Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes



Abstract
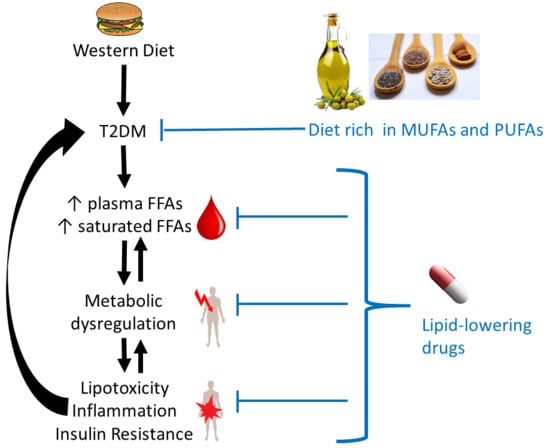
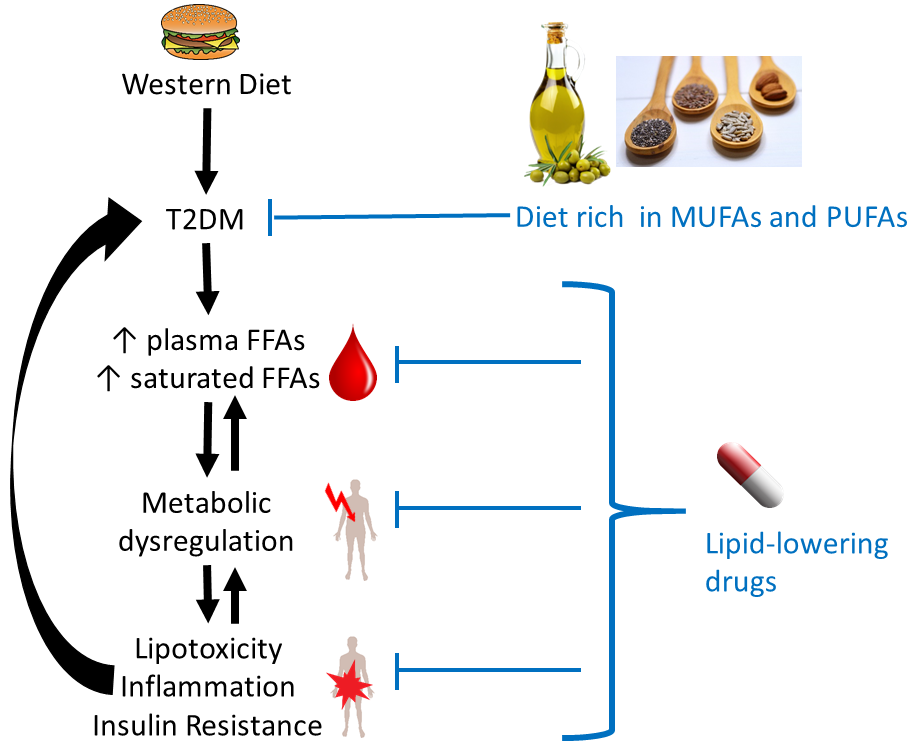
:
1. Introduction
2. FFA Metabolism in Healthy Tissues
2.1. Origins of Plasma FFAs
2.2. Regulation of FFA Metabolism: FFA Receptors
3. Associations between the Intake of Fatty Acids and Occurrence of T2DM
4. Differences in Plasma FFAs in T2DM

{kind=link}
{kind=link}
{kind=link}
| References | Numbers of Subjects | Population | Type of Results | |
|---|---|---|---|---|
| Ctrl. | T2DM | |||
| Clore et al., 2002 [97] | 6 | 6 | BMI- and age-matched; not sex-matched; American cohort | Plasma FFA conc. after a 14 h fast. |
| Yi et al., 2007 [98] | 45 | 78 | Age-, sex- or BMI-matched; Chinese cohort | Plasma FFA conc., fasting not indicated. |
| Liu et al., 2010 [99] | 50 | 53 | Age-and sex-matched; not BMI-matched; Chinese cohort | Plasma FFA conc. after fasting. |
| Grapov et al., 2012 [100] | 12 | 43 | BMI-matched; not age-matched; obese African-American women | Geometric mean of plasma FFA after an overnight fast |
| Lu et al., 2016 [101] | 197 | 197 | Age-and sex-matched; not BMI-matched; Chinese cohort | Trend of plasma FFA, no fasting. |
| Ma et al., 2018-a [102] | 40 | 21 | Age-and BMI-matched; not sex-matched; Kazakh cohort | % total plasma FFA after an 8 h fast. |
| Ma et al., 2018-b [102] | 35 | 39 | Not age-, BMI- or sex-matched; Uyghur cohort | % total plasma FFA after an 8 h fast. |
| FFA Species | Alterations in Plasma FFA Conc. in T2DM | ||||||
|---|---|---|---|---|---|---|---|
| Clore et al., 2002 [97] | Yi et al., 2007 [98] | Liu et al., 2010 [99] | Grapov et al., 2012 [100] | Lu et al., 2016 [101] | Ma et al., 2018-a [102] | Ma et al., 2018-b [102] | |
| Total FFA | ↑ | ↑ | |||||
| Saturated FFA | ↑ | ↑ | |||||
| C6:0, caproic acid | = | ↑ | |||||
| C8:0, caprylic acid | = | = | |||||
| C10:0, capric acid | = | = | |||||
| C12:0, lauric acid | = | = | = | ||||
| C14:0, myristic acid | = | ↑ | ↑ | ↑ | = | = | |
| C15:0, pentadecanoic acid | ↑ | = | = | ||||
| C16:0, palmitic acid | ↑ | ↑ | ↑ | ↑ | ↑ | = | = |
| C18:0, stearic acid | ↑ | ↑ | ↑ | ↑ | ↑ | = | = |
| C19:0, nonadecylic acid | ↑ | ||||||
| C20:0, arachidic acid | ↑ | ↑ | = | = | |||
| C22:0, behenic acid | = | ↓ | |||||
| C24:0, lignoceric acid | = | ↑ | = | ||||
| Unsaturated FFA | ↑ | ||||||
| Monounsaturated FFA | ↑ | ||||||
| C14:1n-9, myristoleic acid | ↓ | ↓ | |||||
| C16:1n-7, palmitoleic acid | = | ↑ | ↑ | ↑↑ | = | = | |
| C16:1n-9, cis-7 hexadecenoic acid | ↑ | ||||||
| C18:1n-9, oleic acid (OA) | ↑ | ↑ | ↑ | ↑ | ↑ | = | = |
| C18:1 trans-n-7, vaccenic acid | ↑ | ↑ | |||||
| C19:1n-9, cis-10 nonadecenoic acid | = | ||||||
| C20:1n-9, gondoic acid | ↑ | ||||||
| C24:1n-9, nervonic acid | ↑ | ||||||
| Total n-7 | ↑ | ||||||
| Total n-9 | ↑ | ||||||
| Polyunsaturated FFA | = | ↑ | |||||
| C18:2n-6, linoleic acid (LA) | = | ↑ | ↑ | ↑ | ↑ | = | = |
| C18:2 trans-n-7 cis-n-9, rumenic acid | ↑ | ||||||
| C18:3n-6, γ-linolenic acid (GLA) | ↑ | ↑ | = | ↓ | ↓ | ||
| C18:3n-3, α-linolenic acid (ALA) | = | = | ↑ | ↑ | = | = | |
| C20:2n-6, eicosadienoic acid | = | = | = | = | |||
| C20:3n-6, dihomo-γ-linolenic acid (DGLA) | ↑ | = | ↓ | ↓ | |||
| C20:4n-6, arachidonic acid (ARA) | = | ↑ | ↑ | = | = | = | |
| C20:5n-3, eicosapentaenoic acid (EPA) | = | ↑ | ↑ | = | = | ↑ | |
| C22:4n-6, adrenic acid | ↑ | ↑ | |||||
| C22:5n-3, docosapentaenoic acid | = | ↑ | ↑ | ||||
| C22:5n-6, osbondic acid | = | = | |||||
| C22:6n-3, docosahexaenoic acid (DHA) | = | ↑ | ↑ | = | = | = | |
| Trans FFA | ↑ | ||||||
| C16:1 trans-n-7, trans-palmitoleic acid | ↑ | ||||||
| C18:2 trans-n-6, linolelaidic acid | ↑ | ||||||
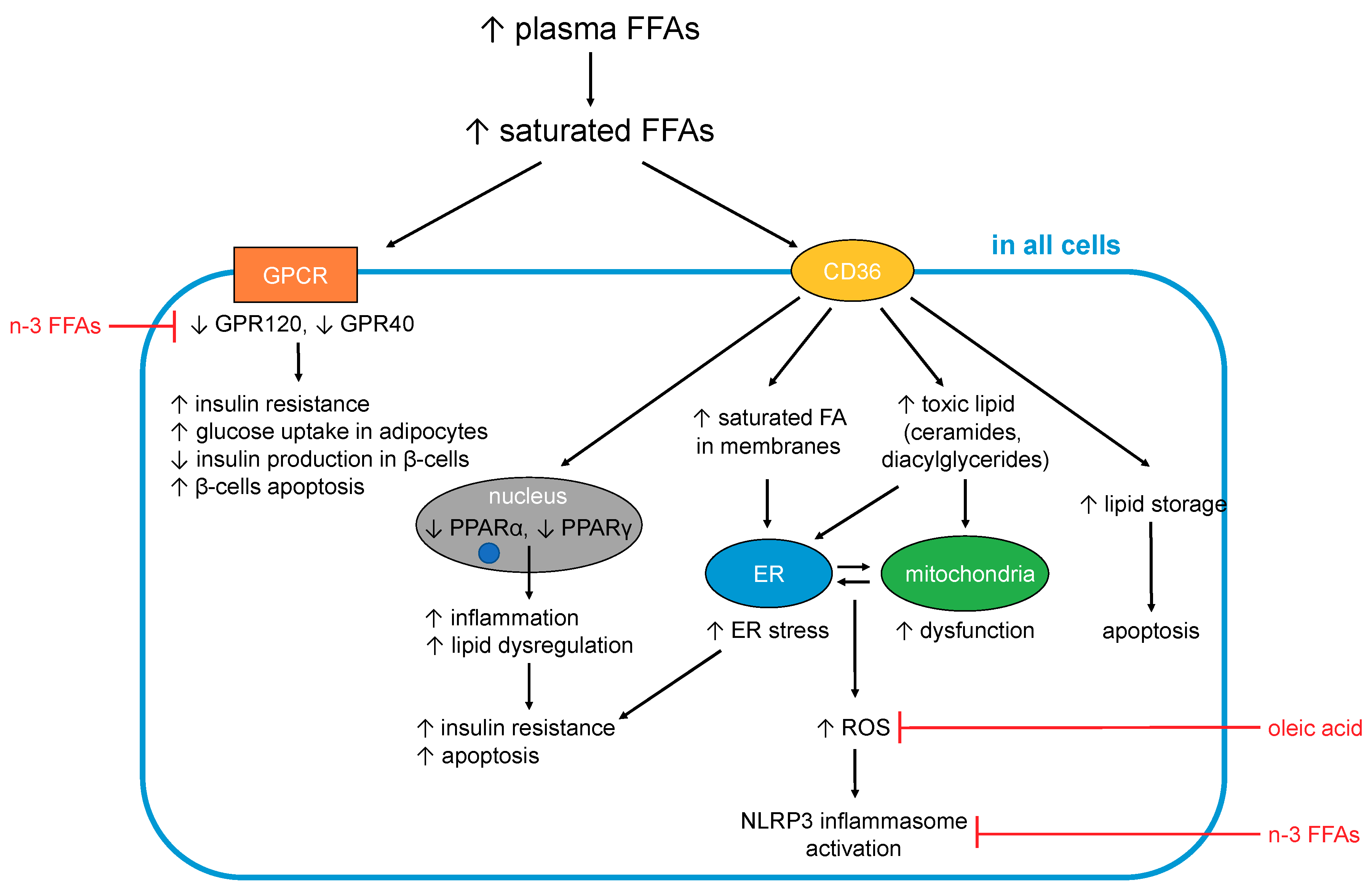
5. FFA Metabolism in T2DM
5.1. FFA-Induced Changes Occuring in All Cells and Tissues
5.1.1. Generation of Toxic Lipids and Lipotoxicity
5.1.2. FFAs and Inflammation
5.1.3. Effects on Cellular Membranes
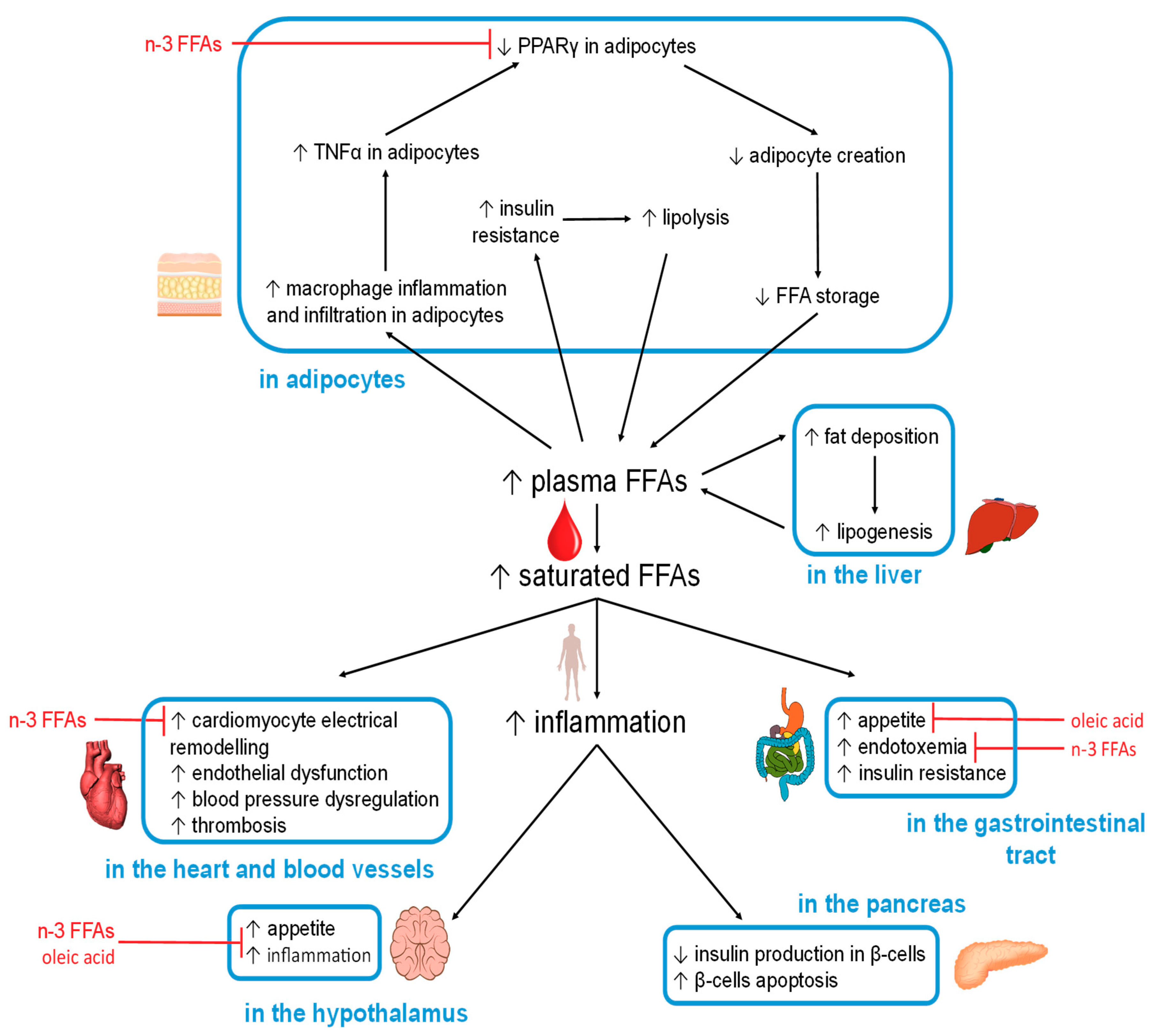
5.2. FFA-Induced Changes in Specific Organs and Tissues
5.2.1. Hypothalamus
5.2.2. Gastrointestinal Tract
5.2.3. Adipocytes
5.2.4. Liver
5.2.5. Pancreas
5.2.6. Heart
5.2.7. Blood Vessels
5.2.8. Skeletal Muscle
6. Effects of Interventions Using Lipid-Lowering Drugs
6.1. Changes in Total Plasma FFA Concentration
| Drug Type | Reference | Subjects | Drug Treatment | Effect |
|---|---|---|---|---|
| Metformin | Pentikäinen et al., 1990 [200] | 24 non-diabetic subjects with hyperlipidaemia | Randomised, double blind, placebo-controlled, crossover study. Metformin 1 g/day or 2 g/day or placebo for 9 weeks. | Unchanged fasting total FFA levels. |
| Landin et al., 1994 [201] | 18 healthy subjects | Randomised, double-blind, placebo-controlled, triple crossover study. Metformin 850 mg twice daily or metoprolol 100 mg/day for 18 weeks. | Reduced fasting total FFA levels. | |
| Lehtovirta et al., 2001 [202] | 40 first-degree relatives of T2DM patients with impaired glucose tolerance | Block-randomised, double-blind, placebo-controlled, parallel group study. Metformin 500 mg twice daily for 6 months. | Unchanged fasting total FFA levels. | |
| Fruehwald-Schultes et al., 2002 [203] | 15 healthy men | Double-blind, placebo-controlled, crossover study. Metformin 850 mg twice daily for 15 days. | Unchanged fasting total FFA levels. | |
| Krysiak et al., 2012 [204] | 58 subjects with impaired fasting glucose | Randomised, placebo-controlled, parallel group study. Simvastatin 40 mg/day + either metformin 1 g thrice daily for 3 months. | Reduced fasting total FFA levels. | |
| Gormsen et al., 2018 [205] | 24 T2DM subjects, 12 healthy subjects | Randomised, placebo-controlled, parallel-group trial. Metformin 1 g twice daily for 3 months. | Unchanged fasting total FFA levels. | |
| Statins | Sahebkar et al., 2016 [206] | Subjects with T2DM, metabolic syndrome and dyslipidaemia | Meta-analysis. Atorvastatin or simvastatin, less than or more than 12 weeks | Reduced total FFA levels. |
| Fibrates | Fenderson et al., 1974 [209] | 4 healthy subjects, 27 subjects with hypolipoproteinaemia | Controlled, parallel group study. Clofibrate 2 g/day for 21 days | Reduced fasting total plasma FFA levels and reduced levels during oral glucose tolerance test in hypolipoproteinaemia subjects but not in healthy subjects. |
| Calvert et al., 1980 [210] | 22 T2DM subjects | Randomised, double-blind, placebo-controlled, crossover study. Clofibrate 1 g twice daily for 12 weeks. | Reduced fasting and 8 h-average total plasma FFA levels. | |
| Jones et al., 1990 [211] | 36 T2DM subjects | Randomised, double-blind, placebo-controlled, parallel group study. Bezafibrate 200 mg thrice daily for 3 months. | Reduced fasting and postprandial total plasma FFA levels. | |
| Alberti et al., 1990 [212] | 20 T2DM subjects | Randomised, double-blind, placebo-controlled, parallel group study. Bezafibrate 200 mg thrice daily for 3 months. | Reduced fasting total plasma FFA levels. | |
| Vuorinen-Markkola et al., 1993 [213] | 20 T2DM subjects | Randomised, double-blind, placebo-controlled, parallel group study. Gemfibrozil 1200 mg/day for 12 weeks. | Unchanged 24 h-average total plasma FFA levels. | |
| Sane et al., 1995 [214] | 20 subjects with hyperinsulinemia and hypertriglyceridemia | Randomised, double-blind, placebo-controlled, parallel group study. Gemfibrozil 1200 mg/day for 12 weeks. | Unchanged 24 h-average total plasma FFA levels and during insulin infusion. | |
| Avogaro et al., 1995 [215] | 18 subjects with hypertriglyceridemia, 11 with T2DM, 7 without T2DM | Randomised, single-blind, placebo-controlled, cross-over study. Gemfibrozil 600 mg twice daily for three months. | Reduced fasting total plasma FFA levels for both populations. Reduced postprandial total FFA levels for the T2DM group. | |
| Jeng et al., 1996 [216] | 24 subjects with hypertriglyceridemia | Randomised, placebo-control, parallel group study. Gemfibrozil 600 mg twice daily for 3 months. | Reduced postprandial total plasma FFA levels. | |
| Avogaro et al., 1999 [217] | 217 T2DM subjects | Randomised, double-bling, placebo-controlled, parallel group study. Gemfibrozil 600 mg twice daily for 20 weeks. | Unchanged fasting total plasma FFA levels. | |
| Mussoni et al., 2000 [218] | 56 subjects with hypertriglyceridemia and glucose intolerance | Randomised, double-blind, placebo-controlled, parallel group study. Gemfibrozil 600 mg twice a day for 5 months. | Increased fasting total plasma FFA levels. | |
| Jonkers et al., 2001 [219] | 17 subjects with hypertriglyceridemia | Randomised, double-blind, placebo-controlled, crossover study. Bezafibrate 400 mg/day for 6 weeks. | Reduced fasting total plasma FFA levels. | |
| Capell et al., 2003 [220] | 11 subjects with hypertriglyceridemia | Randomised, double-blind, placebo-controlled, crossover study. Fenofibrates for 14 days. | Unchanged 24 h-average total plasma FFA levels but reduced levels after heparin infusion. | |
