VKORC1L1, An Enzyme Mediating the Effect of Vitamin K in Liver and Extrahepatic Tissues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Vitamin K, Gamma-Carboxylation and Vitamin K Oxidoreductase
2. VKOR Homologues Are Present in Metazoans, Protists, Bacteria and Plants
3. VKORC1L1 Is A Vertebrate Paralog of VKORC1
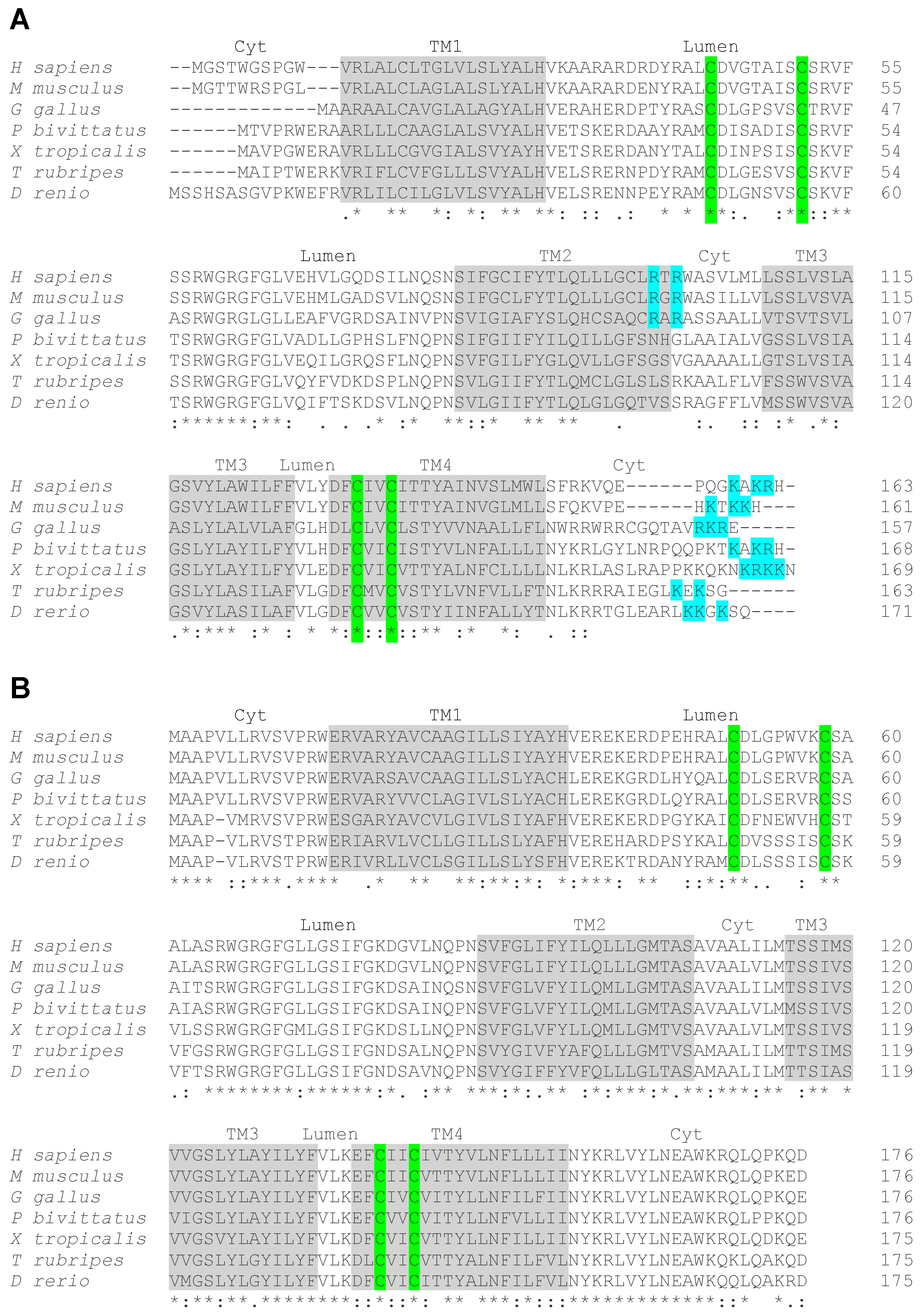
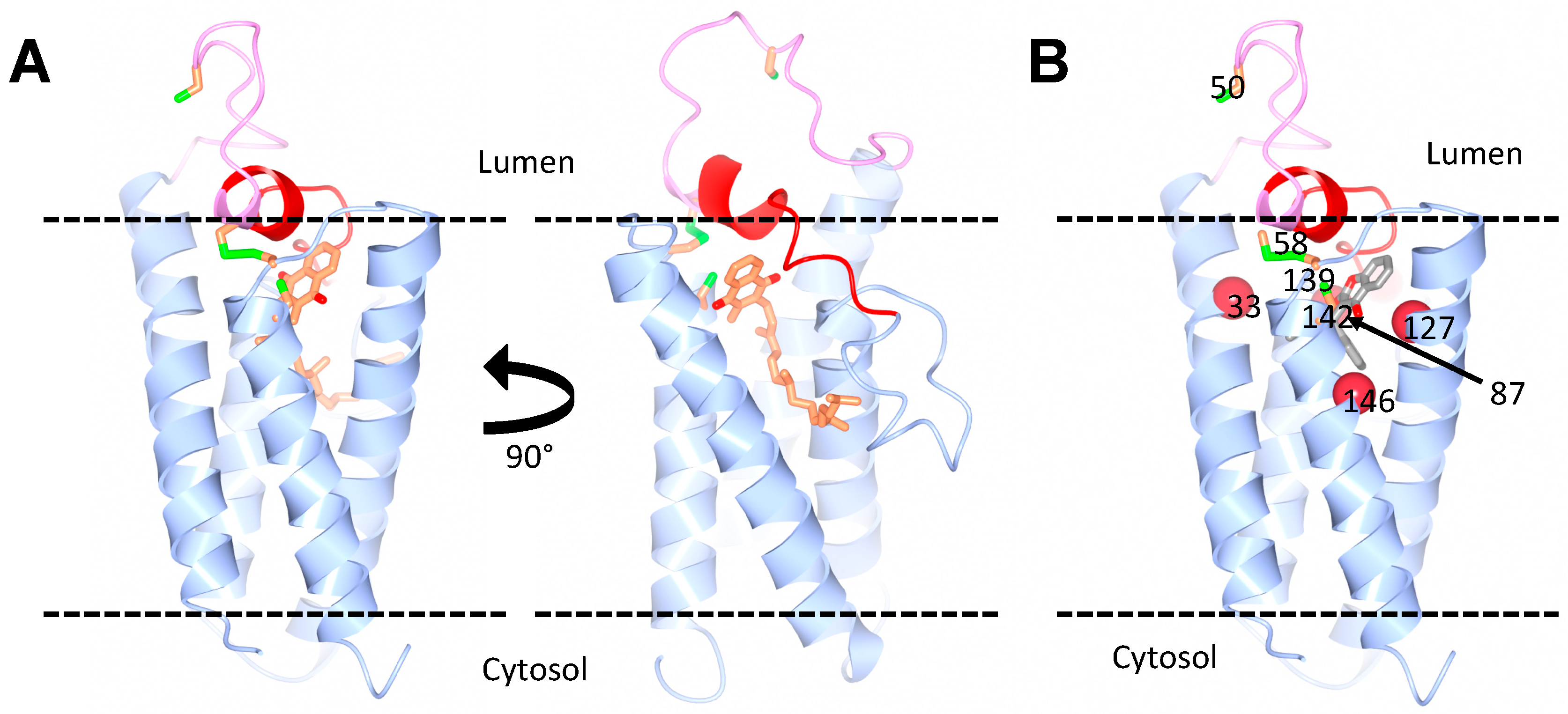
4. Structure of VKORC1 and VKORC1L1
5. Subcellular Localization of VKORC1 and VKORC1L1
6. Evidence That VKORC1L1 Supports Gamma-Carboxylation in Cell Culture Models
7. Expression Pattern of Vkorc1 and Vkorc1l1 in Adult Tissues
8. Evidence That VKORC1L1 Supports Gamma-Carboxylation In Vivo
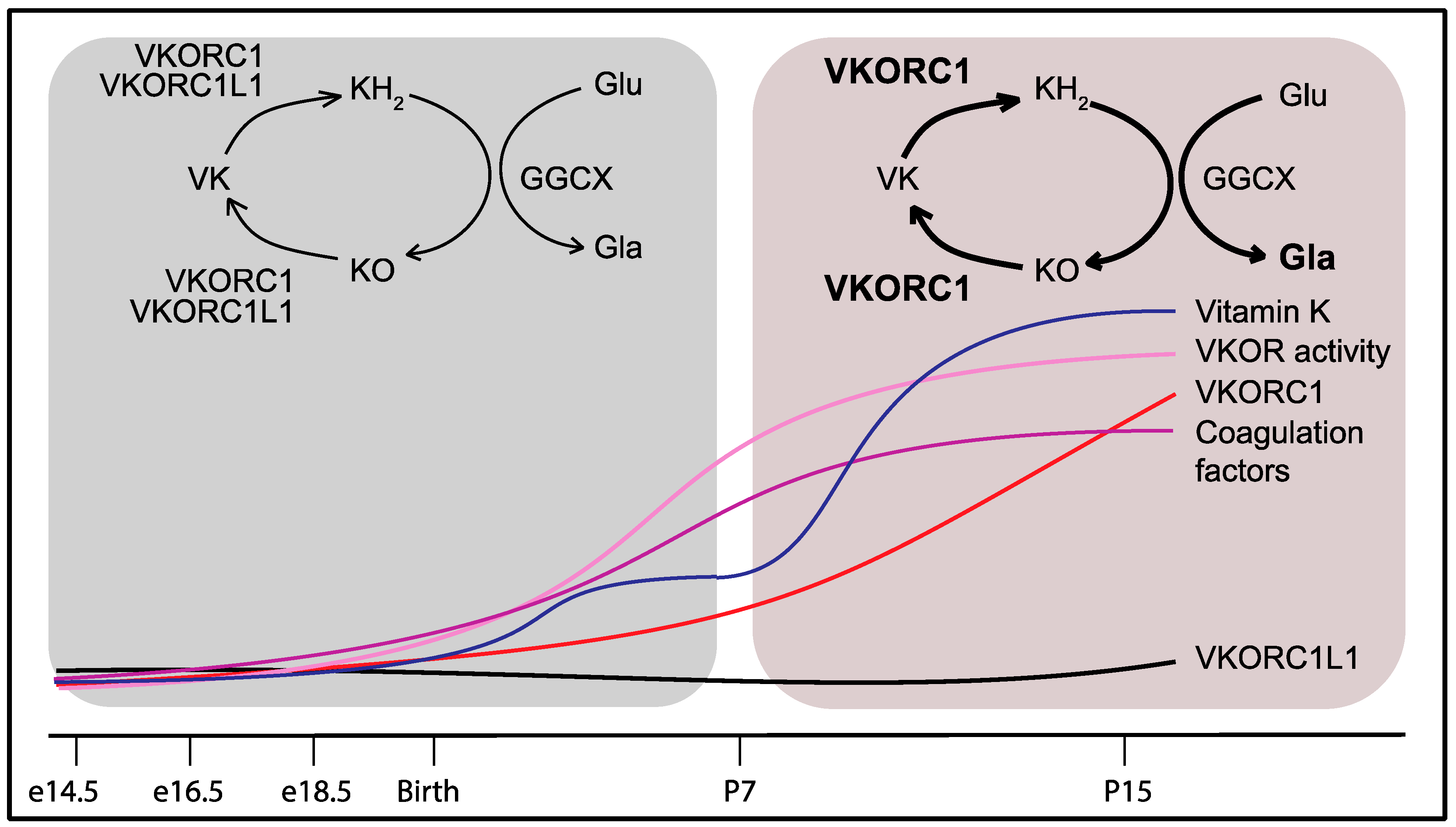
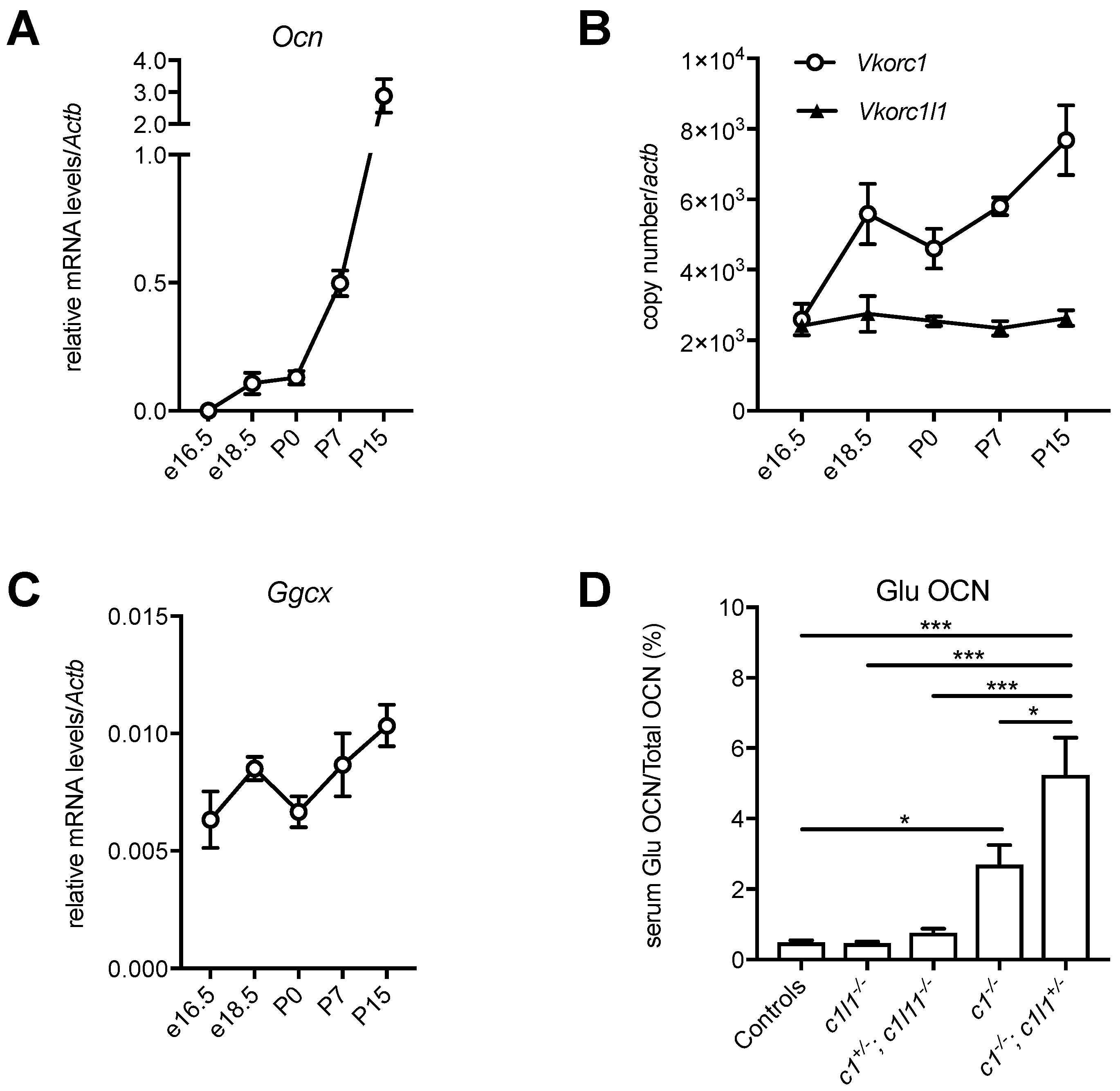
9. Developmental Regulation of the Vitamin K Cycle in the Liver
10. Partial Redundancy between VKORC1 and VKORC1L1 in Osteoblasts during the Perinatal Period
11. VKORC1L1 Is Less Sensitive to Warfarin Than VKORC1 In Vitro
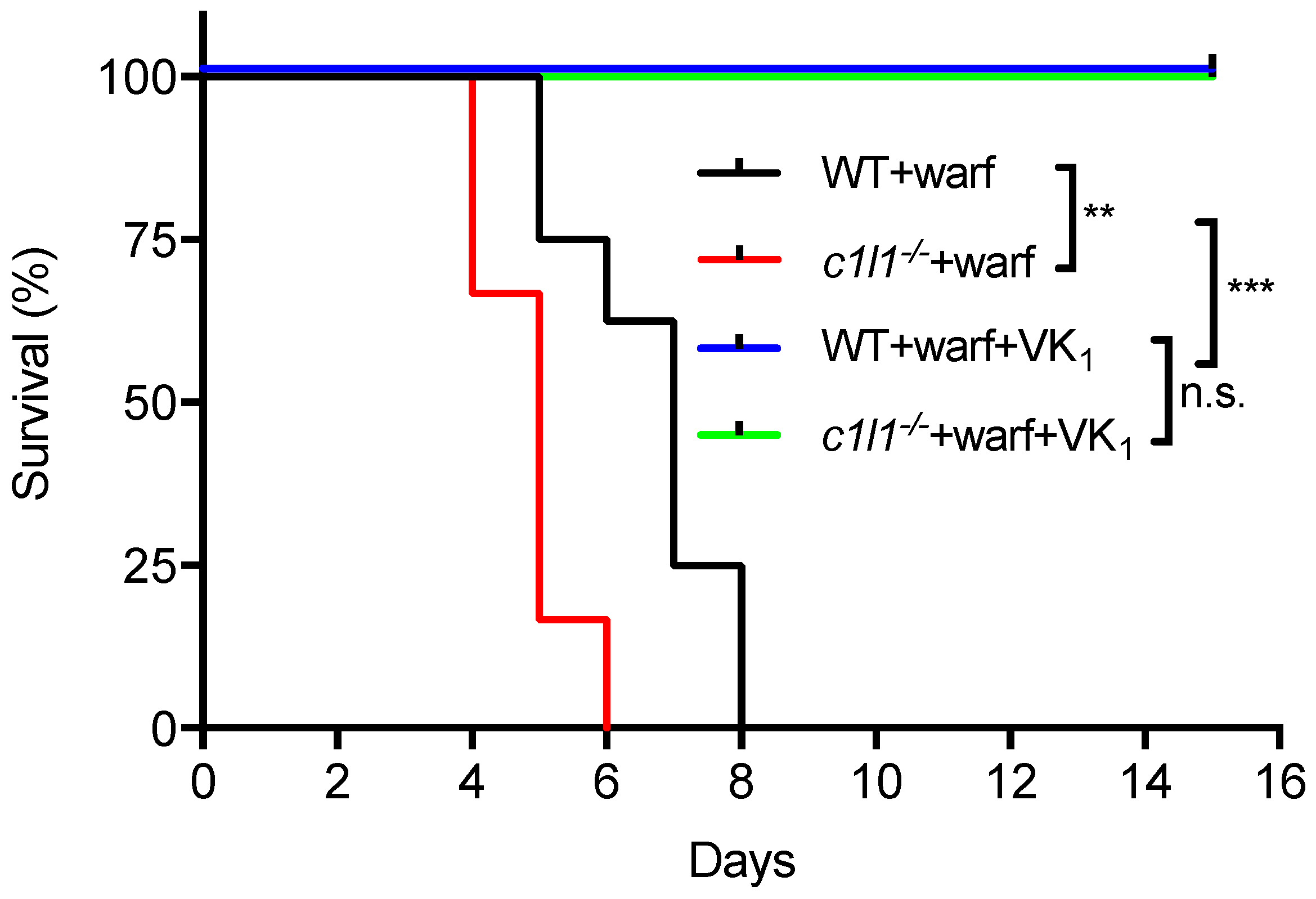
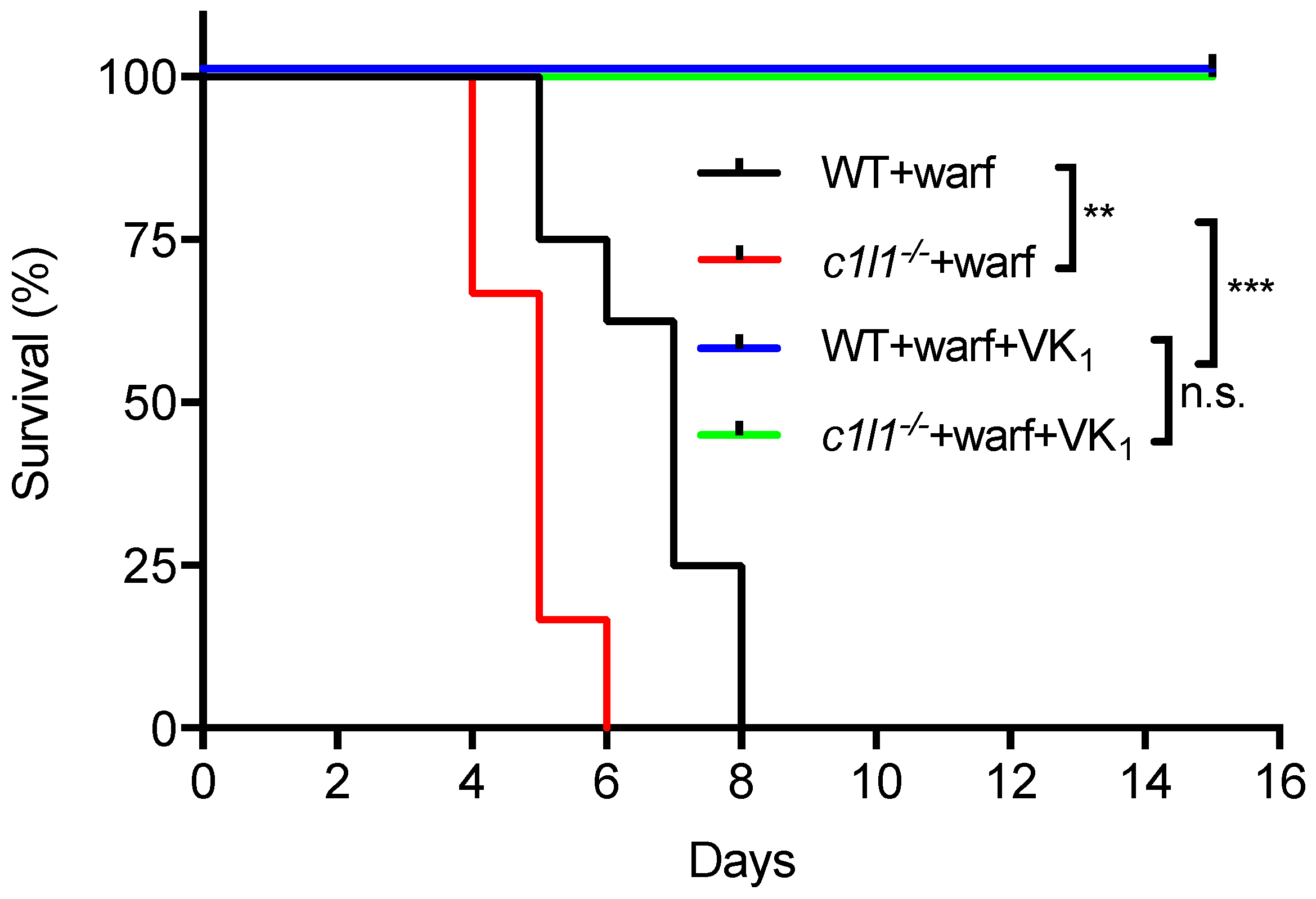
12. VKORC1L1 Is Not the Liver Warfarin Resistant Vitamin K Quinone Reductase
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Furie, B.; Bouchard, B.A.; Furie, B.C. Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood 1999, 93, 1798–1808. [Google Scholar] [PubMed]
- Berkner, K.L. Vitamin K-dependent carboxylation. Vitam. Horm. 2008, 78, 131–156. [Google Scholar] [PubMed]
- Booth, S.L.; Broe, K.E.; Gagnon, D.R.; Tucker, K.L.; Hannan, M.T.; McLean, R.R.; Dawson-Hughes, B.; Wilson, P.W.; Cupples, L.A.; Kiel, D.P. Vitamin K intake and bone mineral density in women and men. Am. J. Clin. Nutr. 2003, 77, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.L.; Vangaveti, V.; Malabu, U.H.; McCulloch, D. The vitamin K-dependent Gla proteins and risk of type 2 diabetes. Diabetologia 2013, 56, 2100–2101. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.S.; Gajic-Veljanoski, O.; Cheung, A.M. Vitamin K and bone health. J. Clin. Densitom. 2013, 16, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Von Kries, R.; Hanawa, Y. Neonatal vitamin K prophylaxis. Report of Scientific and Standardization Subcommittee on Perinatal Haemostasis. Thromb. Haemost. 1993, 69, 293–295. [Google Scholar] [PubMed]
