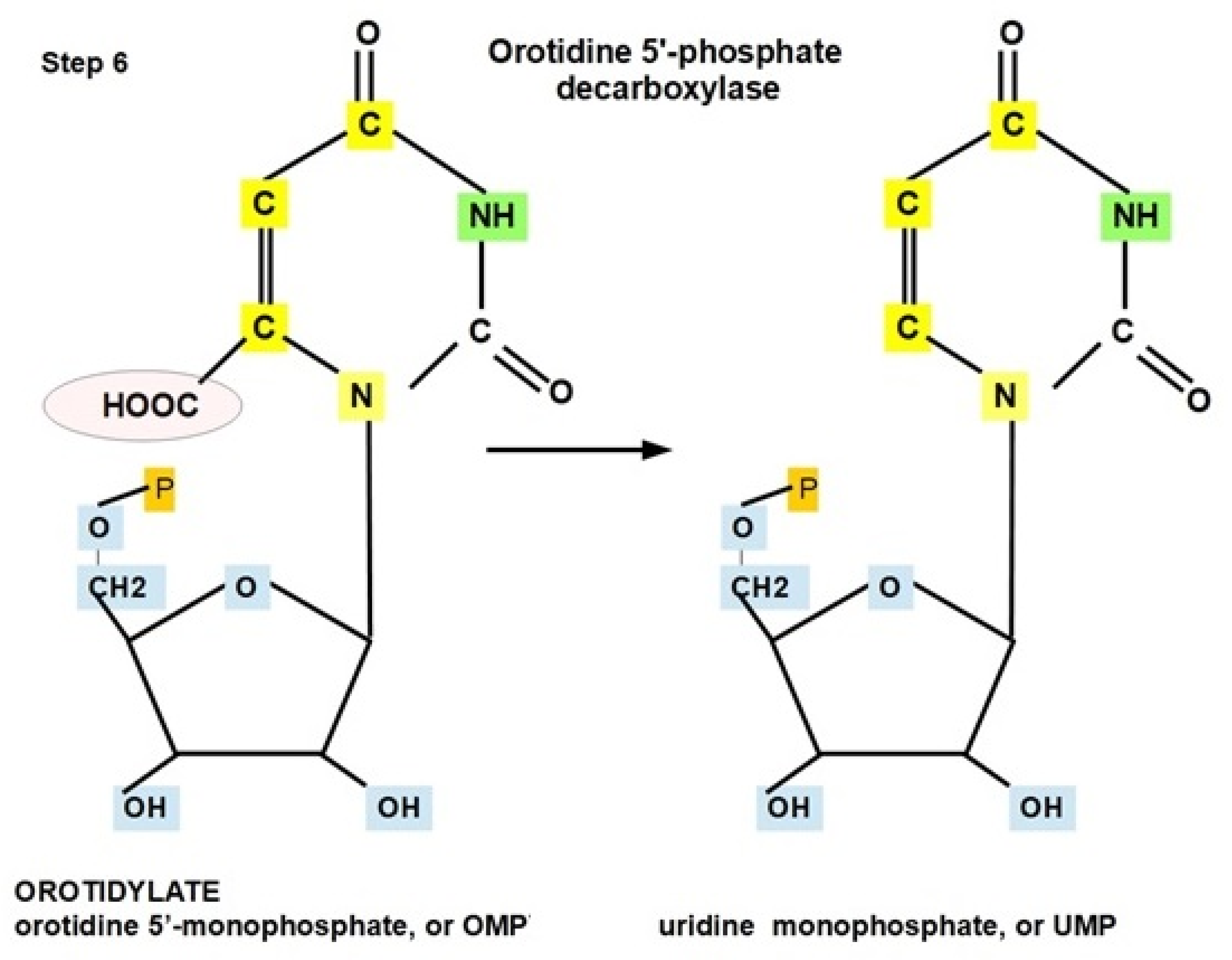
- Step 6:
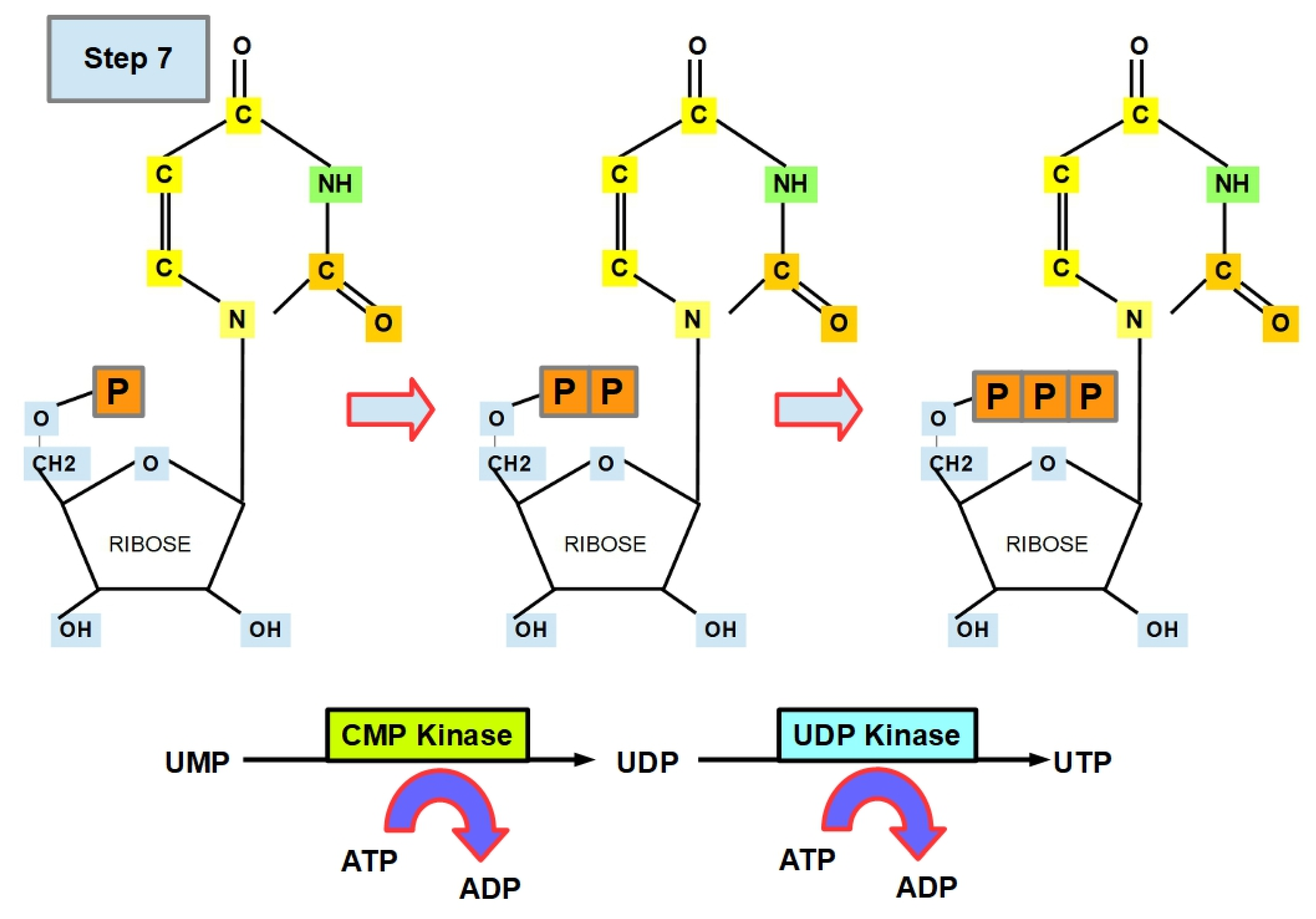
- Step 7 (Uracil Formation):
- Step 7.1:
- Step 7.2:
- Step 8 (Cytosine Formation):
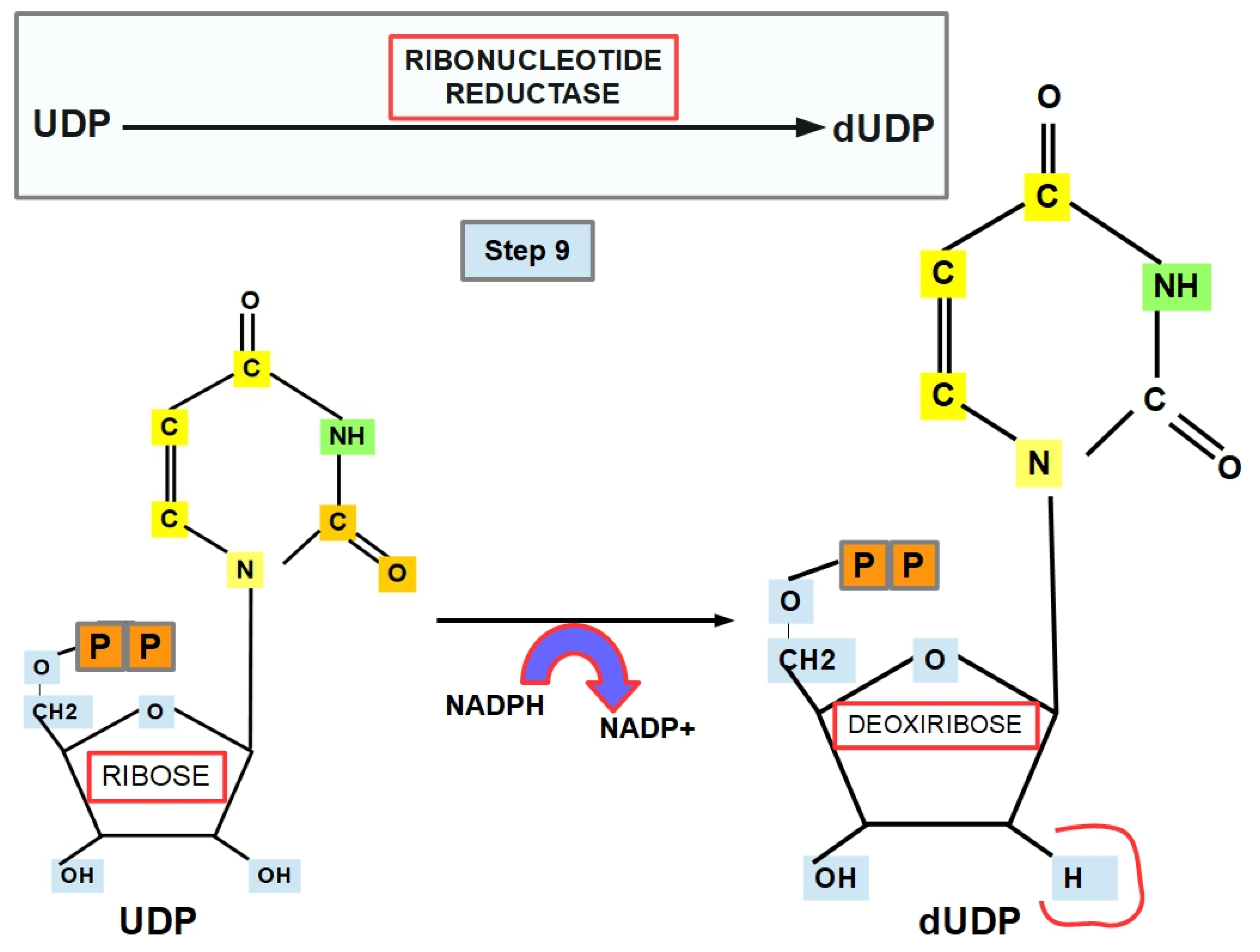
- Step 9 (Thymine Formation):
