
The Role of Myeloperoxidase in Clozapine-Induced Inflammation: A Mechanistic Update for Idiosyncratic Drug-Induced Agranulocytosis


Abstract
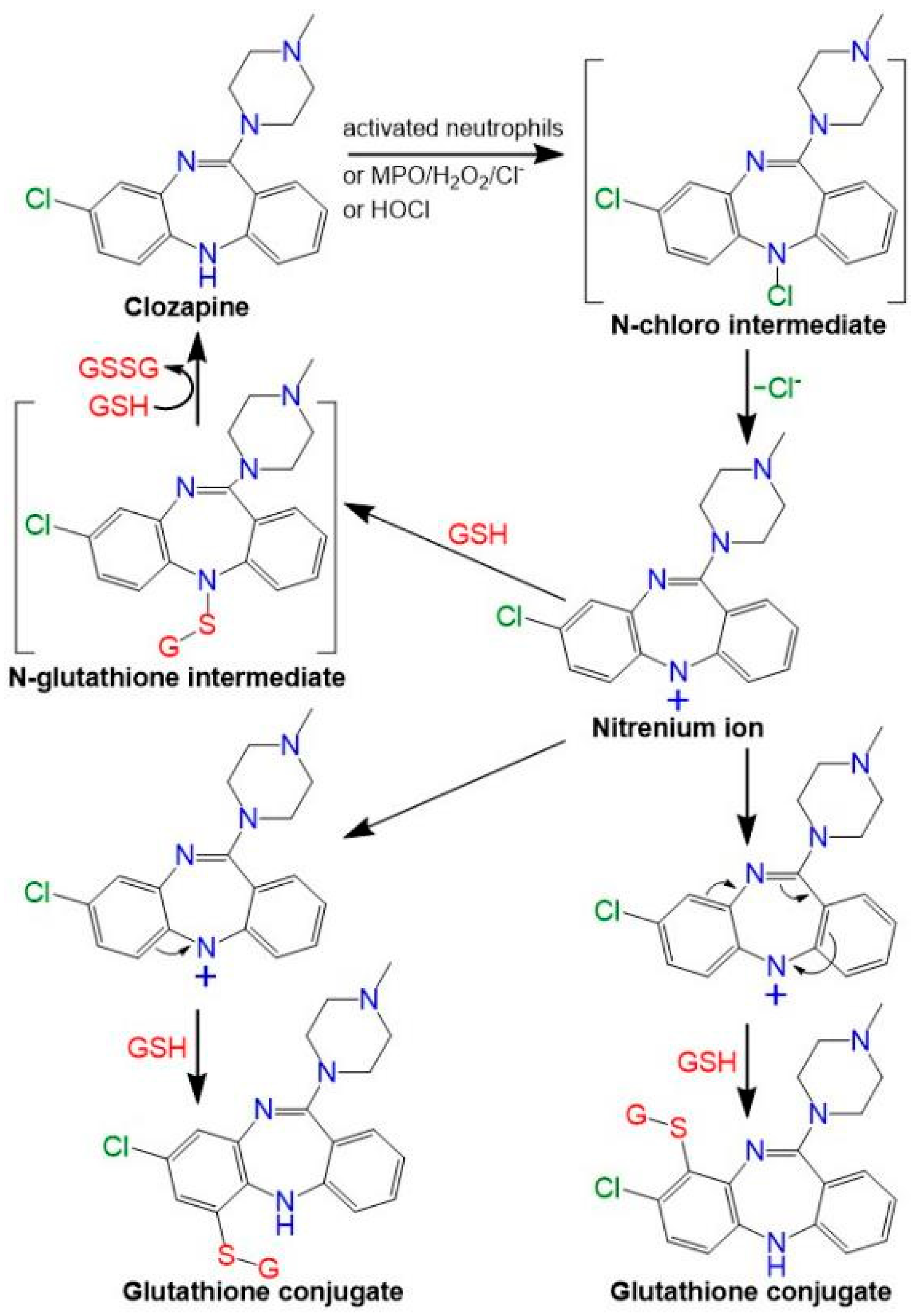
:1. Introduction
2. Results
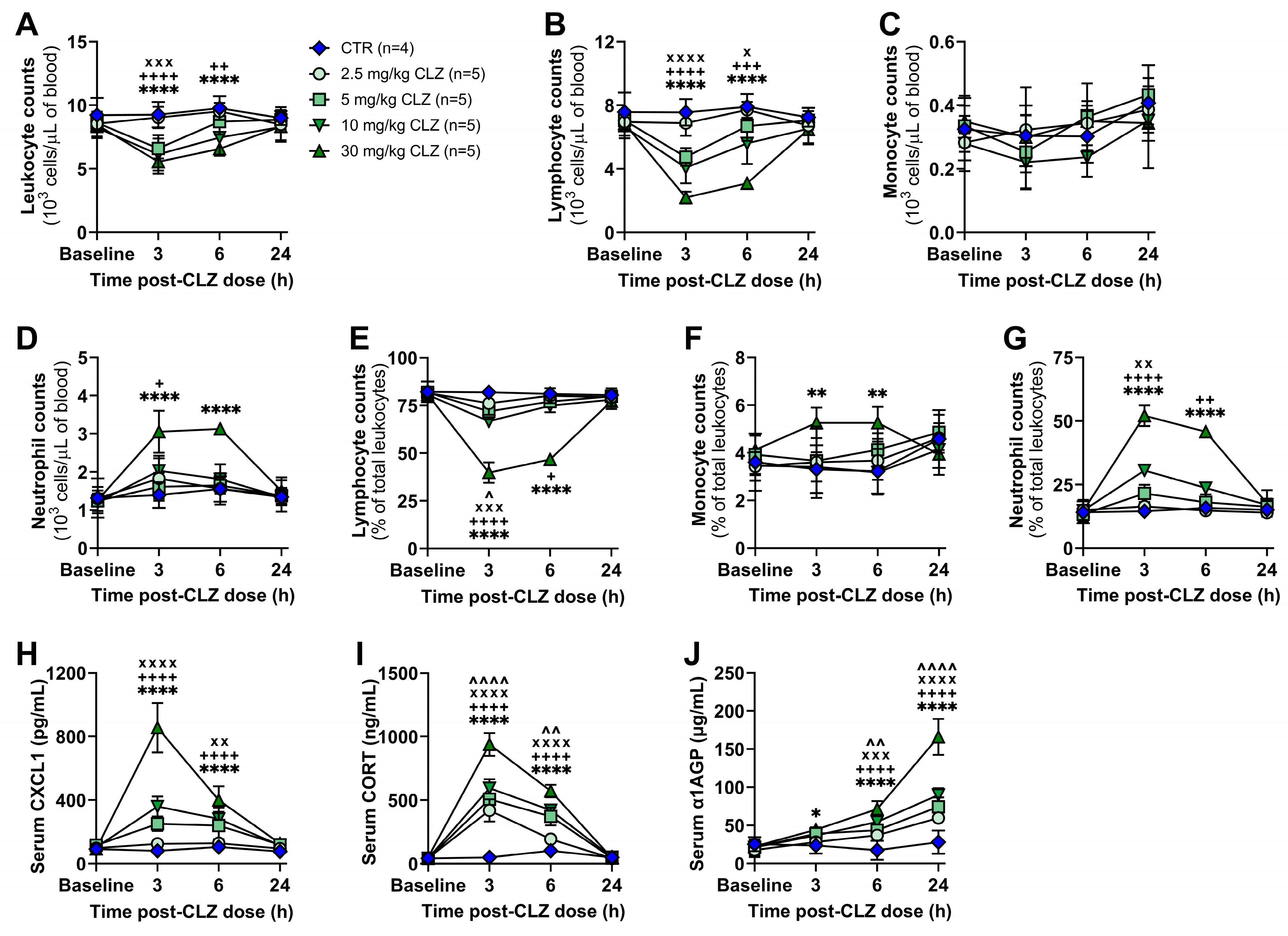
2.1. Clozapine Induces Acute Immune Changes at Subtherapeutic Doses In Vivo
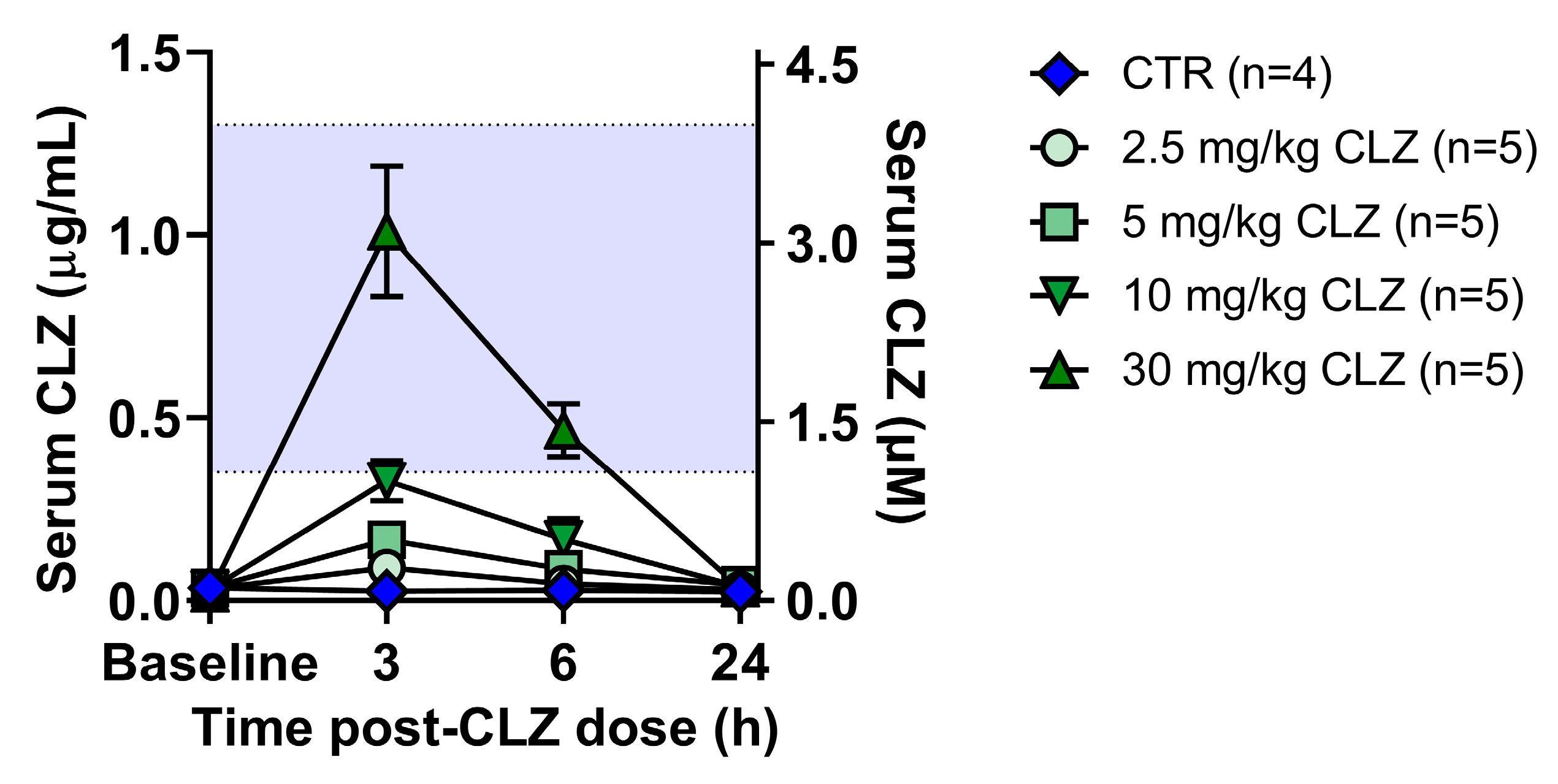
Clozapine Triggers a Rapid and Dose-Related Inflammatory Immune Response in Rats
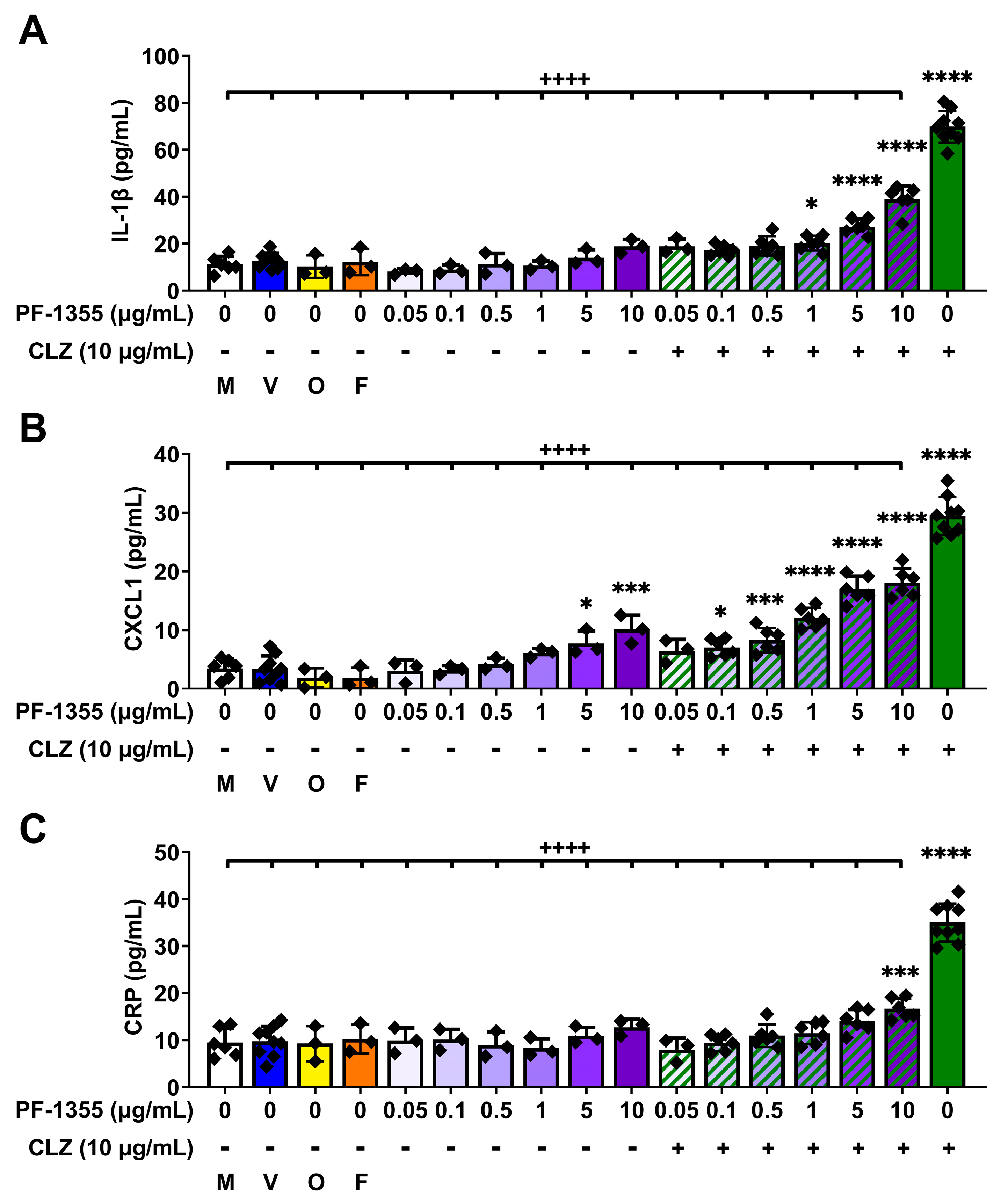
2.2. Myeloperoxidase Inhibition Attenuates Clozapine-Induced Inflammatory Mediator Release In Vitro
2.2.1. Clozapine-Induced Increases in Myeloperoxidase Activity Are Dampened by Myeloperoxidase Inhibition, without Affecting Viability
2.2.2. Clozapine-Induced Increases in Inflammatory Mediator Release Are Dampened by Myeloperoxidase Inhibition In Vitro
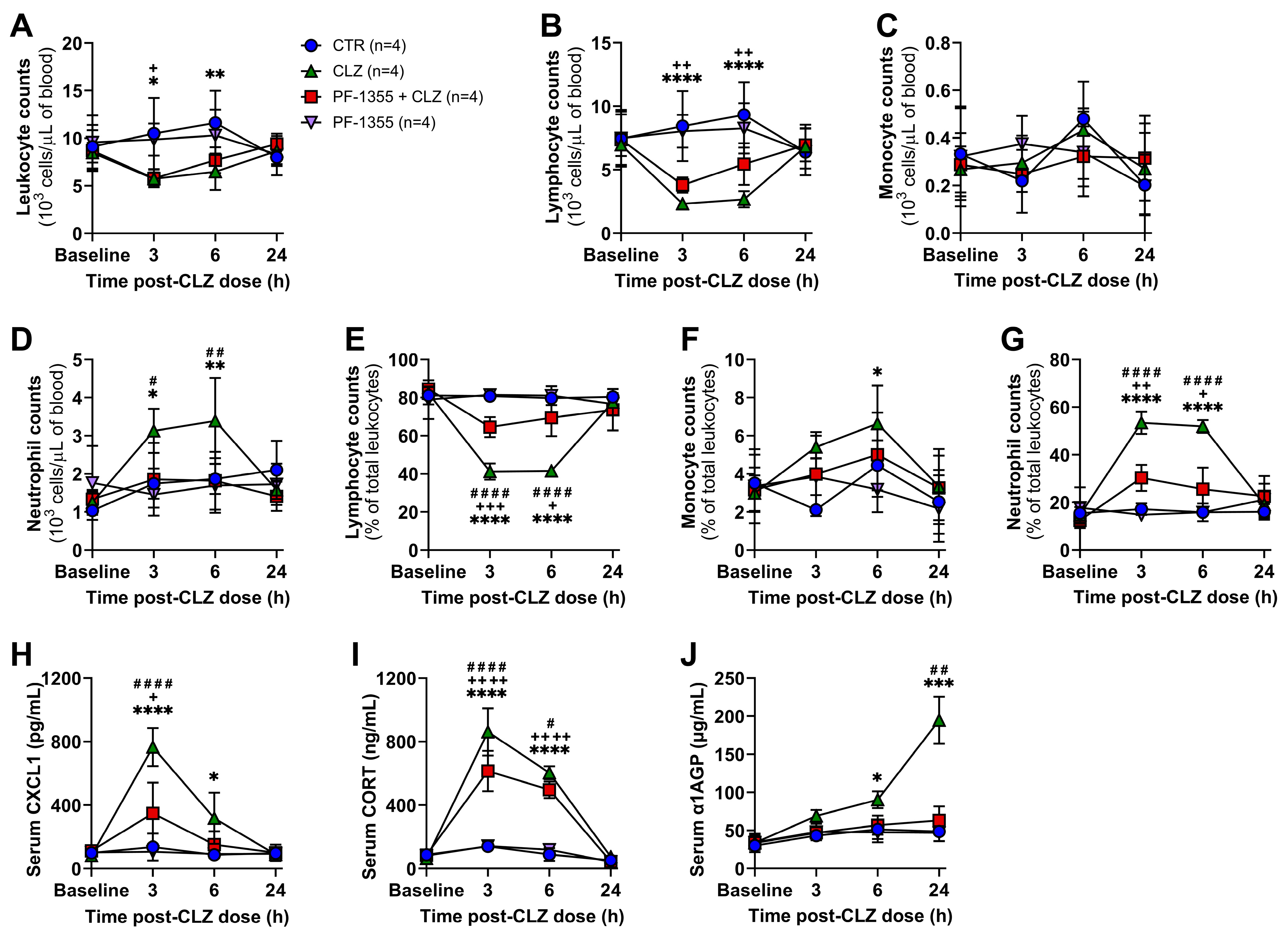
2.3. Myeloperoxidase Inhibition Attenuates Clozapine-Induced Inflammation In Vivo
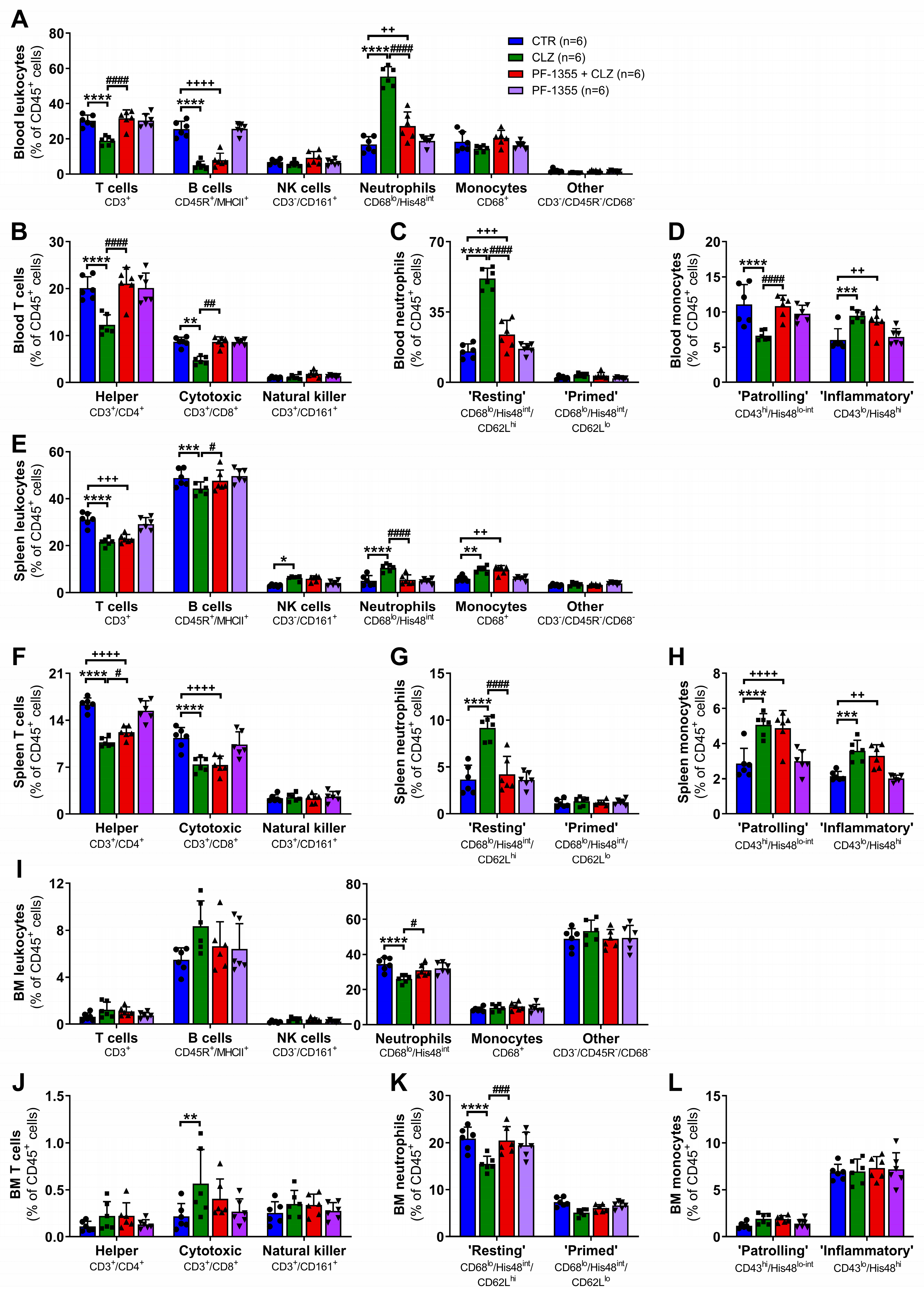
2.3.1. Clozapine-Induced Increases in Neutrophil Mobilization Are Dampened by Myeloperoxidase Inhibition
2.3.2. Clozapine-Induced Neutrophil Chemotaxis Is Dampened by Myeloperoxidase Inhibition
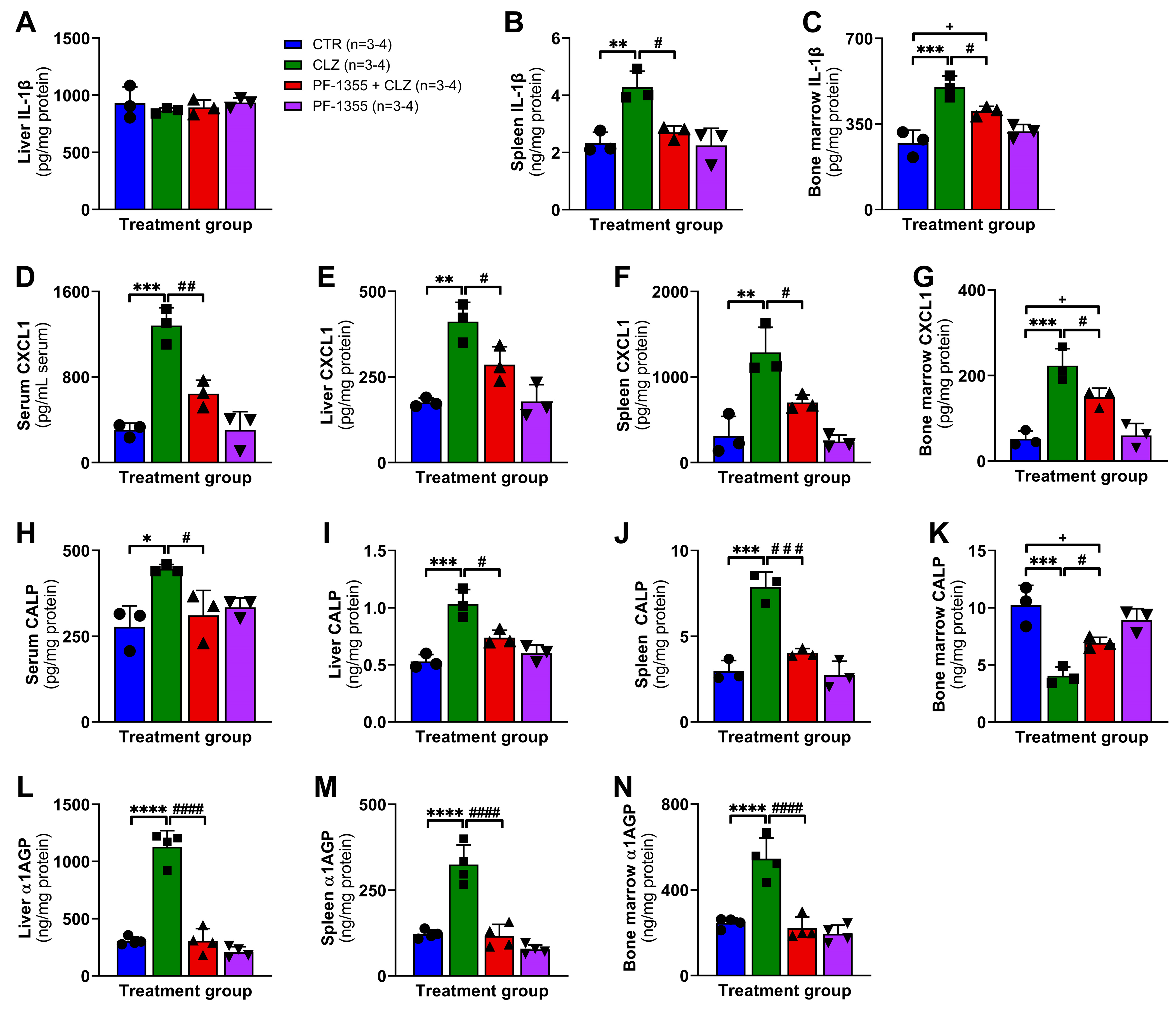
2.3.3. Clozapine-Induced Organ-Specific DAMP and Inflammatory Mediator Release Are Dampened by Myeloperoxidase Inhibition
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture, Maintenance, and Drug Exposure
4.3. Verification of Myeloperoxidase Protein Expression in THP-1 Cells
4.4. Assessment of THP-1 Macrophage Viability
4.5. Measurement of Myeloperoxidase Activity
4.6. Animal Treatment
4.7. Measurement of Serum Clozapine Concentrations
4.8. Tissue Processing and Neutrophil Enrichment
4.9. Measurement of Inflammatory Cytokines and Mediators
4.10. Flow Cytometry
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, C.J. Leucocyte counts in the prevention of drug agranulocytosis. Br. Med. J. 1949, 2, 261–263. [Google Scholar] [CrossRef] [Green Version]
- Andrès, E.; Mourot-Cottet, R. Non-chemotherapy drug-induced neutropenia—An update. Expert Opin. Drug Saf. 2017, 16, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Stepan, A.F.; Walker, D.P.; Bauman, J.; Price, D.A.; Baillie, T.A.; Kalgutkar, A.S.; Aleo, M.D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Zaidi, S. Clozapine: Improvement of Negative Symptoms of Schizophrenia. Cureus 2017, 9, e1973. [Google Scholar] [CrossRef] [Green Version]
- Numata, S.; Umehara, H.; Ohmori, T.; Hashimoto, R. Clozapine Pharmacogenetic Studies in Schizophrenia: Efficacy and Agranulocytosis. Front. Pharmacol. 2018, 9, 1049. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.M.; Rahman, A.; Husain, Z.; Mahmud, S.Z.; Ryan, W.G.; Feldman, J.M. Clozapine: A clinical review of adverse effects and management. Ann. Clin. Psychiatry 2003, 15, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Bogers, J.P.A.M.; Schulte, P.F.J.; Van Dijk, D.; Bakker, B.; Cohen, D. Clozapine Underutilization in the Treatment of Schizophrenia. J. Clin. Psychopharmacol. 