The Impact of Maternal Body Composition and Dietary Fat Consumption upon Placental Lipid Processing and Offspring Metabolic Health



Abstract
:1. Introduction
2. Maternal Obesity and Offspring Metabolic Health
3. Is Maternal BMI an Accurate Predictor of Offspring Metabolic Health?
4. Maternal Dietary Fat Consumption and Offspring Metabolic Health
5. The Impact of Diet and Obesity upon the Placenta
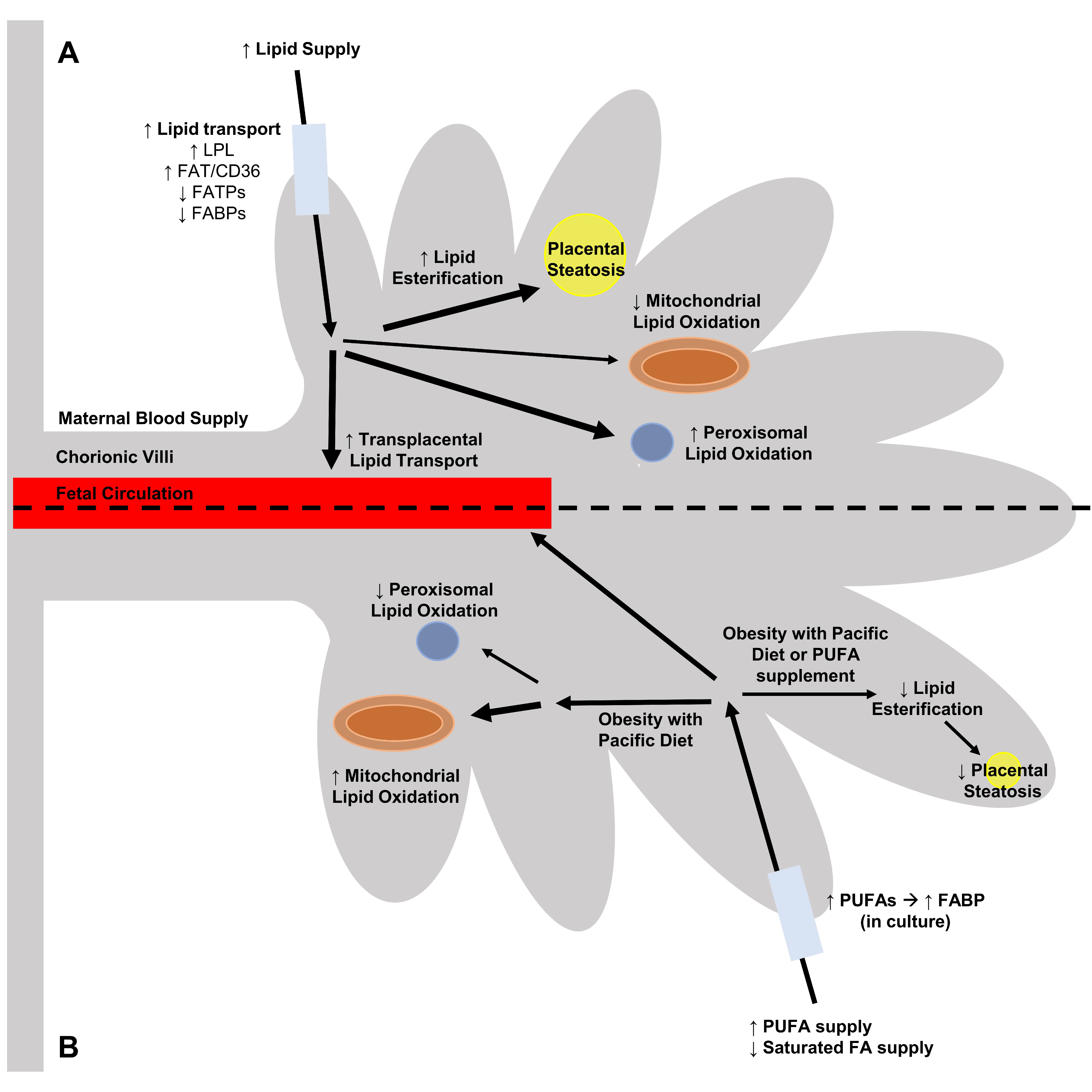
6. Regulation of Placental Lipid Transport in Obesity and the Impact of Dietary Fats
7. Obesity, Diet and Placental Lipid Accumulation
8. Diet and Placental Lipid Oxidation and Acylcarnitine Production in the Obese Environment
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Catalano, P.M.; Tyzbir, E.D.; Roman, N.M.; Amini, S.B.; Sims, E.A. Longitudinal changes in insulin release and insulin resistance in nonobese pregnant women. Am. J. Obstet. Gynecol. 1991, 165, 1667–1672. [Google Scholar] [CrossRef]
- Desoye, G.; Schweditsch, M.O.; Pfeiffer, K.P.; Zechner, R.; Kostner, G.M. Correlation of Hormones with Lipid and Lipoprotein Levels During Normal Pregnancy and Postpartum. J. Clin. Endocrinol. Metab. 1987, 64, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Musial, B.; Vaughan, O.R.; Fernandez-Twinn, D.S.; Voshol, P.; Ozanne, S.E.; Fowden, A.L.; Sferruzzi-Perri, A.N. A Western-style obesogenic diet alters maternal metabolic physiology with consequences for fetal nutrient acquisition in mice. J. Physiol. 2017, 595, 4875–4892. [Google Scholar] [CrossRef] [PubMed]
- Silveira, P.P.; Portella, A.; Goldani, M.Z.; Barbieri, A.M. Developmental origins of health and disease (DOHaD). J. Pediatr. 2007, 83, 494–504. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental Origins of Health and Disease: Brief History of the Approach and Current Focus on Epigenetic Mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Forsdahl, A. Are poor living conditions in childhood and adolescence an important risk factor for arteriosclerotic heart disease? J. Epidemiol. Community Health 1977, 31, 91–95. [Google Scholar] [CrossRef] [Green Version]
- King, J.C. Maternal Obesity, Metabolism, and Pregnancy Outcomes. Annu. Rev. Nutr. 2006, 26, 271–291. [Google Scholar] [CrossRef]
- Wu, G.; Bazer, F.W.; Cudd, T.A.; Meininger, C.J.; Spencer, T.E. Recent Advances in Nutritional Sciences Maternal Nutrition and Fetal. Amino Acids 2004, 134, 2169–2172. [Google Scholar]
- Obesity: Preventing and Managing the Global Epidemic; Report of a WHO Consultation; World Health Organ. Tech.: Geneva, Switzerland, 2000; pp. 1–253.
- McDonald, S.D.; Han, Z.; Mulla, S.; Beyene, J. On behalf of the Knowledge Synthesis Group Overweight and obesity in mothers and risk of preterm birth and low birth weight infants: Systematic review and meta-analyses. BMJ 2010, 341, c3428. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Han, S.; Zhu, J.; Sun, X.; Ji, C.; Guo, X. Pre-Pregnancy Body Mass Index in Relation to Infant Birth Weight and Offspring Overweight/Obesity: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e61627. [Google Scholar] [CrossRef] [Green Version]
- Da Silveira, V.M.F.; Horta, B.L. Peso ao nascer e síndrome metabólica em adultos: Meta-análise. Revista De Saúde Pública 2008, 42, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boney, C.M. Metabolic Syndrome in Childhood: Association with Birth Weight, Maternal Obesity, and Gestational Diabetes Mellitus. Pediatrics 2005, 115, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, R.C. Predicting preschooler obesity at birth: The role of maternal obesity in early pregnancy. Pediatrics 2004, 114, e29–e36. [Google Scholar] [CrossRef] [Green Version]