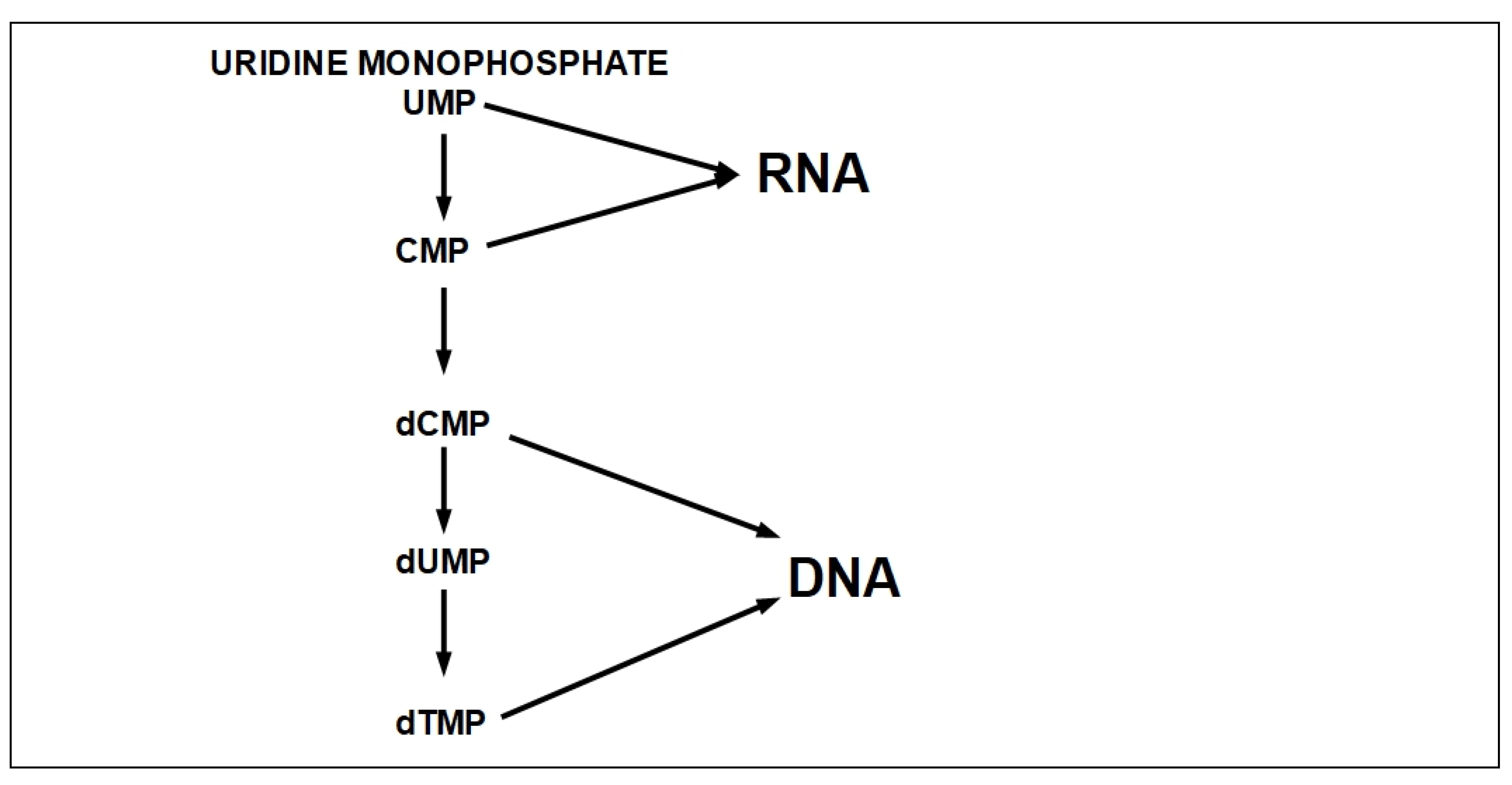
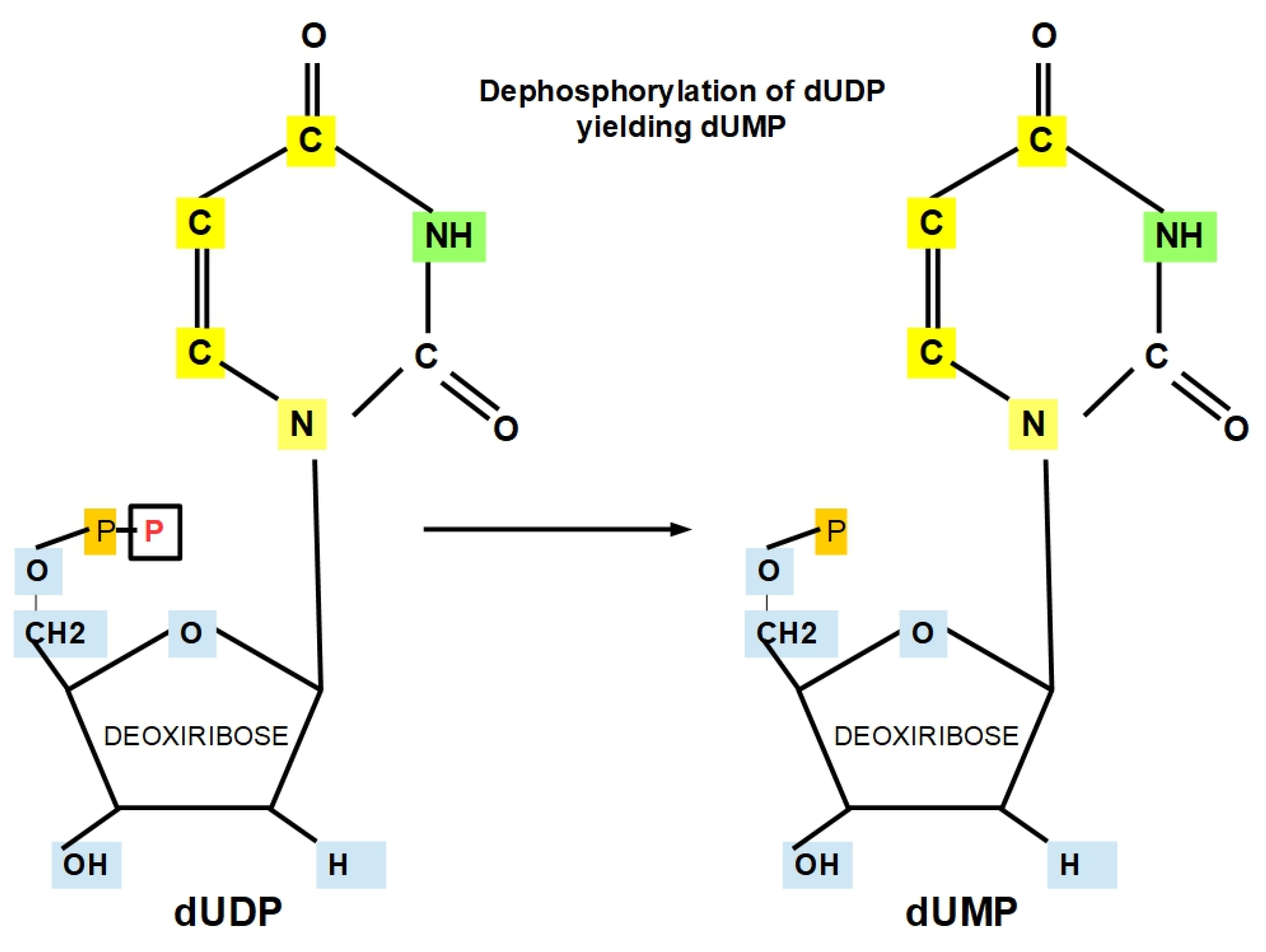
- Step 10:
- Step 11:
3. Discussion
4. Conclusions
- Cancer cells have an increased rate of pyrimidine synthesis, as expected in a cell that must duplicate all its DNA and RNA at an accelerated pace.