2016, 36, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y. Clozapine: Balancing Safety with Superior Antipsychotic Efficacy. Clin. Schizophr. Relat. Psychoses 2012, 6, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Inada, K.; Oshibuchi, H.; Ishigooka, J.; Nishimura, K. Analysis of Clozapine Use and Safety by Using Comprehensive National Data From the Japanese Clozapine Patient Monitoring Service. J. Clin. Psychopharmacol. 2018, 38, 302–306. [Google Scholar] [CrossRef]
- Pick, A.M.; Nystrom, K.K. Nonchemotherapy drug-induced neutropenia and agranulocytosis: Could medications be the culprit? J. Pharm. Pract. 2014, 27, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Curtis, B.R. Non-chemotherapy drug-induced neutropenia: Key points to manage the challenges. Hematol. Am. Soc. Hematol. Educ. Progr. 2017, 2017, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Uetrecht, J. Current understanding of the mechanisms of idiosyncratic drug-induced agranulocytosis. Expert Opin. Drug Metab. Toxicol. 2015, 11, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Li, K.J.; Solomon, H.V.; DeLisi, L.E. Clozapine pharmacogenomics: A review of efficacy, pharmacokinetics, and agranulocytosis. Curr. Opin. Psychiatry 2018, 31, 403–408. [Google Scholar] [CrossRef]
- Legge, S.E.; Walters, J.T. Genetics of clozapine-associated neutropenia: Recent advances, challenges and future perspective. Pharmacogenomics 2019, 20, 279–290. [Google Scholar] [CrossRef]
- Ogese, M.O.; Lister, A.; Jenkins, R.E.; Meng, X.; Alfirevic, A.; Douglas, L.; Mcloughlin, R.; Silva, E.; Park, B.K.; Pirmohamed, M.; et al. Characterization of Clozapine-Responsive Human T Cells. J. Immunol. 2020, 205, 2375–2390. [Google Scholar] [CrossRef]
- Regen, F.; Herzog, I.; Hahn, E.; Ruehl, C.; Le Bret, N.; Dettling, M.; Heuser, I.; Hellmann-Regen, J. Clozapine-induced agranulocytosis: Evidence for an immune-mediated mechanism from a patient-specific in-vitro approach. Toxicol. Appl. Pharmacol. 2017, 316, 10–16. [Google Scholar] [CrossRef]
- Blackman, G.; Lisshammar, J.; Zafar, R.; Pollak, T.; Pritchard, M.; Cullen, A.; Rogers, J.; Carter, B.; Griffiths, K.; Nour, M.; et al. Clozapine Response in Schizophrenia and Hematological Changes. J. Clin. Psychopharmacol. 2021, 41, 19–24. [Google Scholar] [CrossRef]
- Sernoskie, S.C.; Jee, A.; Uetrecht, J.P. The emerging role of the innate immune response in idiosyncratic drug reactionss. Pharmacol. Rev. 2021, 73, 861–896. [Google Scholar] [CrossRef]
- Lobach, A.R.; Uetrecht, J. Clozapine promotes the proliferation of granulocyte progenitors in the bone marrow leading to increased granulopoiesis and neutrophilia in rats. Chem. Res. Toxicol. 2014, 27, 1109–1119. [Google Scholar] [CrossRef]
