
Driving Factors of Microbial Community Abundance and Structure in Typical Forest Soils of Sanjiang Plain, Northeast China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Collection and Physicochemical Characterization
2.2. DNA Extraction
2.3. qPCR
2.4. Illumina MiSeq Sequencing
2.5. Statistical Analyses
2.6. Nucleotide Sequence Accession Numbers
3. Results
3.1. Soil Physicochemical Characterization
3.2. Abundance of Microbial Community in the Three Typical Forest Soils
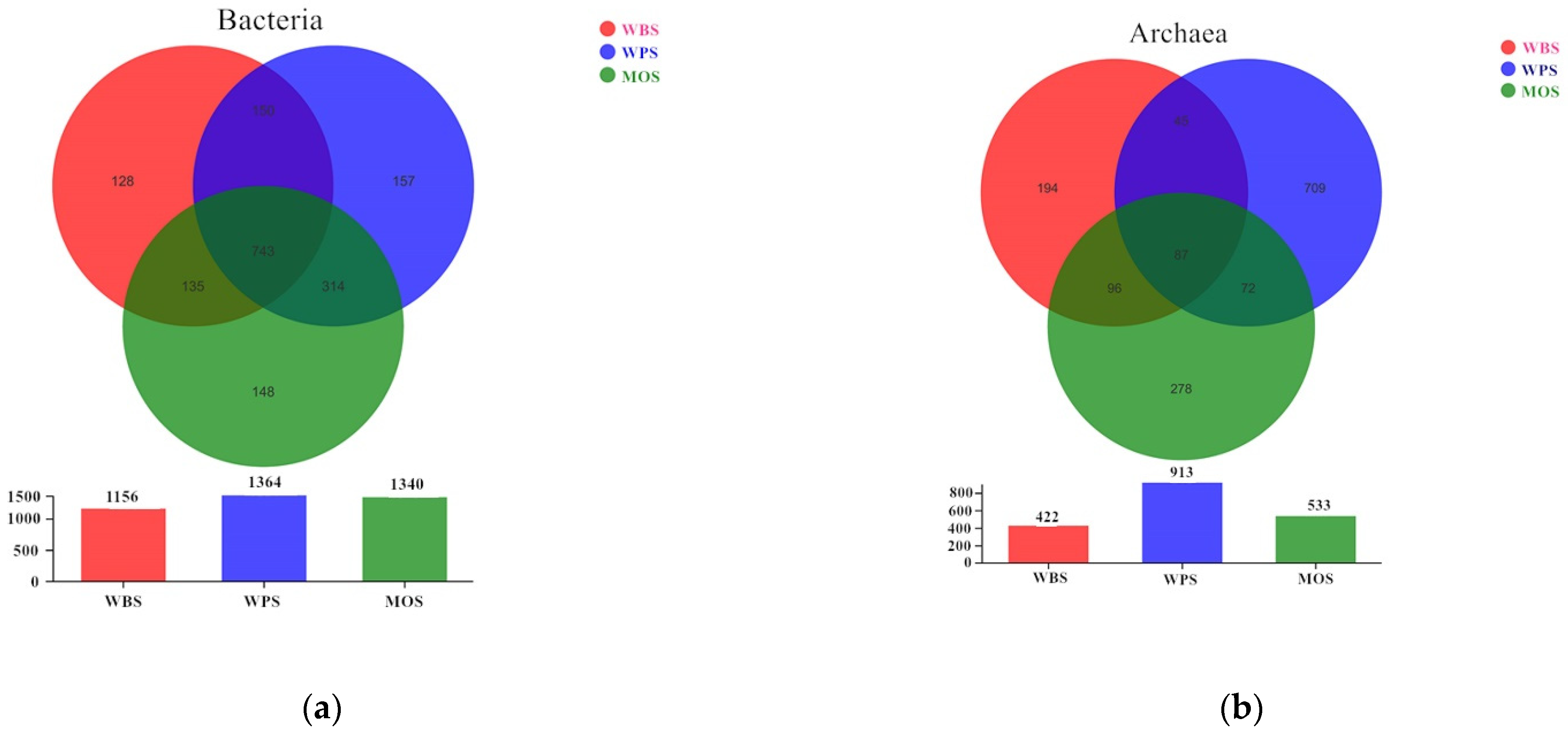
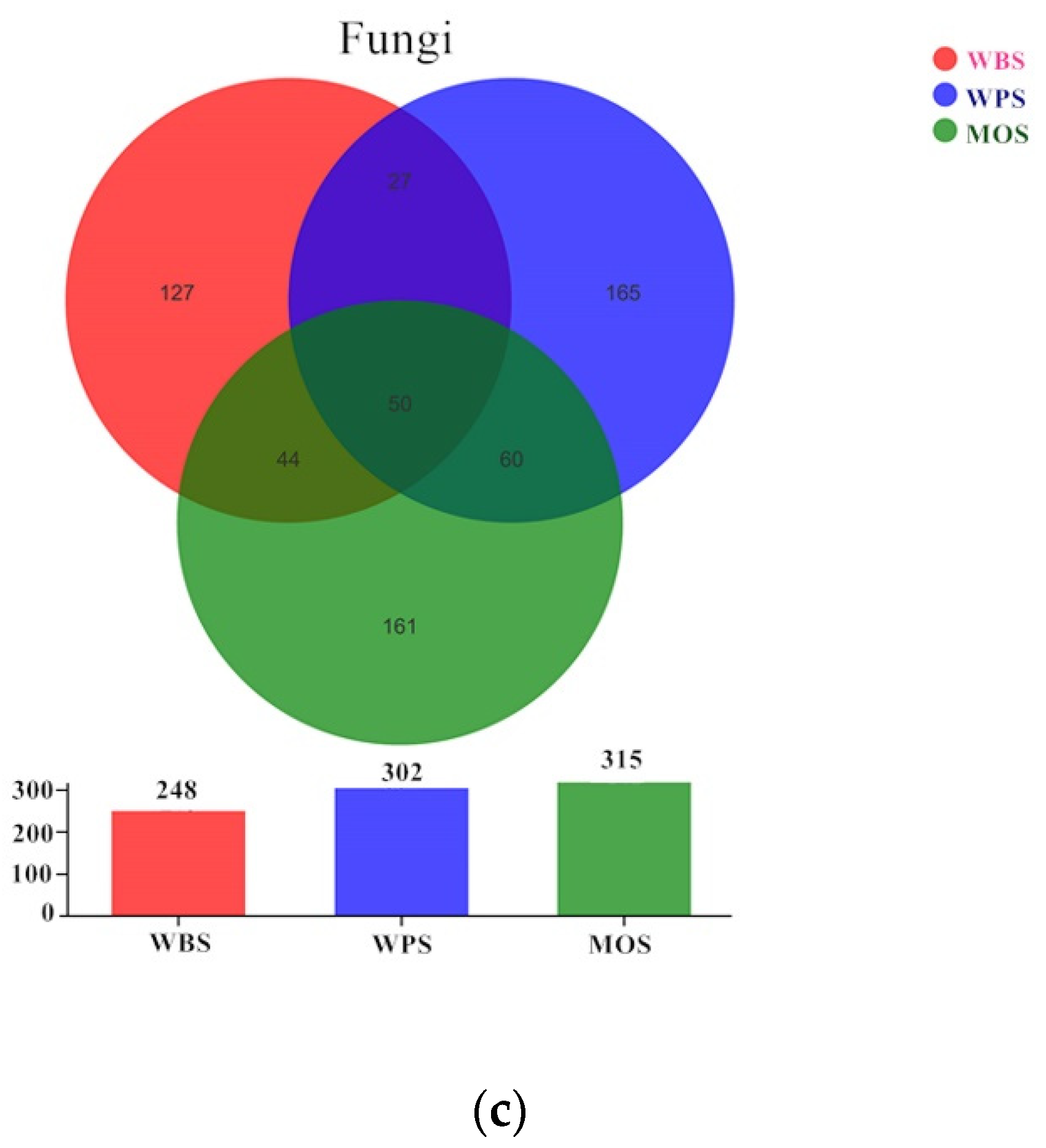
3.3. Community Structure of Microbial Community in the Three Typical Forest Soils
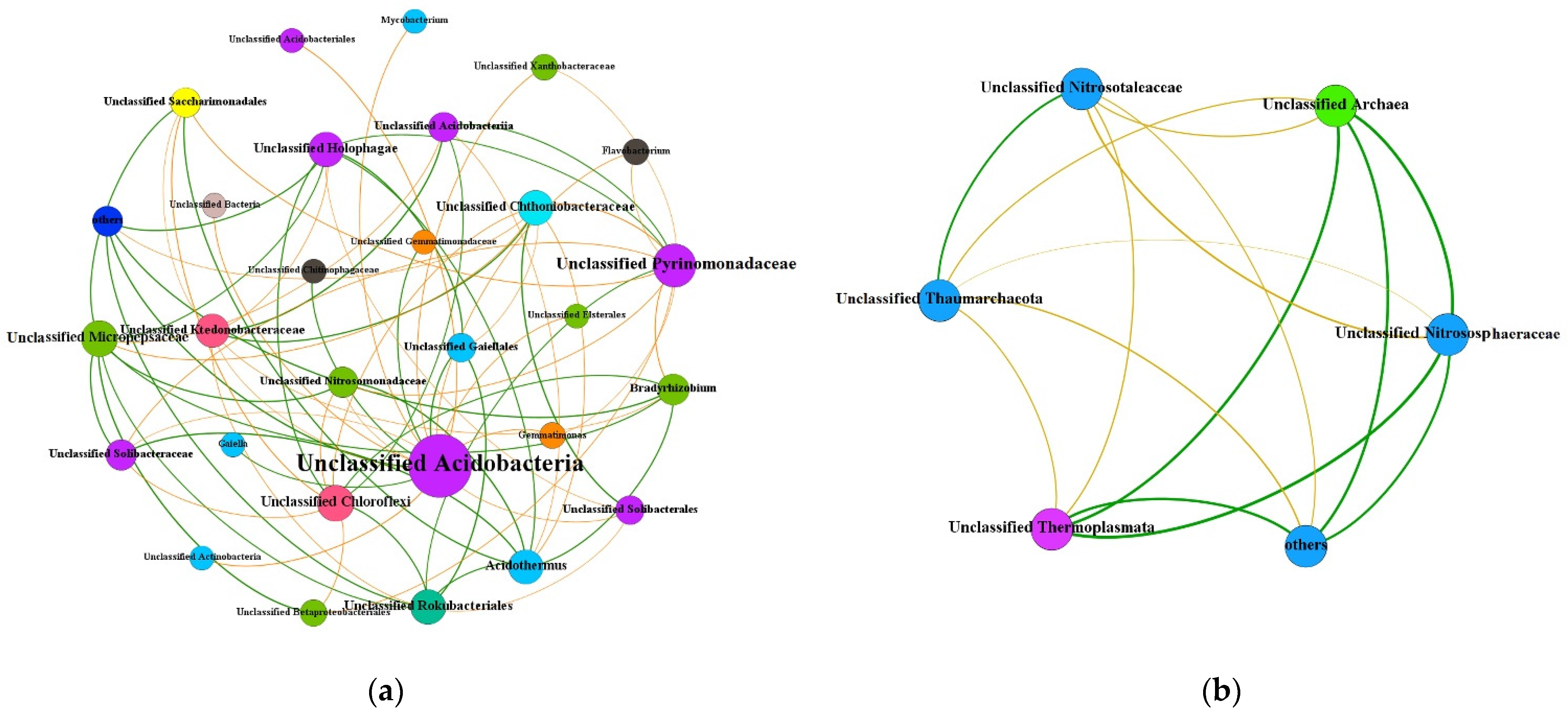
3.4. Co-Occurrence Network Analysis
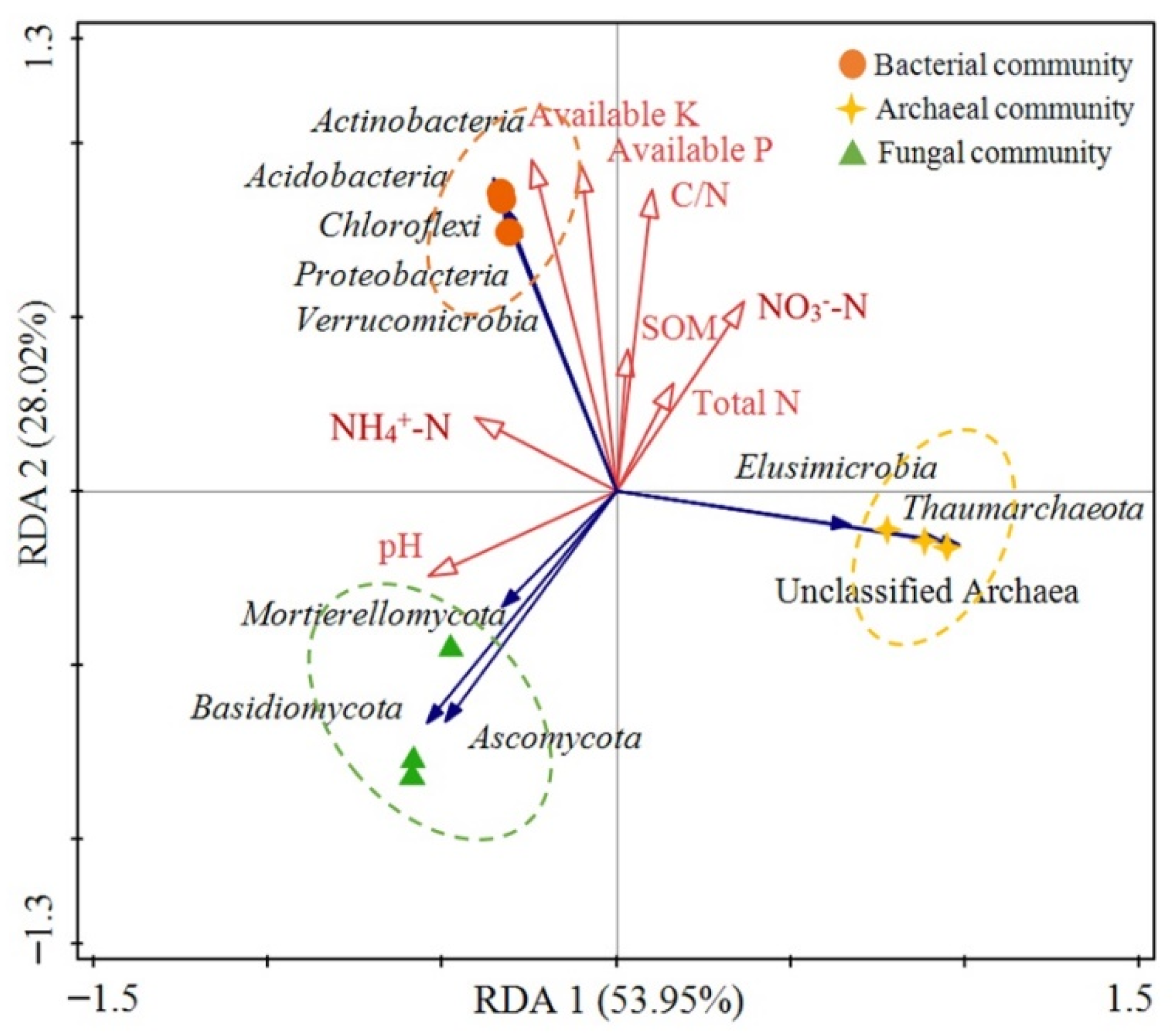
3.5. The Associations between Microbial Community Structure and Environmental Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siles, J.A.; Margesin, R. Abundance and Diversity of Bacterial, Archaeal, and Fungal Communities along an Altitudinal Gradient in Alpine Forest Soils: What are the Driving Factors? Microb. Ecol. 2016, 72, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zeng, Y.; Guo, C.; Bao, Y.; Lu, G.; Reinfelder, J.R.; Dang, Z. Bacterial, Archaeal, and Fungal Community Responses to Acid Mine Drainage-laden Pollution in A Rice Paddy Soil Ecosystem. Sci. Total Environ. 2018, 616, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, J.M.; Sheflin, A.M.; Manter, D.K.; Vivanco, J.M. Manipulating the Soil Microbiome to Increase Soil Health and Plant Fertility. Biol. Fertil. Soils 2012, 48, 489–499. [Google Scholar] [CrossRef]
- Schulz, S.; Brankatschk, R.; Dümig, A.; Kögel-Knabner, I.; Schloter, M.; Zeyer, J. The Role of Microorganisms at Different Stages of Ecosystem Development for Soil Formation. Biogeosciences 2013, 10, 3983–3996. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Tan, Z.; Gong, H.; Huang, Q. Migration and Transformation Mechanisms of Nutrient Elements (N, P, K) within Biochar in Straw-biochar-soil-plant Systems: A Review. ACS Sustain. Chem. Eng. 2018, 7, 22–32. [Google Scholar] [CrossRef]
- Hackl, E.; Pfeffer, M.; Donat, C.; Bachmann, G.; Zechmeister-Boltenstern, S. Composition of the Mmicrobial Communities in the Mineral Soil under Different Types of Natural Forest. Soil Biol. Biochem. 2005, 37, 661–671. [Google Scholar] [CrossRef]