- Stenflo, J.; Fernlund, P.; Egan, W.; Roepstorff, P. Vitamin K dependent modifications of glutamic acid residues in prothrombin. Proc. Natl. Acad. Sci. USA 1974, 71, 2730–2733. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y. Endocrine regulation of energy metabolism by the skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Berkner, K.L.; Harbeck, M.; Lingenfelter, S.; Bailey, C.; Sanders-Hinck, C.M.; Suttie, J.W. Purification and identification of bovine liver gamma-carboxylase. Proc. Natl. Acad. Sci. USA 1992, 89, 6242–6246. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Fregin, A.; Ivaskevicius, V.; Conzelmann, E.; Hortnagel, K.; Pelz, H.J.; Lappegard, K.; Seifried, E.; Scharrer, I.; Tuddenham, E.G.; et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004, 427, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chang, C.Y.; Jin, D.Y.; Lin, P.J.; Khvorova, A.; Stafford, D.W. Identification of the gene for vitamin K epoxide reductase. Nature 2004, 427, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.H.; Huang, T.Y.; Williams, J.; Stafford, D.W. Purified vitamin K epoxide reductase alone is sufficient for conversion of vitamin K epoxide to vitamin K and vitamin K to vitamin KH2. Proc. Natl. Acad. Sci. USA 2006, 103, 19308–19313. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Schulman, S.; Dutton, R.J.; Boyd, D.; Beckwith, J.; Rapoport, T.A. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 2010, 463, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cheng, W.; Fowle Grider, R.; Shen, G.; Li, W. Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer. Nat. Commun. 2014, 5, 3110. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Cui, W.; Zhang, H.; Zhou, F.; Huang, W.; Liu, Q.; Yang, Y.; Li, S.; Bowman, G.R.; Sadler, J.E.; et al. Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat. Struct. Mol. Biol. 2017, 24, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Begovic, E.; Chapman, J.; Putnam, N.H.; Hellsten, U.; Kawashima, T.; Kuo, A.; Mitros, T.; Salamov, A.; Carpenter, M.L.; et al. The Trichoplax genome and the nature of placozoans. Nature 2008, 454, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.M. Genes encoding vitamin-K epoxide reductase are present in Drosophila and trypanosomatid protists. Genetics 2004, 168, 1077–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutton, R.J.; Boyd, D.; Berkmen, M.; Beckwith, J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. USA 2008, 105, 11933–11938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutton, R.J.; Wayman, A.; Wei, J.R.; Rubin, E.J.; Beckwith, J.; Boyd, D. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc. Natl. Acad. Sci. USA 2010, 107, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Stafford, D.W. Mycobacterium tuberculosis vitamin K epoxide reductase homologue supports vitamin K-dependent carboxylation in mammalian cells. Antioxid. Redox. Signal. 2012, 16, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [PubMed]
- Oldenburg, J.; Watzka, M.; Bevans, C.G. VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases? Nutrients 2015, 7, 6250–6280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, S.; Fregin, A.; Hunerberg, M.; Bevans, C.G.; Muller, C.R.; Oldenburg, J. Site-directed mutagenesis of coumarin-type anticoagulant-sensitive VKORC1: Evidence that highly conserved amino acids define structural requirements for enzymatic activity and inhibition by warfarin. Thromb. Haemost. 2005, 94, 780–786. [Google Scholar] [PubMed]
- Jin, D.Y.; Tie, J.K.; Stafford, D.W. The conversion of vitamin K epoxide to vitamin K quinone and vitamin K quinone to vitamin K hydroquinone uses the same active site cysteines. Biochemistry 2007, 46, 7279–7283. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.K.; Nicchitta, C.; Von Heijne, G.; Stafford, D.W. Membrane topology mapping of vitamin K epoxide reductase by in vitro translation/cotranslocation. J. Biol. Chem. 2005, 280, 16410–16416. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Stafford, D.W. Human vitamin K epoxide reductase and its bacterial homologue have different membrane topologies and reaction mechanisms. J. Biol. Chem. 2012, 287, 33945–33955. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Stafford, D.W. Conserved loop cysteines of vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) are involved in its active site regeneration. J. Biol. Chem. 2014, 289, 9396–9407. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cousins, E.; Sandford, G.; Nicholas, J. Human herpesvirus 8 viral interleukin-6 interacts with splice variant 2 of vitamin K epoxide reductase complex subunit 1. J. Virol. 2012, 86, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Chen, X.; Jin, D.Y.; Stafford, D.W.; Pedersen, L.G.; Tie, J.K. Warfarin and vitamin K epoxide reductase: A molecular accounting for observed inhibition. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Sinhadri, B.C.S.; Jin, D.Y.; Stafford, D.W.; Tie, J.K. Vitamin K epoxide reductase and its paralogous enzyme have different structures and functions. Sci. Rep. 2017, 7, 17632. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Van Lith, M.; Mitchell, L.J.; Pringle, M.A.; Inaba, K.; Bulleid, N.J. The membrane topology of vitamin K epoxide reductase is conserved between human isoforms and the bacterial enzyme. Biochem. J. 2016, 473, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, G.; Li, W. Intramembrane Thiol Oxidoreductases: Evolutionary Convergence and Structural Controversy. Biochemistry 2018, 57, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Lacombe, J.; Germain, A.; Oury, F.; Karsenty, G. GGCX and VKORC1 inhibit osteocalcin endocrine functions. J. Cell Biol. 2015, 208, 761–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czogalla, K.J.; Biswas, A.; Rost, S.; Watzka, M.; Oldenburg, J. The Arg98Trp mutation in human VKORC1 causing VKCFD2 disrupts a di-Arginine-based ER retention motif. Blood 2014, 124, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Wallin, R.; Hutson, S. Vitamin K-dependent carboxylation. Evidence that at least two microsomal dehydrogenases reduce vitamin K1 to support carboxylation. J. Biol. Chem. 1982, 257, 1583–1586. [Google Scholar] [PubMed]
