Abstract
Lake basins can behave as accumulators of microplastics released in wastewaters as such or resulting from degradation of larger items before and/or during their journey toward the marine environment as a final sink. A novel multianalytical approach was adopted for the detection and quantification of microplastics with size < 2 mm in the sediments of the volcanic lake of Bracciano, Italy. Simple analytical techniques such as solvent extraction/fractionation (for polyolefins and polystyrene) or depolymerization (for polyethylene terephthalate, PET), along with chromatographic detection (SEC and HPLC), allowed quantitative and qualitative determination of the main synthetic polymer contaminants. In particular, PET microplastic concentrations of 0.8–36 ppm were found, with variability related to the sampling site (exposure to incoming winds and wave action). Proton Nuclear Magnetic Resonance (1H-NMR) and Attenuated Total Reflectance Fourier Transformed InfraRed (ATR-FTIR spectroscopic investigations supported the identification and chemical characterization of plastic fragments and polymer extracts. The average molecular weight of solvent extractable polymers was evaluated from 2D 1H-NMR diffusion ordered spectroscopy (DOSY) experiments. The proposed, easily accessible multianalytical approach can be considered as a useful tool for improving our knowledge on the nature and the concentration of microplastics in sediments, giving insights on the impact of human activities on the health status of aquatic ecosystems.
1. Introduction
Environmental pollution by microplastics (MPs) and nanoplastics (NPs) has raised growing concerns in the last two decades, also as a result of the increasing evidences regarding the direct and indirect negative effects of such contaminants on both marine [1] and soil biota [2,3], and the potential of bioaccumulation and transfer to humans through the food chain. A vast and recent literature accounts for the role of microplastics (those within 0.1–5 mm size being typically considered) as accumulators and carriers of several persistent organic pollutants (POPs) and heavy metals, which can then affect aquatic organisms [4] as well as soils contaminated by MPs, e.g., through sludges from civil wastewater treatment plants [5,6]. The growing number of studies about microplastic pollution in aquatic environments, initially focused on the detection and mapping of floating plastic debris, has expanded more recently to the investigation of those present in sediments [7]. A large fraction of the scientific publications concerning sediments’ analysis deals with marine environments (mainly coastal beaches, estuarine, and sea bottoms) [8], while the research on freshwater sediments (lakes and rivers) is much more limited [9,10,11,12,13]. On the other hand, it has been clearly established that marine pollution from plastic debris originates largely from the transport of wastes produced inland [11,12,13,14,15]. In fact, specific studies on the source of pollution by plastic materials in marine environments have estimated that about 80% of the total mass of plastic waste is originated in urban areas and conveyed to the seas mainly by rivers [16,17,18]. The importance of investigating in depth the extent and nature of microplastic pollution in internal water systems is therefore apparent, with particular attention needed on the different types of polymeric materials as their presence is likely to be directly related to human activities. The results would allow drawing a more accurate picture of the source, extent, and transport of the different kind of microplastics, providing the basis for a correlation with the available data on the distribution of the different polymer types in marine environments on a global scale.
To date, there is no common agreement as to the definition of the term “microplastic” [19]; at the same time, a reference protocol for the qualitative and/or quantitative analysis of the microplastics contained in sediment samples is lacking. The most commonly used strategies for sampling, separation, characterization and quantification of microplastics in sediment samples, whether marine or freshwater, are summarized in Table 1 [8].

Table 1.
Procedures commonly used for the qualitative/quantitative analysis of microplastics contained in sediment samples [8].
The most commonly adopted methodology is based on sorting and counting, possibly associated with purification procedures to remove biogenic organic material [20], and with spectroscopic characterization of the single particles by ATR-FTIR (Attenuated Total Reflectance Fourier Transformed InfraRed) and micro-FTIR or micro-Raman spectroscopies. While the sorting and counting allows to qualify the different types of microplastics by shape, size, color and composition (quite important parameters indeed for understanding the spatial distribution and the interaction with biota), such methodologies are intrinsically inaccurate in terms of quantification of the total amount of plastic particles, as they do not ensure quantitative separation from the inorganic sediment of the smaller and more sticky (oxidatively degraded) polymer fragments, and largely miss the NPs (particles with size < 1 μm), in addition to being more time consuming for a statistically significant evaluation of environmental samples.
Nanoplastics can be expected to be nearly ubiquitous in aquatic environments as they mainly originate from the continuous oxidative degradation of MPs. Unfortunately, devising a suitable analytical protocol for NPs in environmental samples is still an open issue. Their very small size implying a much larger surface to volume ratio, and a surface chemical composition that may differ significantly from that of the virgin polymer, make NPs quite elusive to the collection and isolation from environmental samples, and an accurate qualitative and quantitative analysis very challenging. As a result, knowledge about NPs distribution in the environment is very poor. Little information is available also on the transport rate (both horizontal and vertical) of plastic fragments affecting the travelled distances in both freshwater and marine environment; however, there is clear evidence that the distribution of micro- and nanoplastics cannot be simply connected to the density of the pristine polymer materials due to the interference from hydrodynamic transport and from the fouling that may alter the apparent density of the particles. These factors could determine a selection of plastic materials that remain afloat and may eventually end up on the shores once they reach marine environments, compared to the possibly much larger input from inland waters.
A significant contribution to the plastic litter polluting marine environments is recognized as being the result of inflow of plastic waste from rivers and other waterways collecting the wastewaters and other solid wastes from inland. Semi-closed systems, such as inland lakes, can thus behave as accumulators of plastic debris transported by tributary freshwaters, and particularly of microplastics released in wastewaters as such or resulting from the degradation of larger items before and/or during the journey toward the final sink.
In light of these considerations, the present work deals with a combination of simple analytical protocols and more specialized structural characterizations aiming at providing an accurate and exhaustive qualitative/quantitative determination of the pollution of lake sediments by plastic fragments with average size ranging from 2 mm down to the nanometer. The selected mesh size for the sediment sieving is based on practical criteria of effectiveness in the sieving procedure, and the consideration of microparticles as those with at least one dimension in the sub-millimeter range, including microfibers (typical cross sectional diameter of 10–30 micrometer) with a length that can span in the range from a few hundred micrometer to a few mm. In particular, a procedure based on the selective extraction in organic solvents for the analysis of the total mass of polystyrene and polyolefin (polyethylene and polypropylene) MPs in marine sediments [21,22] has been adopted in combination with a recently developed procedure for the quantitative determination of the total mass of polyethylene terephthalate (PET) microfibers. The latter is based on the hydrolytic depolymerization and quantitative determination of the resulting monomeric terephthalic acid (TPA) [23].
A combination of spectroscopic (ATR FT-IR, micro FT-IR, nuclear magnetic resonance) and liquid chromatography techniques (size exclusion chromatography, SEC, and HPLC) was also exploited to gain better insights on the extent of polymer degradation (molecular weight reduction) and chemical alteration of the macromolecular structure as a result of environmental photo-oxidation. The structural survey using NMR was further implemented by means of translational diffusion (DOSY) measurements to verify whether groups of identifiable signals can be ascribed to the same class of macromolecules in terms of the translational diffusion coefficient (D).