- Lejon, D.P.H.; Chaussod, R.; Ranger, J.; Ranjard, L. Microbial Community Structure and Density under Different Tree Species in an Acid Forest Soil (Morvan, France). Microb. Ecol. 2005, 50, 614–625. [Google Scholar] [CrossRef]
- Yang, F.; Wu, J.; Zhang, D.; Chen, Q.; Zhang, Q.; Cheng, X. Soil Bacterial Community Composition and Diversity in Relation to Edaphic Properties and Plant Traits in Grasslands of Southern China. Appl. Soil Ecol. 2018, 128, 43–53. [Google Scholar] [CrossRef]
- Yu, S.; Ehrenfeld, J.G. Relationships among Plants, Soils and Microbial Communities along A Hydrological Gradient in the New Jersey Pinelands, USA. Ann. Bot. 2010, 105, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ye, D.; Wang, X.; Settles, M.L.; Wang, J.; Hao, Z.; Zhou, L.; Dong, P.; Jiang, Y.; Ma, Z.S. Soil Bacterial Communities of Different Natural Forest Types in Northeast China. Plant Soil 2014, 383, 203–216. [Google Scholar] [CrossRef]
- Cui, Y.; Bing, H.; Fang, L.; Wu, Y.; Yu, J.; Shen, G.; Jiang, M.; Wang, X.; Zhang, X. Diversity Patterns of the Rhizosphere and Bulk Soil Microbial Communities along an Altitudinal Gradient in an Alpine Ecosystem of the Eastern Tibetan Plateau. Geoderma 2019, 338, 118–127. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, X.; Guo, D.; Zhao, J.H.; Yan, L.; Feng, G.Z.; Gao, Q.; Yu, H.; Zhao, L.P. Soil pH is the Primary Factor Driving the Distribution and Function of Microorganisms in Farmland Soils in North-eastern China. Ann. Microbiol. 2019, 69, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Liu, C.; Wang, Q.; Yao, M.; Rui, J.; Zhang, S.; Li, X. Soil Bacterial Community Structure in Chinese Wetlands. Geoderma 2019, 337, 290–299. [Google Scholar] [CrossRef]
- Hu, Y.; Xiang, D.; Veresoglou, S.D.; Chen, F.; Chen, Y.; Hao, Z.; Zhang, X.; Chen, B. Soil Organic Carbon and Soil Structure are Driving Microbial Abundance and Community Composition across the Arid and Semi-arid Grasslands in Northern China. Soil Biol. Biochem. 2014, 77, 51–57. [Google Scholar] [CrossRef]
- Lladó, S.; López–Mondéjar, R.; Baldrian, P. Drivers of Microbial Community Structure in Forest Soils. Appl. Microbiol. Biotechnol. 2018, 102, 4331–4338. [Google Scholar] [CrossRef]
- Tang, M.; Li, L.; Wang, X.; You, J.; Li, J.; Chen, X. Elevational is the main factor controlling the soil microbial community structure in alpine tundra of the Changbai Mountain. Sci. Rep. 2020, 10, 12442. [Google Scholar] [CrossRef]