- Westhofen, P.; Watzka, M.; Marinova, M.; Hass, M.; Kirfel, G.; Muller, J.; Bevans, C.G.; Muller, C.R.; Oldenburg, J. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J. Biol. Chem. 2011, 286, 15085–15094. [Google Scholar] [CrossRef] [PubMed]
- Hammed, A.; Matagrin, B.; Spohn, G.; Prouillac, C.; Benoit, E.; Lattard, V. VKORC1L1, an enzyme rescuing the VKOR activity in some extrahepatic tissues during anticoagulation therapy. J. Biol. Chem. 2013, 288, 28733–28742. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Tie, K.; Stafford, D.W. Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells. J. Thromb. Haemost. 2013, 11, 1556–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czogalla, K.J.; Liphardt, K.; Honing, K.; Hornung, V.; Biswas, A.; Watzka, M.; Oldenburg, J. VKORC1 and VKORC1L1 have distinctly different oral anticoagulant dose-response characteristics and binding sites. Blood Adv. 2018, 2, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Li, S.; Cui, W.; Liu, S.; Liu, Q.; Yang, Y.; Gross, M.; Li, W. Stabilization of warfarin-binding pocket of VKORC1 and VKORL1 by a peripheral region determines their different sensitivity to warfarin inhibition. J. Thromb. Haemost. 2018, 16, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Caspers, M.; Czogalla, K.J.; Liphardt, K.; Muller, J.; Westhofen, P.; Watzka, M.; Oldenburg, J. Two enzymes catalyze vitamin K 2,3-epoxide reductase activity in mouse: VKORC1 is highly expressed in exocrine tissues while VKORC1L1 is highly expressed in brain. Thromb. Res. 2015, 135, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Rishavy, M.A.; Berkner, K.L.; Ferron, M. VKOR paralog VKORC1L1 supports vitamin K-dependent protein carboxylation in vivo. JCI Insight 2018, 3, e96501. [Google Scholar] [CrossRef] [PubMed]
- Fregin, A.; Rost, S.; Wolz, W.; Krebsova, A.; Muller, C.R.; Oldenburg, J. Homozygosity mapping of a second gene locus for hereditary combined deficiency of vitamin K-dependent clotting factors to the centromeric region of chromosome 16. Blood 2002, 100, 3229–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.; Sanchez-Vega, B.; Wu, S.M.; Lanir, N.; Stafford, D.W.; Solera, J. A missense mutation in gamma-glutamyl carboxylase gene causes combined deficiency of all vitamin K-dependent blood coagulation factors. Blood 1998, 92, 4554–4559. [Google Scholar] [PubMed]