- Ng, W.; Kennar, R.; Uetrecht, J. Effect of Clozapine and Olanzapine on Neutrophil Kinetics: Implications for Drug-Induced Agranulocytosis. Chem. Res. Toxicol. 2014, 27, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Sernoskie, S.C.; Lobach, A.R.; Kato, R.; Jee, A.; Weston, J.K.; Uetrecht, J. Clozapine Induces an Acute Proinflammatory Response That Is Attenuated by Inhibition of Inflammasome Signaling: Implications for Idiosyncratic Drug-Induced Agranulocytosis. Toxicol. Sci. 2022, 186, 70–82. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Park, K. Mechanism of clozapine-induced agranulocytosis: Current status of research and implications for drug development. CNS Drugs 1997, 7, 139–158. [Google Scholar] [CrossRef]
- Iverson, S.; Kautiainen, A.; Ip, J.; Uetrecht, J.P. Effect of clozapine on neutrophil kinetics in rabbits. Chem. Res. Toxicol. 2010, 23, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.M.; Austin, G.E. Functional activity of three distinct myeloperoxidase (MPO) promoters in human myeloid cells. Leukemia 2002, 16, 1143–1153. [Google Scholar] [CrossRef] [Green Version]
- Austin, G.E.; Zhao, W.G.; Adjiri, A.; Lu, J.P. Control of myeloperoxidase gene expression in developing myeloid cells. Leuk. Res. 1996, 20, 817–820. [Google Scholar] [CrossRef]
- Lobach, A.R.; Uetrecht, J. Involvement of myeloperoxidase and NADPH oxidase in the covalent binding of amodiaquine and clozapine to neutrophils: Implications for drug-induced agranulocytosis. Chem. Res. Toxicol. 2014, 27, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef]
- Hofstra, A.H.; Uetrecht, J.P. Myeloperoxidase-mediated activation of xenobiotics by human leukocytes. Toxicology 1993, 82, 221–242. [Google Scholar] [CrossRef]
- Liu, Z.C.; Uetrecht, J.P. Clozapine is oxidized by activated human neutrophils to a reactive nitrenium ion that irreversibly binds to the cells. J. Pharmacol. Exp. Ther. 1995, 275, 1476–1483. [Google Scholar]
- Fischer, V.; Haar, J.A.; Greiner, L.; Lloyd, R.V.; Mason, R.P. Possible role of free radical formation in clozapine (clozaril)-induced agranulocytosis. Mol. Pharmacol. 1991, 40, 846–853. [Google Scholar]
- Gardner, I.; Leeder, J.S.; Chin, T.; Zahid, N.; Uetrecht, J.P. A comparison of the covalent binding of clozapine and olanzapine to human neutrophils in vitro and in vivo. Mol. Pharmacol. 1998, 53, 999–1008. [Google Scholar] [PubMed]
- Gardner, I.; Zahid, N.; MacCrimmon, D.; Uetrecht, J.P. A comparison of the oxidation of clozapine and olanzapine to reactive metabolites and the toxicity of these metabolites to human leukocytes. Mol. Pharmacol. 1998, 53, 991–998. [Google Scholar] [PubMed]
- Uetrecht, J.P.; Trager, W. Reactive metabolites. In Drug Metabolism: Chemical and Enzymatic Aspects; Uetrecht, J.P., Trager, W., Eds.; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2007; pp. 145–166. ISBN 9780367453022. [Google Scholar]