The analyses were carried out on sediment samples from two opposite sites of Lake Bracciano in central Italy. The lake, with an area of 57 km2 and maximum depth 172 m, has a circular shape due to its volcanic origin, with no tributary and the only effluent being controlled by an artificial dam. All waters come from precipitations and from the output of a wastewater treatment plant serving a population of about 50,000. Local bills prevent pollution by motorboats and by fertilizers and pesticides that are forbidden in the lake basin, while industrial activities are nearly absent. The samples were collected along the shoreline during a prolonged drought resulting in a hydrographic crisis; as a result, the sandy beach of the retreating shoreline could be considered somehow as representative of both shore and (normally submerged) bottom sediments [24].
2. Materials and Methods
2.1. Sediment Sampling
The collection of sediment samples from Lake Bracciano (42°07′16” N 12°13′55” E) was carried out in May 2017 during a prolonged drought resulting in a water level about 140 cm below the hydrometric zero (163 m above sea level), with consequent retreat of the shoreline by several tens of meters. The sandy sediment samples, collected purposely at that time near the shoreline, are therefore to be considered as indicators of contamination by mixed plastic debris, that is, both those floating and eventually deposited on the shore and those previously deposited on bottom sediments. The sampling sites are located in two beaches, indicated with the prefix BR1 for the site with maximum exposure to prevailing winds and thus higher accumulation of floating debris, and BR2 for the one on the opposite side of the lake (Figure 1a). On each sampling site, three transects (A, B, and C, Figure 1b) across the edges of the three visibly recognizable subsequent storage lines (horizons 1, 2, and 3) at different distances from shoreline were identified.

Figure 1.
(a) Location of the sampling sites. (b) Sampling scheme (BR1 site).
Nine sediment samples were collected at the intersection points within a square area of 50 cm side up to a depth of 5 cm using a metal shovel. Each sand sample was sieved first with a 5 mm mesh screen and freed from larger macroplastic and other biogenic and anthropogenic debris, then separately homogenized and divided into smaller portions, collected in polyethylene bags and closed with metal pins.
2.2. Pre-Treatment of Sediment Samples
Approximately 120–140 g of homogenized and pre-sieved sediment from each sample was placed in a glass crystallizing dish and covered with filter paper to avoid environmental contamination by airborne microplastic during handling. The samples were dried in a ventilated oven at 60 °C for 6 h, weighed, and then sieved with a 2 mm mesh screen to separate the finer sediment containing microplastics from the sieve fraction between 5 and 2 mm. The larger plastic fragments retained in the upper sieve fractions were collected with the aid of an optical stereo microscope for subsequent counting and identification by attenuated total reflectance (ATR) FT-IR spectroscopy.
2.3. Chemicals
Dichloromethane (99.9%, stabilized with amylene, Romil-SpS, Romil Ltd., Cambridge, UK) and xylene (98.5%, Sigma-Aldrich, Merck Life Science S.r.l., Milano, Italy) were used for the extractions of sediment samples. A 1.9 M solution of sodium hydroxide (NaOH, Sigma-Aldrich, 98.0%) in HPLC grade H2O (Sigma-Aldrich) and hexadecyl-tributyl-phosphonium-bromide (TBHDPB, 97%, Sigma-Aldrich) as phase transfer catalyst was used for the hydrolytic depolymerization of PET. Pretreatment of alkaline hydrolyzates of sediment samples was carried out by using dichloromethane, diethyl ether (99.9% Sigma-Aldrich), methanol (MeOH, 99.9%, Sigma-Aldrich), hydrogen peroxide (30% w/v, Panreac, Nova Chimica Srl, Cinisello Balsamo, Italy), and sulfuric acid (95–98% Sigma-Aldrich). The resulting acidic solution was eluted through a reversed-phase SPE cartridge (Macherey-Nagel Chromabond® C18ec, loaded with 500 mg stationary phase, Macherey-Nagel GmbH & Co., Düren, Germany). Methanol (99.9%, Sigma-Aldrich), acetic acid (99.85% Sigma-Aldrich) and HPLC grade H2O (Sigma-Aldrich) were used for the preparation of the HPLC eluent. Chloroform (HPLC grade stabilized with ethanol, Sigma-Aldrich) was used as mobile phase in SEC analysis. NMR analyses were performed on samples in deuterated chloroform (CDCl3, 99.96 %, Sigma-Aldrich). All solvents and reagents were used as purchased without further purifications.
2.4. Analytical Techniques
ATR FT-IR spectra were recorded with a Perkin Elmer (PerkinElmer Italia Spa, Milano, Italy) Spectrum GX spectrometer equipped with a MIRacle TM ATR accessory (angle of incidence 45°) and a germanium crystal. The spectra were recorded over the wavenumber range of 650–4000 cm−1 using a spectral resolution of 4 cm−1. Automatic baseline and smooth corrections were applied after 32 scans. For micro-ATR FT-IR chemical analysis, a Perkin Elmer Spectrum Autoimage System microscope equipped with a germanium ATR crystal was used, and spectra were recorded in the mid-IR region (700–4000 cm−1) using 16 scans at 4 cm−1 spatial resolution. The lateral spatial resolution is limited by the point of contact with the germanium crystal tip (30–40 micron). For each sample, an area of about 4 × 8 mm of sediment from the sieve fraction spread on a glass slide was analyzed and taken as representative of the whole sample. All spectra and images were processed using Spectrum 5.0 software (PerkinElmer).
Size exclusion chromatography (SEC) analyses were performed with an instrument consisting of a Jasco (Jasco Europe S.R.L., Cremella, LC, Italy) PU-2089 Plus four-channel pump, a PL gel (Polymer Laboratories) pre-column packed with polystyrene/divinylbenzene, two in series PL gel MIXED D columns placed in a Jasco CO_2063 column oven, a Jasco RI 2031 Plus refractive index detector, and a Jasco UV-2077 Plus multi-channel UV spectrometer. Chloroform (HPLC grade Sigma-Aldrich) was used as the eluent at 1 mL/min flow rate. For the analysis of PET fragments and reference PET material, the latter were previously dissolved in hexafluoroisopropanol (HFIP) and then diluted in CHCl3 at a 0.05 wt/vol ratio prior to the analysis. ChromNav Jasco software was used for data processing.
HPLC analysis was carried out with a Jasco instrumental assembly consisting of a PU-1580 pump interfaced with a Jasco 1575 UV-Vis detector set at 242 nm. A Jones-Genesis (Jones Chromatography Ltd., Hengoed, UK) Aq column 120 (length 15 cm, internal diameter 4.6 mm, and particle size 4 μm) was used as the stationary phase under isocratic conditions at 0.8 mL/min flow rate of an eluent mixture consisting of HPLC grade water (acidified with 1 wt.% CH3COOH) and MeOH in a 60:40 volume ratio.
The complete procedure of extraction and quantification of the different polymer types is summarized in the scheme of Figure 2.
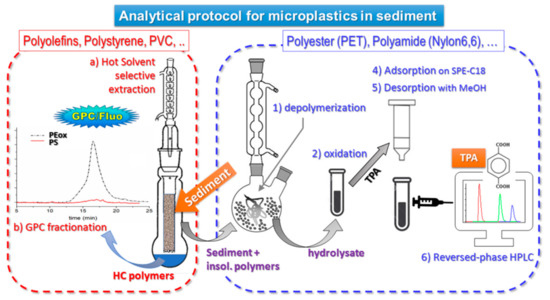
Figure 2.
Analytical protocol for microplastics in sediment [21,23].