- Spohn, G.; Kleinridders, A.; Wunderlich, F.T.; Watzka, M.; Zaucke, F.; Blumbach, K.; Seifried, C.E.; Muller, C.; Paulsson, M.; Bruning, J.C.; et al. VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb. Haemost. 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Sun, H.; Raymond, R.M., Jr.; Furie, B.C.; Furie, B.; Bronstein, M.; Kaufman, R.J.; Westrick, R.; Ginsburg, D. Fatal hemorrhage in mice lacking gamma-glutamyl carboxylase. Blood 2007, 109, 5270–5275. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wu, Q.; Westfield, L.A.; Tuley, E.A.; Lu, D.; Zhang, Q.; Shim, K.; Zheng, X.; Sadler, J.E. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7603–7607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forestier, F.; Daffos, F.; Rainaut, M.; Sole, Y.; Amiral, J. Vitamin K dependent proteins in fetal hemostasis at mid trimester of pregnancy. Thromb. Haemost. 1985, 53, 401–403. [Google Scholar] [PubMed]
- Reverdiau-Moalic, P.; Delahousse, B.; Body, G.; Bardos, P.; Leroy, J.; Gruel, Y. Evolution of blood coagulation activators and inhibitors in the healthy human fetus. Blood 1996, 88, 900–906. [Google Scholar] [PubMed]
- Andrew, M.; Paes, B.; Milner, R.; Johnston, M.; Mitchell, L.; Tollefsen, D.M.; Powers, P. Development of the human coagulation system in the full-term infant. Blood 1987, 70, 165–172. [Google Scholar] [PubMed]
- Dewerchin, M.; Liang, Z.; Moons, L.; Carmeliet, P.; Castellino, F.J.; Collen, D.; Rosen, E.D. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb. Haemost. 2000, 83, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Rahim, S.; Barkhan, P.; Stimmler, L. Plasma vitamin K1 in mothers and their newborn babies. Lancet 1982, 2, 460–463. [Google Scholar] [CrossRef]