- The enzymes participating in pyrimidine synthesis have an optimal efficiency at a pH that is higher than the intracellular pH of normal cells.
- This increased intracellular pH is constantly found in malignant cells as part of the pH paradigm.
- On a theoretical basis, we may assume that lowering the intracellular pH will hamper the efficiency of pyrimidine synthesis and decrease tumor proliferation. There is direct and indirect evidence that intracellular acidification is a valid method for complementing standard treatment schemes.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ehrlich, P. Über den jetzigen Stand der Chemotherapie. Ber. Dtsch. Chem. Ges. 1909, 42, 17–47. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S. Emperor of All Maladies: A Biography of Cancer; Mukherjee, S., Ed.; Scribner: New York, NY, USA, 2010; ISBN1 9781439181713. ISBN2 1439181713. [Google Scholar]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Scott, W.W.; Hodges, C.V. Studies on Prostatic Cancer. III. The Effects of Fever, of Desoxycorticosterone and of Estrogen on Clinical Patients with Metastatic Carcinoma of the Prostate1,2. J. Urol. 1941, 46, 997–1006. [Google Scholar] [CrossRef]
- Farber, S. Some observations on the effect of folic acid antagonists on acute leukemia and other forms of incurable cancer. Blood 1949, 4, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.B.; Elhassan, G.O.; Ibrahim, M.E.; David Polo Orozco, J.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef]
- Johnson, J.D.; Epel, D.; Paul, M. Intracellular pH and activation of sea urchin eggs after fertilisation. Nature 1976, 262, 661–664. [Google Scholar] [CrossRef]
- Johnson, C.H.; Epel, D. Intracellular pH of sea urchin eggs measured by the dimethyloxazolidinedione (DMO) method. J. Cell Biol. 1981, 89, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Dubé, F.; Schmidt, T.; Johnson, C.H.; Epel, D. The hierarchy of requirements for an elevated intracellular pH during early development of sea urchin embryos. Cell 1985, 40, 657–666. [Google Scholar] [CrossRef]
- Lopo, A.; Vacquier, V.D. The rise and fall of intracellular pH of sea urchin eggs after fertilisation. Nature 1977, 269, 590–592. [Google Scholar] [CrossRef]
- Grainger, J.L.; Winkler, M.M.; Shen, S.S.; Steinhardt, R.A. Intracellular pH controls protein synthesis rate in the sea urchin egg and early embryo. Dev. Biol. 1979, 68, 396–406. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Alunni-Fabbroni, M.; Casavola, V.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–2197. [Google Scholar] [CrossRef] [Green Version]
- Fairbanks, L.D.; Bofill, M.; Ruckemann, K.; Simmonds, H.A. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J. Biol. Chem. 1995, 270, 29682–29689. [Google Scholar] [CrossRef] [Green Version]
- Lehninger, A.L.; Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry; Macmillan Learning: Hong Kong, China, 2005. [Google Scholar]
- Tong, X.; Zhao, F.; Thompson, C.B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Fan, T.W.M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [Green Version]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef]
- Lallous, N.; Grande-García, A.; Molina, R.; Ramón-Maiques, S. Expression, purification, crystallization and preliminary X-ray diffraction analysis of the dihydroorotase domain of human CAD. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1341–1345. [Google Scholar] [CrossRef] [Green Version]
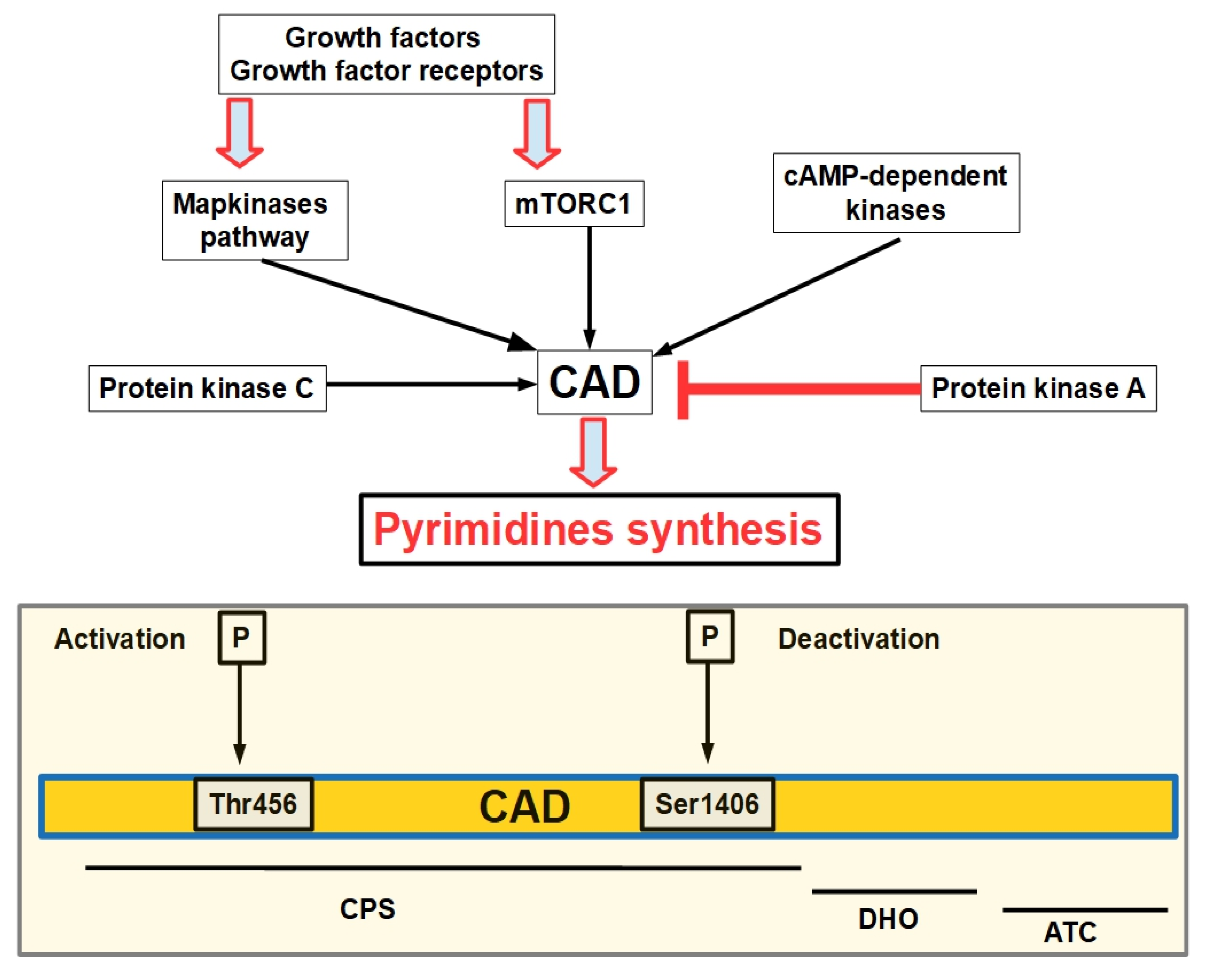
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Graves, L.M.; Guy, H.I.; Kozlowski, P.; Huang, M.; Lazarowski, E.; Pope, R.M.; Collins, M.A.; Dahlstrand, E.N.; Earp, H.S.; Evans, D.R. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 2000, 403, 328–332. [Google Scholar] [CrossRef]
- Carrey, E.A.; Campbell, D.G.; Hardie, D.G. Phosphorylation and activation of hamster carbamyl phosphate synthetase II by cAMP-dependent protein kinase. A novel mechanism for regulation of pyrimidine nucleotide biosynthesis. EMBO J. 1985, 4, 3735–3742. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, T.; Weber, G. Carbamoyl phosphate synthetase (glutamine-hydrolyzing): Increased activity in cancer cells. Science 1981, 212, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.; Reardon, M. Regulation of carbamoyl-phosphate synthase II. Adv. Enzym. Regul. 1986, 25, 65–83. [Google Scholar] [CrossRef]
- Reardon, M.A.; Weber, G. Increased synthesis of carbamoyl-phosphate synthase II (EC 6.3.5.5) in hepatoma 3924A. Cancer Res. 1986, 46, 3673–3676. [Google Scholar]
- Reardon, M.A.; Dixon, J.E.; Weber, G. Increased messenger RNA concentration for carbamoyl-phosphate synthase II in hepatoma 3924A. Biochem. Biophys. Res. Commun. 1987, 147, 494–500. [Google Scholar] [CrossRef]
- Makoff, A.J.; Radford, A. Genetics and biochemistry of carbamoyl phosphate biosynthesis and its utilization in the pyrimidine biosynthetic pathway. Microbiol. Rev. 1978, 42, 307–328. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Cardone, R.A.; Harguindey, S. Na+-H+ exchanger, pH regulation and cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 85–99. [Google Scholar] [CrossRef]
- Bhagavan, N.V.; Ha, C.-E. Nucleotide Metabolism. In Essentials of Medical Biochemistry; Academic Press: Cambridge, MA, USA, 2015; pp. 465–487. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Turner, M.; Belaid, A.; Lam, H.C.; Miller, S.K.; McNamara, M.C.; Baglini, C.; Housden, B.E.; Perrimon, N.; Kwiatkowski, D.J.; et al. mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 2017, 32, 624–638.e5. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Sebolt, J.S.; Aoki, T.; Eble, J.N.; Glover, J.L.; Weber, G. Inactivation by acivicin of carbamoyl-phosphate synthetase II of human colon carcinoma. Biochem. Pharmacol. 1985, 34, 97–100. [Google Scholar] [CrossRef]
- Stieglitz, K.A.; Xia, J.; Kantrowitz, E.R. The First High pH Structure of Escherichia coli Aspartate Transcarbamoylase. Proteins 2009, 74, 318. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, E.; Mossé, H. Aspartate carbamoyltransferase from rat liver. Biochem. J. 1966, 101, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.K.; Inouye, T.; Goldin, A.; Stark, G.R. Antitumor activity of N-(phosphonacetyl)-L-aspartic acid, a transition-state inhibitor of aspartate transcarbamylase. Cancer Res. 1976, 36, 2720–2725. [Google Scholar]
- Lei, Z.; Wang, B.; Lu, Z.; Wang, N.; Tan, H.; Zheng, J.; Jia, Z. New regulatory mechanism-based inhibitors of aspartate transcarbamoylase for potential anticancer drug development. FEBS J. 2020, 287, 3579–3599. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Cooney, D.A.; Vistica, D.T.; Kariya, S.; Johnson, R.K. Mechanisms of sensitivity or resistance of murine tumors to N-(phosphonacetyl)-L-aspartate (PALA). Cancer Treat. Rep. 1979, 63, 1291–1302. [Google Scholar]