| Vega et al., 2003 [221] | 13 men with metabolic syndrome | Randomised, placebo-controlled, crossover study. Fenofibrates 200 mg/day for 8 weeks. | Unchanged fasting total plasma FFA levels and during oral glucose tolerance test. | |
| Li et al., 2011 [222] | 87 obese subjects with hyperinsulinemia but not diabetes, on metformin. | Randomised, double-blind, placebo-controlled, parallel group study. Fenofibrate 200 mg/day for 6 months. | Reduced fasting total plasma FFA levels. | |
| Matsuba et al., 2018 [223] | 27 subjects with hypertriglyceridemia and insulin resistance | Randomised, double-blind, placebo-controlled, parallel group study. Pemafibrate 0.4 mg/day, twice daily for 12 weeks | Reduced fasting total plasma FFA levels. | |
| Ezetimibe | Krysiak et al., 2014 [224] | 39 subjects with hypercholesterolemia and 20 healthy controls | Controlled, parallel group study. Ezetimibe 10 mg/day for 90 days | Reduced fasting total FFA levels in insulin-resistant patients, unchanged in patients without insulin resistance but with hypercholesterolemia. |
| Sugiyama et al., 2015 [225] | 33 T2DM patients, | Randomised, open-label, controlled, parallel group study. Ezetimibe 10 mg/day for 6 months | Reduced fasting total FFA levels. | |
| Nicotinic acid (niacin) | Kelly et al., 2000 [232] | 7 healthy subjects | Randomised, double-blind, placebo-controlled, crossover study. Nicotinic acid 500 mg for 7 days, then 2 g/day for 7 days. | Unchanged fasting total FFA levels. |
| Wang et al., 2000 [231] | 5 healthy women | Single-blind, controlled, parallel group study. Increasing nicotinic acid doses over 1 month up to 1g or placebo. | After 1 month, on-significant elevated fasting total FFA levels, decreased upon taking niacin up to 1 h 30 afterwards, but large rebound from 3 to 6 h afterwards. | |
| Niacin mimetic (hydroxy-carboxylic acid receptor 2 agonist) | Dobbins et al., 2015 [233] | 94 T2DM patients | Randomised, double-blind, placebo-controlled, parallel group study. GSK256073, 5 or 25 mg twice daily or 10 or 50 mg once daily, for 12 weeks. | Reduced fasting total FFA levels at day 2 but less effective or no effect at week 6. |
| Niacin mimetic (GPR109A agonist) | Lauring et al., 2012 [234] | Subjects with mixed dyslipidaemia, 162 for MK-1903 study, 69 for SCH900271 study | Randomised, double-blind, placebo-controlled, parallel group study. MK-1903 150 mg Q8h doses for 4 weeks. Randomised, partially blind, placebo-controlled, parallel-group study. SCH900271 10 mg for 29 days. | MK-1903 reduced fasting total FFA levels at day 1. At day 28 both drugs reduced fasting total FFA levels upon drug intake, but intake induced an immediate rebound which results in elevated 8h-average total FFA levels. |
| Bile acid sequestrants | Vega et al., 2011 [235] | 20 men with metabolic syndrome | Randomised double-blind, placebo-controlled crossover study. Colesevelam 1.875 g twice daily for 8 weeks. | Reduced fasting total FFA levels but increased postprandial total FFA levels. |
| Thiazolidine-diones | Chaiken et al., 1995 [236] | 19 obese T2DM subjects | Randomised, double-blind placebo-controlled, parallel group study. Darglitazone 25 mg/day for 14 days. | Reduced 24 h-average total plasma FFA levels. |
| Buysschaert et al., 1999 [237] | 259 T2DM subjects | Randomized, double-dummy, placebo-controlled, parallel-group study. Troglitazone 100 or 200 mg/day for 16 weeks. | Reduced fasting total plasma FFA levels. | |
| Raskin et al., 2000 [238] | 303 T2DM subjects | Randomised, double-blind, placebo-controlled, parallel group study. Rosiglitazone, 0, 2, 4 or 6 mg twice daily for 2 weeks. | Reduced fasting total plasma FFA levels. | |
| Miyazaki et al., 2001 [239] | 29 T2DM subjects | Randomised double-blind, placebo-controlled, parallel group study. Rosiglitazone 8 mg/day for 12 weeks. | Reduced fasting total plasma FFA levels and reduced level during oral glucose tolerance test. | |
| Kerenyi et al., 2004 [240] | 340 T2DM subjects | Randomised double-blind, placebo-controlled, parallel group study. Rosiglitazone 4 mg twice daily for 26 weeks. | Reduced fasting total plasma FFA levels. | |
| James et al., 2005 [241] | 30 obese and insulin resistant men | Randomised, placebo-controlled, parallel-group study. Metformin 1 g twice daily or rosiglitazone 4 mg twice daily for 8 weeks | Unchanged fasting total plasma FFA levels. | |
| Tan et al., 2005 [242] | 24 T2DM subjects | Randomised, double-blind, placebo-controlled, cross-over study. Rosiglitazone 4 mg twice daily for 12 weeks. | Unchanged fasting total plasma FFA levels, reduced postprandial levels. | |
| Al Majali et al., 2006 [144] | 22 T2DM patients, 10 healthy controls | Randomised, double-blind, placebo-controlled, parallel group study. Pioglitazone 45 mg/day or glibenclamide 5 mg/day | Unchanged fasting or postprandial total plasma FFA levels. | |
| Samaha et al., 2006 [243] | 57 nondiabetic subjects with metabolic syndrome | Randomised, double-blinded, placebo-controlled, parallel group study. Rosiglitazone 8 mg/day for 12 weeks. | Unchanged fasting total plasma FFA levels. | |
| Mittermayer et al., 2007 [244] | 16 healthy men | Randomised, double-blind, placebo-controlled, parallel-group study. Rosiglitazone 8 mg/day for 21 days. | Reduced total plasma FFA levels before and 5 h after lipid infusion. | |
| Miyazaki et al., 2007 [245] | 29 T2DM subjects | Randomised, double-blind, placebo-controlled, parallel group study. Rosiglitazone 8 mg/day for 12 weeks. | Reduced fasting total plasma FFA levels and reduced levels during oral glucose tolerance test. | |
| Krzyzanowska et al., 2007 [246] | 16 healthy men | Randomised, double-blind, placebo-controlled parallel-group study. Rosiglitazone 8 mg/day for 21 days. | Reduced total plasma FFA levels before and 5 h after lipid infusion. | |
| Abbasi et al., 2008 [247] | 37 overweight, nondiabetic, insulin resistant subjects | Randomised, controlled, parallel group study. Fenofibrate 160 mg/day for 12 weeks or rosiglitazone 4 mg once daily for 4 weeks, then 4 mg twice daily for 8 weeks. | Reduced daylong (8 h-average) total plasma FFA levels. | |
| Punthakee et al., 2014 [248] | 190 subjects with impaired fasting glucose or impaired glucose tolerance | Randomised, double-blind, placebo controlled, parallel group study. Rosiglitazone 4mg/day for the first 2 month then 8 mg/day for 3.5 year | Unchanged fasting total plasma FFA levels. | |
| Kim et al., 2014 [249] | 173 subjects with T2DM | Randomised, double-blind, placebo-controlled, parallel-group study. Lobeglitazone 0.5 mg/day for 24 weeks. | Reduced fasting total plasma FFA levels. | |
| n-3 fatty acid | Farsi et al., 2014 [250] | 44 T2DM subjects | Randomised, double-blind, controlled, parallel group study. n-3 soft gels 4 g/day for 10 weeks. | Reduced fasting total plasma FFA levels. |
| ETC-1002 | Thompson et al., 2015 [251] | 56 hypercholesterolemia subjects with statins intolerance | Randomised, double-blind, placebo-controlled, parallel group study. ETC-1002 60 mg/day increased every 2 weeks to 120 mg/day, 180 mg/day and 240 mg/day for a total of 8 weeks. | Unchanged fasting total FFA levels. |
| Combinations of drugs | Gómez-Perez et al., 2002 [252] | 116 T2DM subjects | Randomised, double-blind, placebo-controlled, parallel group study. Metformin 2.5 g/day and placebo, metformin 2.5 g/day and rosiglitazone 2 mg twice daily, or metformin 2.5 g/day and rosiglitazone 4 mg twice daily for 26 weeks. | Reduced fasting total FFA levels. |
| Wagner et al., 2005 [253] | 12 healthy subjects | Randomised, placebo-controlled, incomplete-block, 3-period crossover study. Fenofibrate 201 mg/day, rosiglitazone 4 mg twice daily, or combined fenofibrate 201 mg/day and rosiglitazone 4 mg twice daily. | Reduced fasting total FFA levels. | |
| Boden et al., 2007 [254] | 13 T2DM subjects | Single-blind placebo-controlled, parallel group study. Rosiglitazone 8 mg/day, fenofibrate 160 mg/day or Rosiglitazone 8 mg/day and fenofibrate 160 mg/day for 2 months. | Reduced daily-average total FFA levels. | |
| Plat et al., 2009 [255] | 36 subjects with metabolic syndrome | Randomised, double-blind, placebo-controlled, parallel group study. Simvastatin 10 mg/day, plant stanols 2g/day, or simvastatin 10 mg/day and plant stanols 2 g/day for 9 weeks | Unchanged fasting total FFA levels. | |
| Bays et al., 2011 [256] | 183 subjects with dyslipidaemia | Randomised, double-blind, placebo-controlled, parallel group study. MBX-8025 50 mg/day; MBX-8025 100 mg/day; atorvastatin 20 mg/day; MBX-8025 50 mg/day and atorvastatin 20 mg/day; or MBX-8025 100 mg/day and atorvastatin 20 mg/day for 8 weeks. | Reduced fasting total plasma FFA levels for MBX-8025 50 or 100 mg/day and for MBX-8025 50 mg/day and atorvastatin 20 mg/day. Unchanged levels for atorvastatin alone or with MBX-8025 100 mg/day. | |
| Krysiak et al., 2014 [257] | 65 subjects with hypercholesterolemia | Randomised, not blinded, placebo-controlled, parallel group study. Simvastatin 40 mg/day; ezetimibe 10 mg/day; or simvastatin 40 mg/day and ezetimibe 10 mg/day for 12 weeks. | Reduced fasting total plasma FFA levels. | |