- Balser, T.C.; Firestone, M.K. Linking Microbial Community Composition and Soil Processes in A California Annual Grassland and Mixed-conifer Forest. Biogeochemistry 2005, 73, 395–415. [Google Scholar] [CrossRef]
- Zhou, Y.; Jia, X.; Han, L.; Tian, G.; Kang, S.; Zhao, Y. Spatial Characteristics of the Dominant Fungi and Their Driving Factors in Forest Soils in the Qinling Mountains, China. Catena 2021, 206, 105504. [Google Scholar] [CrossRef]
- Wang, Z.; Song, K.; Ma, W.; Ren, C.; Zhang, B.; Liu, D.; Chen, J.; Song, C. Loss and Fragmentation of Marshes in the Sanjiang Plain, Northeast China, 1954-2005. Wetlands 2011, 31, 945–954. [Google Scholar] [CrossRef]
- Cao, Y.; Tang, C.; Song, X.; Liu, C.; Zhang, Y. Characteristics of Nitrate in Major Rivers and Aquifers of the Sanjiang Plain, China. J. Environ. Monit. 2012, 14, 2624–2633. [Google Scholar] [CrossRef]
- Du, B.; Zheng, Y.; Liu, J.; Mao, D. Threatened Plants in China’s Sanjiang Plain: Hotspot Distributions and Gap Analysis. Sustainability 2018, 10, 194. [Google Scholar] [CrossRef] [Green Version]
- Hernández, M.; Conrad, R.; Klose, M.; Ma, K.; Lu, Y. Structure and Function of Methanogenic Microbial Communities in Soils from Flooded Rice and Upland Soybean Fields from Sanjiang Plain, NE China. Soil Biol. Biochem. 2017, 105, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Frey, B.; Yang, L.; Li, M.H.; Ni, H. Land Use Change Effects on Diversity of Soil Bacterial, Acidobacterial and Fungal Communities in Wetlands of the Sanjiang Plain, North-eastern China. Sci. Rep. 2019, 9, 18535. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, J.; Wu, Y.; Song, Y.; Liu, R.; Cao, Z.; Cui, Y. Shifts of the NirS and NirK Denitrifiers in Different Land Use Types and Seasons in the Sanjiang Plain, China. J. Basic Microbiol. 2019, 59, 1040–1048. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, Y. Effects of Land Use Patterns on the Bacterial Community Structure and Diversity of Wetland Soils in the Sanjiang Plain. J. Soil Sci. Plant Nutr. 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, N.; Wang, H.; Li, J.; Zhong, H.; Dong, H.; Zeng, Z.; Zong, C. Variations in the Diversity of the Soil Microbial Community and Structure under Various Categories of Degraded Wetland in Sanjiang Plain, North-eastern China. Land Degrad. Dev. 2021, 32, 2143–2156. [Google Scholar] [CrossRef]