- Zheng, W.; Warner, R.; Ruggeri, R.; Su, C.; Cortes, C.; Skoura, A.; Ward, J.; Ahn, K.; Kalgutkar, A.; Sun, D.; et al. PF-1355, a Mechanism-Based Myeloperoxidase Inhibitor, Prevents Immune Complex Vasculitis and Anti-Glomerular Basement Membrane Glomerulonephritis. J. Pharmacol. Exp. Ther. 2015, 353, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Pulli, B.; Courties, G.; Tricot, B.; Sebas, M.; Iwamoto, Y.; Hilgendorf, I.; Schob, S.; Dong, A.; Zheng, W.; et al. Myeloperoxidase Inhibition Improves Ventricular Function and Remodeling After Experimental Myocardial Infarction. JACC Basic to Transl. Sci. 2016, 1, 633–643. [Google Scholar] [CrossRef]
- Pollmächer, T.; Fenzel, T.; Mullington, J.; Hinze-Selch, D.; Pollmacher, T.; Fenzel, T.; Mullington, J.; Hinze-Selch, D. The influence of clozapine treatment on plasma granulocyte colony-stimulating (G-CSF) levels. Pharmacopsychiatry 1997, 30, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Pollmacher, T.; Hinze-Selch, D.; Mullington, J. Effects of clozapine on plasma cytokine and soluble cytokine receptor levels. J. Clin. Psychopharmacol. 1996, 16, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Löffler, S.; Löffler-Ensgraber, M.; Fehsel, K.; Klimke, A. Clozapine therapy raises serum concentrations of high sensitive C-reactive protein in schizophrenic patients. Int. Clin. Psychopharmacol. 2010, 25, 101–106. [Google Scholar] [CrossRef]
- De Leon, J.; Ruan, C.-J.; Schoretsanitis, G.; De Las Cuevas, C. A Rational Use of Clozapine Based on Adverse Drug Reactions, Pharmacokinetics, and Clinical Pharmacopsychology. Psychother. Psychosom. 2020, 89, 200–214. [Google Scholar] [CrossRef]
- Lochhead, J.D.; Nelson, M.A.; Schneider, A.L. Risks and Benefits of Rapid Clozapine Titration. Ment. Illn. 2016, 8, 6457. [Google Scholar] [CrossRef]
- Citrome, L.; McEvoy, J.P.; Saklad, S.R. A Guide to the Management of Clozapine-Related Tolerability and Safety Concerns. Clin. Schizophr. Relat. Psychoses 2016, 10, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.L. Modeling and Predicting Stress-Induced Immunosuppression in Mice Using Blood Parameters. Toxicol. Sci. 2004, 83, 101–113. [Google Scholar] [CrossRef]
- Zweiman, B.; Atkins, P.C.; Bedard, P.-M.; Flaschen, S.L.; Lisak, R.P. Corticosteroid effects on circulating lymphocyte subset levels in normal humans. J. Clin. Immunol. 1984, 4, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S. Mechanisms of corticosteroid action on lymphocyte subpopulations. I. Redistribution of circulating T and b lymphocytes to the bone marrow. Immunology 1975, 28, 669. [Google Scholar]
- Marté, J.L.; Toney, N.J.; Cordes, L.; Schlom, J.; Donahue, R.N.; Gulley, J.L. Early changes in immune cell subsets with corticosteroids in patients with solid tumors: Implications for COVID-19 management. J. Immunother. Cancer 2020, 8, e001019. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Kaminker, K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch. Biochem. Biophys. 1962, 96, 465–467. [Google Scholar] [CrossRef]
- Bos, A.; Wever, R.; Roos, D. Characterization and quantification of the peroxidase in human monocytes. Biochim. Biophys. Acta 1978, 525, 37–44. [Google Scholar] [CrossRef]