| Hwang et al., 2019 [258] | 36 T2DM subjects | Randomised, open-label, active-control, parallel group study. Rosuvastatin 20 mg/day or rosuvastatin 5 mg/day and ezetimibe 10 mg/day for 6 weeks. | Reduced fasting total plasma FFA levels. |
6.2. Changes in Specific Plasma FFA Concentrations
| Drug type | Reference | Subjects | Drug Treatment | Types of Results |
|---|---|---|---|---|
| n-3 fatty acid | Conquer et al., 1998 [260] | 22 healthy subjects | Placebo controlled study. Low docosahexaenoate 0.75 g/day or high docosahexaenoate 1.50 g/day for 42 days. | % total serum FFA |
| Conquer et al., 1999 [261] | 19 healthy men | Randomised, controlled, parallel group study. Seal-oil 1 g/day (1.3 g eicosapentaenoate, 1.7 g docosahexaenoate, and 0.8 g docosapentaenoate per day) for 42 days. | Serum FFA conc. | |
| Barre et al., 2016 [262] | 32 T2DM subjects | Randomised, double-blind, controlled, parallel group. Flaxseed oil (60 mg of α-linolenate/ kg/day) for 3 months. | % total serum FFA | |
| Thiazolidinediones | Yi et al., 2007 [98] | 10 subjects with abdominal obesity and T2DM | Rosiglitazone (amount unknown), FFA measured 2, 7, 9 and 14 weeks after, compared to baseline. | Plasma FFA conc. |
| Reconstituted HDL infusions | Drew et al., 2011 [263] | 13 T2DM patients | Randomised, double-blind, placebo-controlled, cross-over study. Reconstituted HDL infusion 80 mg/kg, FFA measured after 4 h, compared to a placebo given to same patients. | Plasma FFA conc. |
| FFA Species | Altered Plasma FFA Conc. in T2DM after Lipid-Lowering Drug Treatment | ||||
|---|---|---|---|---|---|
| Conquer et al. 1998 [260] | Conquer et al. 1999 [261] | Barre et al. 2016 [262] | Yi et al. 2007 [98] | Drew et al. 2011 [263] | |
| Total FFAs | = | ||||
| C12:0, lauric acid | = | = | |||
| C14:0, myristic acid | = | = | = | ↑ | |
| C15:0, pentadecanoic acid | = | ↑ | |||
| C16:0, palmitic acid | = | = | = | = | ↑ |
| C18:0, stearic acid | = | = | = | ↓ (14) | ↑ |
| C19:0, nonadecylic acid | ↑ | ||||
| C20:0, arachidic acid | = | ||||
| C24:0, lignoceric acid | = | ||||
| C14:1n-5, myristoleic acid | = | = | |||
| C16:1n-7, palmitoleic acid | = | = | = | = | |
| C16:1n-9, cis-7 hexadecenoic acid | = | ||||
| C18:1n-9, oleic acid (OA) | = | = | = | = | ↑ |
| C18:1n-7, cis-vaccenic acid | = | ||||
| C18:2n-6, linoleic acid (LA) | = | ↓ | = | ||
| C18:3n-6, γ-linolenic acid (GLA) | ↓ (14) | ||||
| C18:3n-3, α-linolenic acid (ALA) | = | = | ↑ | ↓ (7) | ↑ |
| C20:3n-6, dihomo-γ-linolenic acid (DGLA) | = | = | |||
| C20:4n-6, arachidonic acid (ARA) | = | ↓ | = | = | = |
| C20:5n-3, eicosapentaenoic acid (EPA) | = | ↑ | ↑ | = | = |
| C22:4n-6, adrenic acid | ↓ (2) | ||||
| C22:5 n3, docosapentaenoic acid (DPA) | = | ↑ | = | ↑ | |
| C22:5n-6, osbondic acid | = | ||||
| C22:6n-3, docosahexaenoic acid (DHA) | ↑ | ↑ | = | ↓ (9, 14) | ns |
| C14:1 trans -n-5, trans-myristoleic acid | = | ||||
| C16:1 trans-n-7, trans-palmitoleic acid | = | ||||
| C18:1 trans-n-7, vaccenic acid | = | ||||
| C18:1 trans-n-9, elaidic acid | = | ||||
| C18:2 trans-n-7 cis-n-9, rumenic acid | ↑ | ||||
| C18:2 trans-n-6, linolelaidic acid | ns | ||||
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Wilding, J.P. The importance of free fatty acids in the development of Type 2 diabetes. Diabet. Med. 2007, 24, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmena, R. Type 2 diabetes, dyslipidemia, and vascular risk: Rationale and evidence for correcting the lipid imbalance. Am. Heart J. 2005, 150, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Marz, W. Free fatty acids as a cardiovascular risk factor. Clin. Chem. Lab. Med. 2008, 46, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Amate, L.; Gil, A. Absorption and distribution of dietary fatty acids from different sources. Early Hum. Dev. 2001, 65, S95–S101. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Large, V.; Peroni, O.; Letexier, D.; Ray, H.; Beylot, M. Metabolism of lipids in human white adipocyte. Diabetes Metab. 2004, 30, 294–309. [Google Scholar] [CrossRef]
- Teusink, B.; Voshol, P.J.; Dahlmans, V.E.; Rensen, P.C.; Pijl, H.; Romijn, J.A.; Havekes, L.M. Contribution of fatty acids released from lipolysis of plasma triglycerides to total plasma fatty acid flux and tissue-specific fatty acid uptake. Diabetes 2003, 52, 614–620. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle balance between lipolysis and lipogenesis: A critical point in metabolic homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef]
- Nielsen, T.S.; Jessen, N.; Jorgensen, J.O.; Moller, N.; Lund, S. Dissecting adipose tissue lipolysis: Molecular regulation and implications for metabolic disease. J. Mol. Endocrinol. 2014, 52, R199–R222. [Google Scholar] [CrossRef]
- Carpentier, A.C. Abnormal myocardial dietary fatty acid metabolism and diabetic cardiomyopathy. Can. J. Cardiol. 2018, 34, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Hasegawa, S.; Kasubuchi, M.; Ichimura, A.; Nakajima, A.; Kimura, I. Nutritional signaling via free fatty acid receptors. Int. J. Mol. Sci. 2016, 17, 450. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakatani, Y.; Atsumi, G.I.; Inoue, K.; Kudo, I. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 2017, 37, 121–179. [Google Scholar] [CrossRef] [PubMed]
- Georgiadi, A.; Kersten, S. Mechanisms of gene regulation by fatty acids. Adv. Nutr. 2012, 3, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Dranse, H.J.; Kelly, M.E.; Hudson, B.D. Drugs or diet?—Developing novel therapeutic strategies targeting the free fatty acid family of GPCRs. Br. J. Pharmacol. 2013, 170, 696–711. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef]
- Hara, T.; Ichimura, A.; Hirasawa, A. Therapeutic role and ligands of medium- to long-chain Fatty Acid receptors. Front. Endocrinol. (Lausanne) 2014, 5, 83. [Google Scholar] [CrossRef]
- Christiansen, E.; Watterson, K.R.; Stocker, C.J.; Sokol, E.; Jenkins, L.; Simon, K.; Grundmann, M.; Petersen, R.K.; Wargent, E.T.; Hudson, B.D.; et al. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. Br. J. Nutr. 2015, 113, 1677–1688. [Google Scholar] [CrossRef] [Green Version]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon—Like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Duca, F.A.; Yue, J.T. Fatty acid sensing in the gut and the hypothalamus: In vivo and in vitro perspectives. Mol. Cell. Endocrinol. 2014, 397, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Clim. Assoc. 2010, 121, 206–220. [Google Scholar]
- Yang, M.; Silverstein, R.L. CD36 signaling in vascular redox stress. Free Radic. Biol. Med. 2019, 136, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Martin, H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat. Res. 2010, 690, 57–63. [Google Scholar] [CrossRef]
- Alhazmi, A.; Stojanovski, E.; McEvoy, M.; Garg, M.L. The association between dietary patterns and type 2 diabetes: A systematic review and meta-analysis of cohort studies. J. Hum. Nutr. Diet 2014, 27, 251–260. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef]
- Rice Bradley, B.H. Dietary fat and risk for type 2 diabetes: A review of recent research. Curr. Nutr. Rep. 2018, 7, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Riserus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozaffarian, D. Dietary and policy priorities for cardiovascular disease, diabetes, and obesity: A comprehensive review. Circulation 2016, 133, 187–225. [Google Scholar] [CrossRef] [PubMed]
- Guasch-Ferre, M.; Becerra-Tomas, N.; Ruiz-Canela, M.; Corella, D.; Schroder, H.; Estruch, R.; Ros, E.; Aros, F.; Gomez-Gracia, E.; Fiol, M.; et al. Total and subtypes of dietary fat intake and risk of type 2 diabetes mellitus in the Prevencion con Dieta Mediterranea (PREDIMED) study. Am. J. Clin. Nutr. 2017, 105, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Salas-Salvado, J.; Guasch-Ferre, M.; Diaz-Lopez, A.; Babio, N. Yogurt and diabetes: Overview of recent observational studies. J. Nutr. 2017, 147, 1452S–1461S. [Google Scholar] [CrossRef] [PubMed]
- Mente, A.; Dehghan, M.; Rangarajan, S.; McQueen, M.; Dagenais, G.; Wielgosz, A.; Lear, S.; Li, W.; Chen, H.; Yi, S.; et al. Association of dietary nutrients with blood lipids and blood pressure in 18 countries: A cross-sectional analysis from the PURE study. Lancet Diabetes Endocrinol. 2017, 5, 774–787. [Google Scholar] [CrossRef]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar] [CrossRef]
- Thanopoulou, A.C.; Karamanos, B.G.; Angelico, F.V.; Assaad-Khalil, S.H.; Barbato, A.F.; Del Ben, M.P.; Djordjevic, P.B.; Dimitrijevic-Sreckovic, V.S.; Gallotti, C.A.; Katsilambros, N.L.; et al. Dietary fat intake as risk factor for the development of diabetes: Multinational, multicenter study of the Mediterranean Group for the Study of Diabetes (MGSD). Diabetes Care 2003, 26, 302–307. [Google Scholar] [CrossRef]
- Schwab, U.; Lauritzen, L.; Tholstrup, T.; Haldorssoni, T.; Riserus, U.; Uusitupa, M.; Becker, W. Effect of the amount and type of dietary fat on cardiometabolic risk factors and risk of developing type 2 diabetes, cardiovascular diseases, and cancer: A systematic review. Food Nutr. Res. 2014, 58. [Google Scholar] [CrossRef]