- Heerwagen, M.J.R.; Miller, M.R.; Barbour, L.A.; Friedman, J.E. Maternal obesity and fetal metabolic programming: A fertile epigenetic soil. Am. J. Physiol. Integr. Comp. Physiol. 2010, 299, R711–R722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, L.; Seki, Y.; Vuguin, P.M.; Charron, M.J. Animal models of in utero exposure to a high fat diet: A review. Biochim. Biophys. Acta-Bioenerg. 2013, 1842, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.N.; Woollett, L.A.; Barbour, N.; Prasad, P.D.; Powell, T.L.; Jansson, T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 2008, 23, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef]
- Li, M.; Sloboda, D.M.; Vickers, M.H. Maternal Obesity and Developmental Programming of Metabolic Disorders in Offspring: Evidence from Animal Models. Exp. Diabetes Res. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Elahi, M.M.; Cagampang, F.R.; Mukhtar, D.; Anthony, F.W.; Ohri, S.K.; Hanson, M.A. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br. J. Nutr. 2009, 102, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Howie, G.J.; Sloboda, D.M.; Kamal, T.; Vickers, M.H. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J. Physiol. 2008, 587, 905–915. [Google Scholar] [CrossRef]
- Samuelsson, A.-M.; Matthews, P.A.; Argenton, M.; Christie, M.; McConnell, J.M.; Jansen, E.H.M.; Piersma, A.H.; Ozanne, S.E.; Fernandez-Twinn, D.S.; Remacle, C.; et al. Diet-Induced Obesity in Female Mice Leads to Offspring Hyperphagia, Adiposity, Hypertension, and Insulin Resistance: A Novel Murine Model of Developmental Programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.M.; Zimmet, P.; Shaw, J.E. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Rkhzay-Jaf, J.; O’Dowd, J.F.; Stocker, C.J. Maternal Obesity and the Fetal Origins of the Metabolic Syndrome. Curr. Cardiovasc. Risk Rep. 2012, 6, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, N.M.; Rule, D.C.; Tuersunjiang, N.; Nathanielsz, P.W.; Ford, S.P. Maternal Obesity in Sheep Increases Fatty Acid Synthesis, Upregulates Nutrient Transporters, and Increases Adiposity in Adult Male Offspring after a Feeding Challenge. PLoS ONE 2015, 10, e0122152. [Google Scholar] [CrossRef] [PubMed]
- Philp, L.K.; Muhlhausler, B.S.; Janovská, A.; Wittert, G.A.; Duffield, J.A.; McMillen, I.C. Maternal overnutrition suppresses the phosphorylation of 5′-AMP-activated protein kinase in liver, but not skeletal muscle, in the fetal and neonatal sheep. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R1982–R1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuersunjiang, N.; Odhiambo, J.F.; Long, N.M.; Shasa, D.R.; Nathanielsz, P.W.; Ford, S.P. Diet reduction to requirements in obese/overfed ewes from early gestation prevents glucose/insulin dysregulation and returns fetal adiposity and organ development to control levels. Am. J. Physiol. Metab. 2013, 305, E868–E878. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Pound, L.D.; Comstock, S.M.; Grove, K.L. Consumption of a Western-style diet during pregnancy impairs offspring islet vascularization in a Japanese macaque model. Am. J. Physiol. Metab. 2014, 307, E115–E123. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC) Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 2016, 387, 1377–1396. [CrossRef] [Green Version]
- Women in Canada: A Gender-Based Statistical Report (89-503-X); 2012–2013 Canadian Health Measures Survey; Statistics Canada: Ottawa, ON, Canada, 2013.