- Ruiz-Ramos, A.; Velázquez-Campoy, A.; Grande-García, A.; Moreno-Morcillo, M.; Ramón-Maiques, S. Structure and Functional Characterization of Human Aspartate Transcarbamoylase, the Target of the Anti-tumoral Drug PALA. Structure 2016, 24, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Baillon, J.; Guichard, M.; Malaise, E.P.; Hervé, G. Kinetic parameters of aspartate transcarbamylase in human normal and tumoral cell lines. Cancer Res. 1983, 43, 2277–2282. [Google Scholar]
- O’Dwyer, P.J. The role of low-dose PALA in biochemical modulation. Pharmacol. Ther. 1990, 48, 371–380. [Google Scholar] [CrossRef]
- Balbaa, M.; Abdel-Megeed, M.; Diab, T.; Mansour, H. Inhibition of mammalian aspartate transcarbamylase by quinazolinone derivatives. J. Enzym. Inhib. Med. Chem. 2008, 23, 483–492. [Google Scholar] [CrossRef]
- Bidigare, R.R.; Sander, E.G.; Pettigrew, D.W. Evidence for a pH-dependent isomerization of Clostridium oroticum dihydroorotase. Biochim. Biophys. Acta 1985, 831, 159–160. [Google Scholar] [CrossRef]
- Porter, T.N.; Li, Y.; Raushel, F.M. Mechanism of the dihydroorotase reaction. Biochemistry 2004, 43, 16285–16292. [Google Scholar] [CrossRef] [PubMed]
- Del Caño-Ochoa, F.; Grande-García, A.; Reverte-López, M.; D’Abramo, M.; Ramón-Maiques, S. Characterization of the catalytic flexible loop in the dihydroorotase domain of the human multi-enzymatic protein CAD. J. Biol. Chem. 2018, 293, 18903–18913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmer, J.P.; Kelly, R.E.; Rinker, A.G.; Zimmermann, B.H.; Scully, J.L.; Kim, H.; Evans, D.R. Mammalian dihydroorotase: Nucleotide sequence, peptide sequences, and evolution of the dihydroorotase domain of the multifunctional protein CAD. Proc. Natl. Acad. Sci. USA 1990, 87, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopherson, R.I.; Jones, M.E. The effects of pH and inhibitors upon the catalytic activity of the dihydroorotase of multienzymatic protein pyr1-3 from mouse Ehrlich ascites carcinoma. J. Biol. Chem. 1980, 255, 3358–3370. [Google Scholar] [CrossRef]
- Christopherson, R.I.; Jones, M.E. Interconversion of carbamayl-L-aspartate and L-dihydroorotate by dihydroorotase from mouse Ehrlich ascites carcinoma. J. Biol. Chem. 1979, 254, 12506–12512. [Google Scholar] [CrossRef]
- Chen, K.F.; Lai, Y.Y.; Sun, H.S.; Tsai, S.J. Transcriptional repression of human cad gene by hypoxia inducible factor-1α. Nucleic Acids Res. 2005, 33, 5190. [Google Scholar] [CrossRef]
- Zhao, J.; Tian, M.; Zhang, S.; Delfarah, A.; Gao, R.; Rao, Y.; Savas, A.C.; Lu, A.; Bubb, L.; Lei, X.; et al. Deamidation Shunts RelA from Mediating Inflammation to Aerobic Glycolysis. Cell Metab. 2020, 31, 937–955.e7. [Google Scholar] [CrossRef]
- Ding, C.; He, J.; Liao, W.; Chen, J.; Li, J.; He, G.; Jia, Z.; Zhang, Y.; Luo, J.; Dai, X.; et al. Regulation of WNT/β-catenin signaling by carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) in colorectal cancer cell|Semantic Scholar. Int. J. Clin. Exp. Med. 2017, 10, 16243–16253. [Google Scholar]
- Guan, H.H.; Huang, Y.H.; Lin, E.S.; Chen, C.J.; Huang, C.Y. Structural basis for the interaction modes of dihydroorotase with the anticancer drugs 5-fluorouracil and 5-aminouracil. Biochem. Biophys. Res. Commun. 2021, 551, 33–37. [Google Scholar] [CrossRef]
- Kinder, D.H.; Frank, S.K.; Ames, M.M. Analogs of carbamyl aspartate as inhibitors of dihydroorotase: Preparation of boronic acid transition-state analogs and a zinc chelator carbamylhomocysteine. J. Med. Chem. 2002, 33, 819–823. [Google Scholar] [CrossRef]
- Mohsen, A.W.A.; Rigby, S.E.J.; Jensen, K.F.; Munro, A.W.; Scrutton, N.S. Thermodynamic basis of electron transfer in dihydroorotate dehydrogenase B from Lactococcus lactis: Analysis by potentiometry, EPR spectroscopy, and ENDOR spectroscopy. Biochemistry 2004, 43, 6498–6510. [Google Scholar] [CrossRef]
- Rowland, P.; Nørager, S.; Jensen, K.F.; Larsen, S. Structure of dihydroorotate dehydrogenase B: Electron transfer between two flavin groups bridged by an iron-sulphur cluster. Structure 2000, 8, 1227–1238. [Google Scholar] [CrossRef]
- Argyrou, A.; Washabaugh, M.W.; Pickart, C.M. Dihydroorotate dehydrogenase from Clostridium oroticum is a class 1B enzyme and utilizes a concerted mechanism of catalysis. Biochemistry 2000, 39, 10373–10384. [Google Scholar] [CrossRef]
- Cheleski, J.; Rocha, J.R.; Pinheiro, M.P.; Wiggers, H.J.; Da Silva, A.B.F.; Nonato, M.C.; Montanari, C.A. Novel insights for dihydroorotate dehydrogenase class 1A inhibitors discovery. Eur. J. Med. Chem. 2010, 45, 5899–5909. [Google Scholar] [CrossRef]
- Kahler, A.E.; Nielsen, F.S.; Switzer, R.L. Biochemical characterization of the heteromeric Bacillus subtilis dihydroorotate dehydrogenase and its isolated subunits. Arch. Biochem. Biophys. 1999, 371, 191–201. [Google Scholar] [CrossRef]
- Fagan, R.L.; Nelson, M.N.; Pagano, P.M.; Palfey, B.A. Mechanism of flavin reduction in class 2 dihydroorotate dehydrogenases. Biochemistry 2006, 45, 14926–14932. [Google Scholar] [CrossRef]
- Liu, S.; Neidhardt, E.A.; Grossman, T.H.; Ocain, T.; Clardy, J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.W. Dihydroorotate dehydrogenase (Neurospora). Methods Enzymol. 1978, 51, 63–69. [Google Scholar] [CrossRef]
- Fagan, R.L.; Palfey, B.A. Roles in binding and chemistry for conserved active site residues in the class 2 dihydroorotate dehydrogenase from Escherichia coli. Biochemistry 2009, 48, 7169–7178. [Google Scholar] [CrossRef] [Green Version]
- Zameitat, E.; Pierik, A.J.; Zocher, K.; Löffler, M. Dihydroorotate dehydrogenase from Saccharomyces cerevisiae: Spectroscopic investigations with the recombinant enzyme throw light on catalytic properties and metabolism of fumarate analogues. FEMS Yeast Res. 2007, 7, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Krungkrai, J.; Cerami, A.; Henderson, G.B. Purification and characterization of dihydroorotate dehydrogenase from the rodent malaria parasite Plasmodium berghei. Biochemistry 1991, 30, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Bader, B.; Knecht, W.; Fries, M.; Löffler, M. Expression, purification, and characterization of histidine-tagged rat and human flavoenzyme dihydroorotate dehydrogenase. Protein Expr. Purif. 1998, 13, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Rawls, J.; Knecht, W.; Diekert, K.; Lill, R.; Löffler, M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur. J. Biochem. 2000, 267, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Leban, J.; Vitt, D. Human dihydroorotate dehydrogenase inhibitors, a novel approach for the treatment of autoimmune and inflammatory diseases. Arzneimittelforschung 2011, 61, 66–72. [Google Scholar] [CrossRef]