- Al Rifai, O.; Chow, J.; Lacombe, J.; Julien, C.; Faubert, D.; Susan-Resiga, D.; Essalmani, R.; Creemers, J.W.; Seidah, N.G.; Ferron, M. Proprotein convertase furin regulates osteocalcin and bone endocrine function. J. Clin. Investig. 2017, 127, 4104–4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Hanna, T.; Suda, N.; Karsenty, G.; Ducy, P. Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a. Diabetes 2014, 63, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Karsenty, G.; Ferron, M. In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice. Mol. Metab. 2013, 2, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Ducy, P.; Karsenty, G. An ELISA-based method to quantify osteocalcin carboxylation in mice. Biochem. Biophys. Res. Commun. 2010, 397, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ufer, M. Comparative pharmacokinetics of vitamin K antagonists: Warfarin, phenprocoumon and acenocoumarol. Clin. Pharmacokinet. 2005, 44, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Akpa, B.S.; Ayee, M.A.; Boullerne, A.I.; Braun, D.; Brodsky, S.V.; Gidalevitz, D.; Hauck, Z.; Kalinin, S.; Kowal, K.; et al. The emerging threat of superwarfarins: History, detection, mechanisms, and countermeasures. Ann. N. Y. Acad. Sci. 2016, 1374, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Rishavy, M.A.; Hallgren, K.W.; Wilson, L.; Singh, S.; Runge, K.W.; Berkner, K.L. Warfarin alters vitamin K metabolism: A surprising mechanism of VKORC1 uncoupling necessitates an additional reductase. Blood 2018, 131, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Watzka, M.; Geisen, C.; Bevans, C.G.; Sittinger, K.; Spohn, G.; Rost, S.; Seifried, E.; Muller, C.R.; Oldenburg, J. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: Insights into improved patient diagnosis and treatment. J. Thromb. Haemost. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Hodroge, A.; Longin-Sauvageon, C.; Fourel, I.; Benoit, E.; Lattard, V. Biochemical characterization of spontaneous mutants of rat VKORC1 involved in the resistance to antivitamin K anticoagulants. Arch. Biochem. Biophys. 2011, 515, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Namba, S.; Yamaoka-Tojo, M.; Hashikata, T.; Ikeda, Y.; Kitasato, L.; Hashimoto, T.; Shimohama, T.; Tojo, T.; Takahira, N.; Masuda, T.; et al. Long-term warfarin therapy and biomarkers for osteoporosis and atherosclerosis. BBA. Clin. 2015, 4, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantisattamo, E.; Han, K.H.; O’Neill, W.C. Increased vascular calcification in patients receiving warfarin. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M.; Tripepi, G.; Noale, M.; Plebani, M.; Zaninotto, M.; Piccoli, A.; Naso, A.; Miozzo, D.; Giannini, S.; Avolio, M.; et al. Prevalence of vertebral fractures, vascular calcifications, and mortality in warfarin treated hemodialysis patients. Curr. Vasc. Pharmacol. 2015, 13, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Wallin, R. Vitamin K antagonism of coumarin anticoagulation. A dehydrogenase pathway in rat liver is responsible for the antagonistic effect. Biochem. J. 1986, 236, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, B.O.; Turbyfill, J.L.; Bledsoe, P.J.; Jaiswal, A.K.; Stafford, D.W. Assessment of the contribution of NAD(P)H-dependent quinone oxidoreductase 1 (NQO1) to the reduction of vitamin K in wild-type and NQO1-deficient mice. Biochem. J. 2013, 456, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Williamson, M.K.; Haba, T.; Dell, R.B.; Jee, W.S. Excessive mineralization with growth plate closure in rats on chronic warfarin treatment. Proc. Natl. Acad. Sci. USA 1982, 79, 7734–7738. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1400–1407. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacombe, J.; Ferron, M. VKORC1L1, An Enzyme Mediating the Effect of Vitamin K in Liver and Extrahepatic Tissues. Nutrients 2018, 10, 970. https://doi.org/10.3390/nu10080970
Lacombe J, Ferron M. VKORC1L1, An Enzyme Mediating the Effect of Vitamin K in Liver and Extrahepatic Tissues. Nutrients. 2018; 10(8):970. https://doi.org/10.3390/nu10080970
Chicago/Turabian StyleLacombe, Julie, and Mathieu Ferron. 2018. "VKORC1L1, An Enzyme Mediating the Effect of Vitamin K in Liver and Extrahepatic Tissues" Nutrients 10, no. 8: 970. https://doi.org/10.3390/nu10080970
APA StyleLacombe, J., & Ferron, M. (2018). VKORC1L1, An Enzyme Mediating the Effect of Vitamin K in Liver and Extrahepatic Tissues. Nutrients, 10(8), 970. https://doi.org/10.3390/nu10080970