- Wesolowski, S.R.; Mulligan, C.M.; Janssen, R.C.; Baker, P.R.; Bergman, B.C.; D’Alessandro, A.; Nemkov, T.; MacLean, K.N.; Jiang, H.; Dean, T.A.; et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Mol. Metab. 2018, 18, 25–41. [Google Scholar] [CrossRef]
- Zambrano, E.; Martínez-Samayoa, P.M.; Rodríguez-González, G.L.; Nathanielsz, P.W. Dietary intervention prior to pregnancy reverses metabolic programming in male offspring of obese rats. J. Physiol. 2010, 588, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Sasson, I.E.; Vitins, A.P.; Mainigi, M.A.; Moley, K.H.; Simmons, R.A. Pre-gestational vs gestational exposure to maternal obesity differentially programs the offspring in mice. Diabetologia 2014, 58, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borengasser, S.J.; Kang, P.; Faske, J.; Gomez-Acevedo, H.; Blackburn, M.L.; Badger, T.M.; Shankar, K. High Fat Diet and In Utero Exposure to Maternal Obesity Disrupts Circadian Rhythm and Leads to Metabolic Programming of Liver in Rat Offspring. PLoS ONE 2014, 9, e84209. [Google Scholar] [CrossRef] [Green Version]
- Borengasser, S.J.; Faske, J.; Kang, P.; Blackburn, M.L.; Badger, T.M.; Shankar, K. In utero exposure to prepregnancy maternal obesity and postweaning high-fat diet impair regulators of mitochondrial dynamics in rat placenta and offspring. Physiol. Genom. 2014, 46, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Swanson, A.; David, A. Animal models of fetal growth restriction: Considerations for translational medicine. Placenta 2015, 36, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.L.; Botting, K.J.; Darby, J.R.; David, A.L.; Dyson, R.M.; Gatford, K.L.; Gray, C.; Herrera, E.A.; Hirst, J.J.; Kim, B.; et al. Guinea pig models for translation of the developmental origins of health and disease hypothesis into the clinic. J. Physiol. 2018, 596, 5535–5569. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, P.C.; Chicaybam, G.; Ramos-Filho, D.M.; Dos Santos, R.M.A.R.; Mairink, C.; Sardinha, F.L.C.; El-Bacha, T.; Galina, A.; Tavares-Do-Carmo, M.D.G. Maternal intake of trans-unsaturated or interesterified fatty acids during pregnancy and lactation modifies mitochondrial bioenergetics in the liver of adult offspring in mice. Br. J. Nutr. 2017, 118, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.-M.; Li, Y.; Ning, H.; Wang, C.; Liu, J.; Sun, C. High dietary intake of medium-chain fatty acids during pregnancy in rats prevents later-life obesity in their offspring. J. Nutr. Biochem. 2011, 22, 791–797. [Google Scholar] [CrossRef]
- Zhu, M.; Du, M.; Nathanielsz, P.W.; Ford, S. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 2010, 31, 387–391. [Google Scholar] [CrossRef]
- Salati, J.A.; Roberts, V.H.; Schabel, M.C.; Lo, J.O.; Kroenke, C.D.; Lewandowski, K.S.; Lindner, J.R.; Grove, K.L.; Frias, A.E. Maternal high-fat diet reversal improves placental hemodynamics in a nonhuman primate model of diet-induced obesity. Int. J. Obes. 2018, 43, 906–916. [Google Scholar] [CrossRef]
- Gademan, M.; Vermeulen, M.; Oostvogels, A.J.J.M.; Roseboom, T.J.; Visscher, T.L.S.; Van Eijsden, M.; Twickler, M.T.B.; Vrijkotte, T.G.M. Maternal Prepregancy BMI and Lipid Profile during Early Pregnancy Are Independently Associated with Offspring’s Body Composition at Age 5–6 Years: The ABCD Study. PLoS ONE 2014, 9, e94594. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.E.; Stolinski, M.; Smith, R.D.; Murphy, J.L.; AWootton, S. Effect of fatty acid chain length and saturation on the gastrointestinal handling and metabolic disposal of dietary fatty acids in women. Br. J. Nutr. 1999, 81, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensink, R.P.; Zock, P.; Kester, A.D.M.; Katan, M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: A meta-analysis of 60 controlled trials. Am. J. Clin. Nutr. 2003, 77, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Delarue, J.; Le Foll, C.; Corporeau, C.; Lucas, D. N-3 long chain polyunsaturated fatty acids: A nutritional tool to prevent insulin resistance associated to type 2 diabetes and obesity? Reprod. Nutr. Dev. 2004, 44, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, Y.B.; Hein, G.; Chicco, A. Metabolic Syndrome: Effects of n-3 PUFAs on a Model of Dyslipidemia, Insulin Resistance and Adiposity. Lipids 2007, 42, 427–437. [Google Scholar] [CrossRef]