- Greene, S.; Watanabe, K.; Braatz-Trulson, J.; Lou, L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem. Pharmacol. 1995, 50, 861–867. [Google Scholar] [CrossRef]
- Zhu, S.; Yan, X.; Xiang, Z.; Ding, H.-F.; Cui, H. Leflunomide Reduces Proliferation and Induces Apoptosis in Neuroblastoma Cells In Vitro and In Vivo. PLoS ONE 2013, 8, e71555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Chu, M. Leflunomide: A promising drug with good antitumor potential. Biochem. Biophys. Res. Commun. 2018, 496, 726–730. [Google Scholar] [CrossRef]
- Chen, S.F.; Perrella, F.W.; Behrens, D.L.; Papp, L.M. Inhibition of dihydroorotate dehydrogenase activity by brequinar sodium. Cancer Res. 1992, 52, 3521–3527. [Google Scholar]
- Arteaga, C.L.; Brown, T.D.; Kuhn, J.G.; Shen, H.S.; O’Rourke, T.J.; Beougher, K.; Brentzel, H.J.; Von Hoff, D.D.; Weiss, G.R. Phase I clinical and pharmacokinetic trial of Brequinar sodium (DuP 785; NSC 368390). Cancer Res. 1989, 49, 4648–4653. [Google Scholar]
- Maroun, J.; Ruckdeschel, J.; Natale, R.; Morgan, R.; Dallaire, B.; Sisk, R.; Gyves, J. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Cancer Chemother. Pharmacol. 1993, 32, 64–66. [Google Scholar] [CrossRef] [Green Version]
- Cody, R.; Stewart, D.; DeForni, M.; Moore, M.; Dallaire, B.; Azarnia, N.; Gyves, J.W. Multicenter phase II study of brequinar sodium in patients with advanced breast cancer. Am. J. Clin. Oncol. 1993, 16, 526–528. [Google Scholar] [CrossRef]
- Natale, R.; Wheeler, R.; Moore, M.; Dallaire, B.; Lynch, W.; Carlson, R.; Grillo-lopez, A.; Gyves, L. Multicenter phase II trial of brequinar sodium in patients with advanced melanoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1992, 3, 659–660. [Google Scholar] [CrossRef]
- Moore, M.; Maroun, J.; Robert, F.; Natale, R.; Neidhart, J.; Dallaire, B.; Sisk, R.; Gyves, J. Multicenter phase II study of brequinar sodium in patients with advanced gastrointestinal cancer. Investig. New Drugs 1993, 11, 61–65. [Google Scholar] [CrossRef]
- Peters, G.J. Re-evaluation of Brequinar sodium, a dihydroorotate dehydrogenase inhibitor. Nucleosides Nucleotides Nucleic Acids 2018, 37, 666–678. [Google Scholar] [CrossRef]
- Sykes, D.B.; Kfoury, Y.S.; Mercier, F.E.; Wawer, M.J.; Law, J.M.; Haynes, M.K.; Lewis, T.A.; Schajnovitz, A.; Jain, E.; Lee, D.; et al. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 2016, 167, 171–186.e15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-C.; Sun, J.-M.; Li, X.-H.; Yang, X.-F.; Zhang, Y.-C.; Xin, Y. Role of PTEN and MMP-7 expression in growth, invasion, metastasis and angiogenesis of gastric carcinoma. Pathol. Int. 2003, 53, 659–666. [Google Scholar] [CrossRef]
- Peters, G.J. Antipyrimidine effects of five different pyrimidine de novo synthesis inhibitors in three head and neck cancer cell lines. Nucleosides Nucleotides Nucleic Acids 2018, 37, 329–339. [Google Scholar] [CrossRef]
- Cuthbertson, C.R.; Guo, H.; Kyani, A.; Madak, J.T.; Arabzada, Z.; Neamati, N. The Dihydroorotate Dehydrogenase Inhibitor Brequinar Is Synergistic with ENT1/2 Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1242–1252. [Google Scholar] [CrossRef]
- Dorasamy, M.S.; AB, A.; Nellore, K.; Wong, P.F. Synergistic inhibition of melanoma xenografts by Brequinar sodium and Doxorubicin. Biomed. Pharmacother. 2019, 110, 29–36. [Google Scholar] [CrossRef]
- Sykes, D.B. The emergence of dihydroorotate dehydrogenase (DHODH) as a therapeutic target in acute myeloid leukemia. Expert Opin. Ther. Targets 2018, 22, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef]
- Okuda, K.; Tatematsu, T.; Yano, M.; Nakamae, K.; Yamada, T.; Kasugai, T.; Nishida, T.; Sano, M.; Moriyama, S.; Haneda, H.; et al. The relationship between the expression of thymidylate synthase, dihydropyrimidine dehydrogenase, orotate phosphoribosyltransferase, excision repair cross-complementation group 1 and class III β-tubulin, and the therapeutic effect of S-1 or carboplatin plus paclitaxel in non-small-cell lung cancer. Mol. Clin. Oncol. 2018, 9, 21–29. [Google Scholar] [CrossRef]
- Sakurai, Y.; Sakamoto, K.; Sugimoto, Y.; Yoshida, I.; Masui, T.; Tonomura, S.; Inaba, K.; Shoji, M.; Nakamura, Y.; Uyama, I.; et al. Orotate phosphoribosyltransferase levels measured by a newly established enzyme-linked immunosorbent assay in gastric carcinoma. Cancer Sci. 2006, 97, 492–498. [Google Scholar] [CrossRef]
- Choi, I.S.; Lee, H.S.; Lee, K.W.; Kim, H.; Kim, K.H.; Kim, Y.J.; Kim, J.H.; Kim, W.H.; Lee, J.S. Biomarker analysis in patients with advanced gastric cancer treated with S-1 plus cisplatin chemotherapy: Orotate phosphoribosyltransferase expression is associated with treatment outcomes. Med. Oncol. 2011, 28, 991–998. [Google Scholar] [CrossRef]
- Ichikawa, W.; Uetake, H.; Shirota, Y.; Yamada, H.; Takahashi, T.; Nihei, Z.; Sugihara, K.; Sasaki, Y.; Hirayama, R. Both gene expression for orotate phosphoribosyltransferase and its ratio to dihydropyrimidine dehydrogenase influence outcome following fluoropyrimidine-based chemotherapy for metastatic colorectal cancer. Br. J. Cancer 2003, 89, 1486–1492. [Google Scholar] [CrossRef] [Green Version]
- Sparreboom, A.; Loos, W.J.; de Jonge, M.J.A.; Verweij, J. Clinical trial design: Incorporation of pharmacokinetic, pharmacodynamic, and pharmacogenetic principles. In Anticancer Drug Development; Elsevier: Amsterdam, The Netherlands, 2002; pp. 329–351. [Google Scholar]
- Mizutani, Y.; Wada, H.; Fukushima, M.; Yoshida, O.; Nakanishi, H.; Li, Y.N.; Miki, T. Prognostic significance of orotate phosphoribosyltransferase activity in bladder carcinoma. Cancer 2004, 100, 723–731. [Google Scholar] [CrossRef]
- Akahoshi, K.; Ban, D.; Kuboki, R.; Oba, A.; Ono, H.; Mitsunori, Y.; Kudo, A.; Tanaka, S.; Tanabe, M. Orotate phosphoribosyltransferase as a predictor of benefit from S-1 adjuvant chemotherapy for cholangiocarcinoma patients. J. Gastroenterol. Hepatol. 2019, 34, 1108–1115. [Google Scholar] [CrossRef]
- Yan, D.; Ryu, M.; An, E.; Park, Y.; Na, H.; Ma, J.; Tian, Y.; Cecchi, F.; Hembrough, T.; Kang, Y.; et al. Predicting survival benefit of capecitabine plus cisplatin in patients with metastatic gastric cancer patients using quantitative proteomics. Ann. Oncol. 2018, 29, viii51. [Google Scholar] [CrossRef]
- Ashton, R.W.; Strauss, R.S.; Chung, S.H.; Sloan, D.L. Orotate phosphoribosyltransferase from yeast: Studies of the structure of the pyrimidine substrate binding site. Arch. Biochem. Biophys. 1989, 272, 421–432. [Google Scholar] [CrossRef]