- Zhang, C.; Robinson, D.; Wang, J.; Liu, J.; Liu, X.; Tong, L. Factors Influencing ‘Farmers’ Willingness to Participate in the Conversion of Cultivated Land to Wetland Program in Sanjiang National Nature Reserve, China. Environ. Manag. 2011, 47, 107–120. [Google Scholar] [CrossRef]
- Zhou, Z.Q.; Liu, T. The Current Status, Threats and Protection Way of Sanjiang Plain Wetland, Northeast China. J. For. Res. 2005, 16, 148–152. [Google Scholar]
- Lin, Y.; Bhattacharyya, A.; Campbell, A.N.; Nico, P.S.; Pett-Ridge, J.; Silver, W.L. Phosphorus fractionation responds to dynamic redox conditions in A humid tropical forest soil. J. Geophys. Res. Biogeosci. 2018, 123, 3016–3027. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, S.; Lu, D.; Chen, X.; Feng, W.; Wang, Y.; Zhao, S.; Wang, H. Analysis of Mechanisms of Soil Potassium-holding Capacity Using Different Soils of China. Agron. J. 2022, 114, 825–834. [Google Scholar] [CrossRef]
- Schaub, I.; Baum, C.; Schumann, R.; Karsten, U. Effects of an Early Successional Biological Soil Crust from a Temperate Coastal Sand Dune (NE Germany) on Soil Elemental Stoichiometry and Phosphatase Activity. Microb. Ecol. 2019, 77, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Paasonen-Kivekäs, M.; Yli-Halla, M. A Comparison of Nitrogen and Carbon Reserves in Acid Sulphate and Non Acid Sulphate Soils in Western Finland. Agric. Food Sci. 2005, 14, 57–69. [Google Scholar] [CrossRef]
- Zeng, Q.; An, S. Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities using Different Primer Sets on the Loess Plateau. Microorganisms 2021, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate Estimation of Fungal Diversity and Abundance through Improved lineage-specific Primers Optimized for Illumina Amplicon Sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Si, P.; Liu, E.; Yan, C.; Zhang, Z.; Zhang, Y. Influence of Film Mulching on Soil Microbial Community in a Rainfed Region of Northeastern China. Sci. Rep. 2017, 7, 8468. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Li, M.M.; Zhao, G. Thiocyanate Increases the Nitrous Oxide Formation Process through Modifying the Soil Bacterial Community. J. Sci. Food Agric. 2022, 102, 2321–2329. [Google Scholar] [CrossRef]
- Xue, S.W.; Huang, C.; Tian, Y.X.; Li, Y.B.; Li, J.; Ma, Y.L. Synergistic Effect of rhamnolipids and inoculation on the bioremediation of petroleum-contaminated soils by bacterial consortia. Curr. Microbiol. 2020, 77, 997–1005. [Google Scholar] [CrossRef]