- Lee, Y.; Reilly, B.; Tan, C.; Wang, P.; Aziz, M. Extracellular CIRP Induces Macrophage Extracellular Trap Formation Via Gasdermin D Activation. Front. Immunol. 2021, 12, 5505. [Google Scholar] [CrossRef]
- Siraki, A.G. The many roles of myeloperoxidase: From inflammation and immunity to biomarkers, drug metabolism and drug discovery. Redox Biol. 2021, 46, 102109. [Google Scholar] [CrossRef]
- Rashid, M.H.; Babu, D.; Tran, N.; Reiz, B.; Siraki, A.G. Neutrophil Myeloperoxidase-Mediated N-Demethylation of Quetiapine Leads to N-Desalkylquetiapine, a Pharmacologically Active Cytochrome P450 Metabolite. Chem. Res. Toxicol. 2022, 35, 1001–1010. [Google Scholar] [CrossRef]
- Weston, J.K.; Uetrecht, J. Activation of Inflammasomes by Agents Causing Idiosyncratic Skin Reactions: A Possible Biomarker. Chem. Res. Toxicol. 2014, 27, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Imano, H.; Kato, R.; Ijiri, Y.; Hayashi, T. Activation of inflammasomes by tyrosine kinase inhibitors of vascular endothelial growth factor receptor: Implications for VEGFR TKIs-induced immune related adverse events. Toxicol. Vitr. 2021, 71, 105063. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Ijiri, Y.; Hayashi, T. Amiodarone, Unlike Dronedarone, Activates Inflammasomes via Its Reactive Metabolites: Implications for Amiodarone Adverse Reactions. Chem. Res. Toxicol. 2021, 34, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Ijiri, Y.; Hayashi, T.; Uetrecht, J. Reactive metabolite of gefitinib activates inflammasomes: Implications for gefitinib-induced idiosyncratic reaction. J. Toxicol. Sci. 2020, 45, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Uetrecht, J. Supernatant from Hepatocyte Cultures with Drugs That Cause Idiosyncratic Liver Injury Activates Macrophage Inflammasomes. Chem. Res. Toxicol. 2017, 30, 1327–1332. [Google Scholar] [CrossRef]
- Kato, R.; Ijiri, Y.; Hayashi, T.; Uetrecht, J. The 2-Hydroxyiminostilbene metabolite of carbamazepine or the supernatant from incubation of hepatocytes with carbamazepine activates inflammasomes: Implications for carbamazepine-induced hypersensitivity reactions. Drug Metab. Dispos. 2019, 47, 1093–1096. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Barnett-Vanes, A.; Sharrock, A.; Birrell, M.A.; Rankin, S. A Single 9-Colour Flow Cytometric Method to Characterise Major Leukocyte Populations in the Rat: Validation in a Model of LPS-Induced Pulmonary Inflammation. PLoS ONE 2016, 11, e0142520. [Google Scholar] [CrossRef] [Green Version]
- Bongers, S.H.; Chen, N.; van Grinsven, E.; van Staveren, S.; Hassani, M.; Spijkerman, R.; Hesselink, L.; Lo Tam Loi, A.T.; van Aalst, C.; Leijte, G.P.; et al. Kinetics of Neutrophil Subsets in Acute, Subacute, and Chronic Inflammation. Front. Immunol. 2021, 12, 2386. [Google Scholar] [CrossRef]