- Robinson, J.L.; Arson, I. Nucleotide Inhibition of Post-Orotate Pyrimidine Synthesis Pathway Enzymes of Bovine Mammary Tissue. J. Dairy Sci. 1974, 57, 1410–1413. [Google Scholar] [CrossRef]
- Traut, T.W.; Jones, M.E. Inhibitors of orotate phosphoribosyl-transferase and orotidine-5′-phosphate decarboxylase from mouse Ehrlich ascites cells: A procedure for analyzing the inhibition of a multi-enzyme complex. Biochem. Pharmacol. 1977, 26, 2291–2296. [Google Scholar] [CrossRef]
- Donini, S.; Ferraris, D.M.; Miggiano, R.; Massarotti, A.; Rizzi, M. Structural investigations on orotate phosphoribosyltransferase from Mycobacterium tuberculosis, a key enzyme of the de novo pyrimidine biosynthesis. Sci. Rep. 2017, 7, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchi, M.; Mizuno, H.; Kawai, Y.; Tsuboi, T.; Sumi, S.; Okajima, K.; Hodgson, M.E.; Ogawa, H.; Wada, Y. Molecular cloning of the human UMP synthase gene and characterization of point mutations in two hereditary orotic aciduria families. Am. J. Hum. Genet. 1997, 60, 525–539. [Google Scholar]
- Gabrielsen, B.; Kirsi, J.J.; Kwong, C.D.; Carter, D.A.; Krauth, C.A.; Hanna, L.K.; Huggins, J.W.; Monath, T.P.; Kefauver, D.F.; Blough, H.A.; et al. In vitro and in vivo antiviral (RNA) evaluation of orotidine 5′-monophosphate decarboxylase inhibitors and analogues including 6-azauridine-5′-(ethyl methoxyalaninyl)phosphate (a 5′-monophosphate prodrug). Antivir. Chem. Chemother. 1994, 5, 209–220. [Google Scholar] [CrossRef]
- Kotra, L.; Meza-Avina, M.; Wei, L.; Buhendwa, M.; Poduch, E.; Bello, A.; Pai, E. Inhibition of orotidine 5′-monophosphate decarboxylase and its therapeutic potential. Mini Rev. Med. Chem. 2008, 8, 239–247. [Google Scholar] [CrossRef]
- Smiley, J.A.; Paneth, P.; O’Leary, M.H.; Bell, J.B.; Jones, M.E. Investigation of the enzymatic mechanism of yeast orotidine-5′-monophosphate decarboxylase using 13C kinetic isotope effects. Biochemistry 1991, 30, 6216–6223. [Google Scholar] [CrossRef]
- Appleby, T.C.; Kinsland, C.; Begley, T.P.; Ealick, S.E. The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc. Natl. Acad. Sci. USA 2000, 97, 2005–2010. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Johansson, M.; Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 2008, 283, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.-Y.; Dutschman, G.E.; Lam, W.; Jiang, Z.; Cheng, Y.-C. Characterization of human UMP/CMP kinase and its phosphorylation of D- and L-form deoxycytidine analogue monophosphates. Cancer Res. 2002, 62, 1624–1631. [Google Scholar]
- Van Rompay, A.R.; Johansson, M.; Karlsson, A. Phosphorylation of deoxycytidine analog monophosphates by UMP-CMP kinase: Molecular characterization of the human enzyme. Mol. Pharmacol. 1999, 56, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, J.A.C.; Roy, B.; Topalis, D.; Périgaud, C.; Deville-Bonne, D. Enantio-selectivity of human nucleoside monophosphate kinases. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1375–1379. [Google Scholar] [CrossRef]
- Liu, N.Q.; De Marchi, T.; Timmermans, A.; Trapman-Jansen, A.M.A.C.; Foekens, R.; Look, M.P.; Smid, M.; Van Deurzen, C.H.M.; Span, P.N.; Sweep, F.C.G.J.; et al. Prognostic significance of nuclear expression of UMP-CMP kinase in triple negative breast cancer patients. Sci. Rep. 2016, 6, 32027. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.M.A.; Di Molfetta, D.; Greco, M.R.; Koltai, T.; Alfarouk, K.O.; Reshkin, S.J.; Cardone, R.A. Tumor Microenvironment Features and Chemoresistance in Pancreatic Ductal Adenocarcinoma: Insights into Targeting Physicochemical Barriers and Metabolism as Therapeutic Approaches. Cancers 2021, 13, 6135. [Google Scholar] [CrossRef]
- Chen, S.; Wang, X.; Ye, X.; Ma, D.; Chen, C.; Cai, J.; Fu, Y.; Cheng, X.; Chen, Y.; Gong, X.; et al. Identification of Human UMP/CMP Kinase 1 as Doxorubicin Binding Target Using Protein Microarray. SLAS Discov. 2017, 22, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Apte, U. Galactosamine. In Encyclopedia of Toxicology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 689–690. ISBN 9780123864543. [Google Scholar]
- Preiss, J. Biochemistry and Molecular Biology of Glycogen Synthesis in Bacteria and Mammals and Starch Synthesis in Plants. In Comprehensive Natural Products II; Elsevier: Amsterdam, The Netherlands, 2010; Volume 6, pp. 429–491. ISBN 9780080453828. [Google Scholar]
- Hubbard, J.A.; Binder, D.K. Gliotransmitters. In Astrocytes and Epilepsy; Academic Press: Cambridge, MA, USA, 2016; pp. 53–73. [Google Scholar] [CrossRef]
- Parks, R.E.E.; Aganwal, R.P.P. Nucleoside Diphosphokinases. Enzymes 1973, 8, 307–333. [Google Scholar] [CrossRef]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef]
- Tee, Y.T.; Chen, G.D.; Lin, L.Y.; Ko, J.L.; Wang, P.H. Nm23-H1: A metastasis-associated gene. Taiwan J. Obstet. Gynecol. 2006, 45, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Boissan, M.; Lacombe, M.L. NM23, an example of a metastasis suppressor gene. Bull. Cancer 2012, 99, 431–440. [Google Scholar] [CrossRef]
- Lam, S.C.T.; Packham, M.A. Isolation and kinetic studies of nucleoside diphosphokinase from human platelets and effects of cAMP phosphodiesterase inhibitors. Biochem. Pharmacol. 1986, 35, 4449–4455. [Google Scholar] [CrossRef]
- Zhu, M.; Sun, W.; Wang, Y.; Meng, J.; Zhang, D.; Guo, T.; Ouyang, P.; Ying, H.; Xie, J. Engineered cytidine triphosphate synthetase with reduced product inhibition. Protein Eng. Des. Sel. 2014, 27, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.C.; Kizaki, H.; Weber, G.; Morris, H.P. Increased CTP synthetase activity in cancer cells. Nature 1978, 271, 71–73. [Google Scholar] [CrossRef]
- Kassel, K.M.; Au, D.R.; Higgins, M.J.; Hines, M.; Graves, L.M. Regulation of Human Cytidine Triphosphate Synthetase 2 by Phosphorylation. J. Biol. Chem. 2010, 285, 33727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschuur, A.C.; Van Gennip, A.H.; Leen, R.; Muller, E.J.; Elzinga, L.; Voûte, P.A.; Van Kuilenburg, A.B.P. Cyclopentenyl cytosine inhibits cytidine triphosphate synthetase in paediatric acute non-lymphocytic leukaemia: A promising target for chemotherapy. Eur. J. Cancer 2000, 36, 627–635. [Google Scholar] [CrossRef]
- Richards, N.G.J.; Humkey, R.N.; Li, K.; Meyer, M.E.; Córdova de Sintjago, T.C. Tunnels and Intermediates in the Glutamine-Dependent Amidotransferases. In Comprehensive Natural Products II Chemistry and Biology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 8, pp. 161–230. [Google Scholar] [CrossRef]