- Swedish Council on Health Technology Assessment. Dietary Treatment of Diabetes: A Systematic Review; Swedish Council on Health Technology Assessment: Stockholm, Sweden, 2010. [Google Scholar]
- De Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schunemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [PubMed]
- Imamura, F.; Micha, R.; Wu, J.H.; de Oliveira Otto, M.C.; Otite, F.O.; Abioye, A.I.; Mozaffarian, D. Effects of saturated fat, polyunsaturated fat, monounsaturated fat, and carbohydrate on glucose-insulin homeostasis: A systematic review and meta-analysis of randomised controlled feeding trials. PLoS Med. 2016, 13, e1002087. [Google Scholar] [CrossRef] [PubMed]
- Kinsell, L.W.; Walker, G.; Michaels, G.D.; Olson, F.E. Dietary fats and the diabetic patient. N. Engl. J. Med. 1959, 261, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.C.; Smith, S.R.; Champagne, C.M.; Most, M.M.; Lefevre, M.; DeLany, J.P.; Denkins, Y.M.; Rood, J.C.; Veldhuis, J.; Bray, G.A. Effects of diets enriched in saturated (palmitic), monounsaturated (oleic), or trans (elaidic) fatty acids on insulin sensitivity and substrate oxidation in healthy adults. Diabetes Care 2002, 25, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Ericson, U.; Hellstrand, S.; Brunkwall, L.; Schulz, C.A.; Sonestedt, E.; Wallstrom, P.; Gullberg, B.; Wirfalt, E.; Orho-Melander, M. Food sources of fat may clarify the inconsistent role of dietary fat intake for incidence of type 2 diabetes. Am. J. Clin. Nutr. 2015, 101, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Soedamah-Muthu, S.S.; de Goede, J. Dairy consumption and cardiometabolic diseases: Systematic review and updated meta-analyses of prospective cohort studies. Curr. Nutr. Rep. 2018, 7, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, H.E.; Koeller, E.; Greer, N.; MacDonald, R.; Kane, R.; Wilt, T.J. Effects on health outcomes of a Mediterranean Diet with no restriction on fat intake: A systematic review and meta-analysis. Ann. Intern. Med. 2016, 165, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, L.; Ding, E.L.; Malik, V.S.; de Goede, J.; Geleijnse, J.M.; Soedamah-Muthu, S.S. Consumption of dairy foods and diabetes incidence: A dose-response meta-analysis of observational studies. Am. J. Clin. Nutr. 2016, 103, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A. Yogurt and dairy product consumption to prevent cardiometabolic diseases: Epidemiologic and experimental studies. Am. J. Clin. Nutr. 2014, 99, 1235S–1242S. [Google Scholar] [CrossRef]
- Gao, D.; Ning, N.; Wang, C.; Wang, Y.; Li, Q.; Meng, Z.; Liu, Y.; Li, Q. Dairy products consumption and risk of type 2 diabetes: Systematic review and dose-response meta-analysis. PLoS ONE 2013, 8, e73965. [Google Scholar] [CrossRef]
- Aune, D.; Norat, T.; Romundstad, P.; Vatten, L.J. Dairy products and the risk of type 2 diabetes: A systematic review and dose-response meta-analysis of cohort studies. Am. J. Clin. Nutr. 2013, 98, 1066–1083. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sun, Q.; Giovannucci, E.; Mozaffarian, D.; Manson, J.E.; Willett, W.C.; Hu, F.B. Dairy consumption and risk of type 2 diabetes: 3 cohorts of US adults and an updated meta-analysis. BMC Med. 2014, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Pimpin, L.; Wu, J.H.; Haskelberg, H.; Del Gobbo, L.; Mozaffarian, D. Is butter back? A systematic review and meta-analysis of butter consumption and risk of cardiovascular disease, diabetes, and total mortality. PLoS ONE 2016, 11, e0158118. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.; van der Daphne, A.L.; Grobbee, D.E.; Sluijs, I.; Spijkerman, A.M.; van der Schouw, Y.T. Dietary phylloquinone and menaquinones intakes and risk of type 2 diabetes. Diabetes Care 2010, 33, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Kunutsor, S.; Franco, O.H.; Chowdhury, R. Vitamin D, type 2 diabetes and other metabolic outcomes: A systematic review and meta-analysis of prospective studies. Proc. Nutr. Soc. 2013, 72, 89–97. [Google Scholar] [CrossRef]
- Song, Y.; Wang, L.; Pittas, A.G.; Del Gobbo, L.C.; Zhang, C.; Manson, J.E.; Hu, F.B. Blood 25-hydroxy vitamin D levels and incident type 2 diabetes: A meta-analysis of prospective studies. Diabetes Care 2013, 36, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Santaren, I.D.; Watkins, S.M.; Liese, A.D.; Wagenknecht, L.E.; Rewers, M.J.; Haffner, S.M.; Lorenzo, C.; Hanley, A.J. Serum pentadecanoic acid (15:0), a short-term marker of dairy food intake, is inversely associated with incident type 2 diabetes and its underlying disorders. Am. J. Clin. Nutr. 2014, 100, 1532–1540. [Google Scholar] [CrossRef] [Green Version]
- Forouhi, N.G.; Koulman, A.; Sharp, S.J.; Imamura, F.; Kroger, J.; Schulze, M.B.; Crowe, F.L.; Huerta, J.M.; Guevara, M.; Beulens, J.W.; et al. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident type 2 diabetes: The EPIC-InterAct case-cohort study. Lancet Diabetes Endocrinol. 2014, 2, 810–818. [Google Scholar] [CrossRef]
- Ratnayake, W.M. Concerns about the use of 15:0, 17:0, and trans-16:1n-7 as biomarkers of dairy fat intake in recent observational studies that suggest beneficial effects of dairy food on incidence of diabetes and stroke. Am. J. Clin. Nutr. 2015, 101, 1102–1103. [Google Scholar] [CrossRef]
- Airhart, S.; Cade, W.T.; Jiang, H.; Coggan, A.R.; Racette, S.B.; Korenblat, K.; Spearie, C.A.; Waller, S.; O’Connor, R.; Bashir, A.; et al. A Diet rich in medium-chain fatty acids improves systolic function and alters the lipidomic profile in patients with type 2 diabetes: A pilot study. J. Clin. Endocrinol. Metab. 2016, 101, 504–512. [Google Scholar] [CrossRef]
- Fretts, A.M.; Imamura, F.; Marklund, M.; Micha, R.; Wu, J.H.Y.; Murphy, R.A.; Chien, K.L.; McKnight, B.; Tintle, N.; Forouhi, N.G.; et al. Associations of circulating very-long-chain saturated fatty acids and incident type 2 diabetes: A pooled analysis of prospective cohort studies. Am. J. Clin. Nutr. 2019, 109, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Vessby, B.; Uusitupa, M.; Hermansen, K.; Riccardi, G.; Rivellese, A.A.; Tapsell, L.C.; Nalsen, C.; Berglund, L.; Louheranta, A.; Rasmussen, B.M.; et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU Study. Diabetologia 2001, 44, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Korat, A.A.; Malik, V.; Hu, F.B. Metabolic effects of monounsaturated fatty acid-enriched diets compared with carbohydrate or polyunsaturated fatty acid-enriched diets in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Care 2016, 39, 1448–1457. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Strasser, B.; Hoffmann, G. Effects of monounsaturated fatty acids on glycaemic control in patients with abnormal glucose metabolism: A systematic review and meta-analysis. Ann. Nutr. Metab. 2011, 58, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Belury, M.A.; Cole, R.M.; Snoke, D.B.; Banh, T.; Angelotti, A. Linoleic acid, glycemic control and Type 2 diabetes. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Coelho, O.G.L.; da Silva, B.P.; Rocha, D.; Lopes, L.L.; Alfenas, R.C.G. Polyunsaturated fatty acids and type 2 diabetes: Impact on the glycemic control mechanism. Crit. Rev. Food Sci. Nutr. 2017, 57, 3614–3619. [Google Scholar] [CrossRef] [PubMed]
- O’Mahoney, L.L.; Matu, J.; Price, O.J.; Birch, K.M.; Ajjan, R.A.; Farrar, D.; Tapp, R.; West, D.J.; Deighton, K.; Campbell, M.D. Omega-3 polyunsaturated fatty acids favourably modulate cardiometabolic biomarkers in type 2 diabetes: A meta-analysis and meta-regression of randomized controlled trials. Cardiovasc. Diabetol. 2018, 17, 98. [Google Scholar] [CrossRef] [PubMed]
- Telle-Hansen, V.H.; Gaundal, L.; Myhrstad, M.C.W. Polyunsaturated fatty acids and glycemic control in type 2 diabetes. Nutrients 2019, 11, 1067. [Google Scholar] [CrossRef]
- Abbott, K.A.; Burrows, T.L.; Thota, R.N.; Acharya, S.; Garg, M.L. Do omega-3 PUFAs affect insulin resistance in a sex-specific manner? A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2016, 104, 1470–1484. [Google Scholar] [CrossRef]
- Muley, A.; Muley, P.; Shah, M. ALA, fatty fish or marine n-3 fatty acids for preventing DM?: A systematic review and meta-analysis. Curr. Diabetes Rev. 2014, 10, 158–165. [Google Scholar] [CrossRef]
- Djousse, L.; Gaziano, J.M.; Buring, J.E.; Lee, I.M. Dietary omega-3 fatty acids and fish consumption and risk of type 2 diabetes. Am. J. Clin. Nutr. 2011, 93, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Brostow, D.P.; Odegaard, A.O.; Koh, W.P.; Duval, S.; Gross, M.D.; Yuan, J.M.; Pereira, M.A. Omega-3 fatty acids and incident type 2 diabetes: The Singapore Chinese Health Study. Am. J. Clin. Nutr. 2011, 94, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Imamura, F.; Sharp, S.J.; Koulman, A.; Schulze, M.B.; Zheng, J.; Ye, Z.; Sluijs, I.; Guevara, M.; Huerta, J.M.; et al. Association of plasma phospholipid n-3 and n-6 polyunsaturated fatty acids with type 2 diabetes: The EPIC-InterAct case-cohort study. PLoS Med. 2016, 13, e1002094. [Google Scholar] [CrossRef] [PubMed]