- Diniz, Y.S.; Cicogna, A.C.; Padovani, C.R.; Santana, L.S.; AFaine, L.; Novelli, E.L. Diets rich in saturated and polyunsaturated fatty acids: Metabolic shifting and cardiac health. Nutrition 2004, 20, 230–234. [Google Scholar] [CrossRef]
- Makrides, M. Is there a dietary requirement for DHA in pregnancy? Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 171–174. [Google Scholar] [CrossRef]
- Savard, C.; Lemieux, S.; Weisnagel, S.J.; Fontaine-Bisson, B.; Gagnon, C.; Robitaille, J.; Morisset, A.-S. Trimester-Specific Dietary Intakes in a Sample of French-Canadian Pregnant Women in Comparison with National Nutritional Guidelines. Nutrition 2018, 10, 768. [Google Scholar] [CrossRef] [Green Version]
- Watts, V.; Rockett, H.; Baer, H.J.; Leppert, J.; Colditz, G.A. Assessing Diet Quality in a Population of Low-Income Pregnant Women: A Comparison Between Native Americans and Whites. Matern. Child. Health J. 2006, 11, 127–136. [Google Scholar] [CrossRef]
- Denomme, J.; Stark, K.D.; Holub, B.J. Directly Quantitated Dietary (n-3) Fatty Acid Intakes of Pregnant Canadian Women Are Lower than Current Dietary Recommendations. J. Nutr. 2005, 135, 206–211. [Google Scholar] [CrossRef]
- Siega-Riz, A.M.; Bodnar, L.M.; Savitz, D.A. What are pregnant women eating? Nutrient and food group differences by race. Am. J. Obstet. Gynecol. 2002, 186, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M.; Elias, S.L. Intakes of essential n−6 and n−3 polyunsaturated fatty acids among pregnant Canadian women. Am. J. Clin. Nutr. 2003, 77, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gude, N.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Mele, J.; Muralimanoharan, S.; Maloyan, A.; Myatt, L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am. J. Physiol. Metab. 2014, 307, E419–E425. [Google Scholar] [CrossRef] [Green Version]
- Maloyan, A.; Mele, J.; Muralimanoharan, S.; Myatt, L. Placental metabolic flexibility is affected by maternal obesity. Placenta 2016, 45, 69. [Google Scholar] [CrossRef]
- Kolahi, K.S.; Valent, A.M.; Thornburg, K.L. Cytotrophoblast, Not Syncytiotrophoblast, Dominates Glycolysis and Oxidative Phosphorylation in Human Term Placenta. Sci. Rep. 2017, 7, srep42941. [Google Scholar] [CrossRef] [Green Version]
- Nugent, B.; Bale, T.L. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front. Neuroendocr. 2015, 39, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.; Riley, S.; Reynolds, R.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.; Norman, J.; Denison, F. Placental structure and inflammation in pregnancies associated with obesity. Placenta 2011, 32, 247–254. [Google Scholar] [CrossRef]
- Challier, J.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.; Mouzon, S.H.-D. Obesity in Pregnancy Stimulates Macrophage Accumulation and Inflammation in the Placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Hastie, R.; Lappas, M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta 2014, 35, 673–683. [Google Scholar] [CrossRef]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. 2007, 113, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Schust, D.J. Isolation, purification and in vitro differentiation of cytotrophoblast cells from human term placenta. Reprod. Boil. Endocrinol. 2015, 13, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Figueroa, H.; Chien, E.K.; Ji, H.; Nesbitt, N.L.; Bharathi, S.S.; Goetzman, E. Effects of labor on placental fatty acid β oxidation. J. Matern. Neonatal Med. 2012, 26, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nersisyan, S.A.; Shkurnikov, M.Y.; Knyazev, E.N. Factors Involved in miRNA Processing Change Its Expression Level during Imitation of Hypoxia in BeWo b30 Cells. Dokl. Biochem. Biophys. 2020, 493, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Abaidoo, C.; Warren, M.A.; Andrews, P.W.; Boateng, K.A. A quantitative Assessment of the Morphological Characteristics of BeWo Cells as an in vitro Model of Human Trophoblast Cells. Int. J. Morphol. 2010, 28, 1047–1058. [Google Scholar] [CrossRef]
- Campbell, F.M.; Bush, P.G.; Veerkamp, J.H.; Dutta-Roy, A.K. Detection and cellular localization of plasma membrane-associated and cytoplasmic fatty acid-binding proteins in human placenta. Placenta 1998, 19, 409–415. [Google Scholar] [CrossRef]