NMR measurements were carried out on a Bruker (Bruker Italia S.r.l., Milano, Italy) Advance II DRX400 NMR Spectrometer working at 401.3 MHz for proton Larmor Frequency with a dual channel BBFO probe and gradient coil for pulsed field gradient (PFG) along the z axis (maximum gradient 50 G/cm). Proton 90° pulse length was 11.8 μs at 30 W power and temperature was set to 20 °C (±0.1 °C stability). Here, 1D H NMR spectra were acquired in 64 scans using a standard single 90° pulse sequence with 5 µs of pre-acquisition delay and 2 s for acquisition time. The free induction decay (FID) have been Fourier transformed with a 1 Hz line-broadening and baseline was corrected automatically. Then, 1D 13C-1H decoupled NMR spectra were acquired in 18,000 scans with a standard single 30° pulse, inverted gated proton decoupling, and pulse sequence. Translational diffusion measurements have been acquired with a standard Stimulated Echo (STE) pulse sequence [25,26] with bipolar gradients to minimize distortions on the line shapes of the obtained spectra. Decay curve were acquired in 16 steps increasing the gradient from 2.5 G/cm to 45 G/cm with a PFG of 2 ms length and a diffusion time fixed at 150 ms. Each spectrum of the array has been acquired in 64 scans with a 2 s acquisition time and a recycle delay of 5 s.
Diffusion coefficients corresponding to the signals on the Fourier transformed spectra were calculated by fitting the decay of the integral areas as a function of the square of the gradient strength, all the other parameters being fixed, according to the following equation:
where γ is the proton gyromagnetic ration, δ is the PFG length, G the gradient intensity, Δ the diffusion time and D the diffusion coefficient along the gradient direction. Longitudinal and transverse relaxation contribution to the signal decay are kept constant as a result of δ and Δ being constant. The results obtained have been reported in a diffusion ordered spectroscopy (DOSY) map for data analysis [27]. A further analysis of the NMR measurements has been done in order to obtain the hydrodynamic radius, Rg, of the macromolecules corresponding to each DOSY signals, by using the well-known Stokes-Einstein relationship:
where D and η are the measured translational diffusion coefficient and the viscosity, respectively. A dynamic viscosity of 0.5 MPa·s was considered for the CDCl3 solvent at 20 °C.
3. Results and Discussion
3.1. Isolation and Characterization of Microplastics
The individual sediment samples pre-sieved at 5 mm and mechanically homogenized were divided into 120–140 g aliquots and further sieved at 2 mm to obtain two fractions each: sieve fraction 1, containing plastic fragments with size between 5 and 2 mm, and fraction 2, containing all microplastics smaller than 2 mm. On selected samples representative of both sampling sites and of the horizons farther apart from the shoreline (B and C), and thus less heavily influenced by erratic action of surf water from the waves, the two fractions obtained from one of the aliquots were analyzed by optical microscopy for the detection and identification of microplastics. The fraction 2 obtained from another equivalent aliquot of each sample was subjected to sequential extractions in refluxing organic solvents followed by alkaline hydrolysis as previously described [23], for separate quantitative determination of the extractable hydrocarbon polymers (polystyrene and polyolefins), and of the TPA monomer deriving from the hydrolytic depolymerization of PET.
The isolated plastic fragments were individually weighed on a 0.1 mg precision scale and identified by ATR and micro FT-IR spectroscopy.
As shown in Table 2, the following five types of synthetic polymers could be identified; low density polyethylene (LDPE), polypropylene (PP), polystyrene (PS), polyethylene terephthalate (PET), and traces of acrylics (fibers of acrylonitrile copolymers, AN); no polyamide fibers were detected in the sieve fraction. The polyolefin fragments presented high levels of oxidation, as shown by the presence of intense IR absorptions from aliphatic carbonyl groups (1600–1800 cm−1) and hydroxyl groups. Representative ATR spectra are reported in Figures S1–S6 of the Supporting Information.
Table 2.
Classification of the synthetic polymer fragments (fn) with size between 5 and 2 mm, isolated from fraction 1 of selected representative sediment samples.
As the sediment samples were collected from hydrographic levels only exceptionally not submerged by water, it is reasonable to expect that a significant fraction of the microplastic particles be the result of long term accumulation in bottom sediments, and thus their exposure to photo-oxidizing environmental conditions might have been less extensive that in the case of the floating fragments more recently deposited ashore. In fact, studies concerning the oxidation/degradation processes of plastic materials have shown different intensities of degradation phenomena for MPs exposed to direct sunlight and higher temperatures (as in sandy beaches) as opposed to those for which exposure to photo-oxidative stress is presumably more limited (fragments floating or deposited in submerged sediments of aquifers) [28]. This is particularly true for polyolefins, as they are generally much most sensitive to environmental oxidation/degradation than PET and PS (we consider here only the polymer types that could actually be detected as larger plastics fragments), and may also end up in bottom sediments as a result of vertical transport promoted by the increased density consequent to fouling.
Several smaller particles (size < 2 mm) with the appearance of synthetic polymer fragments could also be detected in the fraction 2 of a few samples with the assistance of a stereo microscope. These particles were analyzed by micro-ATR/FT-IR for unambiguous identification. Based on the recorded spectra most of them could be associated to biogenic polymers (cellulose and lignocellulosic fibres, chitin, etc.), with only a few fibrous fragments being identified as synthetic polymers, mostly PET. As an example, the identification of polymer type for the fragments collected from 123 g of the BR1C3 sediment sample is reported in Table 3:
Table 3.
Classification by type of polymer of the fragments isolated in the passing fraction (size < 2 mm).
It has been reported that the simple separation (manual or by density) of single fragments, even if combined with FT-IR characterization, does not ensure a reliable identification of the polymeric matrix in such environmental samples [21,23]. This may be due to several reasons, the main ones being related to the variable levels of degradation of the polymer and the surface contamination by inorganic and biofouling. Thus, quantitative analyses based on particle separation, identification, and counting may turn out to be of limited statistical value as a result of biogenic material being mistaken for synthetic polymers (and vice-versa) and of incomplete recovery and/or detection of the smaller particles from the environmental samples.
Figure 3 reports the identification of fragments f3 and f1 from sample BR1C3. The FT-IR spectrum of fragment f3 (see Figure 3b) indicates that the main component is calcium carbonate, as shown by the presence of its characteristic absorptions at 3300 (OH stretching), 1637 (O-H bending), 1412 (symmetrical CO32− stretching), and 713 (CO32− deformation in plane) cm−1. The C-H stretching bands at 2929 and 2852 cm−1 are attributable to methylene groups from some contamination by either biogenic or synthetic organic material. The ATR FT-IR spectrum of fragment f1 (Figure 3d) presents the characteristic bands of cellulose at 3300, 2890, 1426, 1162, 1018, 1018 and 989 cm−1, related to O-H stretching; C-C stretching; C-H, C-O, and C-O-C pyranose ring vibration; and β-glycosidic linkage. Additional examples of identification of biogenic particles by micro-ATR FTIR spectra can be found in Figure S7 of the Supporting Information).