- Jovanovski, E.; Li, D.; Thanh Ho, H.V.; Djedovic, V.; Ruiz Marques, A.C.; Shishtar, E.; Mejia, S.B.; Sievenpiper, J.L.; de Souza, R.J.; Duvnjak, L.; et al. The effect of alpha-linolenic acid on glycemic control in individuals with type 2 diabetes: A systematic review and meta-analysis of randomized controlled clinical trials. Medicine 2017, 96, e6531. [Google Scholar] [CrossRef] [PubMed]
- Julius, U. Fat modification in the diabetes diet. Exp. Clin. Endocrinol. Diabetes 2003, 111, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Q.; Qiu, Y.; Mu, Y.; Zhang, X.J.; Liu, L.; Hou, X.H.; Zhang, L.; Xu, X.N.; Ji, A.L.; Cao, R.; et al. A high ratio of dietary n-3/n-6 polyunsaturated fatty acids improves obesity-linked inflammation and insulin resistance through suppressing activation of TLR4 in SD rats. Nutr. Res. 2013, 33, 849–858. [Google Scholar] [CrossRef]
- Innes, J.K.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Yakoob, M.Y.; Shi, P.; Willett, W.C.; Rexrode, K.M.; Campos, H.; Orav, E.J.; Hu, F.B.; Mozaffarian, D. Circulating biomarkers of dairy fat and risk of incident diabetes mellitus among men and women in the United States in two large prospective cohorts. Circulation 2016, 133, 1645–1654. [Google Scholar] [CrossRef]
- Mozaffarian, D.; de Oliveira Otto, M.C.; Lemaitre, R.N.; Fretts, A.M.; Hotamisligil, G.; Tsai, M.Y.; Siscovick, D.S.; Nettleton, J.A. trans-Palmitoleic acid, other dairy fat biomarkers, and incident diabetes: The Multi-Ethnic Study of Atherosclerosis (MESA). Am. J. Clin. Nutr. 2013, 97, 854–861. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Cao, H.; King, I.B.; Lemaitre, R.N.; Song, X.; Siscovick, D.S.; Hotamisligil, G.S. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am. J. Clin. Nutr. 2010, 92, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Tardy, A.L.; Morio, B.; Chardigny, J.M.; Malpuech-Brugere, C. Ruminant and industrial sources of trans-fat and cardiovascular and diabetic diseases. Nutr. Res. Rev. 2011, 24, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Salmeron, J.; Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.A.; Rimm, E.B.; Willett, W.C. Dietary fat intake and risk of type 2 diabetes in women. Am. J. Clin. Nutr. 2001, 73, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Micha, R.; Mozaffarian, D. Trans fatty acids: Effects on cardiometabolic health and implications for policy. Prostaglandins Leukot. Essent. Fat. Acids 2008, 79, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H.; Imamura, F.; Korat, A.A.; Murphy, R.; Tintle, N.; Bassett, J.; Chen, J.; Kroger, J.; Forouhi, N.; Schulze, M.; et al. Trans fatty acid biomarkers and incident type 2 diabetes: Pooled analysis from 10 prospective cohort studies in the Fatty Acids and Outcome Research Consortium (FORCE) (OR33-02-19). Curr. Dev. Nutr. 2019, 3, 02–19. [Google Scholar] [CrossRef]
- Salas-Salvado, J.; Guasch-Ferre, M.; Lee, C.H.; Estruch, R.; Clish, C.B.; Ros, E. Protective effects of the Mediterranean Diet on type 2 diabetes and metabolic syndrome. J. Nutr. 2016, 146, 920S–927S. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, M.; Wessman, Y.; Almgren, P.; Groop, L. High levels of nonesterified fatty acids are associated with increased familial risk of cardiovascular disease. Arter. Thromb. Vasc. Biol. 2000, 20, 1588–1594. [Google Scholar] [CrossRef]
- Salgin, B.; Ong, K.K.; Thankamony, A.; Emmett, P.; Wareham, N.J.; Dunger, D.B. Higher fasting plasma free fatty acid levels are associated with lower insulin secretion in children and adults and a higher incidence of type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, Y.; Cederberg, H.; Vangipurapu, J.; Kangas, A.J.; Soininen, P.; Kuusisto, J.; Uusitupa, M.; Ala-Korpela, M.; Laakso, M. Glycerol and fatty acids in serum predict the development of hyperglycemia and type 2 diabetes in Finnish men. Diabetes Care 2013, 36, 3732–3738. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.M.; English, D.R.; O’Dea, K.; Sinclair, A.J.; Makrides, M.; Gibson, R.A.; Giles, G.G. Plasma phospholipid and dietary fatty acids as predictors of type 2 diabetes: Interpreting the role of linoleic acid. Am. J. Clin. Nutr. 2007, 86, 189–197. [Google Scholar] [CrossRef]
- Mandal, S.; Causevic, A.; Dzudzevic-Cancar, H.; Semiz, S. Free Fatty Acid Profile in Type 2 Diabetic Subjects with Different Control of Glycemia. In CMBEBIH 2017. IFMBE Proceedings; Badnjevic, A., Ed.; Springer: Singapore, 2017; Volume 62. [Google Scholar]
- Razquin, C.; Toledo, E.; Clish, C.B.; Ruiz-Canela, M.; Dennis, C.; Corella, D.; Papandreou, C.; Ros, E.; Estruch, R.; Guasch-Ferre, M.; et al. Plasma lipidomic profiling and risk of type 2 diabetes in the PREDIMED trial. Diabetes Care 2018, 41, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Bakan, E.; Yildirim, A.; Kurtul, N.; Polat, M.F.; Dursun, H.; Cayir, K. Effects of type 2 diabetes mellitus on plasma fatty acid composition and cholesterol content of erythrocyte and leukocyte membranes. Acta Diabetol. 2006, 43, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Leekumjorn, S.; Cho, H.J.; Wu, Y.; Wright, N.T.; Sum, A.K.; Chan, C. The role of fatty acid unsaturation in minimizing biophysical changes on the structure and local effects of bilayer membranes. Biochim. Biophys. Acta 2009, 1788, 1508–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djousse, L.; Biggs, M.L.; Lemaitre, R.N.; King, I.B.; Song, X.; Ix, J.H.; Mukamal, K.J.; Siscovick, D.S.; Mozaffarian, D. Plasma omega-3 fatty acids and incident diabetes in older adults. Am. J. Clin. Nutr. 2011, 94, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clore, J.N.; Allred, J.; White, D.; Li, J.; Stillman, J. The role of plasma fatty acid composition in endogenous glucose production in patients with type 2 diabetes mellitus. Metabolism 2002, 51, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; He, J.; Liang, Y.; Yuan, D.; Gao, H.; Zhou, H. Simultaneously quantitative measurement of comprehensive profiles of esterified and non-esterified fatty acid in plasma of type 2 diabetic patients. Chem. Phys. Lipids 2007, 150, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Guan, C.; Li, K.; Wang, C.; Feng, R.; Sun, C. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2817–2825. [Google Scholar] [CrossRef]
- Grapov, D.; Adams, S.H.; Pedersen, T.L.; Garvey, W.T.; Newman, J.W. Type 2 diabetes associated changes in the plasma non-esterified fatty acids, oxylipins and endocannabinoids. PLoS ONE 2012, 7, e48852. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, Y.; Ong, C.N.; Subramaniam, T.; Choi, H.W.; Yuan, J.M.; Koh, W.P.; Pan, A. Metabolic signatures and risk of type 2 diabetes in a Chinese population: An untargeted metabolomics study using both LC-MS and GC-MS. Diabetologia 2016, 59, 2349–2359. [Google Scholar] [CrossRef]
- Ma, X.L.; Meng, L.; Li, L.L.; Ma, L.N.; Mao, X.M. Plasma free fatty acids metabolic profile among Uyghurs and Kazaks with or without type 2 diabetes based on GC-MS. Exp. Clin. Endocrinol. Diabetes 2018, 126, 604–611. [Google Scholar] [CrossRef]
- Yary, T.; Voutilainen, S.; Tuomainen, T.P.; Ruusunen, A.; Nurmi, T.; Virtanen, J.K. Serum n-6 polyunsaturated fatty acids, Delta 5- and Delta 6-desaturase activities, and risk of incident type 2 diabetes in men: The Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2016, 103, 1337–1343. [Google Scholar] [CrossRef]
- Andersson-Hall, U.; Carlsson, N.G.; Sandberg, A.S.; Holmang, A. Circulating linoleic acid is associated with improved glucose tolerance in women after gestational diabetes. Nutrients 2018, 10, 1629. [Google Scholar] [CrossRef] [PubMed]
- Cabout, M.; Alssema, M.; Nijpels, G.; Stehouwer, C.D.A.; Zock, P.L.; Brouwer, I.A.; Elshorbagy, A.K.; Refsum, H.; Dekker, J.M. Circulating linoleic acid and alpha-linolenic acid and glucose metabolism: The Hoorn Study. Eur. J. Nutr. 2017, 56, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.B.; Kastenmuller, G.; Shin, S.Y.; Petersen, A.K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013, 62, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B. What is lipotoxicity? Adv. Exp. Med. Biol. 2017, 960, 197–220. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293S–301S. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Kasubuchi, M.; Nakajima, A.; Kimura, I. Anti-Inflammatory and insulin-sensitizing effects of free fatty acid receptors. Handb. Exp. Pharmacol. 2017, 236, 221–231. [Google Scholar] [PubMed]
- Perona, J.S. Membrane lipid alterations in the metabolic syndrome and the role of dietary oils. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- British Heart Foundation. BHF Factsheet—UK 2018; British Heart Foundation: London, UK, 2018. [Google Scholar]
- Diabetes UK. Facts and Stats Update January 2019. Available online: https://www.diabetes.org.uk/resources-s3/2019-02/1362B_Facts%20and%20stats%20Update%20Jan%202019_LOW%20RES_EXTERNAL.pdf (accessed on 18 March 2019).