- Larqué, E.; Demmelmair, H.; Klingler, M.; De Jonge, S.; Bondy, B.; Koletzko, B. Expression pattern of fatty acid transport protein-1 (FATP-1), FATP-4 and heart-fatty acid binding protein (H-FABP) genes in human term placenta. Early Hum. Dev. 2006, 82, 697–701. [Google Scholar] [CrossRef]
- Haggarty, P.; Ashton, J.; Joynson, M.; Abramovich, D.R.; Page, K. Effect of Maternal Polyunsaturated Fatty Acid Concentration on Transport by the Human Placenta. Biol. Neonate 1999, 75, 350–359. [Google Scholar] [CrossRef]
- Duttaroy, A.K.; Basak, S. Maternal dietary fatty acids and their roles in human placental development. Prostaglandins Leukot. Essent. Fat. Acids 2020, 155, 102080. [Google Scholar] [CrossRef]
- Dubé, E.; Gravel, A.; Martin, C.; Desparois, G.; Moussa, I.; Ethier-Chiasson, M.; Forest, J.-C.; Giguère, Y.; Masse, A.; Lafond, J. Modulation of Fatty Acid Transport and Metabolism by Maternal Obesity in the Human Full-Term Placenta1. Biol. Reprod. 2012, 87. [Google Scholar] [CrossRef]
- Segura, M.T.; Demmelmair, H.; Krauss-Etschmann, S.; Nathan, P.; Dehmel, S.; Padilla, M.C.; Rueda, R.; Koletzko, B.; Campoy, C. Maternal BMI and gestational diabetes alter placental lipid transporters and fatty acid composition. Placenta 2017, 57, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.; Filis, P.; Soffientini, U.; Bellingham, M.; O’Shaughnessy, P.J.; Fowler, P.A. Placental transporter localization and expression in the Human: The importance of species, sex, and gestational age differences†. Biol. Reprod. 2017, 96, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Sánchez, A.; Demmelmair, H.; Parrilla, J.J.; Koletzko, B.; Larqué, E. Mechanisms involved in the selective transfer of long chain polyunsaturated fatty acids to the fetus. Front. Genet. 2011, 2, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, K.A.R.; Johnsen, G.M.; Staff, A.C.; Duttaroy, A.K. Long-chain Polyunsaturated Fatty Acid Transport across Human Placental Choriocarcinoma (BeWo) Cells. Placenta 2009, 30, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Leroy, C.; Tobin, K.A.R.; Basak, S.; Cathrine Staff, A.; Duttaroy, A.K. Fatty acid-binding protein3 expression in BeWo cells, a human placental choriocarcinoma cell line. Prostaglandins Leukot. Essent. Fat. Acids 2017, 120, 1–7. [Google Scholar] [CrossRef]
- Chassen, S.S.; Ferchaud-Roucher, V.; Palmer, C.; Li, C.; Jansson, T.; Nathanielsz, P.W.; Powell, T.L. Placental fatty acid transport across late gestation in a baboon model of intrauterine growth restriction. J. Physiol. 2020, JP279398. [Google Scholar] [CrossRef]
- Larqué, E.; Krauss-Etschmann, S.; Campoy, C.; Hartl, D.; Linde, J.; Klingler, M.; Demmelmair, H.; Caño, A.; Gil, A.; Bondy, B.; et al. Docosahexaenoic acid supply in pregnancy affects placental expression of fatty acid transport proteins. Am. J. Clin. Nutr. 2006, 84, 853–861. [Google Scholar] [CrossRef]
- Szabo, A.J.; de Lellis, R.; Grimaldi, R.D. Triglyceride synthesis by the human placenta. Am. J. Obstet. Gynecol. 1973, 115, 257–262. [Google Scholar] [CrossRef]
- Pathmaperuma, A.N.; Maña, P.; Cheung, S.N.; Kugathas, K.; Josiah, A.; Koina, M.E.; Broomfield, A.; Delghingaro-Augusto, V.; Ellwood, D.A.; Dahlstrom, J.E. Fatty acids alter glycerolipid metabolism and induce lipid droplet formation, syncytialisation and cytokine production in human trophoblasts with minimal glucose effect or interaction. Placenta 2010, 31, 230–239. [Google Scholar] [CrossRef]
- Perazzolo, S.; Hirschmugl, B.; Wadsack, C.; Desoye, G.; Lewis, R.M.; Sengers, B.G. The influence of placental metabolism on fatty acid transfer to the fetus. J. Lipid Res. 2017, 58, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Kolahi, K.; Louey, S.; Varlamov, O.; Thornburg, K. Real-time tracking of BODIPY-C12 long-chain fatty acid in human term placenta reveals unique lipid dynamics in cytotrophoblast cells. PLoS ONE 2016, 11, e0153522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margariti, E.; Deutsch, M.; Manolakopoulos, S.; Kaflri, G.; Tiniakos, D.; Papatheodoridis, G.V. Non-alcoholic fatty liver disease (nafld) may develop in patients with normal body mass index (BMI). J. Hepatol. 2011, 54, S340. [Google Scholar] [CrossRef]