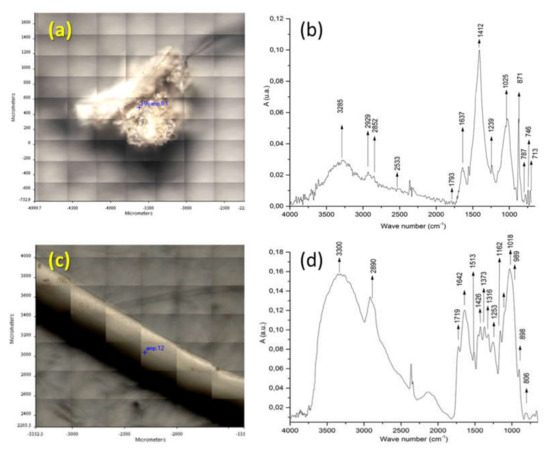
Figure 3.
(a) Image of analyzed surface of fragment f3 from the BR1C3 (see Table 2) sediment sample. (b) ATR FT-IR of fragment f3 of the BR1C3 sediment sample. (c) Image of analyzed surface of fragment f1 of the BR1C3 sediment sample. (d) ATR FT-IR of fragment f1 of the BR1C3 sediment sample.
3.2. Analysis of Polymeric Components Soluble in Dichloromethane
The extraction of the sediments with refluxing dichloromethane allows recovery of PS and of the more heavily degraded polyolefins, in addition to other vinyl polymers (e.g., polyvinyl chloride, PVC) and uncrosslinked silicones (e.g., polydimethylsiloxane) as well as biogenic materials (fatty acids, cutins, low molecular weight terpenoids, etc.). The amount of dichloromethane extractable fraction determined by gravimetric analysis was averaged over all the subsamples from each one of the two sampling sites, BR1 and BR2 (typically 2–3 replicates for each subsample, with the exclusion of the C samples closer to the shoreline).
As reported in Table 4, very small amounts of extractable fraction were obtained (60–75 mg/kg sediment), with limited internal variations among different subsamples, and variability inconsistent with the horizon of the sampling point. All the dichloromethane extracts were then analyzed by means of spectroscopic and chromatographic techniques, namely FT-IR, 1H-NMR, and SEC, as discussed in the following paragraphs.
Table 4.
Gravimetric analysis of the extractable fractions in dichloromethane.
3.2.1. Characterization of Dichloromethane Extracts
The recorded FT-IR spectra of all dichloromethane extracts from the BR1 sediment samples show substantially the same main absorption bands. As a representative example, the spectrum of the BR1A3 extract (shown in Figure 4) presents the characteristic bands of aliphatic hydrocarbons with a large fraction of methylene groups (CH2 asymmetric and symmetric stretching at 2923 and 2851 cm−1, respectively; bending at 1463 cm−1), as in linear alkyl chains, but also a not negligible number of methyl groups (asymmetric CH3 stretching at 2952 cm−1 and bending at 1377 cm−1) that, along with the absorptions from shorter methylene sequences (two weak absorptions due to CH2 rocking for oligomethylene sequences with n > 4 methylene units at 721 cm−1), suggests that the observed hydrocarbon fraction includes a not negligible fraction of small acyclic hydrocarbon molecules and/or highly branched or highly functionalized (oxidized) species. The presence of the latter is confirmed by the multiple absorptions observed at 1700–1735 cm−1 (carbonyl stretching from carboxyl, ketone, and ester groups; C-O-C stretching modes in the 1100–1300 cm−1 range), while the broad absorption at 3370–3390 cm−1 associated with symmetric O-H stretching may be indicative of the presence of hydroxylated species along with some contribution from absorbed moisture. The spectra from the extracts of about a half of the samples also show a small shoulder at 3020–3050 cm−1 (symmetric stretching of aromatic C-H), weak absorptions around 1600 cm−1 (symmetric stretching of the aromatic ring, superimposed to the absorptions from residual water present in the sample), and small peak at about 700 cm−1 (out of plane aromatic C-H deformation), suggesting the presence of some PS microplastics in the corresponding samples. Analytical bands indicative of the presence of PVC (CH out-of-plane bending at 1255 cm−1 and C-Cl symmetric stretching at 616 cm−1) were never clearly detected. On the other hand, many samples presented absorption bands typical of silicones (polydimethylsiloxane and its parent species) at 1261 cm−1 (CH3-Si-CH3 in-plane scissoring), 1095 and 1022 cm−1 (Si-O-Si symmetric stretching), and 804 cm−1 (out-of-plane CH3 bending and Si-C symmetric stretching). A representative selection of the FT-IR spectra from the BR1 extracts can be found in Figures S8–S10 of the Supporting Information.
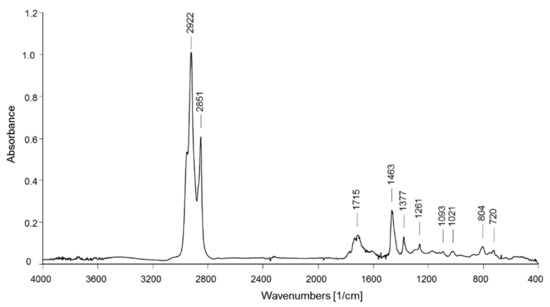
Figure 4.
FT-IR spectrum of the BR1A3 sample extracted in dichloromethane.
The FT-IR spectra of the dichloromethane extracts from the samples of the BR2 site do not show any significant difference with respect to those of the BR1 site. A single worth mentioning exception is the extract from sample BR2B3, showing the characteristic absorptions of an aliphatic polyester (carbonyl stretching of ester groups at 1740 cm−1, C-O and C-O-C stretching vibrations at 1260, 1182, 1144, and 1063 cm−1, Figure S11 of Supporting Information).
Figure 5 shows the 1H-NMR spectrum of the extract in dichloromethane of the BR1A3 extract. In addition to the signals between 0.5 and 2.0 ppm, associated to aliphatic hydrocarbon structures, the small signals between 2.2 and 2.8 ppm indicate the presence of protons on carbon atoms (methyl, methylene and/or methine) in alpha position of ketone or carboxylic groups, in agreement with the results of the FT-IR analyses. Additional weak signals between 3.7 and 4.3 ppm can be associated with protons on carbon atoms bound to ester oxygen (methyl or methylene protons) and/or alcoholic (methylene and methine protons) atoms. Finally, all analyzed samples present a signal at about 0.09–0.1 ppm attributable to the protons of the methyl groups of polysiloxanes, in agreement with the already highlighted presence of silicon-based polymers based on the FT-IR analysis of dichloromethane extracts; the source of low molecular weight uncrosslinked polysiloxane pollutants is likely to be the inflow of civil and domestic wastewaters, as polysiloxanes are present in numerous healthcare and personal care products as well as in some industrial formulations and lubricants.
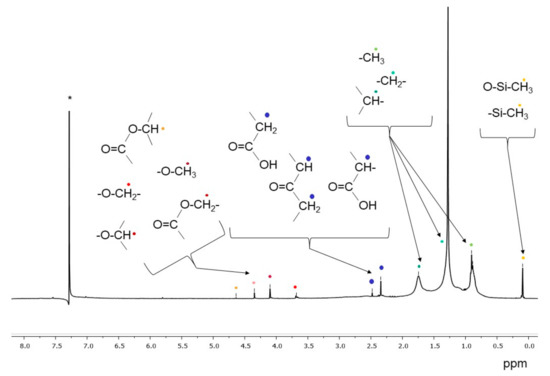
Figure 5.
1H NMR spectrum of the BR1A3 sample extracted in dichloromethane. (* singlet at 7.26 ppm from CHCl3 traces in CDCl3).