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef]
- Lin, N.; Chen, H.; Zhang, H.; Wan, X.; Su, Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine 2012, 42, 107–117. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front. Endocrinol. (Lausanne) 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Tang, H.; Zhuo, S.; Zang, Y.Q.; Le, Y. Regulation of fasting fuel metabolism by toll-like receptor 4. Diabetes 2010, 59, 3041–3048. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Coll, T.; Eyre, E.; Rodriguez-Calvo, R.; Palomer, X.; Sanchez, R.M.; Merlos, M.; Laguna, J.C.; Vazquez-Carrera, M. Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J. Biol. Chem. 2008, 283, 11107–11116. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Hatzinikolas, G.; Weir, J.M.; Janovska, A.; McAinch, A.J.; Game, P.; Meikle, P.J.; Wittert, G.A. Insulin-stimulated glucose uptake and pathways regulating energy metabolism in skeletal muscle cells: The effects of subcutaneous and visceral fat, and long-chain saturated, n-3 and n-6 polyunsaturated fatty acids. Biochim. Biophys. Acta 2011, 1811, 468–475. [Google Scholar] [CrossRef]
- Tumova, J.; Andel, M.; Trnka, J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Martins, A.R.; Nachbar, R.T.; Gorjao, R.; Vinolo, M.A.; Festuccia, W.T.; Lambertucci, R.H.; Cury-Boaventura, M.F.; Silveira, L.R.; Curi, R.; Hirabara, S.M. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: Importance of the mitochondrial function. Lipids Health Dis. 2012, 11, 30. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids: Preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef]
- Wong, S.W.; Kwon, M.J.; Choi, A.M.; Kim, H.P.; Nakahira, K.; Hwang, D.H. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J. Biol. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef]
- Cullberg, K.B.; Larsen, J.O.; Pedersen, S.B.; Richelsen, B. Effects of LPS and dietary free fatty acids on MCP-1 in 3T3-L1 adipocytes and macrophages in vitro. Nutr. Diabetes 2014, 4, e113. [Google Scholar] [CrossRef]
- Legrand-Poels, S.; Esser, N.; L’Homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef]
- Shen, L.; Yang, Y.; Ou, T.; Key, C.C.; Tong, S.H.; Sequeira, R.C.; Nelson, J.M.; Nie, Y.; Wang, Z.; Boudyguina, E.; et al. Dietary PUFAs attenuate NLRP3 inflammasome activation via enhancing macrophage autophagy. J. Lipid Res. 2017, 58, 1808–1821. [Google Scholar] [CrossRef] [Green Version]
- Flachs, P.; Rossmeisl, M.; Kopecky, J. The effect of n-3 fatty acids on glucose homeostasis and insulin sensitivity. Physiol. Res. 2014, 63 (Suppl. 1), S93–S118. [Google Scholar]
- Kirwan, A.M.; Lenighan, Y.M.; O’Reilly, M.E.; McGillicuddy, F.C.; Roche, H.M. Nutritional modulation of metabolic inflammation. Biochem. Soc. Trans. 2017, 45, 979–985. [Google Scholar] [CrossRef]
- Chen, X.H.; Stein, P.; Steer, R.A.; Scholl, T.O. Individual free fatty acids have unique associations with inflammatory biomarkers, insulin resistance and insulin secretion in healthy and gestational diabetic pregnant women. BMJ Open Diab. Res. Care 2019, 7, e000632. [Google Scholar] [CrossRef] [Green Version]
- Pilon, M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016, 15, 167. [Google Scholar] [CrossRef] [Green Version]
- Kroger, J.; Jacobs, S.; Jansen, E.H.; Fritsche, A.; Boeing, H.; Schulze, M.B. Erythrocyte membrane fatty acid fluidity and risk of type 2 diabetes in the EPIC-Potsdam study. Diabetologia 2015, 58, 282–289. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [Green Version]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.; Saad, M.J.; et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE 2009, 4, e5045. [Google Scholar] [CrossRef]
- Tripathi, Y.B.; Pandey, V. Obesity and endoplasmic reticulum (ER) stresses. Front. Immunol. 2012, 3, 240. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef]
- Al Majali, K.; Cooper, M.B.; Staels, B.; Luc, G.; Taskinen, M.R.; Betteridge, D.J. The effect of sensitisation to insulin with pioglitazone on fasting and postprandial lipid metabolism, lipoprotein modification by lipases, and lipid transfer activities in type 2 diabetic patients. Diabetologia 2006, 49, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Cintra, D.E.; Ropelle, E.R.; Moraes, J.C.; Pauli, J.R.; Morari, J.; Souza, C.T.; Grimaldi, R.; Stahl, M.; Carvalheira, J.B.; Saad, M.J.; et al. Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS ONE 2012, 7, e30571. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Fu, J.; Astarita, G.; Li, X.; Gaetani, S.; Campolongo, P.; Cuomo, V.; Piomelli, D. The lipid messenger OEA links dietary fat intake to satiety. Cell. Metab. 2008, 8, 281–288. [Google Scholar] [CrossRef]
- Martinez de Ubago, M.; Garcia-Oya, I.; Perez-Perez, A.; Canfran-Duque, A.; Quintana-Portillo, R.; Rodriguez de Fonseca, F.; Gonzalez-Yanes, C.; Sanchez-Margalet, V. Oleoylethanolamide, a natural ligand for PPAR-alpha, inhibits insulin receptor signalling in HTC rat hepatoma cells. Biochim. Biophys. Acta 2009, 1791, 740–745. [Google Scholar] [CrossRef]
- Chaudhri, O.B.; Field, B.C.; Bloom, S.R. Gastrointestinal satiety signals. Int. J. Obes. (Lond.) 2008, 32 (Suppl. 7), S28–S31. [Google Scholar] [CrossRef] [Green Version]
- Field, B.C.; Chaudhri, O.B.; Bloom, S.R. Bowels control brain: Gut hormones and obesity. Nat. Rev. Endocrinol. 2010, 6, 444–453. [Google Scholar] [CrossRef]
- Owyang, C.; Logsdon, C.D. New insights into neurohormonal regulation of pancreatic secretion. Gastroenterology 2004, 127, 957–969. [Google Scholar] [CrossRef]
- Hayes, M.R.; Covasa, M. Dorsal hindbrain 5-HT3 receptors participate in control of meal size and mediate CCK-induced satiation. Brain Res. 2006, 1103, 99–107. [Google Scholar] [CrossRef]
- Arruda, A.P.; Milanski, M.; Coope, A.; Torsoni, A.S.; Ropelle, E.; Carvalho, D.P.; Carvalheira, J.B.; Velloso, L.A. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 2011, 152, 1314–1326. [Google Scholar] [CrossRef]
- Cheung, G.W.; Kokorovic, A.; Lam, C.K.; Chari, M.; Lam, T.K. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell. Metab. 2009, 10, 99–109. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. In Annual Review of Medicine; Caskey, C.T., Ed.; Annual Reviews: Palo Alto, CA, USA, 2011; Volume 62, pp. 361–380. [Google Scholar]
- Burcelin, R.; Serino, M.; Chabo, C.; Blasco-Baque, V.; Amar, J. Gut microbiota and diabetes: From pathogenesis to therapeutic perspective. Acta Diabetol. 2011, 48, 257–273. [Google Scholar] [CrossRef]
- Santos-Marcos, J.A.; Perez-Jimenez, F.; Camargo, A. The role of diet and intestinal microbiota in the development of metabolic syndrome. J. Nutr. Biochem. 2019, 70, 1–27. [Google Scholar] [CrossRef]
- De Bandt, J.P.; Waligora-Dupriet, A.J.; Butel, M.J. Intestinal microbiota in inflammation and insulin resistance: Relevance to humans. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 334–340. [Google Scholar] [CrossRef]
- Medina-Vera, I.; Sanchez-Tapia, M.; Noriega-Lopez, L.; Granados-Portillo, O.; Guevara-Cruz, M.; Flores-Lopez, A.; Avila-Nava, A.; Fernandez, M.L.; Tovar, A.R.; Torres, N. A dietary intervention with functional foods reduces metabolic endotoxaemia and attenuates biochemical abnormalities by modifying faecal microbiota in people with type 2 diabetes. Diabetes Metab. 2019, 45, 122–131. [Google Scholar] [CrossRef]
- Bailey, M.A.; Holscher, H.D. Microbiome-mediated effects of the Mediterranean Diet on inflammation. Adv. Nutr. 2018, 9, 193–206. [Google Scholar] [CrossRef]
- Adachi, K.; Sugiyama, T.; Yamaguchi, Y.; Tamura, Y.; Izawa, S.; Hijikata, Y.; Ebi, M.; Funaki, Y.; Ogasawara, N.; Goto, C.; et al. Gut microbiota disorders cause type 2 diabetes mellitus and homeostatic disturbances in gut-related metabolism in Japanese subjects. J. Clin. Biochem. Nutr. 2019, 64, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Abdul Rahim, M.B.H.; Chilloux, J.; Martinez-Gili, L.; Neves, A.L.; Myridakis, A.; Gooderham, N.; Dumas, M.-E.J.A.D. Diet-induced metabolic changes of the human gut microbiome: Importance of short-chain fatty acids, methylamines and indoles. Acta Diabetol. 2019, 56, 493–500. [Google Scholar] [CrossRef]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Backhed, F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell. Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Halbleib, M.; Ahmad, F.; Manganiello, V.C.; Greenberg, A.S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes 2002, 51, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell. 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Ye, J. Regulation of PPARgamma function by TNF-alpha. Biochem. Biophys. Res. Commun. 2008, 374, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, X.Z.; Powis, S.H.; Fernando, R.; Mon, W.Y.; Wheeler, D.C.; Moorhead, J.F.; Varghese, Z. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: Evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 2005, 67, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Gannon, N.P.; Conn, C.A.; Vaughan, R.A. Dietary stimulators of GLUT4 expression and translocation in skeletal muscle: A mini-review. Mol. Nutr. Food Res. 2015, 59, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, F.B.; Takeda, D.; Natu, V.; Sztalryd, C. Insulin regulates lipoprotein lipase activity in rat adipose cells via wortmannin—and rapamycin-sensitive pathways. Metabolism 1998, 47, 555–559. [Google Scholar] [CrossRef]
- Garfinkel, A.G.; Nilsson-ehle, P.; Schotz, M.C. Regulation of lipoprotein lipase. Induction by insulin. Biochim. Biophys. Acta 1976, 424, 264–273. [Google Scholar] [CrossRef]
- Chabowski, A.; Coort, S.L.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Insulin stimulates fatty acid transport by regulating expression of FAT/CD36 but not FABPpm. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E781–E789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, K.; Sarkadi-Nagy, E.; Duncan, R.E.; Ahmadian, M.; Sul, H.S. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1–G4. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.F.; Klein, S. Whole body and abdominal lipolytic sensitivity to epinephrine is suppressed in upper body obese women. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1144–E1152. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Firneisz, G. Non-alcoholic fatty liver disease and type 2 diabetes mellitus: The liver disease of our age? World J. Gastroenterol. 2014, 20, 9072–9089. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Kraegen, E.W.; Clark, P.W.; Jenkins, A.B.; Daley, E.A.; Chisholm, D.J.; Storlien, L.H. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 1991, 40, 1397–1403. [Google Scholar] [CrossRef]
- Wei, D.; Li, J.; Shen, M.; Jia, W.; Chen, N.; Chen, T.; Su, D.; Tian, H.; Zheng, S.; Dai, Y.; et al. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic beta-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes 2010, 59, 471–478. [Google Scholar] [CrossRef]