- Calabuig-Navarro, V.; Haghiac, M.; Minium, J.; Glazebrook, P.; Ranasinghe, G.C.; Hoppel, C.; Hauguel de-Mouzon, S.; Catalano, P.; O’Tierney-Ginn, P. Effect of Maternal Obesity on Placental Lipid Metabolism. Endocrinol. 2017, 158, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Calabuig-Navarro, V.; Puchowicz, M.; Glazebrook, P.; Haghiac, M.; Minium, J.; Catalano, P.; Hauguel de Mouzon, S.; O’Tierney-Ginn, P. Effect of ω-3 supplementation on placental lipid metabolism in overweight and obese women. Am. J. Clin. Nutr. 2016, 103, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Cetin, I.; Parisi, F.; Berti, C.; Mandò, C.; Desoye, G. Placental fatty acid transport in maternal obesity. J. Dev. Orig. Health Dis. 2012, 3, 409–414. [Google Scholar] [CrossRef]
- Gázquez, A.; Uhl, O.; Ruíz-Palacios, M.; Gill, C.; Patel, N.; Koletzko, B.; Poston, L.; Larqué, E. Placental lipid droplet composition: Effect of a lifestyle intervention (UPBEAT) in obese pregnant women. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2018, 1863, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Lim, W.; Bazer, F.W.; Song, G. Down-regulation of stearoyl-CoA desaturase-1 increases susceptibility to palmitic-acid-induced lipotoxicity in human trophoblast cells. J. Nutr. Biochem. 2018, 54, 35–47. [Google Scholar] [CrossRef]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA Desaturase Is Required to Produce Active, Lipid-Modified Wnt Proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef] [Green Version]
- Strakovsky, R.S.; Pan, Y.-X. A Decrease in DKK1, a WNT Inhibitor, Contributes to Placental Lipid Accumulation in an Obesity-Prone Rat Model1. Biol. Reprod. 2012, 86. [Google Scholar] [CrossRef]
- Alvarado, F.L.; Calabuig-Navarro, V.; Haghiac, M.; Puchowicz, M.; Tsai, P.-J.S.; O’Tierney-Ginn, P. Maternal obesity is not associated with placental lipid accumulation in women with high omega-3 fatty acid levels. Placenta 2018, 69, 96–101. [Google Scholar] [CrossRef]
- Middleton, P.; Gomersall, J.C.; Gould, J.F.; Shepherd, E.; Olsen, S.F.; Makrides, M. Omega-3 fatty acid addition during pregnancy. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vinding, R.K.; Stokholm, J.; Sevelsted, A.; Chawes, B.L.; Bønnelykke, K.; Barman, M.; Jacobsson, B.; Bisgaard, H. Fish Oil Supplementation in Pregnancy Increases Gestational Age, Size for Gestational Age, and Birth Weight in Infants: A Randomized Controlled Trial. J. Nutr. 2019, 149, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Shekhawat, P.; Bennett, M.J.; Sadovsky, Y.; Nelson, D.M.; Rakheja, D.; Strauss, A.W. Human placenta metabolizes fatty acids: Implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1098–E1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oey, N.A.; Den Boer, M.E.J.; Ruiter, J.P.N.; Wanders, R.J.A.; Wanders, R.J.A.; Duran, M.; Waterham, H.R.; Boer, K.; van der Post, J.A.M.; Wijburg, F.A.; et al. High activity of fatty acid oxidation enzymes in human placenta: Implications for fetal-maternal disease. J. Inherit. Metab. Dis. 2003, 26, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Bennett, M.J.; Foster, B.M.; Domiati-Saad, R.; Rogers, B.B. Evidence for Fatty Acid Oxidation in Human Placenta, and the Relationship of Fatty Acid Oxidation Enzyme Activities with Gestational Age. Placenta 2002, 23, 447–450. [Google Scholar] [CrossRef]
- Boyle, K.E.; Patinkin, Z.W.; Shapiro, A.L.B.; Bader, C.; Vanderlinden, L.; Kechris, K.; Janssen, R.C.; Ford, R.J.; Smith, B.K.; Steinberg, G.R.; et al. Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants. Mol. Metab. 2017, 6, 1503–1516. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T. Peroxisomal beta-oxidation enzymes. Cell Biochem. Biophys. 2000, 32, 63–72. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.-Y.; Zheng, D.; Hickner, R.C.; Brault, J.J.; Cortright, R.N. Peroxisomal gene and protein expression increase in response to a high-lipid challenge in human skeletal muscle. Metabolism 2019, 98, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.H.; Radloff, J.F.; Hull, F.E.; Sweeley, C.C. Incomplete fatty acid oxidation by ischemic heart: Beta-hydroxy fatty acid production. Am. J. Physiol. 1980, 239, H257–H265. [Google Scholar] [CrossRef] [PubMed]