- Endrizzi, J.A.; Kim, H.; Anderson, P.M.; Baldwin, E.P. Mechanisms of Product Feedback Regulation and Drug Resistance in Cytidine Triphosphate Synthetases from the Structure of a CTP-Inhibited Complex. Biochemistry 2005, 44, 13491. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.J.; Loiselle, D.; Haystead, T.A.; Graves, L.M. Human cytidine triphosphate synthetase 1 interacting proteins. Nucleosides. Nucleotides Nucleic Acids 2008, 27, 850–857. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Alqahtani, S.S.; Alshahrani, S.; Morgenstern, J.; Supuran, C.T.; Reshkin, S.J. The possible role of methylglyoxal metabolism in cancer. J. Enzym. Inhib. Med. Chem. 2021, 36, 2010–2015. [Google Scholar] [CrossRef]
- Eklund, H.; Uhlin, U.; Färnegårdh, M.; Logan, D.T.; Nordlund, P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 2001, 77, 177–268. [Google Scholar] [CrossRef]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2014, 34, 2011–2021. [Google Scholar] [CrossRef]
- Su, Y.F.; Wu, T.F.; Ko, J.L.; Tsai, H.T.; Tee, Y.T.; Chien, M.H.; Chou, C.H.; Lin, W.L.; Low, H.Y.; Chou, M.Y.; et al. The Expression of Ribonucleotide Reductase M2 in the Carcinogenesis of Uterine Cervix and Its Relationship with Clinicopathological Characteristics and Prognosis of Cancer Patients. PLoS ONE 2014, 9, e91644. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.; Warden, C.D.; Huang, Y.; Loera, S.; Xue, L.; Zhang, S.; Chu, P.; Zheng, S.; Yen, Y. Prognostic and therapeutic significance of ribonucleotide reductase small subunit M2 in estrogen-negative breast cancers. BMC Cancer 2014, 14, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuno, M.F.; Cerqueira, S.A.; Ramos, M.J. Ribonucleotide Reductase: A Critical Enzyme for Cancer Chemotherapy and Antiviral Agents. Recent Pat. Anticancer Drug Discov. 2007, 2, 11–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, T.; Hino, R.; Uozaki, H.; Maeda, D.; Ushiku, T.; Shinozaki, A.; Sakatani, T.; Fukayama, M. Expression of ribonucleotide reductase M2 subunit in gastric cancer and effects of RRM2 inhibition in vitro. Hum. Pathol. 2010, 41, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Jiang, W.; Jiang, J.; Wang, Y.; Kim, R.; Liu, X.; Liu, X. Predictive and prognostic roles of ribonucleotide reductase M1 in resectable pancreatic adenocarcinoma. Cancer 2013, 119, 173–181. [Google Scholar] [CrossRef]
- Volante, M.; Terzolo, M.; Fassnacht, M.; Rapa, I.; Germano, A.; Sbiera, S.; Daffara, F.; Sperone, P.; Scagliotti, G.; Allolio, B.; et al. Ribonucleotide reductase large subunit (RRM1) gene expression may predict efficacy of adjuvant mitotane in adrenocortical cancer. Clin. Cancer Res. 2012, 18, 3452–3461. [Google Scholar] [CrossRef] [Green Version]
- Souglakos, J.; Boukovinas, I.; Taron, M.; Mendez, P.; Mavroudis, D.; Tripaki, M.; Hatzidaki, D.; Koutsopoulos, A.; Stathopoulos, E.; Georgoulias, V.; et al. Ribonucleotide reductase subunits M1 and M2 mRNA expression levels and clinical outcome of lung adenocarcinoma patients treated with docetaxel/gemcitabine. Br. J. Cancer 2008, 98, 1710–1715. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, P.; Voevodskaya, N.; Klotz, L.O.; Brenneisen, P.; Gräslund, A.; Sies, H. Loss of the tyrosyl radical in mouse ribonucleotide reductase by (-)-epicatechin. Biochem. Biophys. Res. Commun. 2005, 326, 614–617. [Google Scholar] [CrossRef]
- Elford, H.L.; Van’t Riet, B.; Wampler, G.L.; Lin, A.L.; Elford, R.M. Regulation of ribonucleotide reductase in mammalian cells by chemotherapeutic agents. Adv. Enzym. Regul. 1980, 19, 151–168. [Google Scholar] [CrossRef]
- Holmgren, A.; Sengupta, R. The use of thiols by ribonucleotide reductase. Free Radic. Biol. Med. 2010, 49, 1617–1628. [Google Scholar] [CrossRef]
- Petrelli, R.; Meli, M.; Vita, P.; Torquati, I.; Ferro, A.; Vodnala, M.; D’Alessandro, N.; Tolomeo, M.; Del Bello, F.; Kusumanchi, P.; et al. From the covalent linkage of drugs to novel inhibitors of ribonucleotide reductase: Synthesis and biological evaluation of valproic esters of 3′-C-methyladenosine. Bioorg. Med. Chem. Lett. 2014, 24, 5304–5309. [Google Scholar] [CrossRef]
- Lammers, M.; Follmann, H. The ribonucleotide reductases—A unique group of metalloenzymes essential for cell proliferation. In Inorganic Elements in Biochemistry; Springer: Berlin/Heidelberg, Germany, 1983; pp. 27–91. [Google Scholar]
- Zhu, L.; Zhou, B.; Chen, X.; Jiang, H.; Shao, J.; Yen, Y. Inhibitory mechanisms of heterocyclic carboxaldehyde thiosemicabazones for two forms of human ribonucleotide reductase. Biochem. Pharmacol. 2009, 78, 1178–1185. [Google Scholar] [CrossRef]
- Sigmond, J.; Kamphuis, J.A.E.; Laan, A.C.; Hoebe, E.K.; Bergman, A.M.; Peters, G.J. The synergistic interaction of gemcitabine and cytosine arabinoside with the ribonucleotide reductase inhibitor triapine is schedule dependent. Biochem. Pharmacol. 2007, 73, 1548–1557. [Google Scholar] [CrossRef]
- Sato, A.; Bacon, P.E.; Cory, J.G. Studies on the differential mechanisms of inhibition of ribonucleotide reductase by specific inhibitors of the non-heme iron subunit. Adv. Enzym. Regul. 1984, 22, 231–241. [Google Scholar] [CrossRef]
- Krishnan, K.; Prathiba, K.; Jayaprakash, V.; Basu, A.; Mishra, N.; Zhou, B.; Hu, S.; Yen, Y. Synthesis and ribonucleotide reductase inhibitory activity of thiosemicarbazones. Bioorg. Med. Chem. Lett. 2008, 18, 6248–6250. [Google Scholar] [CrossRef]
- Holmgren, A. Regulation of ribonucleotide reductase. Curr. Top. Cell. Regul. 1981, 19, 47–76. [Google Scholar] [CrossRef]
- Smith, P.; Zhou, B.; Ho, N.; Yuan, Y.C.; Su, L.; Tsai, S.C.; Yen, Y. 2.6 A X-ray crystal structure of human p53R2, a p53-inducible ribonucleotide reductase. Biochemistry 2009, 48, 11134–11141. [Google Scholar] [CrossRef] [Green Version]
- Gropper, S.S.; Smith, J.L.; Carr, T.P. Advanced Nutrition and Human Metabolism, 8th ed.; Cengage Learning: Boston, MA, USA, 2021; ISBN1 0357450108. ISBN2 9780357450109. [Google Scholar]
- Lockshin, A.; Moran, R.G.; Danenberg, P.V. Thymidylate synthetase purified to homogeneity from human leukemic cells. Proc. Natl. Acad. Sci. USA 1979, 76, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.; Brown, M.; McNulty, C.; Valentine, J.; Annesley, A.; Galloway, S.; Welch, I.; West, C.M.; Pritchard, S. A pilot study to investigate the role of thymidylate synthase as a marker of prognosis for neoadjuvant chemotherapy in gastric and gastro-oesophageal junction adenocarcinoma. Gastroenterol. Res. Pract. 2013, 2013, 502153. [Google Scholar] [CrossRef] [Green Version]
- Squires, M.H.; Fisher, S.B.; Fisher, K.E.; Patel, S.H.; Kooby, D.A.; El-Rayes, B.F.; Staley, C.A.; Farris, A.B.; Maithel, S.K. Differential expression and prognostic value of ERCC1 and thymidylate synthase in resected gastric adenocarcinoma. Cancer 2013, 119, 3242–3250. [Google Scholar] [CrossRef] [Green Version]