Additional 1H-NMR spectra of representative dichloromethane extracts are reported in the Supporting Information (Figures S12 and S13).
The dichloromethane extracts of BR1B3 and BR2B3 were further analysed by means of 13C NMR and 1H DOSY NMR experiments. For this purpose, a careful investigation was carried out by high resolution 1H-NMR spectroscopy in order to obtain DOSY (Diffusion Ordered Spectroscopy) maps. NMR experiments enable to identify both polymers and their degradation products, as reported in other studies concerning microplastics [29,30,31,32], thus confirming hypothetical degradation mechanisms. In the present case, both extracts of BR1B3 and BR2B3 samples were also analysed by 1D 13C-NMR spectroscopy.
The dichloromethane extract of BR1B3 was selected for these characterizations as it was considered, based on the previous spectroscopic analyses, as representative of those from all the analysed sediment samples, with the exception of the aforementioned BR2B3. While the 1H-NMR spectrum of BR1B3 extract revealed a fairly high complexity, the most intense signals are in the region of aliphatic protons (Figure 6a). Among them, a singlet signal at 0.09 ppm and two broad signals at 0.904 and 1.277 were observed. The first one can be assigned to polydimethylsiloxane (PDMS) methyl protons of residues, similarly to other 1H-NMR spectra (Figure 5 and Figures S12 and S13 in the Supporting Information). In the aromatic region, low intensity signals ascribable to PS fragments can be observed (Figure 6a). In order to assign the more relevant and intense 1H NMR signals, 1D and 2D NMR correlation spectra, 13C NMR (not reported here) and DOSY NMR experiments were performed. In Figure 6, the DOSY maps obtained for the two samples (BR1B3 and BR2B3 dichloromethane extracts) are reported.
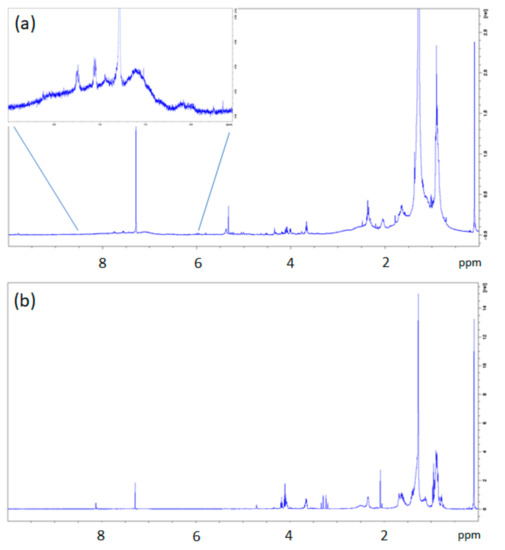
Figure 6.
1H-NMR spectra of dichloromethane extracts from BR1B3 (a) and BR2B3 (b) samples. The inset in BR1B3 (a) shows an enlargement of the aromatic region with small signals ascribable to polystyrene (PS) fragments.
The analysis of the 1H-NMR DOSY map of dichloromethane extracts from BR1B3 (Figure 7a) suggests the presence of inter correlation of proton signals at 0.904 and 1.277 ppm, with a similar value of diffusion coefficient (D = 5.2E−10 m2/s), measured at 25 °C, obtained by using Equation (1) (see Section 2.4). The corresponding hydrodynamic radius, obtained by applying Equation (2), is of approximately 8.2 Å, ascribable to residues of aliphatic polymers of synthetic origin. In accordance, the 13C NMR spectrum (Figure S14 in the Supporting Information) revealed the presence of signals typical of unsaturated aliphatic chains corresponding to the methyl group of PDMS (1 ppm), as well as signals at 14.1, 22.7, 29.6, and 31.9 ppm, which are ascribable to linear n-alkanes. The presence of linear chains was also supported by the lack of CH (methine) groups’ signals.
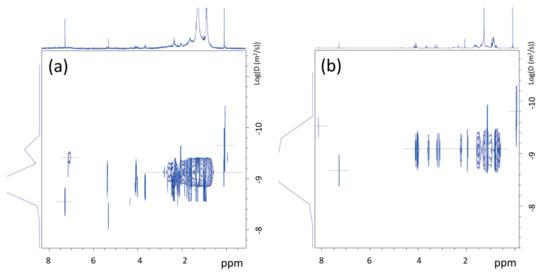
Figure 7.
1H-NMR DOSY spectra of dichloromethane extracts from BR1B3 (a) and BR2B3 (b), measured at T = 25 °C.
In the case of dichloromethane extract obtained from BR2B3 sediment sample, the 1H-NMR spectrum (see Figure 6b) shows a greater complexity than in the BR1B3 sediment extract. In addition to the signals observed in the BR1B3 sample (Figure 6a), groups of signals compatible with substituted aliphatic groups (between 3 and 5 ppm) are also present in comparable intensity, in addition to a signal at about 8 ppm typical of para-disubstituted benzene protons (Figure 6b). This is also supported by the 13C NMR spectrum (Figure S15 in the Supporting Information) showing two net signals at 129.7 ppm and 133.8 ppm (ring benzene) and in the area of carboxylic carbonyls (between 165 and 175 ppm). 1H-13C 2D NMR correlation spectroscopy of this sample (not reported here) allowed us to identify the nature of the carbonyl signals. Indeed, the signal at 165.3 ppm is compatible with the presence of aliphatic-co-aromatic esters, whereas signals at 169.4, 169.5 and 173.5 ppm can be attributed to aliphatic esters. The translational diffusion measurements (1H NMR DOSY) of the BR2B3 dichloromethane extract (see Figure 7b) show diffusion coefficients corresponding to molecules of hydrodynamic radius between 7 and 5 Å compatible with small molecules produced by degradation processes. This aspect is further discussed in the following sections.
3.2.2. Semi-Quantitative Determination of PS Content in Dichloromethane Extracts by SEC Analysis with Differential Refractometric (RI) and UV Double Detector.
The SEC profiles of all nine BR1 samples show similar features, with an exclusive or prevalent Gaussian-like monomodal distribution centered at about retention time r.t. = 16 min, corresponding to low molecular weight species, both in the traces recorded with the RI detector and in those recorded with the UV detector set at 260 nm (see Figure S16 in Supporting Information). In addition to the main fraction, the tracks of the chromatograms from at least one subsample of each horizon line present an additional weak signal centered at about r.t. = 10.5 min, corresponding to a high molecular weight species. The molecular weights of the high molecular weight (MW) fraction in the DCM extracts of the subsamples, when such fraction could be detected by SEC, are listed in Table 5 (the similar r.t. for this fraction in some of the chromatograms is due to a molecular weight distribution above the exclusion limit of the columns).
Table 5.
PS content and average molecular weights of PS fraction as determined by SEC on dichloromethane extracts from BR1 and BR2 sites.