- Zhang, S.; Xia, T.; Wang, J.; Zhao, Y.; Xie, X.; Wei, Z.; Zhang, X.; Song, C.; Song, X. Role of Bacillus Inoculation in Rice Straw Composting and Bacterial Community Stability after Inoculation: Unite Resistance or Individual Collapse. Bioresour. Technol. 2021, 337, 125464. [Google Scholar] [CrossRef]
- Wu, C.; Wu, H.; Liu, D.; Han, G.; Zhao, P.; Kang, Y. Crab Bioturbation Significantly Alters Sediment Microbial Composition and Function in an Intertidal Marsh. Estuar. Coast. Shelf Sci. 2021, 249, 107116. [Google Scholar] [CrossRef]
- Zhou, W.; Han, G.; Liu, M.; Zeng, J.; Liang, B.; Liu, J.; Qu, R. Determining the Distribution and Interaction of Soil Organic Carbon, Nitrogen, pH and Texture in Soil Profiles: A Case Study in the Lancangjiang River Basin, Southwest China. Forests 2020, 11, 532. [Google Scholar] [CrossRef]
- Deng, L.; Shangguan, Z. Afforestation Drives Soil Carbon and Nitrogen Changes in China. Land Degrad. Dev. 2017, 28, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Pietri, J.A.; Brookes, P.C. Relationships between Soil pH and Microbial Properties in a UK Arable Soil. Soil Biol. Biochem. 2008, 40, 1856–1861. [Google Scholar] [CrossRef]
- Kemmitt, S.J.; Wright, D.; Goulding, K.W.; Jones, D.L. pH Regulation of Carbon and Nitrogen Dynamics in two Agricultural Soils. Soil Biol. Biochem. 2006, 38, 898–911. [Google Scholar] [CrossRef]
- Zeng, Q.; An, S.; Liu, Y.; Wang, H.; Wang, Y. Biogeography and the Driving Factors Affecting Forest Soil Bacteria in an Arid Area. Sci. Total Environ. 2019, 680, 124–131. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Trivedi, P.; Osanai, Y.; Liu, Y.R.; Hamonts, K.; Jeffries, T.C.; Singh, B.K. Carbon Content and Climate Variability Drive Global Soil Bacterial Diversity Patterns. Ecol. Monogr. 2016, 86, 373–390. [Google Scholar] [CrossRef]
- Blaud, A.; Phoenix, G.K.; Osborn, A.M. Variation in Bacterial, Archaeal and Fungal Community Structure and Abundance in High Arctic Tundra Soil. Polar Biol. 2015, 38, 1009–1024. [Google Scholar] [CrossRef]
- Mundra, S.; Kjønaas, O.J.; Morgado, L.N.; Krabberød, A.K.; Ransedokken, Y.; Kauserud, H. Soil Depth Matters: Shift in Composition and Inter-Kingdom Co-occurrence Patterns of Microorganisms in Forest Soils. FEMS Microbiol. Ecol. 2021, 97, fiab022. [Google Scholar] [CrossRef]