- Acosta-Montano, P.; Garcia-Gonzalez, V. Effects of dietary fatty acids in pancreatic beta cell metabolism, implications in homeostasis. Nutrients 2018, 10, 393. [Google Scholar] [CrossRef]
- Palomer, X.; Salvado, L.; Barroso, E.; Vazquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, inflammation, and arrhythmias: Role for Interleukin-6 molecular mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.P.; Blindauer, C.A.; Kassaar, O.; Khazaipoul, S.; Martin, E.M.; Sadler, P.J.; Stewart, A.J. Allosteric modulation of zinc speciation by fatty acids. Biochim. Biophys. Acta 2013, 1830, 5456–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassaar, O.; Schwarz-Linek, U.; Blindauer, C.A.; Stewart, A.J. Plasma free fatty acid levels influence Zn2+-dependent histidine-rich glycoprotein–heparin interactions via an allosteric switch on serum albumin. J. Thromb. Haemost. 2015, 13, 101–110. [Google Scholar] [CrossRef]
- Vu, T.T.; Fredenburgh, J.C.; Weitz, J.I. Zinc, an important cofactor in haemostasis and thrombosis. Thomb. Haemost. 2013, 109, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Sanjeevi, N.; Freeland-Graves, J.; Beretvas, S.N.; Sachdev, P.K. Trace element status in type 2 diabetes: A meta-analysis. J. Clin. Diagn. Res. 2018, 12, OE01–OE08. [Google Scholar] [CrossRef]
- Fung, E.B.; Gildengorin, G.; Talwar, S.; Hagar, L.; Lal, A. Zinc status affects glucose homeostasis and insulin secretion in patients with thalassemia. Nutrients 2015, 7, 4296–4307. [Google Scholar] [CrossRef]
- Tanka-Salamon, A.; Komorowicz, E.; Szabo, L.; Tenekedjiev, K.; Kolev, K. Free fatty acids modulate thrombin mediated fibrin generation resulting in less stable clots. PLoS ONE 2016, 11, e0167806. [Google Scholar] [CrossRef]
- Patti, A.M.; Giglio, R.V.; Papanas, N.; Rizzo, M.; Rizvi, A.A. Future perspectives of the pharmacological management of diabetic dyslipidemia. Expert Rev. Clin. Pharmacol. 2019, 12, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Rye, K.A. New era of lipid-lowering drugs. Pharmacol. Rev. 2016, 68, 458–475. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Huttunen, K.M.; Mateusiak, L.; Mikiciuk-Olasik, E.; Sikora, J. Is Metformin a perfect drug? Updates in pharmacokinetics and pharmacodynamics. Curr. Pharmacol. Des. 2017, 23, 2532–2550. [Google Scholar] [CrossRef] [PubMed]
- Pentikainen, P.J.; Voutilainen, E.; Aro, A.; Uusitupa, M.; Penttila, I.; Vapaatalo, H. Cholesterol lowering effect of metformin in combined hyperlipidemia: Placebo controlled double blind trial. Ann. Med. 1990, 22, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Landin, K.; Tengborn, L.; Smith, U. Metformin and metoprolol CR treatment in non-obese men. J. Intern. Med. 1994, 235, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Lehtovirta, M.; Forsen, B.; Gullstrom, M.; Haggblom, M.; Eriksson, J.G.; Taskinen, M.R.; Groop, L. Metabolic effects of metformin in patients with impaired glucose tolerance. Diabet. Med. 2001, 18, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Fruehwald-Schultes, B.; Oltmanns, K.M.; Toschek, B.; Sopke, S.; Kern, W.; Born, J.; Fehm, H.L.; Peters, A. Short-term treatment with metformin decreases serum leptin concentration without affecting body weight and body fat content in normal-weight healthy men. Metabolism 2002, 51, 531–536. [Google Scholar] [CrossRef]
- Krysiak, R.; Okopien, B. Lymphocyte-suppressing and systemic anti-inflammatory effects of high-dose metformin in simvastatin-treated patients with impaired fasting glucose. Atherosclerosis 2012, 225, 403–407. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sondergaard, E.; Christensen, N.L.; Jakobsen, S.; Nielsen, E.H.T.; Munk, O.L.; Tolbod, L.P.; Jessen, N.; Nielsen, S. Metformin does not affect postabsorptive hepatic free fatty acid uptake, oxidation or resecretion in humans: A 3-month placebo-controlled clinical trial in patients with type 2 diabetes and healthy controls. Diabetes Obes. Metab. 2018, 20, 1435–1444. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendia, L.E.; Pedone, C.; Ferretti, G.; Nachtigal, P.; Bo, S.; Derosa, G.; Maffioli, P.; Watts, G.F. Statin therapy and plasma free fatty acids: A systematic review and meta-analysis of controlled clinical trials. Br. J. Clin. Pharmacol. 2016, 81, 807–818. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Plutzky, J. Inflammation in diabetes mellitus: Role of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma agonists. Am. J. Cardiol. 2007, 99, 27B–40B. [Google Scholar] [CrossRef] [PubMed]
- Fenderson, R.W., Jr.; Sekowski, I.; Mohan, C.; Deutsch, S.; Benjamin, F.; Samuel, P. Effect of clofibrate on plasma glucose and serum immunoreactive insulin in patients with hyperlipoproteinemia. Am. J. Clin. Nutr. 1974, 27, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Calvert, G.D.; Blight, L.; Franklin, J.; Oliver, J.; Wise, P.; Gallus, A.S. The effects of clofibrate on plasma glucose, lipoproteins, fibrinogen, and other biochemical and haematological variables in patients with mature onset diabetes mellitus. Eur. J. Clin. Pharmacol. 1980, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.R.; Swai, A.; Taylor, R.; Miller, M.; Laker, M.F.; Alberti, K.G. Lowering of plasma glucose concentrations with bezafibrate in patients with moderately controlled NIDDM. Diabetes Care 1990, 13, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Jones, I.R.; Laker, M.F.; Swai, A.B.; Taylor, R. Effect of bezafibrate on metabolic profiles in non-insulin-dependent diabetes mellitus. J. Cardiovasc. Pharmacol. 1990, 16 (Suppl. 9), S21–S24. [Google Scholar] [CrossRef]
- Vuorinen-Markkola, H.; Yki-Jarvinen, H.; Taskinen, M.R. Lowering of triglycerides by gemfibrozil affects neither the glucoregulatory nor antilipolytic effect of insulin in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993, 36, 161–169. [Google Scholar] [CrossRef]
- Sane, T.; Knudsen, P.; Vuorinen-Markkola, H.; Yki-Jarvinen, H.; Taskinen, M.R. Decreasing triglyceride by gemfibrozil therapy does not affect the glucoregulatory or antilipolytic effect of insulin in nondiabetic subjects with mild hypertriglyceridemia. Metabolism 1995, 44, 589–596. [Google Scholar] [CrossRef]
- Avogaro, A.; Beltramello, P.; Marin, R.; Zambon, S.; Bonanome, A.; Biffanti, S.; Confortin, L.; Manzato, E.; Crepaldi, G.; Tiengo, A. Insulin action and glucose metabolism are improved by gemfibrozil treatment in hypertriglyceridemic patients. Atherosclerosis 1995, 113, 117–124. [Google Scholar] [CrossRef]
- Jeng, C.Y.; Sheu, W.H.; Fuh, M.M.; Shieh, S.M.; Chen, Y.D.; Reaven, G.M. Gemfibrozil treatment of endogenous hypertriglyceridemia: Effect on insulin-mediated glucose disposal and plasma insulin concentrations. J. Clin. Endocrinol. Metab. 1996, 81, 2550–2553. [Google Scholar] [CrossRef]
- Avogaro, A.; Piliego, T.; Catapano, A.; Miola, M.; Tiengo, A. The effect of gemfibrozil on lipid profile and glucose metabolism in hypertriglyceridaemic well-controlled non-insulin-dependent diabetic patients. For the Gemfibrozil Study Group. Acta Diabetol. 1999, 36, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mussoni, L.; Mannucci, L.; Sirtori, C.; Pazzucconi, F.; Bonfardeci, G.; Cimminiello, C.; Notarbartolo, A.; Scafidi, V.; Bittolo Bon, G.; Alessandrini, P.; et al. Effects of gemfibrozil on insulin sensitivity and on haemostatic variables in hypertriglyceridemic patients. Atherosclerosis 2000, 148, 397–406. [Google Scholar] [CrossRef]
- Jonkers, I.J.; de Man, F.H.; van der Laarse, A.; Frolich, M.; Gevers Leuven, J.A.; Kamper, A.M.; Blauw, G.J.; Smelt, A.H. Bezafibrate reduces heart rate and blood pressure in patients with hypertriglyceridemia. J. Hypertens. 2001, 19, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Capell, W.H.; DeSouza, C.A.; Poirier, P.; Bell, M.L.; Stauffer, B.L.; Weil, K.M.; Hernandez, T.L.; Eckel, R.H. Short-term triglyceride lowering with fenofibrate improves vasodilator function in subjects with hypertriglyceridemia. Arter. Thromb. Vasc. Biol. 2003, 23, 307–313. [Google Scholar] [CrossRef]
- Vega, G.L.; Cater, N.B.; Hadizadeh, D.R., 3rd; Meguro, S.; Grundy, S.M. Free fatty acid metabolism during fenofibrate treatment of the metabolic syndrome. Clin. Pharmacol. 2003, 74, 236–244. [Google Scholar] [CrossRef]
- Li, X.M.; Li, Y.; Zhang, N.N.; Xie, Y.H.; Shi, Y.Q. Combination therapy with metformin and fenofibrate for insulin resistance in obesity. J. Int. Med. Res. 2011, 39, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Matsuba, I.; Matsuba, R.; Ishibashi, S.; Yamashita, S.; Arai, H.; Yokote, K.; Suganami, H.; Araki, E. Effects of a novel selective peroxisome proliferator-activated receptor-alpha modulator, pemafibrate, on hepatic and peripheral glucose uptake in patients with hypertriglyceridemia and insulin resistance. J. Diabetes Investig. 2018, 9, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, R.; Zmuda, W.; Okopien, B. The effect of ezetimibe on adipose tissue hormones in patients with isolated hypercholesterolemia. Pharmacol. Rep. 2014, 66, 442–447. [Google Scholar] [CrossRef]
- Sugiyama, S.; Jinnouchi, H.; Hieshima, K.; Kurinami, N.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; Matsui, K.; Jinnouchi, T. A pilot study of ezetimibe vs. atorvastatin for improving peripheral microvascular endothelial function in stable patients with type 2 diabetes mellitus. Lipids Health Dis. 2015, 14, 37. [Google Scholar] [CrossRef]
- Zhang, Y.; Schmidt, R.J.; Foxworthy, P.; Emkey, R.; Oler, J.K.; Large, T.H.; Wang, H.; Su, E.W.; Mosior, M.K.; Eacho, P.I.; et al. Niacin mediates lipolysis in adipose tissue through its G-protein coupled receptor HM74A. Biochem. Biophys. Res. Commun. 2005, 334, 729–732. [Google Scholar] [CrossRef]
- Galescu, O.A.; Crocker, M.K.; Altschul, A.M.; Marwitz, S.E.; Brady, S.M.; Yanovski, J.A. A pilot study of the effects of niacin administration on free fatty acid and growth hormone concentrations in children with obesity. Pediatr. Obes. 2018, 13, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Torrens, S.L.; Areta, J.L.; Parr, E.B.; Hawley, J.A. Carbohydrate dependence during prolonged simulated cycling time trials. Eur. J. Appl. Physiol. 2016, 116, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.H.; Vlazny, D.; Smailovic, A.; Miles, J.M. Intravenous niacin acutely improves the efficiency of dietary fat storage in lean and obese humans. Diabetes 2012, 61, 3172–3175. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.H.; Qamar, A.; Gadi, R.; Lilly, S.; Goel, H.; Hampson, J.; Mucksavage, M.L.; Nathanson, G.A.; Rader, D.J.; Dunbar, R.L. Extended-release niacin acutely suppresses postprandial triglyceridemia. Am. J. Med. 2012, 125, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Basinger, A.; Neese, R.A.; Christiansen, M.; Hellerstein, M.K. Effects of nicotinic acid on fatty acid kinetics, fuel selection, and pathways of glucose production in women. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E50–E59. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Lawson, J.A.; Campbell, L.V.; Storlien, L.H.; Jenkins, A.B.; Whitworth, J.A.; O’Sullivan, A.J. Effects of nicotinic acid on insulin sensitivity and blood pressure in healthy subjects. J. Hum. Hypertens. 2000, 14, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbins, R.; Byerly, R.; Gaddy, R.; Gao, F.; Mahar, K.; Napolitano, A.; Ambery, P.; Le Monnier de Gouville, A.C. GSK256073 acutely regulates NEFA levels via HCA2 agonism but does not achieve durable glycaemic control in type 2 diabetes. A randomised trial. Eur. J. Pharmacol. 2015, 755, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Lauring, B.; Taggart, A.K.; Tata, J.R.; Dunbar, R.; Caro, L.; Cheng, K.; Chin, J.; Colletti, S.L.; Cote, J.; Khalilieh, S.; et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci. Transl. Med. 2012, 4, 148ra115. [Google Scholar] [CrossRef]