The chromatographic profiles of the extracts from the BR2 site (see Figure S18 in Supporting Information) are quite different from those from the BR1 site. Both the RI and UV traces relative to the main fraction, still centered at r.t. = 16 min and assigned to low MW species, show a polymodal distribution, probably associated with poorly resolved well-defined oligomers and/or low MW (probably biogenic) species. Moreover, all the analysed BR2 extracts, with the exception of BR2B2, present the additional small peak at about r.t. = 10.5 min (in the case of BR2B3 a weaker and broader signal is observed at 10.5 > r.t. > 12, indicative of a fraction quantitatively comparable to that of the other samples, but with molecular weight distribution within the exclusion limit of the columns). Both the BR1 and the BR2 extracts show the same feature of the high MW fraction, that is, a positive detection at 260 nm and no detection at 340 nm (Figure 8, and Figure S19 in the Supporting Information), indicating that such high MW fraction consist mainly of polystyrene, its UV absorption ranging between 210–300 nm.
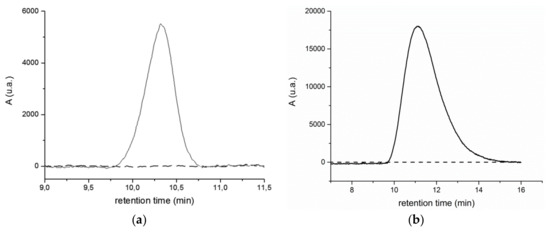
Figure 8.
(a) UV 260 nm (red line) and UV 340 nm (black line) chromatographic profiles of the BR2A2 sample extracted in dichloromethane and (b) UV 260 nm (solid line) and UV 340 nm (black line) chromatographic profiles of reference PS standard solution (1 mg/mL).
In the case of BR2B3 dichloromethane extract, a high molecular weight fraction only detectable in the RI trace of the SEC chromatogram could not be attributed to PS since no signal could be detected at the corresponding retention time in the UV trace of the chromatogram. Based on the characterizations carried out on this sample, the high molecular weight fraction observed in the SEC analyses could be attributed to a predominantly aliphatic polyester. In fact, both the FT-IR (Figure S11 in Supporting Information) and the high-resolution 1H NMR spectra in Figure 6 suggest the intensity of the signals ascribed to ester-type carbonyl groups was much higher than in the corresponding spectra of all other extracts. This evidence is further supported by the fact that both polystyrene and aliphatic polyesters are soluble in the extracting dichloromethane solvent.
3.3. Analysis of Polymeric Components Soluble in Xylene
Extraction with refluxing xylene of the sediment samples previously extracted with DCM was performed on samples BR1B3 and BR2B3, taken as representative of the horizon farther away from the swash zone for each sampling site. A modified version of a previously reported procedure [21] was adopted to improve the recovery of the extracted high molecular weight polyolefins (polyethylene and polypropylene). With this purpose, after the consecutive extraction of three aliquots about 50 g each of sediment with 200 mL xylene, the obtained xylene solution was concentrated to about 20 mL by distilling off the excess xylene, then 30–50 mL methanol was added to precipitate the polymers that could be recovered by filtration on PVDF 0.22 μm, without loss possibly caused by the formation of a polymer film on the walls of the extractor. The solid filtered off was dried and weighed. An unexpectedly small extractable fraction was found in both samples, namely 22.8 ppm and 14.6 ppm in BR1B3 and BR2B3, respectively.
The small amount (a few mg) of filtered solids were analyzed directly on the PVDF filter membrane by micro-ATR/FTIR. In both samples, the spectra recorded preliminarily on single spots clearly distinguishable by microscopic observation from the underlying filter presented the expected features typical of mildly oxidized polyolefin materials such as polyethylene and polypropylene.
Figure 9 reports a typical spectrum from the extract of sample BR2B3 and the spectral correlation map recorded from an area of 4x8 mm, using as reference the average spectrum obtained from the individual spots. The spectral map shows a good homogeneity of the material on the filter, the heterogeneity being possibly associated with different degrees of oxidation of the polyolefin mix.
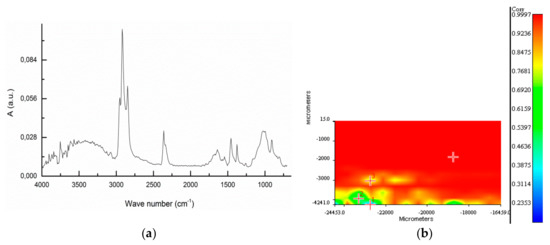
Figure 9.
BR2B3 extract: (a) ATR FT-IR spectrum averaged over the whole 4 × 8 mm area analyzed; (b) Correlation map referred to the average spectrum for the analyzed surface.
In Figure 10 the results of the analyses performed on the BR2B3 extract are reported. In this case, due to the poorer coverage of the filter by the extracted material, the micro-ATR mapping of a 7 × 4 mm area was carried out by subtracting the typical spectrum of the often dominant PVDF background from those previously recorded. Again, the spectra of the selected spots exhibit strong characteristic absorption bands at 2917, 2851, and 1460 and 1370 cm−1 assigned to stretching and deformation vibration mode of methyl and methylene groups of polyolefins.
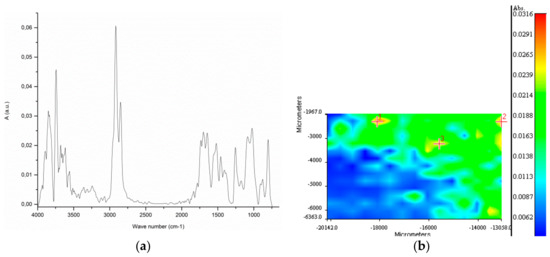
Figure 10.
BR2B3 sample (a) ATR FT-IR spectrum representative of points 1, 2, and 3 of the map on the right. (b) Correlation map for the analyzed surface, referred to the representative spectrum on the left.
3.4. Analysis of the Total PET Content in the Sediment Samples
To quantify the PET content in the sediment samples a recently developed analytical method was adopted [23], which provides for a high sensitivity determination of the total mass of PET by means of total alkaline hydrolysis of the polymer to the corresponding monomers, ethylene glycol and terephthalic acid (TPA), followed by quantitative analysis of the latter by reversed phase HPLC. For this purpose, the residues of the sediment samples previously subjected to sequential extraction with dichloromethane and xylene, were united according to the respective horizon (e.g., samples BR1A1, BR1B1 and BR1C1 were combined and mechanically homogenized to obtain the medium composite sample BR1-H1; the same procedure was adopted to produce the medium composite samples BR1-H2 and BR1-H3), then treated with hot aqueous NaOH 1.9 M in a 1:3 vol/wt ratio in the presence of a phase transfer catalyst, and the hydrolyzate was purified according to the previously reported procedure to remove most of the TPA contaminants before HPLC analysis (see ref. 17 for the experimental details). For the quantitative determination of TPA by HPLC a linear calibration in the 0.2–1.6 ppm range was built using pure TPA as the standard (see Figure S20 in Supporting Information). The results listed in Table 6 are obtained from at least three replicates of each medium composite sample. In Table 7 the results of the analyses performed on the samples from the BR2 site are reported. In this case the data are referred to the single subsamples, analyzed in three replicates, to account for the much larger differences of PET concentration in the various subsamples within the same sampling site.
Table 6.
Average PET concentrations at different sampling horizons from the BR1 site.
Table 7.
Average PET concentrations in individual subsamples from the BR2 site.
The set of data indicates the presence of a significant contamination by PET-based microplastics in both sites. In particular, the level of contamination in the BR1 site appears to be rather uniform in the 3.4–4.0 ppm (mg PET/kg sediment) range, with moderate differences in the measured concentrations both among the replicates (subsamples drawn from the same medium composite sample), as shown by the relatively small standard deviations, and among the three medium composite samples from different horizons.