- Rutkowsky, J.M.; Knotts, T.A.; Ono-Moore, K.D.; McCoin, C.S.; Huang, S.; Schneider, D.; Singh, S.; Adams, S.H.; Hwang, D.H. Acylcarnitines activate proinflammatory signaling pathways. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1378–E1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.R.; Boyle, K.E.; Koves, T.R.; Ilkayeva, O.R.; Muoio, D.M.; Houmard, J.A.; Friedman, J.E. Metabolomic analysis reveals altered skeletal muscle amino acid and fatty acid handling in obese humans. Obesity 2015, 23, 981–988. [Google Scholar] [CrossRef]
- Baker, P.R.; Patinkin, Z.; Shapiro, A.L.; De La Houssaye, B.A.; Woontner, M.; Boyle, K.E.; Vanderlinden, L.; Dabelea, D.; Friedman, J.E. Maternal obesity and increased neonatal adiposity correspond with altered infant mesenchymal stem cell metabolism. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [Green Version]
- Turer, A.T.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; van der Westhuizen, J.; Mathew, J.P.; Schwinn, D.A.; Glower, D.D.; Newgard, C.B.; Podgoreanu, M.V. Metabolomic profiling reveals distinct patterns of myocardial substrate use in humans with coronary artery disease or left ventricular dysfunction during surgical ischemia/reperfusion. Circulation 2009, 119, 1736–1746. [Google Scholar] [CrossRef] [Green Version]
- Thiele, I.G.I.; Niezen-Koning, K.E.; van Gennip, A.H.; Aarnoudse, J.G. Increased Plasma Carnitine Concentrations in Preeclampsia. Obstet. Gynecol. 2004, 103, 876–880. [Google Scholar] [CrossRef]
- Koster, M.P.H.; Vreeken, R.J.; Harms, A.C.; Dane, A.D.; Kuc, S.; Schielen, P.C.J.I.; Hankemeier, T.; Berger, R.; Visser, G.H.A.; Pennings, J.L.A. First-Trimester Serum Acylcarnitine Levels to Predict Preeclampsia: A Metabolomics Approach. Dis. Markers 2015, 2015, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, K.; Donovan, B.; Fleener, D.; Bedell, B.; Borowski, K. Pregnancy-Related Changes of Amino Acid and Acylcarnitine Concentrations: The Impact of Obesity. Am. J. Perinatol. Rep. 2016, 6, e329–e336. [Google Scholar] [CrossRef] [Green Version]
- Hellmuth, C.; Lindsay, K.L.; Uhl, O.; Buss, C.; Wadhwa, P.D.; Koletzko, B.; Entringer, S. Association of maternal prepregnancy BMI with metabolomic profile across gestation. Int. J. Obes. 2017, 41, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampey, B.P.; Freemerman, A.J.; Zhang, J.; Kuan, P.-F.; Galanko, J.A.; O’Connell, T.M.; Ilkayeva, O.R.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; et al. Metabolomic Profiling Reveals Mitochondrial-Derived Lipid Biomarkers That Drive Obesity-Associated Inflammation. PLoS ONE 2012, 7, e38812. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Animal Model | Dietary Fat (% Caloric Intake) | Pre-Gestational Obesity | Pre-Conception Diet Exposure | Gestational Diet Exposure | Maternal Diet Reversal | Offspring Weaning | Reference |
|---|---|---|---|---|---|---|---|
| C57/B6 mice | 60% High fat diet (HFD) 25% fat control diet | HFD-induced obesity | 10–12-week HFD exposure before pregnancy | HFD maintained through pregnancy and lactation | Yes—2-cell stage embryo transfer | Weaned onto control diet | Sasson [34] |
| C57/B6 mice | 45% HFD 10% fat control diet | HFD-induced obesity | Diet commenced at 4 weeks; breeding at 10 weeks | HFD through pregnancy | No | Randomly assigned HFD or control diet | Elahi [20] |
| C57/BL6 mice | 32% HFD 11% fat control diet | HFD-induced obesity | 8-week pre-conception HFD-exposure | HFD through pregnancy | No | Fetal collections | Jones [17] |
| C57/B6 mice | 16% HFD control diet 3% fat | Diet-induced obesity | 6-week diet exposure pre-conception | HFD maintained through pregnancy and weaning | No | Pups weaned onto standard chow | Samuelsson [22] |
| C57/B6 mice | High trans-fat diet (6% partially hydrogenated vegetable oil + 1% soybean oil) 7% soybean oil control diet | No pre-pregnancy obesity | No HFD exposure pre-conception | High trans-fat diet through pregnancy and weaning only | No | Weaned onto control diet | de Velasco [39] |
| Sprague-Dawley Rats | 60% HFD 24% fat control diet | HFD-induced obesity | HFD commenced Postnatal day (PND) 24; breeding PDN 120 | HFD throughout pregnancy | No | Weaned onto control diet | Srinivasan [18] |
| Sprague-Dawley Rats | 140% overfeeding model | Overfeeding-induced obesity | 3-week overfeeding prior to conception | Overfeeding discontinued during pregnancy | Yes—dams switched to control feeding through pregnancy and lactation | Randomly weaned onto control (17% fat) or HFD (45% fat) | Borengasser [35] |