- Yrlid, U.; Jenkins, C.D.; MacPherson, G.G. Relationships between Distinct Blood Monocyte Subsets and Migrating Intestinal Lymph Dendritic Cells In Vivo under Steady-State Conditions. J. Immunol. 2006, 176, 4155–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprenkeler, E.G.G.; Zandstra, J.; van Kleef, N.D.; Goetschalckx, I.; Verstegen, B.; Aarts, C.E.M.; Janssen, H.; Tool, A.T.J.; van Mierlo, G.; van Bruggen, R.; et al. S100A8/A9 Is a Marker for the Release of Neutrophil Extracellular Traps and Induces Neutrophil Activation. Cells 2022, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Polkowska-Pruszyńska, B.; Gerkowicz, A.; Rawicz-Pruszyński, K.; Krasowska, D. The Role of Fecal Calprotectin in Patients with Systemic Sclerosis and Small Intestinal Bacterial Overgrowth (SIBO). Diagnostics 2020, 10, 587. [Google Scholar] [CrossRef] [PubMed]
- Jarlborg, M.; Courvoisier, D.S.; Lamacchia, C.; Martinez Prat, L.; Mahler, M.; Bentow, C.; Finckh, A.; Gabay, C.; Nissen, M.J. Serum calprotectin: A promising biomarker in rheumatoid arthritis and axial spondyloarthritis. Arthritis Res. Ther. 2020, 22, 105. [Google Scholar] [CrossRef]
- Jukic, A.; Bakiri, L.; Wagner, E.F.; Tilg, H.; Adolph, T.E. Calprotectin: From biomarker to biological function. Gut 2021, 70, 1978–1988. [Google Scholar] [CrossRef]
- Stříž, I.; Trebichavský, I. Calprotectin-a Pleiotropic Molecule in Acute and Chronic Inflammation. Physiol. Res 2004, 53, 245–253. [Google Scholar] [CrossRef]
- Fischer-Cornelssen, K.A. Fluperlapine in 104 schizophrenic patients. Open multicenter trial. Arzneimittelforschung. 1984, 34, 125–130. [Google Scholar]
- Uetrecht, J.P. Metabolism of clozapine by neutrophils. Possible implications for clozapine-induced agranulocytosis. Drug Saf. 1992, 7 (Suppl. S1), 51–56. [Google Scholar] [CrossRef]
- Zhu, X.; Li, J.; Liu, F.; Uetrecht, J.P. Involvement of T Helper 17 Cells in D-Penicillamine-Induced Autoimmune Disease in Brown Norway Rats. Toxicol. Sci. 2011, 120, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Uetrecht, J.P.; Zahid, N. N-Chlorination and oxidation of procainamide by myeloperoxidase: Toxicological implications. Chem. Res. Toxicol. 1991, 4, 218–222. [Google Scholar] [CrossRef]
- Rubin, R.; Uetrecht, J.; Zahid, N. Metabolism of Procainamide to a Hydroxylamine by Human Neutrophils and Mononuclear Leukocytes. Chem. Res. Toxicol. 1988, 1, 74–78. [Google Scholar] [CrossRef]
- Tingle, M.D.; Jewell, H.; Maggs, J.L.; O’Neill, P.M.; Park, B.K. The bioactivation of amodiaquine by human polymorphonuclear leucocytes in vitro: Chemical mechanisms and the effects of fluorine substitution. Biochem. Pharmacol. 1995, 50, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- McGirr, L.G.; Jatoe, S.D.; O’Brien, P.J. Myeloperoxidase catalysed cooxidative metabolism of methimazole: Oxidation of glutathione and NADH by free radical intermediates. Chem. Biol. Interact. 1990, 73, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.; Uetrecht, J. How Reactive Metabolites Induce an Immune Response That Sometimes Leads to an Idiosyncratic Drug Reaction. Chem. Res. Toxicol. 2017, 30, 295–314. [Google Scholar] [CrossRef]