On the other hand, the level of contamination in the BR2 site appears to be affected by a strong variability both within the subsamples and among the different sampling horizons. As a possible explanation for such differences one could take into account the prevailing winds in Lake Bracciano; site BR1, in which a more uniform PET microplastic distribution was detected, finds itself most frequently down wind, being therefore more often exposed to incoming currents and wave action that are likely to result in a more uniform transport of PET microplastics, with respect to the downwind BR2 site. Differences of this type have been found in other studies aimed at the determination of MPs in Italian lakes [33,34].
4. Conclusions
The multianalytical approach adopted in this work allowed the identification and, in the case of PET, the accurate quantification of the pollution from microplastics (passing fraction from a sieve with 2 mm mesh size) in sediment samples from Lake Bracciano, Italy. ATR FT-IR spectroscopy allowed the identification of small particles as fragments of biogenic origin in several cases in which particles singled out from the sieve fraction had been preliminarily (visually) identified as synthetic plastic debris, thus confirming the difficulty to effectively separate microplastics from the rest of the debris without a huge waste of time and resources. The adopted analytical methodology, instead, was based on either solvent extraction and quantification by gravimetry (for polyolefins) or chromatography with UV detection (for polystyrene) in the case of solvent soluble polymers, while depolymerization and HPLC analysis of its TPA monomer concentration for PET, insoluble in common organic solvents. High resolution 1H NMR spectra of microplastics’ extracts in dichloromethane obtained from different sampling sites were of help in confirming the chemical nature of plastic-base materials. Moreover, 2D 1H-NMR DOSY experiments on the dichloromethane extracts allowed us to better understand the level of plastic degradation of different samples.
The concentration of PS (0.1–1.5 ppm in about a half of the subsamples) and polyolefin (in the 10–20 ppm range for the two analyzed subsamples, one for each sampling site) is possibly a lower threshold, given the fact that the sediment samples were taken from a beach site formerly included in the lake bottom area, in which low density microplastic debris are less likely to end up. On the other hand, the contamination by PET, detected in all subsamples at concentrations between 1.8 and 13.6 ppm, is likely to be the result of the deposition of microfibers washed out from domestic and industrial textile washing. Finally, the widespread presence of polysiloxanes and, in one subsample, of an aliphatic or mixed aliphatic-aromatic polyester could be detected.
Supplementary Materials
The following are available online at https://www.mdpi.com/2071-1050/12/3/878/s1. Figure S1: ATR spectrum of the BR1B3-f1 fragment; Table S1: Characteristic IR absorptions of polyethylene; Figure S2: ATR spectrum of the BR2C2-f3 fragment; Table S2: Characteristic IR absorptions of polypropylene; Figure S3: ATR spectrum of the BR2B2-f1 fragment; Figure S4: ATR spectrum of the BR2C2-f1 fragment; Table S3. Characteristic IR absorptions of polyethylene terephthalate; Figure S5: ATR spectrum of the BR1C3-f2 fragment; Table S4: Characteristic IR absorptions of polystyrene; Figure S6: (a) Optical microscopy image showing the spots of the surface of fragment BR1C3-f4 analysed by micro-ATR; (b) micro-ATR FTIR of spot 5-1 (isolated black fiber); (c) micro-ATR FTIR of spot 5-2 (white fibers); Figure S7: Biogenic fragment from BR1C3: (a) optical microscopy image; (b) micro-ATR FTIR; Figure S8: FT-IR spectra of BR1A1 (green line), BR1B1 (red line) and BR1C1 (black line) extracts in dichloromethane; Figure S9: FT-IR spectra of BR1A2 (red line), BR1B2 (black line) and BR1C2 (green line) extracts in dichloromethane; Figure S10: FT-IR spectra of BR1A3 (red line) and BR1B3 (blue line) and BR1C3 (green line) extracts in dichloromethane; Figure S11: FT-IR spectrum of the BR2B3 sample extracted in in dichloromethane; Figure S12: 1H-NMR spectrum of the BR1B1 sample extracted in dichloromethane; Figure S13: 1H-NMR spectrum of the BR1C3 sample extracted in dichloromethane; Figure S14: 13C-NMR spectrum of the BR1B3 sample extracted in dichloromethane; Figure S15: 13C-NMR spectrum of the BR2B3 sample extracted in dichloromethane; Figure S16: SEC profiles as recorded by Refractive index (a) and UV (λ = 260 nm) (b) detectors from DCM extracts of BR1A1 sample; Figure S17: Calibration line recorded with UV detector (260 nm) for SEC analysis; Figure S18: SEC profiles as recorded by (a) Refractive index, and (b) UV (λ = 260 nm) detectors from DCM extracts of BR2A2 sample; Figure S19: UV 260 nm (red line) and UV 340 nm (black line) SEC profiles of DCM extracts from: (a) BR1A1; (b) BR1B3; (c) BR2A3; (d) BR2C2; Figure S20: Experimental calibration line for HPLC quantitative analysis of TPA.
Author Contributions
Conceptualization, V.C. and A.C.; methodology, A.C., V.D., L.P., and A.P.; investigation, A.C, S.B., V.V., M.C. and V.I.; writing—Original draft preparation, A.C., V.V., A.M., and M.C.; writing—Review and editing, V.C. and V.D.; funding acquisition, V.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Pisa, PRA 2017_17 project, and partially by the Fondazione Cassa di Risparmio di Lucca, Bando “Ricerca” 2020-2021.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Machovsky-Capuska, G.E.; Amiot, C.; Denuncio, P.; Grainger, R.; Raubenheimer, D. A nutritional perspective on plastic ingestion in wildlife. Sci. Total Environ. 2019, 656, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Seijo, A.; Santos, B.; Ferreira da Silva, E.; Cachada, A.; Pereira, R. Low-density polyethylene microplastics as a source and carriers of agrochemicals to soil and earthworms. Environ. Chem. 2019, 16, 8–17. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, Y.; Wang, J.; Zhang, M.; Jia, W.; Qin, X. LDPE microplastic films alter microbial community composition and enzymatic activities in soil. Environ. Pollut. 2019, 254, 112983. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Cho, H.-J.; Kim, E.; Huh, Y.H.; Kim, H.-J.; Kim, B.; Kang, T.; Lee, J.-S.; Jeong, J. Bioaccumulation of polystyrene nanoplastics and their effect on the toxicity of Au ions in zebrafish embryos. Nanoscale 2019, 11, 3173–3185. [Google Scholar] [CrossRef]
- Magni, S.; Binelli, A.; Pittura, L.; Avio, C.G.; Della Torre, C.; Parenti, C.C.; Gorbi, S.; Regoli, F. The fate of microplastics in an Italian Wastewater Treatment Plant. Sci. Total Environ. 2019, 652, 602–610. [Google Scholar] [CrossRef]