| Sprague-Dawley Rats | 140% overfeeding model | Overfeeding-induced obesity | 3-week overfeeding prior to conception | Overfeeding discontinued during pregnancy | Yes—dams switched to control feeding through pregnancy and lactation | Randomly weaned onto control (17% fat) or HFD (45% fat) | Borengasser [36] |
| Wistar Rats | 45% HFD 18% fat control diet | HFD-induced obesity with pre-gestational HFD exposure | Pre-conception HFD—commenced PND 22; breeding at PND 120 Pregnancy and lactation HFD—commenced at breeding and maintained through lactation) | HFD through pregnancy | No | Randomly assigned HFD or control diet | Howie [21] |
| Wistar Rats | 38% HFD-diets 15% fat control diet | No pre-pregnancy obesity | No HFD exposure pre-conception | HFD during pregnancy only; cross-fostered to lean dams during lactation | No | Weaned onto control diet; HFD exposure at 8 weeks | Dong [40] |
| Wistar Rats | 20% lard supplement in HFD 5% fat control diet | HFD-induced obesity | HFD exposure from PND 21 to breeding at PND 120 | HFD maintained through pregnancy and lactation | Yes—diet intervention back to control diet at PND 90 | Not specified | Zambrano [33] |
| Sheep | 155% overfeeding model | No pre-gestational obesity | Overfeeding commenced gestational day 115 | Overfeeding from gestational day 115 to gestation (~day 150) | No | Control diet during lactation and weaning | Philip [26] |
| Sheep | 150% overfeeding model | Overfeeding-induced obesity | 60-day overfeeding exposure before mating | Overfeeding through gestation, control diet during lactation | No | control diet | Long [25] |
| Sheep | 150% overfeeding model | Overfeeding-induced obesity | 60-day overfeeding exposure before mating | Overfeeding until fetal collection | No | Fetal collection | Zhu [41] |
| Sheep | 150% overfeeding model | Overfeeding-induced obesity | 60-day overfeeding exposure before mating | Overfeeding continued through pregnancy (with no intervention) | Yes—150% overfeeding until gestational day 28 (with obesity intervention) | Fetal collection | Tuersunjiang [27] |
| Japanese Macaque | 36% HFD 14% fat control diet | HFD-induced obesity | 4–7-year HFD exposure pre-conception | HFD maintained through to fetal collections at gestational day 130 | Yes—diet reversal 3 months prior to breeding | Fetal collection | Salati [42] |
| Japanese Macaque | 32% HFD 14% fat control diet | HFD-induced obesity | 2–4-year pre-gestational HFD induced obesity | HFD, or diet-reversal through pregnancy | Yes—pre-conception diet reversal on subsequent pregnancy | Weaned onto mothers gestational diet | McCurdy [28] |
| Japanese Macaque | 32% HFD 14% fat control diet | HFD-induced obesity | 4–7-year pre-gestational HFD exposure | HFD, or diet reversal through pregnancy | Yes—switched back to control diet in 5th breeding season | Weaned onto in utero or reverse diet | Pound [29] |
| Japanese Macaque | 32% HFD 14% fat control diet | HFD-induced obesity | 2–9-year pre-conception HFD exposure | HFD, or diet reversal through pregnancy | Yes—switched back to control diet in 9th breeding season | Fetal collections | Wesolowski [32] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Easton, Z.J.W.; Regnault, T.R.H. The Impact of Maternal Body Composition and Dietary Fat Consumption upon Placental Lipid Processing and Offspring Metabolic Health. Nutrients 2020, 12, 3031. https://doi.org/10.3390/nu12103031
Easton ZJW, Regnault TRH. The Impact of Maternal Body Composition and Dietary Fat Consumption upon Placental Lipid Processing and Offspring Metabolic Health. Nutrients. 2020; 12(10):3031. https://doi.org/10.3390/nu12103031
Chicago/Turabian StyleEaston, Zachary J. W., and Timothy R. H. Regnault. 2020. "The Impact of Maternal Body Composition and Dietary Fat Consumption upon Placental Lipid Processing and Offspring Metabolic Health" Nutrients 12, no. 10: 3031. https://doi.org/10.3390/nu12103031
APA StyleEaston, Z. J. W., & Regnault, T. R. H. (2020). The Impact of Maternal Body Composition and Dietary Fat Consumption upon Placental Lipid Processing and Offspring Metabolic Health. Nutrients, 12(10), 3031. https://doi.org/10.3390/nu12103031