- Vega, G.L.; Dunn, F.L.; Grundy, S.M. Effect of colesevelam hydrochloride on glycemia and insulin sensitivity in men with the metabolic syndrome. Am. J. Cardiol. 2011, 108, 1129–1135. [Google Scholar] [CrossRef]
- Chaiken, R.L.; Eckert-Norton, M.; Pasmantier, R.; Boden, G.; Ryan, I.; Gelfand, R.A.; Lebovitz, H.E. Metabolic effects of darglitazone, an insulin sensitizer, in NIDDM subjects. Diabetologia 1995, 38, 1307–1312. [Google Scholar] [CrossRef]
- Buysschaert, M.; Bobbioni, E.; Starkie, M.; Frith, L. Troglitazone in combination with sulphonylurea improves glycaemic control in Type 2 diabetic patients inadequately controlled by sulphonylurea therapy alone. Troglitazone Study Group. Diabet. Med. 1999, 16, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Raskin, P.; Rappaport, E.B.; Cole, S.T.; Yan, Y.; Patwardhan, R.; Freed, M.I. Rosiglitazone short-term monotherapy lowers fasting and post-prandial glucose in patients with type II diabetes. Diabetologia 2000, 43, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Glass, L.; Triplitt, C.; Matsuda, M.; Cusi, K.; Mahankali, A.; Mahankali, S.; Mandarino, L.J.; DeFronzo, R.A. Effect of rosiglitazone on glucose and non-esterified fatty acid metabolism in Type II diabetic patients. Diabetologia 2001, 44, 2210–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerenyi, Z.; Samer, H.; James, R.; Yan, Y.; Stewart, M. Combination therapy with rosiglitazone and glibenclamide compared with upward titration of glibenclamide alone in patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pr. 2004, 63, 213–223. [Google Scholar] [CrossRef]
- James, A.P.; Watts, G.F.; Mamo, J.C. The effect of metformin and rosiglitazone on postprandial lipid metabolism in obese insulin-resistant subjects. Diabetes Obes. Metab. 2005, 7, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.D.; Fielding, B.A.; Currie, J.M.; Humphreys, S.M.; Desage, M.; Frayn, K.N.; Laville, M.; Vidal, H.; Karpe, F. The effects of rosiglitazone on fatty acid and triglyceride metabolism in type 2 diabetes. Diabetologia 2005, 48, 83–95. [Google Scholar] [CrossRef]
- Samaha, F.F.; Szapary, P.O.; Iqbal, N.; Williams, M.M.; Bloedon, L.T.; Kochar, A.; Wolfe, M.L.; Rader, D.J. Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high-density lipoprotein cholesterol and metabolic syndrome. Arter. Thromb. Vasc. Biol. 2006, 26, 624–630. [Google Scholar] [CrossRef]
- Mittermayer, F.; Schaller, G.; Pleiner, J.; Krzyzanowska, K.; Kapiotis, S.; Roden, M.; Wolzt, M. Rosiglitazone prevents free fatty acid-induced vascular endothelial dysfunction. J. Clin. Endocrinol. Metab. 2007, 92, 2574–2580. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Cersosimo, E.; Triplitt, C.; DeFronzo, R.A. Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 2007, 72, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Krzyzanowska, K.; Mittermayer, F.; Krugluger, W.; Roden, M.; Schernthaner, G.; Wolzt, M. Adiponectin concentrations increase during acute FFA elevation in humans treated with rosiglitazone. Horm. Metab. Res. 2007, 39, 769–772. [Google Scholar] [CrossRef]
- Abbasi, F.; Chen, Y.D.; Farin, H.M.; Lamendola, C.; Reaven, G.M. Comparison of three treatment approaches to decreasing cardiovascular disease risk in nondiabetic insulin-resistant dyslipidemic subjects. Am. J. Cardiol. 2008, 102, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Punthakee, Z.; Almeras, N.; Despres, J.P.; Dagenais, G.R.; Anand, S.S.; Hunt, D.L.; Sharma, A.M.; Jung, H.; Yusuf, S.; Gerstein, H.C. Impact of rosiglitazone on body composition, hepatic fat, fatty acids, adipokines and glucose in persons with impaired fasting glucose or impaired glucose tolerance: A sub-study of the DREAM trial. Diabet. Med. 2014, 31, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Kim, D.M.; Woo, J.T.; Jang, H.C.; Chung, C.H.; Ko, K.S.; Park, J.H.; Park, Y.S.; Kim, S.J.; Choi, D.S. Efficacy and safety of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 24-weeks: A multicenter, randomized, double-blind, parallel-group, placebo controlled trial. PLoS ONE 2014, 9, e92843. [Google Scholar] [CrossRef] [PubMed]
- Farsi, P.F.; Djazayery, A.; Eshraghian, M.R.; Koohdani, F.; Saboor-Yaraghi, A.A.; Derakhshanian, H.; Zarei, M.; Javanbakht, M.H.; Djalali, M. Effects of supplementation with omega-3 on insulin sensitivity and non-esterified free fatty acid (NEFA) in type 2 diabetic patients. Arq. Bras. Endocrinol. Metab. 2014, 58, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.D.; Rubino, J.; Janik, M.J.; MacDougall, D.E.; McBride, S.J.; Margulies, J.R.; Newton, R.S. Use of ETC-1002 to treat hypercholesterolemia in patients with statin intolerance. J. Clin. Lipidol. 2015, 9, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Perez, F.J.; Fanghanel-Salmon, G.; Antonio Barbosa, J.; Montes-Villarreal, J.; Berry, R.A.; Warsi, G.; Gould, E.M. Efficacy and safety of rosiglitazone plus metformin in Mexicans with type 2 diabetes. Diabetes Metab. Res. Rev. 2002, 18, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Larson, P.J.; Weiss, S.; Miller, J.L.; Doebber, T.W.; Wu, M.S.; Moller, D.E.; Gottesdiener, K.M. Individual and combined effects of peroxisome proliferator-activated receptor and {gamma} agonists, fenofibrate and rosiglitazone, on biomarkers of lipid and glucose metabolism in healthy nondiabetic volunteers. J. Clin. Pharmacol. 2005, 45, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Homko, C.; Mozzoli, M.; Zhang, M.; Kresge, K.; Cheung, P. Combined use of rosiglitazone and fenofibrate in patients with type 2 diabetes: Prevention of fluid retention. Diabetes 2007, 56, 248–255. [Google Scholar] [CrossRef]
- Plat, J.; Brufau, G.; Dallinga-Thie, G.M.; Dasselaar, M.; Mensink, R.P. A plant stanol yogurt drink alone or combined with a low-dose statin lowers serum triacylglycerol and non-HDL cholesterol in metabolic syndrome patients. J. Nutr. 2009, 139, 1143–1149. [Google Scholar] [CrossRef]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef]
- Krysiak, R.; Zmuda, W.; Okopien, B. The effect of simvastatin-ezetimibe combination therapy on adipose tissue hormones and systemic inflammation in patients with isolated hypercholesterolemia. Cardiovasc. Ther. 2014, 32, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.C.; Jun, J.E.; Jeong, I.K.; Ahn, K.J.; Chung, H.Y. Comparison of the efficacy of Rosuvastatin monotherapy 20 mg with Rosuvastatin 5 mg and Ezetimibe 10 mg combination therapy on lipid parameters in patients with type 2 diabetes mellitus. Diabetes Metab. J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kanda, S.; Nakashima, R.; Takahashi, K.; Tanaka, J.; Ogawa, J.; Ogata, T.; Yachi, M.; Araki, K.; Ohsumi, J. Potent antidiabetic effects of rivoglitazone, a novel peroxisome proliferator-activated receptor-gamma agonist, in obese diabetic rodent models. J. Pharmacol. Sci. 2009, 111, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Conquer, J.A.; Holub, B.J. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J. Lipid Res. 1998, 39, 286–292. [Google Scholar] [PubMed]
- Conquer, J.A.; Cheryk, L.A.; Chan, E.; Gentry, P.A.; Holub, B.J. Effect of supplementation with dietary seal oil on selected cardiovascular risk factors and hemostatic variables in healthy male subjects. Thromb. Res. 1999, 96, 239–250. [Google Scholar] [CrossRef]
- Barre, D.E.; Mizier-Barre, K.A.; Griscti, O.; Hafez, K. Flaxseed oil supplementation manipulates correlations between serum individual mol % free fatty acid levels and insulin resistance in type 2 diabetics. Insulin resistance and percent remaining pancreatic beta-cell function are unaffected. Endocr. Regul. 2016, 50, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Carey, A.L.; Natoli, A.K.; Formosa, M.F.; Vizi, D.; Reddy-Luthmoodoo, M.; Weir, J.M.; Barlow, C.K.; van Hall, G.; Meikle, P.J.; et al. Reconstituted high-density lipoprotein infusion modulates fatty acid metabolism in patients with type 2 diabetes mellitus. J. Lipid Res. 2011, 52, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Poudyal, H.; Brown, L. Should the pharmacological actions of dietary fatty acids in cardiometabolic disorders be classified based on biological or chemical function? Prog. Lipid Res. 2015, 59, 172–200. [Google Scholar] [CrossRef]
- Gao, H.; Geng, T.; Huang, T.; Zhao, Q. Fish oil supplementation and insulin sensitivity: A systematic review and meta-analysis. Lipids Health Dis. 2017, 16, 131. [Google Scholar] [CrossRef]
- Jeromson, S.; Gallagher, I.J.; Galloway, S.D.; Hamilton, D.L. Omega-3 Fatty Acids and Skeletal Muscle Health. Mar. Drugs 2015, 13, 6977–7004. [Google Scholar] [CrossRef]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vazquez-Carrera, M. Palmitic and oleic acid: The Yin and Yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, C.; Schiller, K.; Schulze, M.B. Omega-3 and omega-6 fatty acids and type 2 diabetes. Curr. Diab. Rep. 2013, 13, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Balk, E.M.; Adams, G.P.; Langberg, V.; Halladay, C.; Chung, M.; Lin, L.; Robertson, S.; Yip, A.; Steele, D.; Smith, B.T.; et al. Omega-3 Fatty Acids and Cardiovascular Disease: An Updated Systematic Review. Evid. Rep. Technol. Assess. (Full. Rep.) 2016, 223, 1–1252. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
I. S. Sobczak, A.; A. Blindauer, C.; J. Stewart, A. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. https://doi.org/10.3390/nu11092022
I. S. Sobczak A, A. Blindauer C, J. Stewart A. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients. 2019; 11(9):2022. https://doi.org/10.3390/nu11092022
Chicago/Turabian StyleI. S. Sobczak, Amélie, Claudia A. Blindauer, and Alan J. Stewart. 2019. "Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes" Nutrients 11, no. 9: 2022. https://doi.org/10.3390/nu11092022
APA StyleI. S. Sobczak, A., A. Blindauer, C., & J. Stewart, A. (2019). Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients, 11(9), 2022. https://doi.org/10.3390/nu11092022