- Corradini, F.; Meza, P.; Eguiluz, R.; Casado, F.; Huerta-Lwanga, E.; Geissen, V. Evidence of microplastic accumulation in agricultural soils from sewage sludge disposal. Sci. Total Environ. 2019, 671, 411–420. [Google Scholar] [CrossRef]
- Cauwenberghe, L.V.; Devriese, L.; Galgani, F.; Robbens, J.; Janssen, C.R. Microplastics in sediments: A review of techniques, occurrence and effects. Mar. Environ. Res. 2015, 111, 5–17. [Google Scholar] [CrossRef]
- Hanvey, J.S.; Lewis, P.J.; Lavers, J.L.; Crosbie, N.D.; Pozo, K.; Clarke, B.O. A review of analytical techniques for quantifying microplastics in sediments. Anal. Methods 2017, 9, 1369–1383. [Google Scholar] [CrossRef]
- Faure, F.; Demars, C.; Wieser, O.; Kunz, M.; de Alencastro, L.F. Plastic pollution in Swiss surface waters: Nature and concentrations, interaction with pollutants. Environ. Chem. 2015, 5, 582–591. [Google Scholar] [CrossRef]
- Horton, A.A.; Walton, A.; Spurgeon, D.J.; Lahive, E.; Svendsen, C. Microplastics in freshwater and terrestrial environments: Evaluating the current understanding to identify the knowledge gaps and future research priorities. Sci. Total Environ. 2017, 586, 127–141. [Google Scholar] [CrossRef]
- Gasperi, J.; Dris, R.; Bonin, T.; Rocher, V.; Tassin, B. Assessment of floating plastic debris in surface water along the Seine River. Environ. Pollut. 2014, 195, 163–166. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.; Hoellein, T.J.; Mason, S.A.; Schluep, J.; Kelly, J.J. Microplastic is an Abundant and Distinct Microbial Habitat in an Urban River. Environ. Sci. Technol. 2014, 20, 11863–11871. [Google Scholar] [CrossRef] [PubMed]
- Eerkes-Medrano, D.; Thompson, R.C.; Aldridge, D.C. Microplastics in freshwater systems: A review of the emerging threats, identification of knowledge gaps and prioritisation of research needs. Water Res. 2015, 75, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.; Ramler, D. The discharge of certain amounts of industrial microplastic from a production plant into the River Danube is permitted by the Austrian legislation. Environ. Pollut. 2015, 200, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Zbyszewski, M.; Corcoran, P.L. Distribution and Degradation of Fresh Water Plastic Particles Along the Beaches of Lake Huron, Canada. Water Air Soil Pollut. 2011, 220, 365–372. [Google Scholar] [CrossRef]
- Cable, R.N.; Beletsky, D.; Beletsky, R.; Wigginton, K.; Locke, B.W.; Duhaime, M.B. Distribution and Modelled Transport of Plastic Pollution in the Great Lakes, the World’s Largest Freshwater Resource. Front. Environ. Sci. 2017. [Google Scholar] [CrossRef]
- Hajbane, S.; Pattiaratchi, C.B. Plastic Pollution Patterns in Offshore, Nearshore and Estuarine Waters: A Case Study from Perth, Western Australia. Front. Mar. Sci. 2017. [Google Scholar] [CrossRef]
- Wang, W.; Ndungu, A.W.; Li, Z.; Wang, J. Microplastics pollution in inland freshwaters of China: A case study in urban surface waters of Wuhan, China. Sci. Total Environ. 2017, 575, 1369–1374. [Google Scholar] [CrossRef]
- Hartmann, N.B.; Hüffer, T.; Thompson, R.C.; Hassello, M.; Verschoor, A.; Daugaard, A.E.; Rist, S.; Karlsson, T.; Brennholt, N.; Cole, M.; et al. Are We Speaking the Same Language? Recommendations for a Definition and Categorization Framework for Plastic Debris. Environ. Sci. Technol. 2019, 53, 1039–1047. [Google Scholar] [CrossRef]
- Hurley, R.R.; Lusher, A.L.; Olsen, M.; Nizzetto, L. Validation of a Method for Extracting Microplastics from Complex, Organic-Rich, Environmental Matrices. Environ. Sci. Technol. 2018, 52, 7409–7417. [Google Scholar] [CrossRef]
- Ceccarini, A.; Corti, A.; Erba, F.; Modugno, F.; Nasa, J.L.; Bianchi, S.; Castelvetro, V. The Hidden Microplastics: New Insight and Figures from the Thorough Separation and Characterization of Microplastics and of Their Degradation By-products in Coastal Sediments. Environ. Sci. Technol. 2018, 52, 5634–5643. [Google Scholar] [CrossRef] [PubMed]
- Biver, T.; Bianchi, S.; Carosi, M.R.; Ceccarini, A.; Corti, A.; Manco, E.; Castelvetro, V. Selective determination of poly(styrene) and polyolefin microplastics in sandy beach sediments by gel permeation chromatography coupled with fluorescence detection. Mar. Pollut. Bull. 2018, 136, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Castelvetro, V.; Corti, A.; Bianchi, S.; Ceccarini, A.; Manariti, A.; Vinciguerra, V. Quantification of poly (ethylene terephthalate) micro- and nanoparticle contaminants in marine sediments and other environmental matrices. J. Hazard. Mater. 2020, 385, 121517. [Google Scholar] [CrossRef]
- Bracciano Smart Lake. Available online: https://braccianosmartlake.com/rilevamento-quota-lago/ (accessed on 5 January 2020).
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288. [Google Scholar] [CrossRef]
- Gibbs, S.J.; Johnson, C.S. A PFG NMR experiment for accurate diffusion and flow studies in the presence of eddy currents. J. Magn. Reson. 1991, 93, 395–402. [Google Scholar] [CrossRef]
- Johnson, C.S., Jr. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles, applications. Prog. NMR Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Andrady, A.L. Microplastics in the marine environment. Mar. Pollut. Bull. 2011, 62, 596–1605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Jia, H.Z.; Zhao, S.; Xia, T.J.; Guo, X.T.; Wang, T.C.; Zhu, L.Y. Formation of Environmentally Persistent Free Radicals on Microplastics under Light Irradiation. Environ. Sci. Technol. 2019, 53, 8177–8186. [Google Scholar] [CrossRef]
- Peez, N.; Janiska, M.C.; Imhof, W. The first application of quantitative H-1 NMR spectroscopy as a simple and fast method of identification and quantification of microplastic particles (PE, PET, and PS). Anal. Bioanal. Chem. 2019, 411, 823–833. [Google Scholar] [CrossRef]
- Rodriguez-Seijo, A.; da Costa, J.P.; Rocha-Santos, T.; Duarte, A.C.; Pereira, R. Oxidative stress, energy metabolism and molecular responses of earthworms (Eisenia fetida) exposed to low-density polyethylene microplastics. Environ. Sci. Pollut. Res. 2018, 25, 33599–33610. [Google Scholar] [CrossRef]
- Zaltariov, M.-F.; Bele, A.; Vasiliu, L.; Gradinaru, L.; Vornicu, N.; Racles, C.; Cazacu, M. Assessment of chemicals released in the marine environment by dielectric elastomers useful as active elements in wave energy harvesters. J. Hazard. Mater. 2018, 341, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.K.; Paglialonga, L.; Czech, E.; Tamminga, M. Microplastic pollution in lakes and lake shoreline sediments, A case study on Lake Bolsena and Lake Chiusi (central Italy). Environ. Pollut. 2016, 213, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Sighicelli, M.; Pietrelli, L.; Lecce, F.; Iannilli, V.; Falconieri, M.; Coscia, L.; Di Vito, S.; Nuglio, S.; Zampetti, G. Microplastic pollution in the surface waters of Italian Subalpine Lakes. Environ. Pollut. 2018, 236, 645–651. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).