- Galijasevic, S. The development of myeloperoxidase inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 1–7. [Google Scholar] [CrossRef]
- Lockhart, J.S.; Sumagin, R. Non-Canonical Functions of Myeloperoxidase in Immune Regulation, Tissue Inflammation and Cancer. Int. J. Mol. Sci. 2022, 23, 12250. [Google Scholar] [CrossRef]
- Frangie, C.; Daher, J. Role of myeloperoxidase in inflammation and atherosclerosis. Biomed. Rep. 2022, 16, 53. [Google Scholar] [CrossRef]
- Stamp, L.K.; Khalilova, I.; Tarr, J.M.; Senthilmohan, R.; Turner, R.; Haigh, R.C.; Winyard, P.G.; Kettle, A.J. Myeloperoxidase and oxidative stress in rheumatoid arthritis. Rheumatology 2012, 51, 1796–1803. [Google Scholar] [CrossRef] [Green Version]
- Kisic, B.; Miric, D.; Dragojevic, I.; Rasic, J.; Popovic, L. Role of Myeloperoxidase in Patients with Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2016, 2016, 1069743. [Google Scholar] [CrossRef] [Green Version]
- Chami, B.; Martin, N.J.J.; Dennis, J.M.; Witting, P.K. Myeloperoxidase in the inflamed colon: A novel target for treating inflammatory bowel disease. Arch. Biochem. Biophys. 2018, 645, 61–71. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Maki, R.A. The Role of Myeloperoxidase in Neurodegenerative Disease. In Mammalian Heme Peroxidases: Diverse Roles in Health and Disease; Hawkins, C., Nauseef, W.M., Eds.; CRC Press: Boca Raton, FL, USA, 2021; pp. 249–273. ISBN 9781000450613. [Google Scholar]
- Soubhye, J.; Van Antwerpen, P.; Dufrasne, F. A patent review of myeloperoxidase inhibitors for treating chronic inflammatory syndromes (focus on cardiovascular diseases, 2013–2019). Expert Opin. Ther. Pat. 2020, 30, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: Formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2002, 15, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Prütz, W.A. Hypochlorous Acid Interactions with Thiols, Nucleotides, DNA, and Other Biological Substrates. Arch. Biochem. Biophys. 1996, 332, 110–120. [Google Scholar] [CrossRef]
- Marsche, G.; Heller, R.; Fauler, G.; Kovacevic, A.; Nuszkowski, A.; Graier, W.; Sattler, W.; Malle, E. 2-Chlorohexadecanal Derived From Hypochlorite-Modified High-Density Lipoprotein–Associated Plasmalogen Is a Natural Inhibitor of Endothelial Nitric Oxide Biosynthesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2302–2306. [Google Scholar] [CrossRef] [PubMed]
- Ikeda-Saito, M. A Study of Ligand Binding to Spleen Myeloperoxidase. Proc. Natl. Acad. Sci. USA 1987, 26, 251–259. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef]
- Goldmann, O.; Medina, E. The expanding world of extracellular traps: Not only neutrophils but much more. Front. Immunol. 2013, 3, 420. [Google Scholar] [CrossRef] [Green Version]
- Irizarry-Caro, J.A.; Carmona-Rivera, C.; Schwartz, D.M.; Khaznadar, S.S.; Kaplan, M.J.; Grayson, P.C. Brief Report: Drugs Implicated in Systemic Autoimmunity Modulate Neutrophil Extracellular Trap Formation. Arthritis Rheumatol. 2018, 70, 468–474. [Google Scholar] [CrossRef] [Green Version]