- Santolini, M.; Barabási, A.L. Predicting Perturbation Patterns from the Topology of Biological Networks. Proc. Natl. Acad. Sci. USA 2018, 115, E6375–E6383. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone Taxa as Drivers of Microbiome Structure and Functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Wu, X.; Yang, J.; Ruan, H.; Wang, S.; Yang, Y.; Naeem, I.; Wang, L.; Liu, L.; Wang, D. The Diversity and Co-occurrence Network of Soil Bacterial and Fungal Communities and Their Implications for a New Indicator of Grassland Degradation. Ecol. Indic. 2021, 129, 107989. [Google Scholar] [CrossRef]
- Wei, H.; Peng, C.; Yang, B.; Song, H.; Li, Q.; Jiang, L.; Wei, G.; Wang, K.; Wang, H.; Liu, S.; et al. Contrasting Soil Bacterial Community, Diversity, and Function in Two Forests in China. Front. Microbiol. 2018, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Fierer, N.; Lauber, C.L.; Caporaso, J.G.; Knight, R.; Grogan, P. Soil Bacterial Diversity in the Arctic is not Fundamentally Different from that Found in Other Biomes. Environ. Microbiol. 2010, 12, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based Assessment of Soil pH as A Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The Influence of Soil Properties on the Structure of Bacterial and Fungal Communities across Land-use Types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Azman, A.S.; Othman, I.; Fang, C.M.; Chan, K.G.; Goh, B.H.; Lee, L.H. Antibacterial, Anticancer and Neuroprotective Activities of Rare Actinobacteria from Mangrove Forest Soils. Indian J. Microbiol. 2017, 57, 177–187. [Google Scholar] [CrossRef]
- Xiao, X.; Liang, Y.; Zhou, S.; Zhuang, S.; Sun, B. Fungal Community Reveals Less Dispersal Limitation and Potentially More Connected Network than that of Bacteria in Bamboo Forest Soils. Mol. Ecol. 2018, 27, 550–563. [Google Scholar] [CrossRef]
- Lu, X.; Seuradge, B.J.; Neufeld, J.D. Biogeography of Soil Thaumarchaeota in Relation to Soil Depth and Land Usage. FEMS Microbiol. Ecol. 2017, 93, fiw246. [Google Scholar] [CrossRef]
- Biggs-Weber, E.; Aigle, A.; Prosser, J.I.; Gubry-Rangin, C. Oxygen Preference of Deeply-rooted Mesophilic Thaumarchaeota in Forest Soil. Soil Biol. Biochem. 2020, 148, 107848. [Google Scholar] [CrossRef]
- Feng, H.; Guo, J.; Wang, W.; Song, X.; Yu, S. Soil Depth Determines the Composition and Diversity of Bacterial and Archaeal Communities in A Poplar Plantation. Forests 2019, 10, 550. [Google Scholar] [CrossRef] [Green Version]