- Houghton, P.J.; Houghton, J.A.; Germain, G.; Torrance, P.M. Development and characterization of a human colon adenocarcinoma xenograft deficient in thymidine salvage. Cancer Res. 1987, 47, 2117–2122. [Google Scholar]
- Yoo, B.K.; Emdad, L.; Gredler, R.; Fuller, C.; Dumur, C.I.; Jones, K.H.; Jackson-Cook, C.; Su, Z.Z.; Chen, D.; Saxena, U.H.; et al. Transcription factor Late SV40 Factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 8357–8362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calascibetta, A.; Martorana, A.; Cabibi, D.; Aragona, F.; Sanguedolce, R. Relationship between thymidylate synthase and p53 and response to FEC versus taxane adjuvant chemotherapy for breast carcinoma. J. Chemother. 2011, 23, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Pestalozzi, B.C.; Peterson, H.F.; Gelber, R.D.; Goldhirsch, A.; Gusterson, B.A.; Trihia, H.; Lindtner, J.; Cortés-Funes, H.; Simmoncini, E.; Byrne, M.J.; et al. Prognostic importance of thymidylate synthase expression in early breast cancer. J. Clin. Oncol. 1997, 15, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Rustum, Y.M. Thymidylate synthase: A critical target in cancer therapy? Front. Biosci. 2004, 9, 2467–2473. [Google Scholar] [CrossRef] [PubMed]
- Jason, T.L.H.; Berg, R.W.; Vincent, M.D.; Koropatnick, J. Antisense targeting of thymidylate synthase (TS) mRNA increases TS gene transcription and TS protein: Effects on human tumor cell sensitivity to TS enzyme-inhibiting drugs. Gene Expr. 2007, 13, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, E.; Backus, H.H.J.; Wouters, D.; Ferreira, C.G.; Van Houten, V.M.M.; Brakenhoff, R.H.; Poupon, M.F.; Azzarello, A.; Pinedo, H.M.; Peters, G.J. Changes in the status of p53 affect drug sensitivity to thymidylate synthase (TS) inhibitors by altering TS levels. Br. J. Cancer 2007, 96, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Walla, M.; Wyatt, M.D. Uracil incorporation into genomic DNA does not predict toxicity caused by chemotherapeutic inhibition of thymidylate synthase. DNA Repair 2008, 7, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Cardone, R.A.R.; Alfarouk, K.O.K.; Elliott, R.L.R.; Alqahtani, S.S.S.; Ahmed, S.S.B.M.; Aljarbou, A.A.N.; Greco, M.; Cannone, S.; Reshkin, S.S.J. The Role of Sodium Hydrogen Exchanger 1 in Dysregulation of Proton Dynamics and Reprogramming of Cancer Metabolism as a Sequela. Int. J. Mol. Sci. 2019, 20, 3694. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, L.; Seyfried, T.; Alfarouk, K.O.; Da Veiga Moreira, J.; Fais, S. Out of Warburg effect: An effective cancer treatment targeting the tumor specific metabolism and dysregulated pH. Semin. Cancer Biol. 2017, 43, 134–138. [Google Scholar] [CrossRef]
- Moore, E.C. The Effects of Ferrous Ion and Dithioerythritol on Inhibition by Hydroxyurea of Ribonucleotide Reductase1|Cancer Research|American Association for Cancer Research. Cancer Res. 1969, 29, 291–295. [Google Scholar]
- Moore, E.C.; Reichard, P. Enzymatic synthesis of deoxyribonucleotides. vi. the cytidine diphosphate reductase system from novikoff hepatoma. J. Biol. Chem. 1964, 239, 3453–3456. [Google Scholar] [CrossRef]
- Trovato Salinaro, A.; Pennisi, M.; Di Paola, R.; Scuto, M.; Crupi, R.; Cambria, M.T.; Ontario, M.L.; Tomasello, M.; Uva, M.; Maiolino, L.; et al. Neuroinflammation and Neurohormesis in the Pathogenesis of Alzheimer’s Disease and Alzheimer-Linked Pathologies: Modulation by Nutritional Mushrooms. Immun. Ageing 2018, 15, 8. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, E. Hormesis: Path and Progression to Significance. Int. J. Mol. Sci. 2018, 19, 2871. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ahn, Y.H.; Benjamin, I.J.; Honda, T.; Hicks, R.J.; Calabrese, V.; Cole, P.A.; Dinkova-Kostova, A.T. HSF1-Dependent Upregulation of Hsp70 by Sulfhydryl-Reactive Inducers of the KEAP1/NRF2/ARE Pathway. Chem. Biol. 2011, 18, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Zuehlke, A.D.; Wren, N.; Tenge VJohnson, J.L. Interaction of heat shock protein 90 and the co-chaperone Cpr6 with Ura2, a bifunctional enzyme required for pyrimidine biosynthesis. J. Biol. Chem. 2013, 288, 27406–27414. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Optimal pH | Possible Inhibitor |
|---|---|---|
| Phosphate synthetase II | 7.4 | Acivicin [33] |
| Aspartate transcarbamoylase (ATCase) | pH in bacteria (8.5) | N-(phosphonacetyl)-l-aspartate (PALA) [38] |
| Dihydroorotate dehydrogenase | 8 | Leflunomide [68,69,70] |
| Orotate phosphoribosyltransferase | The forward reaction is 8, the reverse one is 6.5–7.5 | Include xanthosine, uridine 5′-phosphates, cytidine barbiturate, 5-flouro orotate [95,96] |
| Orotidine 5′-phosphate decarboxylase | approx. 7.5 | Pyrazofurin and 6-aza uridine 5′-monophosphate [99,100] |
| Cytidine monophosphate kinase | 7.4 | Gemcitabine [65] |
| Nucleoside-diphosphate kinase | 8 | Theophylline [117] |
| Cytosine triphosphate synthase | 8 | Cyclopentenyl cytosine [121] |
| Ribonucleotide reductase enzyme | 7.5–8 | Cisplatin, chlorambucil, desferrioxamine, gemcitabine, and hydroxyurea [135,136,137,138,139,140,141] |
| Tymidylate synthase | 7.0 and 8.1 | Capecitabine and 5 fluorouracil [154,155,156,157] |
| Ribonucleotide reductase | 7.5 to 8 with a low iron level | Gemcitabine and iron chelators [160] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, S.S.; Koltai, T.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Ahmed, S.B.M.; Molfetta, D.D.; Carvalho, T.M.A.; Cardone, R.A.; Reshkin, S.J.; et al. Role of pH in Regulating Cancer Pyrimidine Synthesis. J. Xenobiot. 2022, 12, 158-180. https://doi.org/10.3390/jox12030014
Alqahtani SS, Koltai T, Ibrahim ME, Bashir AHH, Alhoufie STS, Ahmed SBM, Molfetta DD, Carvalho TMA, Cardone RA, Reshkin SJ, et al. Role of pH in Regulating Cancer Pyrimidine Synthesis. Journal of Xenobiotics. 2022; 12(3):158-180. https://doi.org/10.3390/jox12030014
Chicago/Turabian StyleAlqahtani, Saad Saeed, Tomas Koltai, Muntaser E. Ibrahim, Adil H. H. Bashir, Sari T. S. Alhoufie, Samrein B. M. Ahmed, Daria Di Molfetta, Tiago M. A. Carvalho, Rosa Angela Cardone, Stephan Joel Reshkin, and et al. 2022. "Role of pH in Regulating Cancer Pyrimidine Synthesis" Journal of Xenobiotics 12, no. 3: 158-180. https://doi.org/10.3390/jox12030014
APA StyleAlqahtani, S. S., Koltai, T., Ibrahim, M. E., Bashir, A. H. H., Alhoufie, S. T. S., Ahmed, S. B. M., Molfetta, D. D., Carvalho, T. M. A., Cardone, R. A., Reshkin, S. J., Hifny, A., Ahmed, M. E., & Alfarouk, K. O. (2022). Role of pH in Regulating Cancer Pyrimidine Synthesis. Journal of Xenobiotics, 12(3), 158-180. https://doi.org/10.3390/jox12030014