- De Abreu Costa, L.; Henrique Fernandes Ottoni, M.; Geralda dos Santos, M.; Batista Meireles, A.; Gomes de Almeida, V.; de Fátima Pereira, W.; Alves de Avelar-Freitas, B.; Eustáquio Alvim Brito-Melo, G. Dimethyl Sulfoxide (DMSO) Decreases Cell Proliferation and TNF-α, IFN-γ, and IL-2 Cytokines Production in Cultures of Peripheral Blood Lymphocytes. Molecules 2017, 22, 1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buur-Rasmussen, B.; Brøsen, K. Cytochrome P450 and therapeutic drug monitoring with respect to clozapine. Eur. Neuropsychopharmacol. 1999, 9, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Lund-Johansen, F.; Terstappen, L.W.M.M. Differential surface expression of cell adhesion molecules during granulocyte maturation. J. Leukoc. Biol. 1993, 54, 47–55. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laser | Filter | Marker | Clone | Fluorochrome | Supplier |
|---|---|---|---|---|---|
| - | - | CD32 * | D34-485 | - | BD Pharmingen |
| Violet (407 nm) | 780/60 | CD8a | OX-8 | BV786 | BD Biosciences |
| 710/50 | CD172a | OX-41 | BV711 | ||
| 670/30 | CD161 | 10/78 | BV650 | ||
| 610/20 | CD45RA | OX-33 | BV605 | ||
| 525/50 | Live/Dead | - | e506 | eBioscience | |
| 450/50 | CD62L | HLR1 | BV421 | BD Biosciences | |
| Blue (488 nm) | 710/50 | CD3e | G4.18 | PerCP-eFluor710 | eBioscience |
| 530/30 | His48 | His48 | FITC | BD Pharmingen | |
| Yellow-Green (561 nm) | 780/60 | CD43 | W3/13 | PE-Cy7 | BioLegend |
| 610/20 | CD68 ** | ED1 | Dylight594 | Novus Biologicals | |
| Red (640 nm) | 780/60 | CD45 | OX-1 | APC-Cy7 | BD Biosciences |
| 670/30 | CD11b | WT.5 | APC | ||
| Ultraviolet (355 nm) | 515/30 | RT1B (MHCII) | OX-6 | BUV496 | BD Biosciences |
| 379/28 | CD4 | OX-35 | BUV395 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sernoskie, S.C.; Jee, A.; Uetrecht, J. The Role of Myeloperoxidase in Clozapine-Induced Inflammation: A Mechanistic Update for Idiosyncratic Drug-Induced Agranulocytosis. Int. J. Mol. Sci. 2023, 24, 1243. https://doi.org/10.3390/ijms24021243
Sernoskie SC, Jee A, Uetrecht J. The Role of Myeloperoxidase in Clozapine-Induced Inflammation: A Mechanistic Update for Idiosyncratic Drug-Induced Agranulocytosis. International Journal of Molecular Sciences. 2023; 24(2):1243. https://doi.org/10.3390/ijms24021243
Chicago/Turabian StyleSernoskie, Samantha Christine, Alison Jee, and Jack Uetrecht. 2023. "The Role of Myeloperoxidase in Clozapine-Induced Inflammation: A Mechanistic Update for Idiosyncratic Drug-Induced Agranulocytosis" International Journal of Molecular Sciences 24, no. 2: 1243. https://doi.org/10.3390/ijms24021243
APA StyleSernoskie, S. C., Jee, A., & Uetrecht, J. (2023). The Role of Myeloperoxidase in Clozapine-Induced Inflammation: A Mechanistic Update for Idiosyncratic Drug-Induced Agranulocytosis. International Journal of Molecular Sciences, 24(2), 1243. https://doi.org/10.3390/ijms24021243