- Bastida, F.; Hernández, T.; Albaladejo, J.; García, C. Phylogenetic and Functional Changes in the Microbial Community of Long-term Restored Soils under Semiarid Climate. Soil Biol. Biochem. 2013, 65, 12–21. [Google Scholar] [CrossRef]
- Xu, C.; Du, C.; Jian, J.; Hou, L.; Wang, Z.K.; Wang, Q.; Geng, Z.C. The Interplay of Labile Organic Carbon, Enzyme Activities and Microbial Communities of Two Forest Soils across Seasons. Sci. Rep. 2021, 11, 5002. [Google Scholar] [CrossRef]
- Jiang, P.K.; Xu, Q.F.; Xu, Z.H.; Cao, Z.H. Seasonal Changes in Soil Labile Organic Carbon Pools within A Phyllostachys Praecox Stand under High Rate Fertilization and Winter Mulch in Subtropical China. For. Ecol. Manag. 2006, 236, 30–36. [Google Scholar] [CrossRef]
- Xue, L.; Ren, H.; Li, S.; Leng, X.; Yao, X. Soil Bacterial Community Structure and Co-occurrence Pattern during Vegetation Restoration in Karst Rocky Desertification Area. Front. Microbiol. 2017, 8, 2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, C.A.; Thomas, N.; Robinson, L.; Tate, K.R.; Ross, D.J.; Dando, J.; Singh, B.K. Physiological, Biochemical and Molecular Responses of the Soil Microbial Community after Afforestation of Pastures with Pinus radiata. Soil Biol. Biochem. 2009, 41, 1642–1651. [Google Scholar] [CrossRef]
- Ma, B.; Lv, X.; Cai, Y.; Chang, S.X.; Dyck, M.F. Liming Does not Counteract the Influence of Long–term Fertilization on Soil Bacterial Community Structure and its Co–occurrence Pattern. Soil Biol. Biochem. 2018, 123, 45–53. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Group | Primers | Sequence | PCR Conditions |
|---|---|---|---|
| Bacterial 16S rRNA | 338F | 5′-ACTCCTACGGGAGGCAGCAG-3′ | [33] |
| 806R | 5′-GGACTACHVGGGTWTCTAAT-3′ | ||
| Archaeal 16S rRNA | 524F10extF | 5′-TGYCAGCCGCCGCGGTAA-3′ | [8] |
| Arch958RmodR | 5′-YCCGGCGTTGAVTCCAATT-3′ | ||
| Fungal ITS regions | ITS1F | 5′-CTTGGTCATTTAGAGGAAGTAA-3′ | [34] |
| ITS2R | 5′-GCTGCGTTCTTCATCGATGC-3′ |
| Typical Forest Soils | pH | Available P (mg kg−1) | Available K (mg kg−1) | NH4+-N (mg kg−1) | NO3−-N (mg kg−1) | SOM (g kg−1) | Total N (g kg−1) | C/N |
|---|---|---|---|---|---|---|---|---|
| WBS | 4.92 ± 0.06 a | 33.2 ± 5.82 a | 97.5 ± 13.1 a | 28.25 ± 3.89 a | 8.63 ± 1.85 a | 78.35 ± 6.54 b | 4.81 ± 0.15 c | 9.43 ± 0.49 a |
| WPS | 5.03 ± 0.08 ab | 29.5 ± 5.84 a | 93.5 ± 13.1 a | 25.18 ± 3.51 a | 9.35 ± 1.01 a | 70.29 ± 3.51 ab | 4.20 ± 0.55 b | 9.71 ± 0.14 a |
| MOS | 5.12 ± 0.06 b | 30.6 ± 5.97 a | 98.1 ± 11.6 a | 20.36 ± 3.73 a | 6.76 ± 1.42 a | 62.25 ± 4.68 a | 3.58 ± 0.67 a | 10.12 ± 0.46 a |
| Microbe | Typical Forest Soils | Shannon Index | Simpson Index |
|---|---|---|---|
| Bacteria | WBS | 5.41 ± 0.26 a | 0.0105 ± 0.0008 b |
| WPS | 6.00 ± 0.57 a | 0.0056 ± 0.0007 a | |
| MOS | 5.87 ± 0.41 a | 0.0071 ± 0.0006 a | |
| Archaea | WBS | 2.47 ± 0.13 a | 0.1865 ± 0.0109 b |
| WPS | 3.08 ± 0.23 b | 0.1725 ± 0.0086 b | |
| MOS | 3.39 ± 0.22 b | 0.0879 ± 0.0087 a | |
| Fungi | WBS | 2.96 ± 0.16 b | 0.1254 ± 0.0075 a |
| WPS | 2.15 ± 0.12 a | 0.2872 ± 0.0130 b | |
| MOS | 2.34 ± 0.18 a | 0.2304 ± 0.0101 c |
| Soil Characteristics | Bacterial Abundance | Archaeal Abundance | Fungal Abundance | pH | Available P | Available K | NH4+-N | NO3−-N | SOM | Total N | C/N |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacterial abundance | 1.00 | ||||||||||
| Archaeal abundance | 0.927 ** | 1.00 | |||||||||
| Fungal abundance | 0.918 ** | 0.952 ** | 1.00 | ||||||||
| pH | 0.801 ** | 0.795 * | 0.799 ** | 1.00 | |||||||
| Available P | −0.176 | −0.358 | −0.326 | −0.140 | 1.00 | ||||||
| Available K | −0.236 | −0.316 | −0.084 | −0.140 | 0.917 | 1.00 | |||||
| NH4+-N | −0.363 | −0.244 | −0.161 | −0.301 | 0.235 | 0.235 | 1.00 | ||||
| NO3−-N | −0.461 | −0.373 | −0.359 | −0.625 | 0.362 | 0.362 | 0.523 | 1.00 | |||
| SOM | −0.772 * | −0.750 * | −0.783 * | −0.870 ** | 0.155 | 0.155 | 0.454 | 0.658 | 1.00 | ||
| Total N | −0.755 * | −0.705 * | −0.715 * | −0.899 ** | 0.047 | 0.047 | 0.585 | 0.612 | 0.928 ** | 1.00 | |
| C/N | 0.227 | 0.109 | 0.089 | 0.397 | 0.266 | 0.266 | −0.493 | −0.148 | −0.174 | −0.525 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Hou, X.; Islam, Z.U.; Zhang, Z.; Zhu, B.; Yang, T. Driving Factors of Microbial Community Abundance and Structure in Typical Forest Soils of Sanjiang Plain, Northeast China. Sustainability 2022, 14, 8040. https://doi.org/10.3390/su14138040
Wang C, Hou X, Islam ZU, Zhang Z, Zhu B, Yang T. Driving Factors of Microbial Community Abundance and Structure in Typical Forest Soils of Sanjiang Plain, Northeast China. Sustainability. 2022; 14(13):8040. https://doi.org/10.3390/su14138040
Chicago/Turabian StyleWang, Chunyong, Xintong Hou, Zia UI Islam, Zhenbin Zhang, Bo Zhu, and Tianhao Yang. 2022. "Driving Factors of Microbial Community Abundance and Structure in Typical Forest Soils of Sanjiang Plain, Northeast China" Sustainability 14, no. 13: 8040. https://doi.org/10.3390/su14138040